

Abstract
Cardiolipin (CL) is an anionic phospholipid mainly located in the inner mitochondrial membrane, where it helps regulate bioenergetics, membrane structure, and apoptosis. Localized, phase-segregated domains of CL are hypothesized to control mitochondrial inner membrane organization. However, the existence and underlying mechanisms regulating these mitochondrial domains are unclear. Here, we first isolated detergent-resistant cardiac mitochondrial membranes that have been reported to be CL-enriched domains. Experiments with different detergents yielded only nonspecific solubilization of mitochondrial phospholipids, suggesting that CL domains are not recoverable with detergents. Next, domain formation was investigated in biomimetic giant unilamellar vesicles (GUVs) and newly synthesized giant mitochondrial vesicles (GMVs) from mouse hearts. Confocal fluorescent imaging revealed that introduction of cytochrome c into membranes promotes macroscopic proteolipid domain formation associated with membrane morphological changes in both GUVs and GMVs. Domain organization was also investigated after lowering tetralinoleoyl-CL concentration and substitution with monolyso-CL, two common modifications observed in cardiac pathologies. Loss of tetralinoleoyl-CL decreased proteolipid domain formation in GUVs, because of a favorable Gibbs-free energy of lipid mixing, whereas addition of monolyso-CL had no effect on lipid mixing. Moreover, murine GMVs generated from cardiac acyl-CoA synthetase-1 knockouts, which have remodeled CL acyl chains, did not perturb proteolipid domains. Finally, lowering the tetralinoleoyl-CL content had a stronger influence on the oxidation status of cytochrome c than did incorporation of monolyso-CL. These results indicate that proteolipid domain formation in the cardiac mitochondrial inner membrane depends on tetralinoleoyl-CL concentration, driven by underlying lipid-mixing properties, but not the presence of monolyso-CL.
Keywords: cardiolipin, membrane structure, mitochondria, cytochrome c, lipid vesicle, detergent solubilization, giant mitochondrial vesicles, giant unilamellar vesicles, lipid domains, monolyso-cardiolipin
Introduction
Cardiolipin (CL) 2 is a unique anionic phospholipid that is localized to the inner mitochondrial membrane, where it plays a role in regulating bioenergetics, membrane structure, and apoptosis (1–6). Several mechanisms are proposed to explain how CL regulates inner mitochondrial membrane structure-function. These mechanisms include CL directly binding select enzymes, anchoring cytochrome c, promoting correct cristae morphology, and enhancing supercomplex formation (7–13). One poorly studied hypothesis is that CL facilitates the formation of distinct phase-segregated domains (14, 15). The biological advantage of CL-enriched domains would be to concentrate respiratory protein complexes for enhanced electron channeling, to augment membrane curvature, and to serve as docking platforms for apoptotic signals (16–22).
The limited understanding on the existence of CL-enriched domains comes largely from two- or three-component model membrane studies. Calorimetric studies with binary phospholipid mixtures in the presence of cytochrome c, truncated Bid, or creatine kinase suggested segregation of CL into distinct clusters (18). Studies with Brewster angle microscopy of phosphatidylcholine/phosphatidylethanolamine monolayers containing creatine kinase have also provided direct support for CL domains (23). Furthermore, microscopy studies with biomimetic CL-containing mixtures have demonstrated the unique ability of cytochrome c to regulate membrane morphology and induce CL domain formation (24, 25). CL domains have also been identified biochemically with detergent-extraction methods; similar to approaches used for the study of detergent-resistant “lipid raft” membranes (17).
There are several major gaps in knowledge regarding CL-enriched domain organization. The first major limitation is that many studies have not effectively modeled the inner mitochondrial composition and even relied on lipid raft models (24, 26). Approximately 70% of the inner mitochondrial membrane is accounted for by phosphatidylcholine (40%) and phosphatidylethanolamine (30%), whereas CL makes up about 15–20% of total phospholipids (7). Phosphatidylinositol, phosphatidic acid, and phosphatidylserine account for the remaining phospholipids, with very low levels of cholesterol. Thus, CL domain formation should be studied in the context of inner mitochondrial-mimicking membranes. Second, the underlying mechanistic factors that regulate CL domain formation are not defined. These are essential to establish because in a range of diseases, CL is subject to considerable physiochemical modifications that impair mitochondrial inner membrane structure-function (3, 27–29). For instance, with heart failure and type II diabetes, cardiac CL acyl chains undergo extensive remodeling that could disrupt CL domain formation (30–37). In Barth syndrome, there is a significant loss of cardiac tetralinoleoyl-CL and increased levels of monolyso-CL (MLCL) (27, 38–42).
The first objective of this study was to determine the utility of biochemical detergent extraction to isolate CL domains. Subsequently, we investigated if CL domains can form in the absence or presence of select proteins. These proteins included cytochrome c, creatine kinase, and α-synuclein, which are all associated with mitochondrial membranes. The approach relied on quantitative imaging of biomimetic giant unilamellar vesicles (GUVs) modeling the cardiac inner mitochondrial membrane, and the use of giant mitochondrial vesicles (GMVs) constructed for the first time with phospholipids extracted from rodent cardiac mitochondria. Next, we determined if decreased CL levels, or the accumulation of MLCL, as observed in various diseases, disrupted the organization of proteolipid domains. These studies relied on biomimetic mitochondrial GUVs as well as lipid monolayers mimicking the inner mitochondrial membrane lipid composition to quantify lipid-lipid mixing. In parallel, we constructed GMVs using cardiac tissue harvested from control and acyl-CoA synthetase 1 knockout mice to determine how CL remodeling impacted the organization of native phospholipids and thereby proteolipid domains.
