

Abstract
The orphan nuclear receptor Liver Receptor Homologue-1 (LRH-1) is an emerging drug target for metabolic disorders. The most effective known LRH-1 modulators are phospholipids or synthetic hexahydropentalene compounds. While both classes have micromolar efficacy, they target different portions of the ligand binding pocket and activate LRH-1 through different mechanisms. Guided by crystallographic data, we combined aspects of both ligand classes into a single scaffold, resulting in the most potent and efficacious LRH-1 agonists to date.
Keywords: Nuclear receptor, LRH-1, phospholipid hybrid, metabolic disease, agonist
Liver Receptor Homologue-1 (LRH-1, NR5A2) is an orphan nuclear hormone receptor (NR) that serves as an important hepatic regulator of lipid and glucose metabolism in adults.1 LRH-1 activation increases bile acid biosynthesis2−5 and reverse cholesterol transport,6,7 while diminishing the hepatic acute phase response8 and resolving endoplasmic reticulum stress.9 It has been shown that steatosis (increased fat accumulation in the liver) is decreased by even modest elevation of bile acid levels.10 Because steatosis is tightly correlated with insulin resistance,11,12 LRH-1 has emerged as a promising drug target for metabolic diseases like type II diabetes and nonalcoholic fatty liver disease (NAFLD).13,14 While endogenous LRH-1 ligands are unknown, the receptor binds a variety of phospholipids (PLs) in vitro, which presumably contributes to a low level of constitutive activity.15−17 However, a subset of phosphatidycholines (PCs) have been shown to activate LRH-1 above basal levels.18 Specifically, certain medium-chained, saturated PCs (i.e., diundecanoylphosphatidylcholine (DUPC, PC 11:0/11:0) and dilauroylphosphatidylcholine (DLPC, PC 12:0/12:0)) activate LRH-1 in vitro and in vivo when administered exogenously. In two models of obesity-induced insulin resistance, DLPC treatment decreased hepatic and circulating lipids, increased bile acid production, and improved insulin sensitivity and glucose homeostasis.18 These effects were entirely LRH-1 dependent, as they did not occur in liver-specific LRH-1 knockout mice.
Our recent structural studies with DLPC revealed an unusual ligand binding mode. Unlike typical NR ligands, which are buried deep in the ligand binding pocket, DLPC binds near the solvent accessible surface bridging residues near the mouth of the pocket and leaving unoccupied space behind its alkyl tails.14 DLPC binding is mediated by polar interactions of the phosphate headgroup with a cluster of residues at the mouth of the LRH-1 binding pocket (i.e., G421, Y516, and K520), as well as by hydrophobic interactions of the lipid tails. We demonstrated that DLPC induces specific conformational changes within the LRH-1 ligand binding domain (LBD) that promotes allosteric signaling from the ligand binding pocket to the activation function surface (AFS), the site of coregulator interaction (Figure 1).14,19 DLPC is able to promote recruitment of specific coactivators, suggesting that this signaling network is used to communicate ligand status to the AFS.14
Figure 1.
Structure of LRH-1 bound to PC agonist. Allosteric activation of LRH-1 drives coactivator recruitment and transcriptional activity, resulting in improved metabolic function.
Although DLPC-mediated antidiabetic effects are striking, very high concentrations of this agonist are required to activate LRH-1 (100 μM in vitro and 100 mg/kg twice daily in vivo).18 Further, PCs are not ideal pharmacological agents since they are constantly remodeled,20 prone to hydrolysis,21 and frequently incorporated in membranes where they may impact biological processes influenced by membrane composition and fluidity. Thus, potent and chemically stable small molecules that mimic phospholipid-driven LRH-1 activation could provide powerful tools for further evaluation of LRH-1 biology.
The development of synthetic LRH-1 modulators is difficult, in part, because the LBD is large and highly lipophilic. Accordingly, very few chemical scaffolds have successfully impacted LRH-1 activity.22 The most effective synthetic LRH-1 agonists are aryl-substituted hexahydropentalene (6HP) derivatives developed by Whitby in collaboration with a group at GlaxoSmithKline (GSK). A first-generation agonist from this effort, GSK8470, effectively drives LRH-1-mediated gene transcription but is highly unstable under acidic conditions.23 Guided by an X-ray crystal structure of a GSK8470/LRH-1 LBD complex, extensive synthetic efforts yielded a second-generation agonist, RJW100 (2), which is shown in Figure 2. Importantly, RJW100 retained LRH-1 activity and displayed vastly improved chemical stability.24
Figure 2.

