

Abstract

Solid-state nanopores have emerged as promising platforms for biosensing including diagnostics for disease detection. Here we show nanopore experiments that detect CRISPR-dCas9, a sequence-specific RNA-guided protein system that specifically binds to a target DNA sequence. While CRISPR-Cas9 is acclaimed for its gene editing potential, the CRISPR-dCas9 variant employed here does not cut DNA but instead remains tightly bound at a user-defined binding site, thus providing an excellent target for biosensing. In our nanopore experiments, we observe the CRISPR-dCas9 proteins as local spikes that appear on top of the ionic current blockade signal of DNA molecules that translocate through the nanopore. The proteins exhibit a pronounced blockade signal that allows for facile identification of the targeted sequence. Even at the high salt conditions (1 M LiCl) required for nanopore experiments, dCas9 proteins are found to remain stably bound. The binding position of the target sequence can be read from the spike position along the DNA signal. We anticipate applications of this nanopore-based CRISPR-dCas9 biosensing approach in DNA-typing based diagnostics such as quick disease-strain identification, antibiotic-resistance detection, and genome typing.
Keywords: Nanopores, CRISPR-Cas9, biosensing, diagnostics
While nanopores are most well-known for their use in biophysics experiments and DNA sequencing,1 they also hold great promise as diagnostic sensing devices, due to their high sensitivity, simple readout, ease of device fabrication, and their ability to detect individual disease-related biomarkers. A solid-state nanopore is a nanometer-sized hole that is etched in a thin membrane such as silicon nitride (SiN). Upon application of a voltage across the nanopore in an electrolyte solution, biomolecules such as DNA will translocate through the pore, thus temporarily blocking the passage of ions that leads to a drop in the current, which constitutes the basic sensing signal. This simple and elegant principle enables an easy readout and interpretation of the nanopore signal. Solid-state nanopores feature a number of additional advantages for developing biosensors such as their robustness, sensitivity, versatility, and compatibility with CMOS fabrication processes.2 Indeed, in recent years, solid-state nanopores have been applied to study protein–DNA interactions with proteins such as nucleosomes3 or antibodies4 and for protein-sensing applications of medically relevant proteins such as thrombin.5 The larger hydrodynamic radius of these protein–DNA complexes gives rise to a larger blockade current signal as they transverse the pore. These studies have suggested a genome-mapping type of approach through the monitoring of sequence-specific proteins.6 However, such an approach has remained challenging because suitable strong-DNA-binding proteins are rare due to the high salt concentrations that are required for decent signals in nanopore experiments.
Here we employ the CRISPR-dCas9 protein system for the detection of DNA motifs. In recent years, Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and its CRISPR-associated (Cas) proteins have gained enormous attention for its unprecedented potential as a gene-editing tool. The Cas9 protein is able to bind to a target DNA sequence through a guide RNA (gRNA).7 This gRNA is easily programmable, allowing Cas9 to target any DNA sequence, including DNA motifs that uniquely identify a variety of disease-DNA.8 The CRISPR-Cas9 protein has been shown to be sensitive to even single nucleotide mismatches and has a large hydrodynamic diameter (∼7 nm).9,10 The Cas9 protein can be modified to remove its endonuclease activity (i.e., cleaving activity) while keeping its programmable DNA-binding activity intact.11 This Cas9 variant, known as dCas9, is favorable to be repurposed as a DNA detection tool in the nanopore detection scheme proposed here.
In this Letter, we demonstrate a solid-state nanopore-based detection scheme for probing DNA motifs using CRISPR-dCas9 (Figure 1). Target RNA is preincubated with dCas9 and then mixed with the DNA sample of interest (see Methods). Next, the DNA with bound CRISPR-dCas9 is driven through the solid-state nanopore where the dCas9 protein gives rise to a pronounced feature on top of the DNA signal which allows for facile identification of specific DNA motifs (Figure 1b,c). We demonstrate stable binding of dCas9 to DNA, even in the high salt concentrations of the nanopore buffer. The high salt concentration and the large hydrodynamic radius of the dCas9 boost the nanopore signal and allow for identification of the targeted sequence in easily fabricated large-diameter solid-state nanopores. The binding position of dCas9 along the DNA can be determined from the spike position along each nanopore event. We anticipate applications of the CRISPR-dCas9 detection scheme in DNA-typing diagnostics with solid-state nanopores such as quick disease strain identification, antibiotic resistance detection, and genome typing.