Results
Mitochondrial CL microdomains cannot be isolated with detergent-extraction methods
We first determined if different detergents were equally efficacious at extracting CL-enriched microdomains from rat cardiac mitochondria. Rat myocardial tissue was utilized to ensure a high abundance of material for microdomain isolation. We relied on Triton X-100, Nonidet P-40, and digitonin at various concentrations (0.01, 0.05, and 0.5% w/v). Increasing the concentration of all three detergents led to an overall decrease of phospholipid levels, as measured by TLC, in the insoluble detergent-resistant membrane (DRM) (I) fractions (Fig. 1, A–C). This decrease was paralleled by an increase in the amount of lipids in the detergent-soluble membrane (DSM (S)) fractions (Fig. 1, A–C). The increase in the concentration of Triton X-100 from 0.01 to 0.05% caused a larger reduction in the amount of phospholipids present in the DRM fraction than Nonidet P-40 or digitonin (Fig. 1, A–C). At a concentration of 0.5%, phospholipids in the DRM fraction were appreciably reduced with all three detergents (Fig. 1, A–C).
Figure 1.

Detergent extraction is not an effective tool for isolating cardiac mitochondrial CL-enriched domains. Isolated rat mitochondria were solubilized with (A) Triton X-100, (B) Nonidet P-40, or (C) digitonin at various concentrations (0.01, 0.05, or 0.5% w/v). Images to the right of each plot are representative TLC plates from one experiment. Quantification of protein levels in the DRMs in response to solubilization with (D) Triton X-100, (E) Nonidet P-40, or (F) digitonin at various concentrations (0.01, 0.05, or 0.5% w/v) is shown. DRMs are indicated as the insoluble (I) fraction and DSMs are indicated as the soluble (S) fraction. Data are average ± S.D. from two to three independent experiments.
Both DSM (S) and DRM (I) fractions were assayed for protein content. Triton X-100 and Nonidet P-40 had a higher concentration of protein in their DSM fractions than their DRM fractions, at all detergent concentrations (Fig. 1, D and E). Digitonin had more protein in the DRM fraction than the DSM fraction at lower detergent concentrations, but at 0.5% digitonin, very little protein was found in the DRM fraction (Fig. 1F). Overall, these findings challenge the notion that CL microdomains can be effectively isolated with biochemical detergent extraction. Therefore, we used a more controlled and less invasive model system to study CL microdomains.
The presence of cytochrome c induces proteolipid microdomain formation and morphological changes in GUVs and GMVs
We constructed GUVs, modeling the inner mitochondrial membrane, and GMVs to study CL microdomain organization. GMVs were constructed from native phospholipids extracted from isolated cardiac mitochondria. To visualize GUVs and GMVs, we employed two fluorescent probes, Texas Red DHPE (Texas Red), which is known to report on liquid-disordered domains of lipid membranes (43, 44), and nonyl acridine orange (NAO), which is suggested to report on CL (45, 46). Structures of the fluorescent probes are presented in Fig. S1, A and B.
We first visualized GUVs and GMVs in the absence of cytochrome c (−cyt c) using Texas Red (Fig. 2, A and B, left panels), which revealed a homogenous distribution of the fluorophore throughout the membrane. The addition of 17 μm cyt c to GUVs and GMVs containing Texas Red (Fig. 2, A and B, right panels) revealed fluorophore sequestration and the formation of proteolipid microdomains. The average fluorophore area was calculated using the equatorial cross-sectional view of GUVs and GMVs and was determined to decrease by 1.3–1.4–fold upon the addition of cyt c (Fig. 2, C and D). The formation of phase-segregated domains was accompanied by clear membrane morphological changes with the GUVs and GMVs. To exemplify, the addition of cyt c decreased the average diameter of GUVs and GMVs with Texas Red (Fig. 2, E and F, respectively) by 1.8-fold.
Figure 2.

Cytochrome c-induced proteolipid microdomains form in biomimetic and native cardiac mitochondrial vesicles. A and B, representative images of (A) GUVs and (B) GMVs visualized by Texas Red DHPE (0.1 mol %) in the absence (left panels) or presence (right panels) of 17 μm cyt c. GUVs were composed of (18:0–22:6)PC/(16:0 20:4)PE/(18:2)4CL/DOPI/DOPS/Chol (39.9/30/20/5/3/2 mol %). GMVs were constructed from native phospholipids extracted from isolated murine cardiac mitochondria. The average fluorophore area (C and D) and the average vesicle diameter (E and F) were determined for GUVs and GMVs. Each dot represents a single GUV or GMV. Data are average ± S.D. from a total of 60–85 vesicles analyzed from three to four independent experiments. Asterisks indicate significance from vesicles in the absence of cyt c: ****, p < 0.0001. Scale bars are 10 μm.
We next visualized vesicles using the fluorescent probe NAO. Similar observations to Texas Red were made when GUVs and GMVs were visualized by NAO. In the absence of cyt c, the NAO fluorophore was homogenously distributed throughout the membrane of GUVs and GMVs, and no microdomains were observed (Fig. 3, A and B, left panels). Upon the addition of 17 μm cyt c, strong phase segregation of NAO (Fig. 3, A and B, middle panels) was observed. Quantification revealed that 17 μm cyt c decreased the average fluorophore area by up to 2.2–3.5–fold for GUVs and GMVs visualized by NAO (Fig. 3, C and D, respectively). Similarly, the diameter of the GUVs and GMVs also decreased upon the addition of cyt c by 1.5–2.5 (Fig. 3, E and F, respectively). We conducted co-localization analyses to confirm that the phase-separated domains were forming in regions where cytochrome c was associating. These studies revealed overlap (average Pearson's correlation coefficient of 45.1%) between NAO and an Alexa 647 antibody against cytochrome c (Fig. S2).
Figure 3.