Design of hybrid PL mimics, where two different agonist classes are combined into a single scaffold. In addition to the shown hydrophilic and π-stacking interactions, each ligand makes extensive hydrophobic interactions (omitted for clarity).
Our laboratories recently reported the X-ray crystal structure of LRH-1 bound to RJW100 (PDB 5L11).25 Although RJW100 and GSK8470 bind LRH-1 in the same pocket, we found that their orientations are drastically different. In addition to extensive hydrophobic interactions, the binding pose of RJW100 is largely driven by a water-mediated polar interaction between the side-chain of threonine 352 (T352) and the ligand hydroxyl group (illustrated in Figure 2). Superimposing the cocrystal structures of both RJW100 and DLPC in the LBD reveals direct intersection of the hexyl tail of RJW100 and the Sn1-lauroyl tail of DLPC. We questioned whether the binding features of both ligand classes (i.e., phospholipid interactions with surface residues and 6HP interactions deep in the pocket) could be achieved by a single scaffold, thus achieving greater engagement with the ligand binding pocket. We designed a series of hybrid phospholipid mimics with three distinct regions: the 6HP core of RJW100 to interact with T352, modular alkyl linkers to maintain hydrophobic interactions, and terminal polar groups to engage the PC headgroup binding domain near the mouth of the pocket (Figure 2). Here, we show that this approach resulted in the development of novel LRH-1 agonists with significant improvements over both of the individual scaffolds.
To generate the targeted PL mimics, we employed Whitby’s carbene-interrupted Pauson–Khand (PK) reaction, where zirconocene-mediated reaction of silyl ether-containing 1,1-dibromoalkanes (3a–3h, n = 1–8), enynes (4), and phenylacetylene would deliver the 6HP-containing primary alcohols 5 (Figure 3).26 We developed this specific approach because it would allow for systematic variation of the alkyl linker length, and the primary alcohols could be easily manipulated to a range of polar head groups. This retrosynthetic disconnection is ideal, in part, because it utilizes readily accessible precursors of similar complexity (3 and 4) in the convergent 6HP-forming step.
Figure 3.
Synthetic approach to hybrid compounds.
Whitby showed that, although exo and endo diastereomers of RJW100 are roughly equipotent, the exo diastereomer is a more effective agonist for LRH-1.24 Moreover, we have found that endo-RJW100 is a poor LRH-1 activator in cellular assays.25 As such, we sought reaction conditions to selectively produce the exo isomer. Similar to other PK reactions, Whitby’s system operates through an early transition state, where diastereoselectivity is dictated by the conformational preference of the corresponding enyne starting materials. We prepared a small collection of enynes 4 with different alcohol protecting groups and found that the exo-selectivity increased with relative population of pre-exo conformer (as indicated by JHβ, Hγ),27 where a methoxymethyl (MOM) ether gave the desired exo-6HP diastereomer in 7.2:1 d.r. (Figure 3). In addition to providing diastereocontrol, MOM protection to give 4d was strategically enabling due to its orthogonal reactivity to silyl protecting groups. This allowed for chemoselective manipulation of the primary alcohol. Using exo-selective cyclization conditions, we utilized a series of intermediates 3a–3h to afford the requisite hybrid precursors 5a–5h as diastereomeric mixtures (∼7:1 dr, favoring the desired exo isomers).
To directly examine the effect of combining PC binding elements with those of the 6HP scaffold, we designed a hybrid compound where the RJW100 core was appended to a glycerophosphorylcholine headgroup (Scheme 1). Analysis of the crystallographic data described above indicated that this could be achieved via extension of the RJW100 alkyl chain (along the trajectory of the Sn1 lipid tail) along with truncation of the Sn2 tail (to avoid disruption of 6HP binding). Accordingly, our synthetic efforts, as shown in Scheme 1, began with alcohol 5e to provide the seven required methylene units between the 6HP core and acylglycerol. Ley–Griffith oxidation of 5e efficiently yielded terminal carboxylic acid 6, which participated in a regioselective cobalt-mediated opening of epoxide 7(28) to give ester 8. Sequential acylation and deprotection with tetrabutylammonium fluoride (TBAF) afforded primary alcohol 10. Terminal phosphorylcholine construction commenced with reaction of 10 with 2-chloro-1,3,2-dioxaphospholane 2-oxide in toluene. After filtration and concentration of the reaction mixture, the crude cyclic phospholane was opened with trimethylamine. Finally, acidic cleavage of the MOM-ether and isolation by preparative HPLC gave the hexahydropentalene-phospholipid hybrid 11.