Figure 1.

dCas9-DNA nanopore detection scheme. (a) gRNA is preincubated with dCas9 (labeled in green; not to scale) and then incubated to form a complex with target DNA prior to nanopore detection. (b) Voltage applied across the membrane induces an ionic current through a 20 nm size nanopore aperture and drives dCas9-DNA complexes to translocate through the nanopore. (c) The dCas9 signal appears as an additional blockade event (the sharp spike in the red shaded region) on top of the single-molecule DNA ionic-current signature which shows a constant current blockade except for the start of the event (gray shaded area) where DNA enters the pore in a folded manner.12
CRISPR-dCas9 is found to stably bind to DNA even at the high salt concentrations that are favored for nanopore experiments. Figure 2a shows a clear shift of the band of the dCas9-DNA complex with respect to the DNA control bands in both the Cas9 reaction buffer and 1 M LiCl conditions. Notably most of the DNA gets bound by dCas9, even in the 1 M LiCl condition. The experiment also shows that dCas9 can remain bound over the ∼2.5 h course of the gel experiment. Note that the bands in the 1 M LiCl condition did not run to the same position as the Cas9 reaction buffer because of electrostatic screening in the high-salt conditions. Gel experiments for the full range of salt concentrations from 0.5 M LiCl to 4 M LiCl can be found in the Supporting Information (see SI.4, Figure S-1).
Figure 2.

Stability of dCas9 on DNA at high salt and examples of nanopore current traces. (a) Gel shift assay showing binding of dCas9 in 1 M LiCl nanopore buffer. A clear shift in the DNA band at 1 M LiCl indicates that the dCas9 remains bound on the DNA even at high salt concentrations. The plus (+) sign indicates 1 kbp DNA samples that were incubated with the dCas9 complex. (b) Examples of nanopore current traces for a 20 kb long dsDNA-only control sample. The left trace shows a type 1 event, where the DNA translocated in a linear head-to-tail fashion, while the right trace shows a type 2–1 event, where there is a fold at the start of the translocation. (c) Examples of nanopore current traces for a sample where CRISPR-dCas9 was incubated with DNA. Due to the CRISPR-dCas9 complex that is locally bound along the DNA, additional spikes are seen on top of the DNA events. Left and right traces show type 1 and type 2–1 events, respectively. The dCas9 complex was mixed in a solution of 20 kb dsDNA and incubated at 37 °C for 30 min prior to nanopore experiment. The sample was then diluted to a final concentration of 1 M LiCl.
DNA translocation events in nanopore experiments clearly show the signatures of CRISPR-dCas9 binding. Upon addition of a control sample with bare DNA to the flow cell with a 20 nm SiN nanopore, we observe the typical current trace signals (Figure 2b), namely, a ∼ 2 ms long partial blockade of the ionic current with a blockade current of I0 = 0.12 nA, often starting with a double-blockade current of 2I0 at the start of the current trace that can be attributed to the capture and translocation of a folded ds-DNA at the start of the event.12 Notably, the nanopore events for the CRISPR-dCas9-incubated sample look exactly the same except for the fact that they contain a single sharp spikelike feature on top of the DNA current signature; see Figure 2c. We attribute these spikes along the DNA blockade to the presence of the locally bound dCas9 protein. The localized spikes have a short duration (∼25–60 μs) and a much deeper blockade level (up to 1.5 nA) than the bare dsDNA. Such a large signal is also expected since the dCas9 protein will block a sizable part of the pore volume upon translocating through the nanopore. Though most spikes were sharp, some of them exhibited broadening or more complex features due to sticking events (see SI.7 Figure S-4). The results reported here are mainly obtained from one particular experiment on the same pore with N = 308 events recorded for the dCas9-DNA samples as well as N = 308 events for the DNA-only control sample. Similar results were obtained for three independent other runs with different pores (yielding 30%, 34%, and 33% of events with spikes higher than 3I0).