Proteolipid domain formation in biomimetic and native cardiac mitochondrial vesicles is observed with NAO fluorescence independent of cytochrome c concentration. A and B, representative images of (A) GUVs and (B) GMVs visualized by NAO (0.1 mol %) in the absence (left panels) or presence of 17 μm (middle panels) and 3.8 μm (right panels) cyt c. GUVs were composed of (18:0–22:6)PC/(16:0–20:4)PE/(18:2)4CL/DOPI/DOPS/Chol (39.9/30/20/5/3/2 mol %). GMVs were constructed from native phospholipids extracted from isolated murine cardiac mitochondria. The average fluorophore area (C and D) and the average vesicle diameter (E and F) were determined for GUVs and GMVs. Each dot represents a single GUV or GMV. Data are average ± S.D. from a total of 40–65 vesicles analyzed from three to four independent experiments. Asterisks indicate significance from vesicles in the absence of cyt c: ****, p < 0.0001. Scale bars are 10 μm.
We also tested if cytochrome c-induced lipid microdomain formation depended on protein concentration. The addition of 3.8 μm cyt c promoted lipid microdomain formation as observed by phase segregation of NAO in GUVs and GMVs (Fig. 3, A and B, right panels). The addition of 3.8 μm cyt c to GUVs and GMVs also decreased the average area occupied by NAO (Fig. 3, C and D) as well as the average vesicle diameter (Fig. 3, E and F). Overall, Texas Red and NAO showed similar results (Figs. 2 and 3); therefore, for the subsequent imaging studies we used NAO given that is suggested to be CL specific (45, 46).
Cytochrome c interactions with biomimetic and native mitochondrial lipid monolayers decreases the elasticity modulus
Given that cyt c decreased the diameters of GUVs and GMVs, we hypothesized that cyt c could be promoting changes in the elastic properties of the membrane. Therefore, we constructed biomimetic and native mitochondrial lipid monolayers, in the absence and presence of cyt c, to quantify the elasticity modulus (Cs−1), which is an inverse measure of membrane elasticity. We first present raw surface pressure-area isotherms for both biomimetic (Fig. 4A) and native (Fig. 4B) mitochondrial monolayers in the absence (black lines) and presence (red lines) of 17 μm cyt c. We only tested one concentration of protein because we observed similar effects on microdomain organization with 3.8 and 17 μm cyt c.
Figure 4.

The addition of cytochrome c to biomimetic and native mitochondrial monolayers decreases the elasticity modulus. Monolayers were constructed with (A) biomimetic membranes composed of (18:0–22:6)PC/(16:0–20:4)PE/(18:2)4CL/DOPI/DOPS/Chol (40/30/20/5/3/2 mol %) or (B) native mitochondrial phospholipids from native phospholipids extracted from isolated murine cardiac mitochondria. Pressure-area isotherms were analyzed at a physiologically relevant pressure of 30 mN/m to calculate the elasticity modulus (C and D), in the absence or presence of 17 μm cytochrome c (cyt c). Data are average ± S.D. from three to six independent experiments. Asterisks indicate significance from monolayers in the absence of cyt c: ***, p < 0.001.
The isotherms were used to calculate the elasticity modulus at surface pressures of 30 mN/m. The addition of cyt c to biomimetic (Fig. 4C) and native (Fig. 4D) mitochondrial monolayers decreased the elasticity modulus by nearly 2-fold. Therefore, these results demonstrate that cyt c regulates the elastic properties of biomimetic and native mitochondrial monolayers.
Lowering of (18:2)4CL levels, rather than substitution of (18:2)4CL with MLCL, disrupts proteolipid domain formation in GUVs
In several pathologies, such as cardiac ischemia-reperfusion injury, type 2 diabetes, and Barth syndrome, there is loss of (18:2)4CL content and remodeling of CL acyl chains (27, 35, 47, 48). There is a debate in the literature whether which of these is more consequential (49). Therefore, we studied how a reduction of (18:2)4CL concentration and CL acyl chain composition influenced lipid microdomain organization in GUVs.
In the absence of cyt c, GUVs containing 20 mol % (18:2)4CL (Fig. 5A, left panel), a decrease in (18:2)4CL concentration by 50 mol % (Fig. 5B, left panel), or 15/5 mol % MLCL/(18:2)4CL (Fig. 5C, left panel) displayed no lipid microdomain formation. This was consistent with our previous imaging experiments in which no lipid microdomains were observed in the absence of cyt c. The addition of 3.8 μm cyt c to GUVs containing (18:2)4CL (Fig. 5A, right panel) promoted proteolipid microdomain formation. Upon decreasing (18:2)4CL concentration by 50%, we observed two distinct populations. One GUV population displayed proteolipid domains (Fig. 5B(I), right panel) and another group of GUVs that did not display formation of proteolipid microdomains (Fig. 5B(II), right panel) in the presence of cyt c. The distribution of these populations was determined from a total of 155 individual GUVs, where ∼56% of GUVs displayed domain formation. Conversely, the other 44% of GUVs showed no clear microdomain formation because NAO was homogenously distributed throughout the membrane of these vesicles. Strikingly, inclusion of MLCL promoted strong domain formation (Fig. 5C, right panel). Quantification of the GUVs showed that cyt c lowered the fluorophore area in nearly all cases (Fig. 5, D–F). Similarly, the addition of cyt c decreased the diameter of the GUVs (Fig. 5, G–I).
Figure 5.