Scheme 1. Synthesis and Biological Evaluation of DLPC/RJW100 Hybrid 11 from 5e.

(A) Reagents and conditions: (a) TPAP, NMO, H2O, MeCN, 23 °C; 1 h, 85% yield; (b) i. Co(salen), Et2O, 23 °C; 1 h; ii. DIPEA, neat, 60 °C, 16 h, 24% yield; (c) propionyl chloride, DMAP, THF, 23 °C; 30 min, 77% yield; (d) TBAF, THF, 23 °C; 16 h, 76% yield; (e) 2-chloro-1,3,2-dioxaphospholane 2-oxide, TEA, toluene, 23 °C; 4 h; (f) trimethylamine, MeCN, 90 °C, 16 h; (g) conc. aq. HCl, MeCN, 23 °C; 10 min, purified by preparative HPLC (4% isolated yield over three steps e–g). (B) Ligand-driven LRH-1 activity using a Luciferase reporter assay. Maximum activation value for each ligand (fold vs vehicle) at a single point (30 μM).
We evaluated the efficacy of 11 using a luciferase reporter assay, which measures LRH-1 activity in intact (HeLa) cells. This functional assay utilizes a reporter plasmid in which the gene for luciferase is cloned downstream of a LRH-1 response element. In this way, luciferase is generated in response to LRH-1 transactivation. Maximum LRH-1 activation values for DLPC, RJW100, and 11 at 30 μM ligand concentration are given in Scheme 1B, and dose-dependent increases in luminescence was observed in all cases. EC50 and maximum activation were calculated from the resulting dose–response curves. We found that 11 was considerably more potent than the parent phospholipid DLPC (EC50 of 13 ± 6 μM vs >100 μM for DLPC). The low potency of DLPC in this assay is consistent with previous reports.18 RJW100 was more potent than 11, with an EC50 of 1.1 ± 0.6 μM; however, 11 induced higher levels of LRH-1 activation than either DLPC or RJW100 in these experiments (Scheme 1).
Encouraged by this result, we were motivated to refine the structure of the LRH-1 hybrid agonists. To do this, we aimed to remove the hydrolytic liabilities inherent to the glyceryl esters while retaining the 6HP core and phosphorylcholine elements. Also key to this study was the systematic evaluation of distance between these binding elements. In the overlaid ligand poses of 1 and 2, the linear measured distance between the first methylene unit in the hexyl tail off the 6HP core and the DLPC phosphate is 10.0 Å. However, the ligand binding pocket is fluxional; we have observed a change in the shape of the LRH-1 LBD to accommodate either DLPC or RJW100.25 We anticipated that hybrid ligand would be anchored at either end (by 6HP and polar group), though hydrophobic interactions within the pocket could enforce a nonlinear trajectory of the hybrid tail, underscoring the importance of systematic evaluation of linker length. As such, we targeted a series of hybrid compounds 12a–12h comprising the 6HP core, phosphorylcholine polar group, and alkyl linker with systematically varied lengths. Importantly, this series would enable evaluation of the central hybrid design strategy, and the compounds would be considerably more synthetically accessible.
The synthesis of 12a–12h is summarized in Scheme 2A. Beginning with primary alcohols 5a–5h, transformation to their corresponding cyclic phospholanes followed by ring opening with trimethylamine afforded the crude MOM-protected phosphorylcholines. Direct cleavage of the MOM group with aqueous hydrochloric acid and purification by HPLC gave rise to the exo-compounds 12 with linkers between 6- and 13-atoms long. Evaluation of this series revealed that the improvement to LRH-1 activity that was observed with the hybrid 11 could be retained, in spite of significant structural simplification. Scheme 2B shows LRH-1 activity in the presence of these ligands, where each vertical bar represents activity at a given dose of each compound (concentration increases from 0–30 μM, represented by triangles). Agonism was clearly related to linker length; compounds with shorter linkers (12a–12c, n = 1–3) did not induce luciferase expression at greater levels than vehicle alone, while 12d and 12e (n = 4,5) had only moderate activity at the highest dose. Compounds with longer linkers (12f–12h, n = 6–8) induced robust, dose-dependent LRH-1 activation, presumably through enhanced pocket engagement. Notably, the optimal alkyl linker length is 12-atoms, as demonstrated by the activity of compound 12g, where the maximum LRH-1 activity was significantly higher than RJW100. Maximum LRH-1 activity, driven by 12g, was 2.1-fold over DMSO, and potency was largely retained (EC50 = 5 ± 2 μM). Visual examination of the plot in Scheme 2B reveals that ligand-driven LRH-1 activity is diminished on either side of the optimal linker length. As illustrated in Scheme 2C, the findings presented here are consistent with proposed hydrophobic and hydrophilic interactions outlined in the design scheme. Though tracing the specific conformation of the alkyl chain (i.e., emulating the phospholipid Sn1 or Sn2 pathway) will require structural studies, correlation between length and activation are most consistent with the Sn2 pathway.