The deep spikelike events enable us to clearly separate two different populations, i.e., those with and those without dCas9 bound to the DNA. The two can best be discriminated based on the maximum current blockade of each translocation event. Figure 3c,d displays a scatter plot of the maximum current blockade versus dwell time of each translocation event. Both for the dCas9-DNA sample and for the DNA-only control sample, two clusters can be observed with maxima at ∼0.12 and ∼0.24 nA. These two clusters are due to, respectively, linear head-to-tail translocations of dsDNA, known as type 1 events (cf. Figure 2b left) that show up as the 0.12 nA level, and type 2–1 events (cf. Figure 2b right) where DNA enters the pore in a folded fashion, resulting in twice the blockade level.12 While virtually all events for bare dsDNA have a maximum current blockade level of less than 0.4 nA, the data for the dCas9-DNA sample shows a population with a clearly larger maximum current blockade level.
Figure 3.
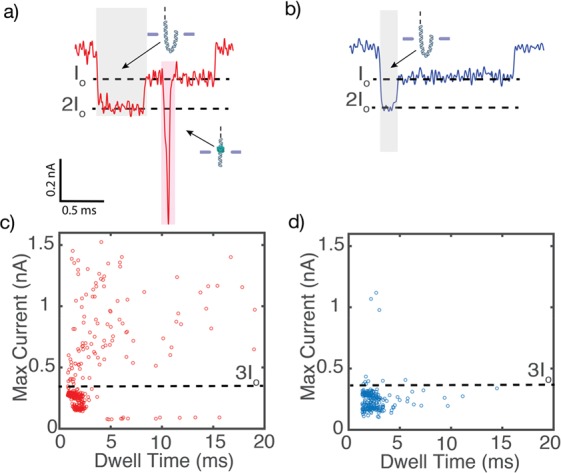
Statistics of nanopore measurements of CRISPR-dCas9 bound to DNA. (a) Typical example of a dCas9-labeled DNA translocation event. The dCas9-labeled DNA displays an additional spike in the current blockade signal that is observed when the DNA-bound protein transverses the pore (shaded in red). A deeper current blockade of DNA at the start of the translocation (shaded in gray) is due to a fold in the DNA as it first enters the pore. (b) Example translocation signal from a typical DNA event. (c,d) Scatter diagrams of the maximum current blockade versus dwell time of each translocation event for the dCas9-DNA sample and the bare DNA-only control sample, respectively. Unlike the bare DNA sample, the maximum current for dCas9-labeled DNA events shows very large values of 0.4–1.5 nA that clearly exceed 3 times the blockade level I0 for bare DNA (3I0 = 0.36 nA).
We observed spikes on a large fraction of the events for the dCas9-DNA sample. Upon using a threshold of 3I0 = 0.36 nA, we largely select only the events that corresponded to dCas9-bound DNA. In this way, we deduce that 35% of the events (108/308) for the dCas9-DNA sample showed these spikes. In the bare DNA-only control sample, this fraction was much lower, ∼5% (10/308). The fraction of spiked events did not reduce during the typical duration of the nanopore experiment of ∼2 h, indicating that the dCas9 remained stably bound to the DNA for extended periods of time even in the high-salt nanopore buffer.