Proteolipid domain organization in biomimetic mitochondrial vesicles is disrupted with a reduction of cardiolipin concentration but not with the inclusion of MLCL. A–C, representative images of biomimetic mitochondrial GUVs containing (A) 20 mol % (18:2)4CL, (B) 50 mol % reduction of (18:2)4CL, or (C) 15/5 mol % MLCL/(18:2)4CL. The vesicles were composed of (18:0–22:6)PC/(16:0–20:4)PE/CL/DOPI/DOPS/Chol and were visualized with 0.1 mol % NAO. GUVs were constructed in the absence or presence of 3.8 μm cytochrome c. The average fluorophore area of NAO (D–F) and the average GUV diameter (G–I) were determined for vesicles in the absence or presence of cyt c. Each dot represents a single GUV. Data are average ± S.D. from a total of 45–155 vesicles analyzed from three to four independent experiments. Asterisks indicate significance from vesicles in the absence of cyt c, unless otherwise noted: **, p < 0.01; ****, p < 0.0001. Scale bars are 10 μm.
A reduction in (18:2)4CL concentration promotes favorable Gibbs-free energy of lipid mixing
Mechanistically, lipid domains are driven by the Gibbs-free energy of lipid mixing. Therefore, we used pressure-area isotherms to calculate the change in Gibbs-free energy between the absence and presence of cyt c. We first present the raw surface pressure-area isotherms for biomimetic mitochondrial monolayers containing (18:2)4CL (Fig. 6A), 50% depletion of (18:2)4CL concentration (Fig. 6B), or MLCL/(18:2)4CL (Fig. 6C) in the absence (black traces) and presence (red traces) of 3.8 μm cyt c. Secondary analyses revealed that a 50% loss of (18:2)4CL significantly shifted the change in Gibbs-free energy of mixing upon cyt c addition toward a more favorable value by 2.5-fold (Fig. 6D). In contrast, inclusion of MLCL had no effect on the thermodynamics of lipid mixing (Fig. 6D).
Figure 6.
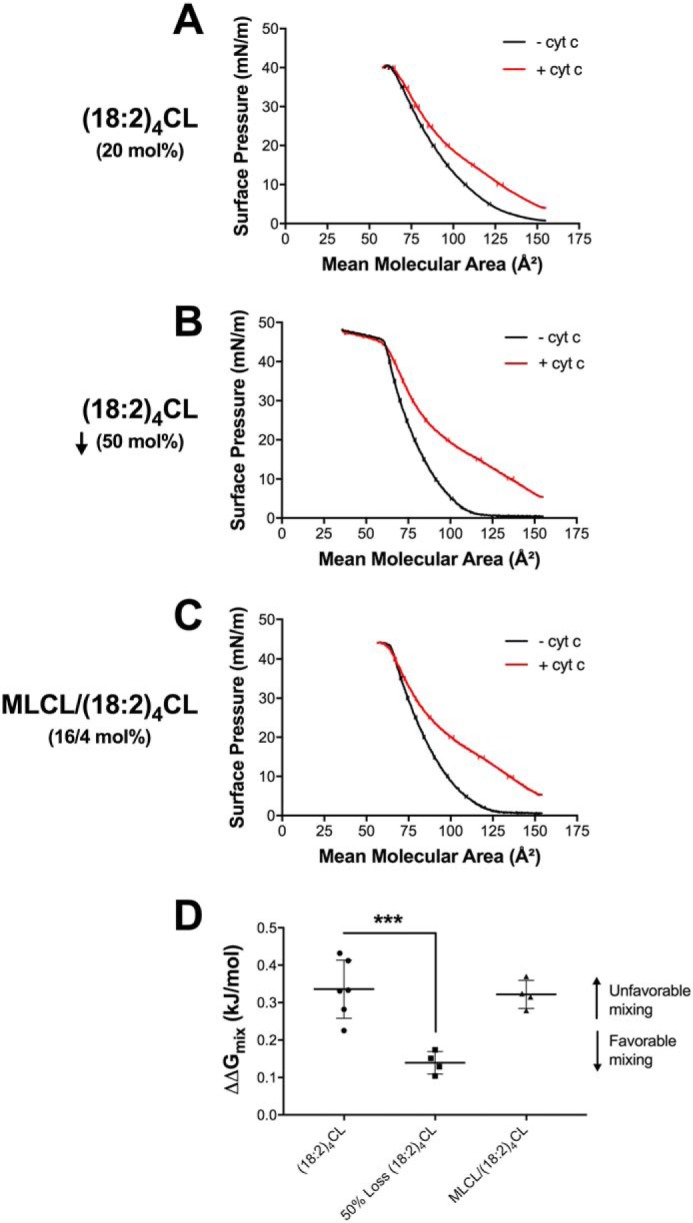
Proteolipid domain organization in biomimetic membranes is disrupted with the loss of cardiolipin concentration due to favorable Gibbs-free energy of mixing. Biomimetic monolayers were constructed containing (A) 20 mol % (18:2)4CL, (B) 50 mol % reduction of (18:2)4CL, or (C) 16/4 mol % MLCL/(18:2)4CL. Biomimetic monolayers were composed of (18:0–22:6)PC/(16:0–20:4)PE/CL/DOPI/DOPS/Chol in the absence or presence of 3.8 μm cytochrome c. The Gibbs-free energy of mixing (D) was calculated at a physiologically relevant surface pressure of 30 mN/m, in the absence and presence of cyt c. The ΔΔG (i.e. ΔG(+)protein − ΔG(−)protein) of lipid mixing (D) is presented where a higher, more positive, ΔΔGmix value indicates unfavorable lipid-lipid mixing. Data are average ± S.D. from four to six independent experiments. Asterisks indicate significance from monolayers containing (18:2)4CL: ***, p < 0.001.
Proteolipid microdomain formation is not disrupted in GMVs synthesized using native mitochondrial phospholipids extracted from cardiac tissue of Acsl1T−/− mice
To further investigate how CL acyl chain composition regulates proteolipid domain organization, we relied on GMVs constructed from cardiac-specific acyl-CoA synthetase knockout mice (Acsl1T−/−). The rationale for this model was that CL acyl chains are notably remodeled, although total CL levels do not change (50). Specifically, previously published LC/MS analyses show that Acsl1T−/− mice have increased incorporation of oleic acid (18:1) into CL at the expense of the loss of linoleic acid (18:2) (50).