Scheme 2. Synthesis and Evaluation of Simplified 6HP-Phosphorylcholine Hybrids 12.
(A) Reagents and conditions: (a) 2-chloro-1,3,2-dioxaphospholane 2-oxide, TEA, toluene, 23 °C; 4 h; (b) trimethylamine, MeCN, 90 °C, 16 h; (c) conc. aq. HCl, MeCN, 23 °C; 10 min. 12–49% over three steps and after purification by preparative HPLC. (B) Luciferase reporter assay demonstrating ligand efficacy, each bar represents activity at a different concentration of designated compound, from right to left: 0.003, 0.03, 0.3, 1.0, 3.0, 10, and 30 μM. Represented as a measure of fold increase in activity over vehicle. (C) Overlay of DLPC and RJW100 in LRH-1 LBD. Number of atoms in linker is counted between the 6HP core and key PC binding element.
Having established that our ligand hybrid design strategy could be used to improve agonist efficacy and that structural simplification is well tolerated, we sought to further optimize our hybrid scaffold. While our most effective phosphorylcholine hybrid (12g) demonstrated that the ester units are unnecessary, removing the choline moiety and synthesizing the directly analogous phosphates would likely hinder further development. Ultimately, phosphates can be poorly cell permeant, and cellular stability can be low due to the action of phosphatases.29 Accordingly, we considered a number of phosphate isosteres. Of these, carboxylic acids were attractive because they are chemically stable and readily accessible from our collection of primary alcohols 5. If successful, phosphate substitution in this manner could illuminate avenues for further scaffold optimization as carboxylic acid isosteres have been extensively studied.30 In addition to improved binding properties, other polar groups would offer the ability to improve the aqueous solubility of this agonist scaffold.
As summarized in Scheme 3A, Ley–Griffith oxidation of each parent alcohol 5a–5h followed by MOM deprotection gave the desired carboxylic acid hybrids as diastereomeric mixtures that were separated by HPLC to afford the pure exo-isomers. Again, we observed highly pronounced length-dependent LRH-1 activation by luciferase expression. While the hybrid compounds with shorter linkers (13a–13d, n = 1–4) did not appear to be better LRH-1 agonists than RJW100, the compounds with longer linkers were significantly more effective (activity of each compound by dose is visually depicted in Scheme 3B). The most efficacious agonist 13g (6HP-CA) effectively drives LRH-1 transcriptional activity (maximum activity = 2.3 ± 0.2 fold over vehicle) with significantly improved potency (EC50 = 0.4 ± 0.4 μM), and length deviation on either side (i.e., shorter or longer linkers) resulted in loss of activity. Dose-response curves for the key compounds in this study are given in Scheme 3C. Although activities of the phosphorylcholine compounds 12 and DLPC do not appear to reach saturation (potentially due to low solubility and membrane incorporation), the synthetic hybrid 13g displayed a sigmoidal activation curve in this concentration range (Table 1).
Scheme 3. Synthesis and Evaluation of Simplified 6HP-Carboxylic Acid Hybrids 13.