We demonstrate that the spikes were indeed due to the bound dCas9 targeting our DNA region of interest by performing a number of different controls (Figure 4a). First, we investigated the translocation behavior of only the dCas9 protein by translocating a sample containing 150 nM of dCas9 (∼5× excess of the concentration used in the dCas9-DNA experiments). This yielded translocation events only in the opposite applied bias (see SI.5 Figure S-2). Also the dCas9-RNA complex translocated in the opposite polarity to the dCas9-RNA-DNA (see SI.5 Figure S-2). Furthermore, we investigated the effect of the gRNA sequence on the binding of dCas9 to the DNA. For this, we incubated dCas9 with gRNA containing a sequence that is not complementary to our 20 kbp DNA. We saw a sharp drop in the percentage of spike events, as compared to the 35% observed when the gRNA and target site on the 20 kbp DNA were complementary in their sequences. We then performed two more experiments where we used a mutated target gRNA sequence: (i) Mut1, where base pair 1 and 3 (as counted from the NGG region) were mutated, and (ii) Mut2, where every alternate base pair was mutated from the target sequence (see SI.2, changes highlighted in yellow). In both cases, we again observed a sharp drop in the percentage of spike events, as expected. Only when the entire target sequence is present, the percentage of spike events was found to rise to a much larger value of ∼35% (Figure 4a).
Figure 4.

Percentage of spike events and dwell times histograms. (a) Percentage of spike events, for various conditions. Left bar shows a much higher number of events with spikes than in all controls where one of the components was lacking or where the guide RNA was mutated or nonfitting to the target DNA sequence. (b) Bar graph showing the percentage of spikes larger than 3I0 for the dCas9-DNA sample and the DNA-only control at three different voltages. At all voltages, the percentage of spike events in the dCas9-DNA sample significantly exceeds the presence of spike events of the DNA-only events (that are due to knots and folds). (c) Histogram of the dwell times of the dCas9-DNA at 100 mV (purple), 200 mV (blue), and 300 mV (red). At 300 and 200 mV, the distribution times are clustered near 0.5 and 1 ms, respectively, whereas a broadening of the dwell times is observed at 100 mV due to interactions with the pore.
The fraction of dCas9 events was measured as a function of applied voltage, see Figure 4b. The percentage of spikes in the dCas9-DNA sample is found to be lower at the higher voltages. Although the current blockade signals are more pronounced at the higher voltages (see SI.11 Figure S-8 for example traces), the translocation speed of the dCas9-DNA molecules is also higher which causes dCas9 spikes to be missed due to the limited bandwidth of the amplifier. Across the entire voltage range, however, the percentage of spikes was always 5–7 times higher than that measured in the DNA-only control sample. The 5% false positives can be attributed to DNA knots which show up as deep blockade events.13 From the work reported by Plesa et al.,13 we expect about 2–5% events with knots at or beyond the 3I0 level for our linear 20 kb dsDNA. Figure 4c shows dwell time histograms of dCas9-DNA events. At the higher applied voltages (cf. the 200 and 300 mV data), we observe, as expected, a narrow dwell-time distribution, indicating that the dCas9-DNA complex flows freely through the pore. At lower voltage, however, a broadening of the distribution is observed for the dCas9-DNA events (e.g., the 100 mV data in Figure 4c), which was not seen for the DNA-only sample (see SI.10 Figure S-7). We attribute this to temporary sticking interactions between the protein and the inner SiN surface of the nanopore, as the dCas9 protein is positively charged in the pH 7.5 buffer (given its isoelectric point of 9), whereas the SiN surface is negatively charged.