Upon the addition of 3.8 μm cyt c, proteolipid microdomain formation was observed in GMVs constructed from control (Fig. 7A) and Acsl1T−/− (Fig. 7B) mice. Proteolipid domains were similar in size between control and Acsl1T−/− GMVs, as measured by the average area occupied by NAO (Fig. 7C). Furthermore, the diameter of the vesicles was also similar between the control and Acsl1T−/− GMVs (Fig. 7D). Taken together, these results suggested that the remodeling of tetralinoleoyl-CL to other acyl chains does not impair the formation of cytochrome c-induced microdomains visualized by NAO fluorescence.
Figure 7.
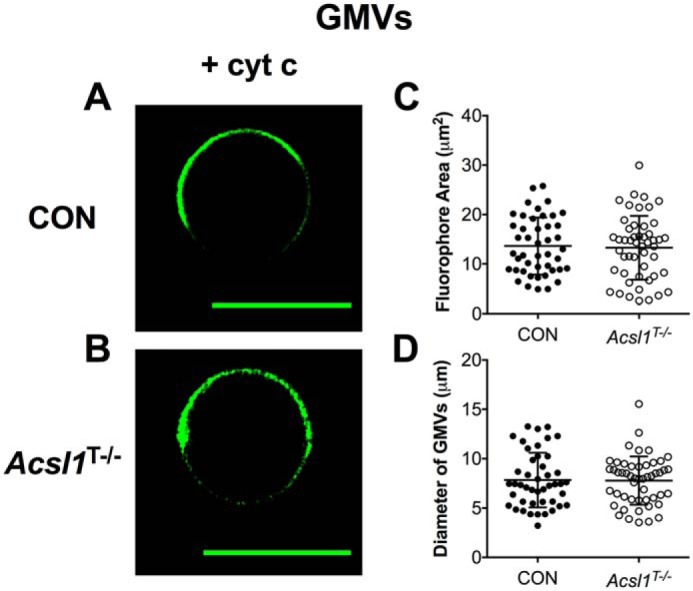
Cytochrome c-induced proteolipid microdomain organization is not disrupted in GMVs constructed from murine Acsl1T−/− cardiac tissue. A and B, representative images of GMVs constructed native mitochondrial phospholipids extracted from control (CON) (A) and Acsl1T−/− (B) murine cardiac tissue. GMVs were visualized with 0.1 mol % NAO in the presence of 3.8 μm cytochrome c. C and D, average fluorophore area (C) and the average GMV diameter (D). Each dot represents a single GMV. Data are average ± S.D. from a total of 45–50 vesicles analyzed from three independent experiments. Scale bars are 10 μm.
We further investigated how the accumulation of oleic acid in CL regulated proteolipid domain formation. The rationale for using the tetraoleoyl-CL species was that oleic acid is heavily accumulated in CL at the expense of linoleic acid in the murine knockout of Acsl1T−/− (50). We specifically constructed biomimetic GUVs containing tetraoleoyl-CL ((18:1)4CL) in the absence and presence of cyt c. In the absence of protein, NAO was homogenously distributed throughout the entire membrane of biomimetic GUVs containing (18:1)4CL (Fig. S3A). Upon addition of cyt c, NAO was sequestered into localized regions of the membrane (Fig. 3A). Similarly, cyt c-induced proteolipid domains were generally smaller in size, as indicated by a decreased average microdomain area and decreased vesicle diameter (Fig. 3, B and C).
Lipid microdomains form in the presence of mitochondrial creatine kinase in GUVs
We subsequently determined if other membrane-associated proteins could promote lipid microdomain formation in biomimetic mitochondrial GUVs. We tested the ability of purified recombinant α-synuclein (Fig. 8A) and mitochondrial creatine kinase (Fig. 8B) to induce proteolipid microdomains in GUVs. We selected α-synuclein (α-syn) because its size is similar to cytochrome c, and mitochondrial creatine kinase (mtCK) because it is previously reported, in simpler model membranes, to bind to CL and promote domain formation (18, 51, 52).
Figure 8.

Lipid microdomain formation depends upon specific lipid-protein interactions. A and B, representative images of biomimetic GUVs in the presence of 3.8 μm (A) α-syn or (B) mtCK. Vesicles were composed of (18:0–22:6)PC/(16:0–20:4)PE/(18:2)4CL/DOPI/DOPS/Chol and were visualized with 0.1 mol % NAO. The average fluorophore area occupied by NAO (C and D) and the average GUV diameter (E and F) were determined for vesicles in the absence and presence of protein. Each dot represents a single GUV. Data are average ± S.D. from a total of 25–90 vesicles analyzed from 3 independent experiments. Asterisks indicate significance from vesicles in the absence of protein: ****, p < 0.0001. Scale bars are 10 μm.
The addition of 3.8 μm α-syn to biomimetic mitochondrial GUVs containing (18:2)4CL did not promote observable microdomains (Fig. 8A) because NAO was homogenously distributed throughout the entire GUV membrane. In contrast, mtCK promoted strong phase segregation of the NAO probe (Fig. 8B). Quantitatively, there was no change in the average area occupied by NAO for α-syn (Fig. 8C), whereas mtCK lowered the fluorophore area of GUVs (Fig. 8D). The diameter of the GUVs was maintained with α-syn (Fig. 8E) and lowered with mtCK (Fig. 8F).