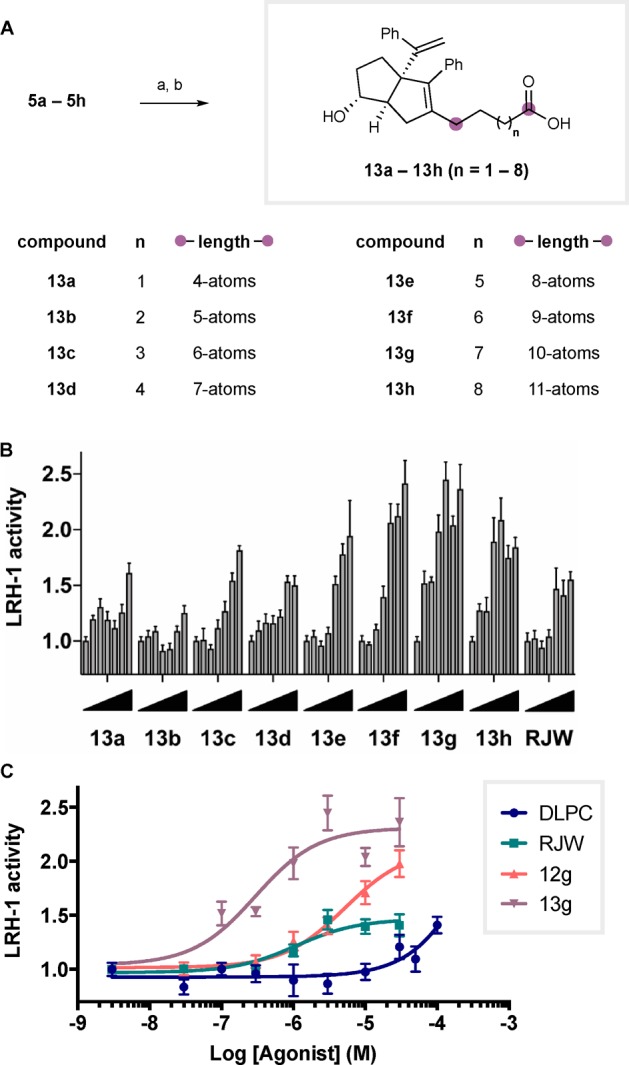
(A) Reagents and conditions: (a) TPAP, NMO, H2O, MeCN, 23 °C; 1 h; (b) conc. aq. HCl, MeCN, 23 °C, 10 min. 40–85% yield over two steps. (B) Luciferase reporter assay demonstrating ligand efficacy, each bar represents activity at a different concentration of designated compound, from right to left: 0.003, 0.03, 0.3, 1.0, 3.0, 10, and 30 μM. Represented as a measure of fold increase in activity over vehicle. (C) Overlaid dose–response curves of indicated ligands.
Table 1. Summarized Activity Data for Hybrid PL Mimics.
| compd | EC50(μM) | max LRH-1 activationa |
|---|---|---|
| RJW100 (1) | 1.1 ± 0.6 | 1.47 ± 0.06 |
| 11 | 13 ± 6 | 2.7 ± 0.3 |
| 12g | 5 ± 2 | 2.1 ± 0.1 |
| 6HC-CA (13g) | 0.4 ± 0.4 | 2.3 ± 0.2 |
Maximum LRH-1 activation was calculated at the top of the resulting dose–response curves, as a fold increase in LRH-1 activity over baseline (vehicle).
In summary, we have described the development of a new and highly effective class of LRH-1 agonists. Guided by structural insights from our laboratories, we have incorporated key binding elements from two discrete ligand classes into a single hybrid scaffold. This ideal was first validated through synthesis of a 6HP-phospholipid hybrid (11), which displayed significantly higher LRH-1 activation than either of the parent structures (RJW100 and DLPC). Simplification of this scaffold and systematic evaluation of various alkyl linkers revealed a clear correlation between linker length and LRH-1 activation, in support of our hypothesis that agonism could be improved through a dual binding mode. Substitution of the terminal phosphate groups with carboxylic acids afforded 6HP-CA (13g), the most highly efficacious and potent LRH-1 agonist that has been reported to date. Efforts to investigate the binding pose of these ligands for further development, probe their mechanism of receptor activation, accurately measure direct LRH-1 binding of these ligands, evaluate and optimize physiochemical properties, and assess their activity in vivo are currently underway in our laboratories.
Acknowledgments
This work was supported in part by the National Institutes of Health under the following awards: T32GM008602 (S.G.M.), F31DK111171 (S.G.M.), R01DK095750 (E.A.O.), R01DK114213 (E.A.O., N.T.J.), and the Emory Catalyst Award (E.A.O., N.T.J).