Interestingly, we can discern the location where the dCas9 protein binds along the DNA molecule as it translocates through the nanopore. For estimating this location, we measure, for each event, the integral of the current blockade up to the dCas9 spike, as well as the integral of the full event. These values are a measure for, respectively, the amount of DNA that has translocated until the dCas9 is encountered and the total amount of DNA. Accordingly, the relative position of the dCas9 along the DNA strand is given by the ratio of these two integral current values. Figure 5 shows a histogram of the dCas9 position obtained from the method thus applied. We observe two peaks, corresponding to the two orientations in which a translocating DNA molecule can enter the nanopore, either first with the end close to the protein-labeled site or, in the other case, leading with the end further away from that site. We fit two Gaussian curves to the histogram and obtained two peaks at 0.31 ± 0.04 and 0.75 ± 0.02 (errors are the standard error of the mean). These peak values match remarkably well with the targeted site of the dCas9 on the DNA at 0.28 (5872/20 678 kbp) or equivalently (counting from the other end) 0.72 (14 806/20 678 kbp). One might ask whether some error might arise from assuming a constant average translocation velocity, as there will be some speed up of DNA velocity toward the end of the translocation.14 However, quantitatively this effect is very small for long molecules. The significant width of the two Gaussian peaks can be explained by residual nanopore-protein interactions occurring during translocation as well as by stochastic fluctuations in the velocity of the translocating molecule that broaden the distribution.14 For the first peak, the folded 2–1 events may furthermore introduce an ambiguity in the localization of the dCas9 peak position in the, relatively rare, cases that the folded region involves a large (>28% of the DNA length) fold at the start of the event.
Figure 5.
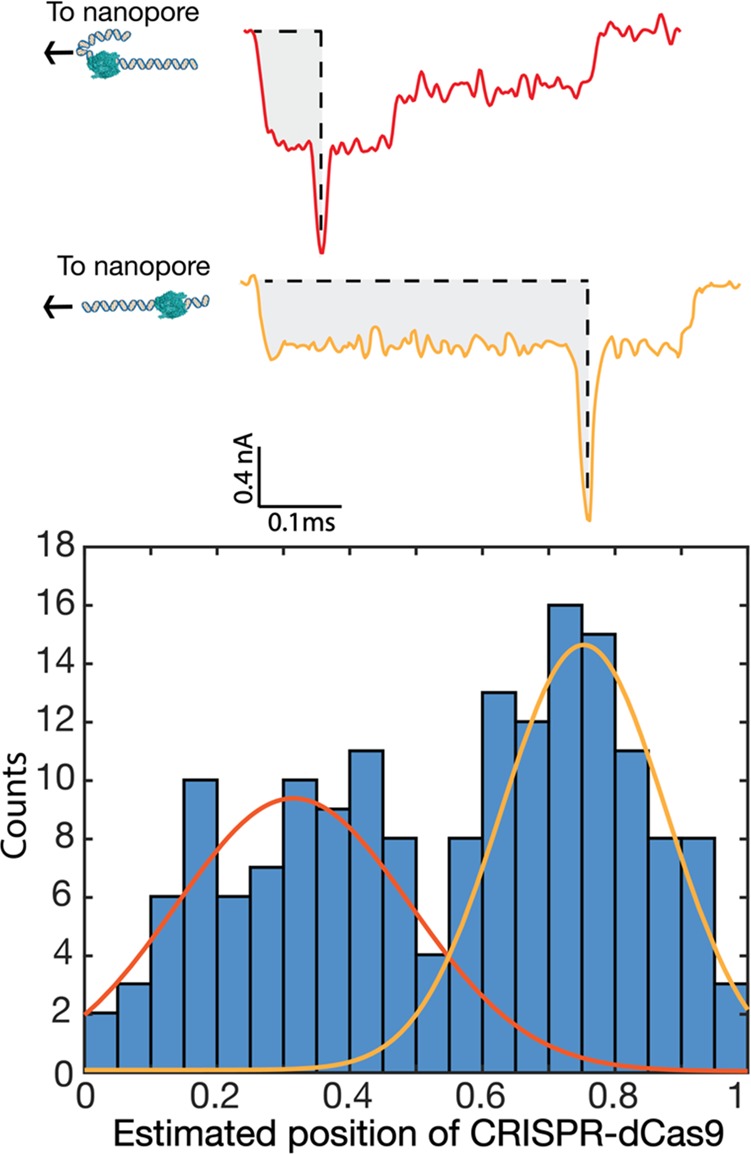
CRISPR-dCas9 binding position as detected in the translocation events. (a) Two examples of events where a dCas9-induced spike is located near the start or end of an event, which can be attributed to the fact that a DNA molecule can enter the pore with either of its two ends. In order to determine the relative binding position of the dCas9 proteins along the DNA, the integral of the current blockade up to the spike position was divided by the total integral of the entire translocation event. (b) Histogram of this fraction, binned into 20 bins. Two peaks are observed at 0.31 ± 0.04 and 0.75 ± 0.02 (where the errors are the standard error of the mean). The two peaks correlate well with the designed positions of the target site of the dCas9 on the DNA which are at 0.28 (5872/20 678 kbp) or 0.72 (14 806/20 678 kbp) for the two orientations.