(18:2)4CL oxidizes the heme cofactor iron-metal center in cytochrome c more effectively than MLCL
Finally, we investigated if the loss of CL concentration had a stronger effect than MLCL on the oxidation of cytochrome c. The rationale for this study was that MLCL may lack the ability to bind in the proper orientation near redox-sensitive cofactors, resulting in dramatic changes in midpoint potentials and impediments to electron transfer (53, 54). By using UV-visual spectroscopy, we monitored the interaction of (18:2)4CL (Fig. 9A) and MLCL (Fig. 9B) with the heme cofactors of cytochrome c.
Figure 9.

The reduction of (18:2)4CL concentration has a greater effect on cytochrome c oxidation than MLCL. A and B, representative normalized UV-visible absorption spectra displaying the oxidation of the heme cofactor iron-metal center in cytochrome c upon increasing concentrations of (18:2)4CL (A) or MLCL (B). Cytochrome c was fully reduced with the addition of ascorbate (90 mm, solid red line). Full spectra (200–700 nm) were obtained at a pH of 7.4 in the presence of glucose oxidase (15 units), glucose (5 mm), and catalase (5 units) to ensure O2 and H2O2 free conditions. Blue shift of the Soret band at 400 nm (data not shown) and decreases in the α-heme bands at 518 and 550 nm (A and B) are characteristic of oxidation of the Fe(II) center to Fe(III) with increasing lipid concentrations. For comparison, cyt c was fully oxidized after the final addition of lipid using 3 mm K3[Fe(CN)6] (dashed red line). C, the relative change in absorbance at 550 nm, defined as the absorbance at a specific lipid concentration/absorbance of the fully reduced complex, was determined and plotted as a function of lipid concentration (μm). Data are average ± S.D. from three independent experiments.
Interactions between CL and the low spin, Fe(II)-heme center of the reduced protein, resulted in oxidation of the metal center to a high spin, Fe(III) center. These changes in cytochrome c's redox properties were dose-dependent as demonstrated by a blue-shift in the Soret bands (400 nm, data not shown) and decrease in α-heme bands (540–560 nm, Fig. 9). Plots of the extent of cytochrome c oxidation versus lipid concentration (Fig. 9C) show that a higher concentration of MLCL (closed circles) was needed to fully oxidize cytochrome c relative to (18:2)4CL (open circles), and a dose-dependent effect of (18:2)4CL concentration on the redox properties of cyt c.
Discussion
Biochemical detergent extraction is ineffective for isolating CL-enriched domains
One major advancement from this study was data demonstrating that detergent extraction was ineffective for isolating and studying CL-enriched domains. We investigated the efficacy of three different detergents for extracting CL-enriched domains because CL domains are reported to be isolatable using nonionic detergents at cold temperatures (17). Triton X-100 was used because it is the preferred detergent for lipid domain extraction (55, 56). Nonidet P-40 was selected due to its nonionic nature and digitonin was relied on given its use for isolation of mitochondrial supercomplexes (57). The analyses revealed that detergent extraction displayed no specificity for CL-enriched domains because all phospholipids and proteins were generally solubilized with increasing detergent concentrations. Thus, similar to conclusions made in the study of plasma membrane lipid rafts, detergent was not an effective tool for studying CL domains (58).
CL-cytochrome c interactions promote domain formation and membrane morphological changes
Given the limitations of detergent extraction, an imaging approach was used to study mitochondrial inner membrane domains. The novelty of our approach was in the combined analysis of native mitochondrial and biomimetic vesicles that represent or model the phospholipid composition of the inner mitochondrial membrane. The observation that proteolipid domains form with cyt c, as measured with the fluorescent probe NAO, was in agreement with the literature. For instance, (14:0)4CL in raft/nonraft mixtures promoted phase separation in the presence of dynamin-related protein 1 (22). The observed morphological changes with CL were also consistent with a previous study that suggested interactions between CL and cytochrome c in raft/nonraft mixtures promoted membrane budding and ultimately formation of a folded structure (24). The positively charged NAO was likely interacting with negatively charged regions and thereby promoting the formation of cyt c multimers associated with structural modifications. Alternatively, the binding of NAO to the membranes may be simply a reflection of ion-ion interactions.
Supporting monolayer studies showed that the addition of cytochrome c was disrupting membrane packing accompanied by changes in the elasticity modulus. The morphological changes were of biological significance because CL domains are hypothesized to form in select regions of the membrane, specifically in areas of high membrane curvature (59). For instance, Renner and Weibel (16) reported that CL domains localized in regions of high curvature induced with microchambers using an Escherichia coli model. Thus, proteolipid domains in the inner mitochondrial membrane may be localized to regions of high curvature induced by strong interactions between cytochrome c and CL.
We did not observe proteolipid domain formation with the intrinsically disordered protein α-synuclein. The rationale for using α-synuclein was based on evidence that it can interact with mitochondria to diminish complex I activity (60). Furthermore, α-syn is known to bind acidic phospholipids although these interactions have been shown to be significantly augmented by the presence of phosphatidylethanolamine, a neutral phospholipid (61). The results with mitochondrial creatine kinase were consistent with previous literature demonstrating the ability of this protein to promote the formation of proteolipid domains (18, 52). Notably, creatine kinase is also hypothesized to localize to cristae sites where CL is enriched (62, 63).
A reduction in tetralinoleoyl-CL concentration but not substitution with MLCL prevents proteolipid domain formation through changes in lipid miscibility
In conditions such as Barth syndrome, cardiovascular diseases, obesity, and diabetes, CL levels are lowered or undergo changes in acyl chain composition. It is hypothesized that such changes may be detrimental to mitochondrial function. To exemplify, in cardiac ischemia-reperfusion injury, total (18:2)4CL content is selectively depleted (32, 48, 64). Therefore, our data provide a potential mechanism by which mitochondrial function may become impaired. That is, perhaps a reduction in total (18:2)4CL content disrupts formation of mitochondrial proteolipid domains. Furthermore, the loss of CL in ischemia is also associated with the loss of cyt c, which triggers apoptotic signaling cascades (65, 66). Thus, formation of CL-cyt c domains may be a critical mechanistic link and perhaps a therapeutic target for preventing the robust apoptosis observed with cardiac ischemia-reperfusion injury (65, 66).