Glossary
ABBREVIATIONS
- LRH-1
Liver Receptor Homologue-1
- NR
nuclear receptor
- NAFLD
nonalcoholic fatty liver disease
- PL
phospholipid
- PC
phosphatidylcholine
- DLPC
dilauroylphosphatidylcholine
- DUPC
diundecanoylphosphatidylcholine
- LBD
ligand binding domain
- AFS
activation function surface
- 6HP
hexahydropentalene
- PK
Pauson–Khand
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00361.
Detailed chemical syntheses, biological procedures, purity information, spectral data for key compounds, and additional activity data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Stein S.; Schoonjans K. Molecular Basis for the Regulation of the Nuclear Receptor LRH-1. Curr. Opin. Cell Biol. 2015, 33, 26–34. 10.1016/j.ceb.2014.10.007. [DOI] [PubMed] [Google Scholar]
- Goodwin B.; Jones S. A.; Price R. R.; Watson M. A.; McKee D. D.; Moore L. B.; Galardi C.; Wilson J. G.; Lewis M. C.; Roth M. E.; Maloney P. R.; Willson T. M.; Kliewer S. A. A Regulatory Cascade of the Nuclear Receptors FXR, SHP-1, and LRH-1 Represses Bile Acid Biosynthesis. Mol. Cell 2000, 6 (3), 517–526. 10.1016/S1097-2765(00)00051-4. [DOI] [PubMed] [Google Scholar]
- Lu T. T.; Makishima M.; Repa J. J.; Schoonjans K.; Kerr T. A.; Auwerx J.; Mangelsdorf D. J. Molecular Basis for Feedback Regulation of Bile Acid Synthesis by Nuclear Receptors. Mol. Cell 2000, 6 (3), 507–515. 10.1016/S1097-2765(00)00050-2. [DOI] [PubMed] [Google Scholar]
- Mataki C.; Magnier B. C.; Houten S. M.; Annicotte J.-S.; Argmann C.; Thomas C.; Overmars H.; Kulik W.; Metzger D.; Auwerx J.; Schoonjans K. Compromised Intestinal Lipid Absorption in Mice with a Liver-Specific Deficiency of Liver Receptor Homolog 1. Mol. Cell. Biol. 2007, 27 (23), 8330–8339. 10.1128/MCB.00852-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y.-K.; Schmidt D. R.; Cummins C. L.; Choi M.; Peng L.; Zhang Y.; Goodwin B.; Hammer R. E.; Mangelsdorf D. J.; Kliewer S. A. Liver Receptor Homolog-1 Regulates Bile Acid Synthesis but is not Essential for Feedback Regulation of Bile Acid Synthesis. Mol. Endocrinol. 2008, 22 (6), 1345–1356. 10.1210/me.2007-0565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoonjans K.; Annicotte J. S.; Huby T.; Botrugno O. A.; Fayard E.; Ueda Y.; Chapman J.; Auwerx J. Liver Receptor Homolog 1 Controls the Expresion of the Scavenger Receptor Class B Type I. EMBO Rep. 2002, 3 (12), 1181–1187. 10.1093/embo-reports/kvf238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein S.; Oosterveer M. H.; Mataki C.; Xu P.; Lemos V.; Havinga R.; Dittner C.; Ryu D.; Menzies K. J.; Wang X.; Perino A.; Houten S. M.; Melchior F.; Schoonjans K. SUMOylation-Dependent LRH-1/PROX1 Interaction Promotes Atherosclerosis by Decreasing Hepatic Reverse Cholesterol Transport. Cell Metab. 2014, 20 (4), 603–613. 10.1016/j.cmet.2014.07.023. [DOI] [PubMed] [Google Scholar]
- Venteclef N.; Jakobsson T.; Steffensen K. R.; Treuter E. Metabolic Nuclear Receptor Signaling and the Inflammatory Acute Phase Response. Trends Endocrinol. Metab. 2011, 22 (8), 333–343. 10.1016/j.tem.2011.04.004. [DOI] [PubMed] [Google Scholar]