In summary, we have demonstrated identification of CRISPR-dCas9 bounded DNA sequences at the single-molecule level in a nanopore. The binding of dCas9 to dsDNA is stable even in the high-salt conditions (1 M LiCl) that enable a high signal-to-noise ratio, which is advantageous for accurate nanopore detection. The large hydrodynamic size of the dCas9 complex produces clear spikes that can be readily distinguished from the current traces of bare DNA. We showed that the location of the dCas9 binding site along the DNA can also be detected, enabling identification of the DNA site that was used to bind the dCas9 complex through the sequence-programmable gRNA. A significant population of the protein remains bound to the DNA for the duration of the nanopore experiment (as large as 35% in 2 h), allowing facile determination of the possible presence of the targeted DNA sequences in the sample that is probed.
Looking ahead, the development of a nanopore-based diagnostic tool for direct DNA detection of disease-inducing agents presents key advantages over traditional serological methods based on the detection of antibodies, proteins, and biomarkers emanating from infectious agents. The presence of such biomarkers can differ strongly depending on the progression and stage of the disease,15 while the DNA of the causative agents remains present throughout. In the scheme proposed here, the use of dCas9 opens new avenues for DNA-sequence-sensing capabilities with large-diameter solid-state nanopores that can be scaled up and mass-produced.16 The resiliency of the dCas9 protein to the high-salt concentrations (whereas most DNA-binding proteins detach from DNA at high salinity) makes it a particularly fitting candidate as a sequence-specific protein that can be detected through the read-out capabilities of solid-state nanopores.
In recent years, other nanopore-based DNA mapping techniques have been proposed as alternatives to the more conventional PCR-based (polymerase chain reaction amplification) DNA-detection method, which require thermocycling. These proposed nanopore techniques rely on the use of functionalized surfaces or complementary DNA labels.17−21 Compared to these approaches, our detection scheme has multiple advantages. First, the ease of programmability of the gRNA enables versatile detection of a wide range of targets as well as multiplexing. Other nanopore approaches require extensive preparation of either the sample (design, folding, and attachment of DNA origami labels) or the nanopore surface (cleaning and (re)functionalization of the nanopore), and procedures will have to be repeated between each experimental run for the detection of different analytes. Furthermore, the dCas9 protein can target and search dsDNA directly without any temperature cycling step or additional enzyme for unzipping the DNA. This significantly speeds up the sample processing time, and may be of sue for straightforward point-of-care diagnostics. The ease of approach of our proposed dCas9 detection scheme over the other techniques makes nanopore sensing of DNA targets using CRISPR-dCas9 a promising platform for disease detection and diagnostics. Indeed, we envision the use of such nanopore sensing with CRISPR-dCas9 for the fast detection and identification of DNA motifs of medically relevant DNA targets.