The result that MLCL did not perturb proteolipid domain organization does not preclude other mechanisms by which MLCL may exert its effects on mitochondrial bioenergetics. For instance, MLCL is not as tightly bound as CL to respiratory proteins, which occurs in response to CL degradation in Barth syndrome (39). Furthermore, increasing levels of MLCL are likely to influence membrane properties outside of microdomain formation such as viscosity and permeability. Thus, it is plausible that MLCL could impair respiratory enzyme activities and electron channeling. Future studies will be needed to investigate how MLCL disrupts differing aspects of membrane biophysical organization and thereby protein-lipid and protein-protein interactions.
There are several emerging studies suggesting that CL acyl chain composition is not a major driver of mitochondrial membrane structure-function. Our results with MLCL on microdomain formation and cytochrome c oxidation status were consistent with these previous studies. For example, in a yeast model, remodeled and unremodeled CL were indistinguishable when analyzing oxidative phosphorylation activity (49). Similarly, CL content varies between differing tissues, which further argues against the notion that CL acyl chain composition is a key regulator of bioenergetics function. In addition, restoration of WT CL levels in differing disease models does not rescue mitochondrial functional responses. For instance, in aging or in a previous study with the heart-specific acyl-CoA 1 synthetase knockout, restoration of (18:2)4CL did not improve key mitochondrial end points (50, 67).
It is important to point out that in some cases of extreme CL remodeling, the acyl chains may be major drivers of changes in mitochondrial bioenergetics. We previously demonstrated that dietary docosahexaenoic acid promoted robust CL remodeling, which lowered cardiac respiratory enzyme activities through changes in protein-CL binding and formation of membrane microdomains (68).
Although our data suggest that acyl chain composition may not be a major driver of mitochondrial microdomains, additional studies are needed to understand how CL oxidation products influence mitochondrial inner membrane physical properties. The use of the 18:1-containing CL would be a limitation given that it is not prone to generating downstream second messengers. Future studies will need to specifically investigate how oxidation products, which are known to influence bioenergetics, could regulate mitochondrial structure and thereby function (69).
Conclusion
The data demonstrate that CL-specific microdomains cannot be effectively isolated via classical biochemical detergent extraction techniques. More importantly, membrane domains were formed in GUVs and newly constructed GMVs with cytochrome c. These domains were sensitive to a reduction in CL concentration but not substitution with MLCL. At a molecular level, the impairment in domain formation with the loss of tetralinoleoyl-CL was due to changes in the Gibbs-free energy of lipid mixing. Collectively, the results have strong implications for the ongoing debate about CL concentration and acyl chain composition and their role in regulating membrane biophysical organization.
Experimental procedures
Materials
(18:1)4CL, 1,2-di-(9Z-octadecenoyl)-sn-glycero-3-phosphocholine (DOPC), 1-stearoyl-2-docosahexaenoyl-sn-glycero-3-phosphocholine (18:0–22:6 PC), 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphoethanolamine (16:0–20:4 PE),1,2-dioleoyl-sn-glycero-3-phospho-(1′-myo-inositol) (DOPI), 1,2-dioleoyl-sn-glycero-3-phospho-l-serine (DOPS), bovine heart cardiolipin ((18:2)4CL)), bovine heart MLCL, and cholesterol (Chol) were purchased from Avanti Polar Lipids (Alabaster, AL). The fluorescent probes Texas Red DHPE and NAO were purchased from Life Technologies. Equine heart cyt c was obtained from Sigma. The mouse monoclonal cytochrome c primary antibody conjugated with Alexa Fluor 647 was purchased from Novus Biologicals (Littleton, CO).
All other reagents and solvents (HPLC grade) were purchased from Sigma or Fisher Scientific. Lipid mixtures were handled under low light conditions and fresh stocks of lipid mixtures were used for each experiment to minimize oxidation.
Isolation of mitochondria from rodent cardiac tissue
Mitochondrial isolations were performed on ice and all buffers and instruments were pre-chilled to 4 °C. All animal experiments were conducted in accordance with guidelines established by the Guide for Care and Use of Laboratory Animals and with prior approval by the University's Animal Care and Use Committee. Adult male C57BL/6J mice we purchased from Jackson Laboratories and were housed on a 12:12-h light:dark cycle and had unrestricted access to standard chow and water. Adult male Sprague-Dawley rats (150–250 g) were obtained from Charles River and were housed on a 12:12-h light:dark cycle and had unrestricted access to standard chow and water. Acsl1T−/− mice were maintained as previously described (50). One animal was sacrificed per isolation and cardiac tissue was collected immediately following euthanasia. Mice were anesthetized by isoflurane. Rats were anesthetized with ketamine/xylazine (90 mg/kg of ketamine, 10 mg/kg of xylazine, i.p.) and after the absence of animal eye-blink, toe pinch, and righting reflexes, rats were sacrificed. Cardiac tissue was excised and immediately placed in pre-chilled mitochondrial isolation medium (MIM) containing 20 mm HEPES (pH 7.2), sucrose (300 mm), and EDTA (1 mm). Rodent cardiac tissue was manually minced for 5 min and subjected to homogenization (7 strokes) with a Teflon Potter glass homogenizer. The homogenate was centrifuged at 500 × g for 5 min and the decanted supernatant was centrifuged again at 500 × g for 5 min. The supernatant was decanted and centrifuged at 10,000 × g for 15 min, and the resulting pellet was washed with MIM prior to centrifugation at 12,000 × g for an additional 15 min. The resulting mitochondrial pellet(s) were resuspended in either 300 μl of MIM (for rat cardiac mitochondria) or in 150 μl of MIM (for murine cardiac mitochondria) and frozen at −80 °C. Total mitochondrial protein concentrations were determined prior to freezing using a BCA protein assay (ThermoScientific, La Jolla, CA).