- Mamrosh J. L.; Lee J. M.; Wagner M.; Stambrook P. J.; Whitby R. J.; Sifers R. N.; Wu S.; Tsai M.; Demayo F. J.; Moore D. D. Nuclear Receptor LRH-1/NR5A2 is Required and Targetable for Liver Endoplasmic Reticulum Stress Resolution. eLife 2014, 3, e01694. 10.7554/eLife.01694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M.; Houten S. M.; Wang L.; Moschetta A.; Mangelsdorf D. J.; Heyman R. A.; Moore D. D.; Auwerx J. Bile Acids Lower Triglyceride Levels via a Pathway involving FXR, SHP, and SREBP-1c. J. Clin. Invest. 2004, 113 (10), 1408–1418. 10.1172/JCI21025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusi K. Nonalcoholic Fatty Liver Disease in Type 2 Diabetes Mellitus. Curr. Opin. Endocrinol., Diabetes Obes. 2009, 16 (2), 141–149. 10.1097/MED.0b013e3283293015. [DOI] [PubMed] [Google Scholar]
- Savage D. B.; Petersen K. F.; Shulman G. I. Disordered Lipid Metabolism and the Pathogenesis of Insulin Resistance. Physiol. Rev. 2007, 87 (2), 507–520. 10.1152/physrev.00024.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. M.; Lee Y. K.; Mamrosh J. L.; Busby S. A.; Griffin P. R.; Pathak M. C.; Ortlund E. A.; Moore D. D. A Nuclear-Receptor-Dependent Phosphatidylcholine Pathway with Antidiabetic Effects. Nature 2011, 474 (7352), 506–510. 10.1038/nature10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musille P. M.; Pathak M.; Lauer J. L.; Hudson W. H.; Griffin P. R.; Ortlund E. A. Antidiabetic Phospholipid-Nuclear Receptor Complex Reveals the Mechanism for Phospholipid-Driven Gene Regulation. Nat. Struct. Mol. Biol. 2012, 19 (5), 532–S2. 10.1038/nsmb.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortlund E. A.; Lee Y.; Solomon I. H.; Hager J. M.; Safi R.; Choi Y.; Guan Z.; Tripathy A.; Raetz C. R. H.; McDonnell D. P.; Moore D. D.; Redinbo M. R. Modulation of Human Nuclear Receptor LRH-1 Activity by Phospholipids and SHP. Nat. Struct. Mol. Biol. 2005, 12 (4), 357–363. 10.1038/nsmb910. [DOI] [PubMed] [Google Scholar]
- Krylova I. N.; Sablin E. P.; Moore J.; Xu R. X.; Waitt G. M.; MacKay J. A.; Juzumiene D.; Bynum J. M.; Madauss K.; Montana V.; Lebedeva L.; Suzawa M.; Williams J. D.; Williams S. P.; Guy R. K.; Thornton J. W.; Fletterick R. J.; Willson T. M.; Ingraham H. A. Structural Analyses Reveal Phosphatidyl Inositols as Ligands for the NR5 Orphan Receptors SF-1 and LRH-1. Cell 2005, 120 (3), 343–355. 10.1016/j.cell.2005.01.024. [DOI] [PubMed] [Google Scholar]
- Sablin E. P.; Blind R. D.; Uthayaruban R.; Chiu H. J.; Deacon M.; Das D.; Ingraham H. A.; Fletterick R. J. Structure of Liver Receptor Homolog-1 (NR5A2) with PIP3 Hormone Bound in the Ligand Binding Pocket. J. Struct. Biol. 2016, 192 (3), 342–348. 10.1016/j.jsb.2015.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. M.; Lee Y. K.; Mamrosh J. L.; Busby S. A.; Patrick R.; Pathak M. C.; Ortlund E. A.; Moore D. D. Antidiabetic Actions of a Phosphatidylcholine Ligand for Nuclear Receptor LRH-1. Nature 2011, 474 (7352), 506–510. 10.1038/nature10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musille P. M.; Kossmann B. R.; Kohn J. A.; Ivanov I.; Ortlund E. A. Unexpected Allosteric Network Contributes to LRH-1 Co-Regulator Selectivity. J. Biol. Chem. 2016, 291 (3), 1411–1426. 10.1074/jbc.M115.662874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald J. I.; Sprecher H. Phospholipid Fatty Acid Remodeling in Mammalian Cells. Biochim. Biophys. Acta, Lipids Lipid Metab. 1991, 1084 (2), 105–121. 10.1016/0005-2760(91)90209-Z. [DOI] [PubMed] [Google Scholar]
- Ridgway N. D. The Role of Phosphatidylcholine and Choline Metabolites to Cell Proliferation and Survival. Crit. Rev. Biochem. Mol. Biol. 2013, 48 (1), 20–38. 10.3109/10409238.2012.735643. [DOI] [PubMed] [Google Scholar]