Methods
gRNA-dCas9 Complex Assembly
We assembled gRNA, dCas9 and DNA in a 1× Cas9 Nuclease Reaction Buffer (New England Biolabs, 20 mM HEPES, 100 mM NaCl, 5 mM MgCl2, 0.1 mM EDTA (pH 6.5 @ 25 °C) in a molar ratio of 100:10:1 (RNA/dCas9/DNA). Excess ratios of dCas9 were used to ensure maximum binding of the protein to DNA. For the gel electrophoresis experiments, 1 kbp of DNA was incubated with the dCas9-gRNA complex. Full details on the gRNA sequence, DNA plasmids, and dCas9-gRNA complex assembly protocol can be found in the Supporting Information.
dCas9 Binding Assay
The binding affinity of the dCas9 protein to the DNA in 1 M LiCl was first verified using 6% retardation polyacrylamide gel electrophoresis (PAGE). A volume of 10 μL of sample was diluted with 5 μL of 5% glycerol to aid with the loading of the sample into the gel. In all experiments, the sample was first prepared in 1× Cas9 reaction buffer and then diluted to the final salt concentration prior to the gel experiments. Two samples of dCas9-DNA complex were prepared, one with 1 M LiCl (buffered with 10 mM Tris-HCl, 1 mM EDTA, pH 8) and one in 1× Cas9 Nuclease Reaction Buffer (Cas9 reaction buffer) which served as a positive control. For the DNA-only control samples, DNA with the same concentration (300 nM) was diluted in the same salt conditions and loaded into the lane parallel to the respective dCas9-DNA complex lane. The gel was run at 120 V for 2 h and stained with ethidium bromide for imaging.
Nanopore Experiment
We used TEM-drilled 20 nm diameter SiN nanopores for the experiments. The SiN membrane containing the nanopore was loaded in a PEEK (Polyether ether ketone) flow cell. We used Ag/AgCl electrodes and an Axopatch 200B amplifier (Molecular Devices) for current detection. The traces were recorded at 100 kHz and further low pass filtered at 20 kHz for the data at 100 mV and 50 kHz for the data taken from 200 to 300 mV with the Transanalyzer Matlab package.22
For the nanopore experiments, the dCas9-DNA complex, prepared in 1X reaction buffer, was diluted to a final concentration of 1.5 ng/μL in 1 M LiCl solution (buffered with 10 mM Tris-HCl, 1 mM EDTA, pH 8). Approximately 40 μL of the solution was pipetted to the negatively biased cis compartment of the flow cell. The complex was electrophoretically driven through the nanopore with a bias voltage of 100 mV (or otherwise, as indicated in the text) and detected by measuring changes in the current flowing through the pore. Following the experiment, the flow cell and nanopore were repeatedly flushed with 1 M LiCl solution. Blank traces were recorded to ensure that no residual sample remained prior to the DNA-only control experiment that was carried out in the same nanopore. The total measurement time was typically 1–2 h for each sample. During this time, we observed no discernible decrease in the event rate.
Acknowledgments
We acknowledge useful discussions with Alessio Fragasso, Daniel Verschueren, and Xin Shi. This work was supported by the European Research Council Advanced Grant SynDiv (No. 669598) and The Netherlands Organisation for Scientific Research (NWO/OCW), as part of the NanoFront and Basyc programs.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.nanolett.8b02968.