- For other synthetic scaffolds that have been reported to modulate LRH-1, see:; a de Jesus Cortez F.; Suzawa M.; Irvy S.; Bruning J. M.; Sablin E.; Jacobson M. P.; Fletterick R. J.; Ingraham H. A.; England P. M. Disulfide-Trapping Identifies a New, Effective Chemical Probe for Activating the Nuclear Receptor Human LRH-1 (NR5A2). PLoS One 2016, 11 (7), e0159316. 10.1371/journal.pone.0159316. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lalit M.; Gangwal R. P.; Dhoke G. V.; Damre M. V.; Khandelwal K.; Sangamwar A. T. J. Mol. Struct. 2013, 1049, 315–325. 10.1016/j.molstruc.2013.06.035. [DOI] [Google Scholar]; c Benod C.; Carlsson J.; Uthayaruban R.; Hwang P.; Irwin J. J.; Doak A. K.; Schoichet B. K.; Sablin E. P.; Fletterick R. J. Structure-Based Discovery of Antagonists of Nuclear Receptor LRH-1. J. Biol. Chem. 2013, 288 (27), 19830–19844. 10.1074/jbc.M112.411686. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Corzo C. A.; Mari Y.; Chang M. R.; Khan T.; Kuruvilla D.; Nuhant P.; Kuman N.; West G. M.; Duckett D. R.; Roush W. R.; Griffin P. R. Antiproliferation Activity of a Small Molecule Repressor of Liver Receptor Homolog 1. Mol. Pharmacol. 2015, 87, 296–304. 10.1124/mol.114.095554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitby R. J.; Dixon S.; Maloney P. R.; Delerive P.; Goodwin B. J.; Parks D. J.; Willson T. M. Identification of Small Molecule Agonists of the Orphan Nuclear Receptors Liver Receptor Homolog-1 and Steroidogenic Factor-1. J. Med. Chem. 2006, 49, 6652. 10.1021/jm060990k. [DOI] [PubMed] [Google Scholar]
- Whitby R. J.; Stec J.; Blind R. D.; Dixon S.; Leesnitzer L. M.; Orband-Miller L. A.; Williams S. P.; Willson T. M.; Xu R.; Zuercher W. J.; Cai F.; Ingraham H. A. Small Molecule Agonists of the Orphan Nuclear Receptors Steroidogenic Factor-1 (SF-1, NR5A1) and Liver Receptor Homologue-1 (LRH-1, NR5A2). J. Med. Chem. 2011, 54 (7), 2266. 10.1021/jm1014296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mays S. G.; Okafor C. D.; Whitby R. J.; Goswami D.; Stec J.; Flynn A. R.; Dugan M. C.; Jui N. T.; Griffin P. R.; Ortlund E. A. Crystal Structures of the Nuclear Receptor, Liver Receptor Homolog 1, Bound to Synthetic Agonists. J. Biol. Chem. 2016, 291 (49), 25281–25291. 10.1074/jbc.M116.753541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stec J.; Thomas E.; Dixon S.; Whitby R. J. Tandem Insertion of Halocarbenoids and Lithium Acetylides in Zirconacycles: A Novel Rearrangement to Zirconium Alkenylidenates by β-Addition to an Alkynyl Zirconecene. Chem. - Eur. J. 2011, 17, 4896–4904. 10.1002/chem.201002962. [DOI] [PubMed] [Google Scholar]
- Adrio J.; Rivero M. R.; Carretero J. C. Endo-Selective Intramolecular Pauson-Khand Reactions of Gamma-Oxygenated-Alpha,Beta-Unsaturated Phenylsulfones. Chem. - Eur. J. 2001, 7 (11), 2435–2448. . [DOI] [PubMed] [Google Scholar]
- Jacobsen E. N.; Kakiuchi F.; Konsler R. G.; Larrow J. F.; Tokunaga M. Enantioselective Catalytic Ring Opening of Epoxides with Carboxylic Acids. Tetrahedron Lett. 1997, 38 (5), 773–776. 10.1016/S0040-4039(96)02414-8. [DOI] [Google Scholar]
- Rye C. S.; Baell J. B. Phosphate Isosteres in Medicinal Chemistry. Curr. Med. Chem. 2005, 12 (26), 3127–3141. 10.2174/092986705774933452. [DOI] [PubMed] [Google Scholar]
- Ballatore C.; Huryn D. M.; Smith A. B. III Carboxylic Acid (Bio)Isosteres in Drug Design. ChemMedChem 2013, 8, 385–395. 10.1002/cmdc.201200585. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.