Information on the guide RNA sequence, gRNA folding, Cas9 complex assembly, gel assays, and control nanopore experiments performed (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Bayley H. Nanopore sequencing: from imagination to reality. Clin. Chem. 2015, 61, 25–31. 10.1373/clinchem.2014.223016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker C. Solid-state nanopores. Nat. Nanotechnol. 2007, 2, 209–215. 10.1038/nnano.2007.27. [DOI] [PubMed] [Google Scholar]
- Soni G. V.; Dekker C. Detection of Nucleosomal Substructures using Solid-State Nanopores. Nano Lett. 2012, 12, 3180–3186. 10.1021/nl301163m. [DOI] [PubMed] [Google Scholar]
- Plesa C.; Ruitenberg J. W.; Witteveen M. J.; Dekker C. Detection of individual proteins bound along DNA using solid-state nanopores. Nano Lett. 2015, 15, 3153. 10.1021/acs.nanolett.5b00249. [DOI] [PubMed] [Google Scholar]
- Sze J. Y. Y.; Ivanov A. P.; Cass A. E. G.; Edel J. B. Single molecule multiplexed nanopore protein screening in human serum using aptamer modified DNA carriers. Nat. Commun. 2017, 8, 1552. 10.1038/s41467-017-01584-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howorka S.; Siwy Z. Nanopores and Nanochannels: From Gene Sequencing to Genome Mapping. ACS Nano 2016, 10, 9768–9771. 10.1021/acsnano.6b07041. [DOI] [PubMed] [Google Scholar]
- Sternberg S. H.; Redding S.; Jinek M.; Greene E. C.; Doudna J. A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 2014, 507, 62–67. 10.1038/nature13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abudayyeh O. O.; et al. RNA targeting with CRISPR–Cas13. Nature 2017, 550, 280–284. 10.1038/nature24049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haurwitz R. E.; et al. Sequence- and Structure-Specific RNA Processing by a CRISPR Endonuclease. Science (Washington, DC, U. S.) 2010, 329, 1355–1358. 10.1126/science.1192272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H.; et al. Multiplexed labeling of genomic loci with dCas9 and engineered sgRNAs using CRISPRainbow. Nat. Biotechnol. 2016, 34, 528–530. 10.1038/nbt.3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi L. S.; et al. Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell 2013, 152, 1173–1183. 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm A. J.; Chen J. H.; Zandbergen H. W.; Dekker C. Translocation of double-strand DNA through a silicon oxide nanopore. Phys. Rev. E 2005, 71, 051903. 10.1103/PhysRevE.71.051903. [DOI] [PubMed] [Google Scholar]
- Plesa C.; et al. Direct observation of DNA knots using a solid-state nanopore. Nat. Nanotechnol. 2016, 11, 1093–1097. 10.1038/nnano.2016.153. [DOI] [PubMed] [Google Scholar]
- Plesa C.; van Loo N.; Ketterer P.; Dietz H.; Dekker C. Velocity of DNA during Translocation through a Solid-State Nanopore. Nano Lett. 2015, 15, 732–737. 10.1021/nl504375c. [DOI] [PubMed] [Google Scholar]
- Fierz W.Basic Problems of Serological Laboratory Diagnosis. In Molecular Diagnosis of Infectious Diseases; Decler J.; Reischl U., Eds.; Humana Press, 2004; pp 393–428; DOI: 10.1385/1-59259-679-7:393. [DOI] [PubMed] [Google Scholar]
- Verschueren D. V.; Yang W.; Dekker C. Lithography-based fabrication of nanopore arrays in freestanding SiN and graphene membranes. Nanotechnology 2018, 29, 145302. 10.1088/1361-6528/aaabce. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei R.; Pedone D.; Zürner A.; Döblinger M.; Rant U. Fabrication of Metallized Nanopores in Silicon Nitride Membranes for Single-Molecule Sensing. Small 2010, 6, 1406–1414. 10.1002/smll.201000253. [DOI] [PubMed] [Google Scholar]
- Rant U.; et al. Switchable DNA interfaces for the highly sensitive detection of label-free DNA targets. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 17364–17369. 10.1073/pnas.0703974104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; et al. DNA origami-based shape IDs for single-molecule nanomechanical genotyping. Nat. Commun. 2017, 8, 14738. 10.1038/ncomms14738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong J.; Zhu J.; Keyser U. F. Single molecule based SNP detection using designed DNA carriers and solid-state nanopores. 436. Chem. Commun. 2017, 53, 436. 10.1039/C6CC08621G. [DOI] [PubMed] [Google Scholar]
- Hernández-Ainsa S.; et al. DNA Origami Nanopores for Controlling DNA Translocation. ACS Nano 2013, 7, 6024–6030. 10.1021/nn401759r. [DOI] [PubMed] [Google Scholar]
- Plesa C.; Dekker C. Data analysis methods for solid-state nanopores. Nanotechnology 2015, 26, 084003. 10.1088/0957-4484/26/8/084003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.