Extraction of rodent cardiac mitochondrial detergent-resistant (DRM) and detergent-soluble (DSM) membranes and analyses
Experimental procedures were modified from an established protocol for isolating cardiolipin-enriched DRMs (17, 70). Briefly, 1.0 mg of isolated mitochondria was incubated in a Beckham Coulter Centrifuge Tube (355635) on ice for 10 min using an extraction buffer containing 25 mm HEPES (pH 7.5), 150 mm sodium chloride, 10 μg/ml of aprotinin and various concentrations of Triton X-100, digitonin, or Nonidet P-40 detergents. The solution was centrifuged in an Optima Max-XP Ultracentrifuge with a TLA 100.4 rotor at 20,000 × g for 3 min at 4 °C. The detergent-soluble fraction was extracted from the centrifuge tube and the detergent-resistant pellet was resuspended in a solubilization buffer containing 50 mm Tris-HCl (pH 8.8), 1% SDS, and 5 mm EDTA. DNA was sheared from the detergent-resistant fraction by passage through a 1.00-ml syringe and 20½-gauge needle. The fractions were then transferred to a fresh glass tube for lipid extraction. TLC was conducted on DRM and DSM fractions as previously described (53). Protein levels in the fractions were also quantified using a standard BCA assay kit.
Extraction of native phospholipids from isolated rodent cardiac mitochondria
Experimental procedures were modified from the Bligh and Dyer method (71). Frozen aliquots of rodent cardiac mitochondria were thawed and transferred into a glass test tube. For every 10 mg/ml of mitochondria, samples were resuspended in 10 ml of 70/30 (v/v) CHCl3/MeOH (0.05% BHT) solution and vortexed for 45 s. Upon vortex mixing, 2.5 ml of 0.88% KCl was added to each sample and vortexed for an additional 45 s. The resulting mixture was centrifuged at 2000 rpm for 10 min at room temperature (23 °C). Upon separation of phases, the bottom layer (organic) was aspirated and dried via a gentle stream of N2 gas. The extract was resuspended in 2.0 ml of chloroform and a second extraction was performed. Upon recovering the lipid-containing organic layer, the sample was aspirated and dried via a gentle stream of N2 gas prior to being resuspended in HPLC grade chloroform (final concentration 0.1–0.25 μmol/25 μl). Total phosphorous analysis of extracted native mitochondrial phospholipids was conducted as previously described (31).
Construction of GUVs and GMVs
GUVs were constructed by co-dissolving lipids (39.9 mol % 18:0–22:6 PC, 30.0 mol % 16:0–20:4 PE, 20 mol % (18:2)4CL, 5 mol % DOPI, 3 mol % DOPS, 2 mol % Chol), and either the fluorescent probe Texas Red DHPE or NAO (0.1 mol %) in chloroform (0.5 mg/ml). The concentration of PC, PE, CL, PI, PS, and Chol approximated ratios from shotgun lipidomic analyses (data not shown) and were consistent with the literature (7). For select GUV experiments in which CL's content and acyl chain composition was altered, (18:2)4CL concentration was decreased 50% by mass or replaced with MLCL. GMVs were constructed by co-dissolving extracted native mitochondrial phospholipids in HPLC-grade chloroform. Roughly 10 μg of total lipid was spread onto the conductive side of an indium tin oxide-coated glass slide. The lipid-coated slide was subjected to dark vacuum for 1–2 h to remove excess solvent. Once the lipid film was dried, an electroformation chamber was assembled as described (72). GUVs and GMVs were constructed by electroformation at room temperature as previously described (26, 72, 73). Once electroformation was complete, vesicles were extracted from the chamber using a 20½-gauge needle/syringe and allowed to equilibrate briefly prior to microscopy sample preparation. For select experiments, the addition of cyt c (3.8–17 μm final concentration), as well as other proteins, to GUVs and GMVs occurred immediately prior to imaging. Samples were drawn into a rectangular capillary tube (VitroTubes) and mounted onto a glass microscope slide as previously shown (68, 74).
Confocal microscopy and image analysis
Imaging was conducted with an Olympus FV1000 Confocal Microscope using a ×60/1.35NA oil immersion objective (Olympus, Waltham, MA) and a Zeiss LSM 880 with Airyscan using a ×63/1.40NA oil immersion objective (Carl Zeiss Microscopy, Thornwood, NY). The Texas Red DHPE probe and Alexa 594 secondary antibody were excited by a diode laser at 559 nm, whereas the NAO probe was excited with an argon laser at 488 nm. A diode laser at 633–635 nm excited the Alexa 647 primary antibody used for cyt c immunofluorescence.
All acquired images were of GUV equatorial cross-sections. Lipid microdomain and colocalization analysis was conducted using NIH ImageJ software (74). After background subtraction, the average area occupied by the fluorophore was determined by adjusting the image threshold and analyzing particles that were ≥0.1 μm2. We selected this threshold, because an area <0.1 μm2 resulted in particles/domains that could not be accurately measured. The average vesicle diameter was determined by measuring the horizontal distance between the membrane edges. For colocalization measurements, we determined the fraction of cytochrome c colocalized with NAO using the Coloc 2 and JACoP plugins. Colocalization was quantified after background subtraction using Pearson's correlation coefficient.
Construction and analysis of biomimetic and native mitochondrial monolayers
Biomimetic mitochondrial monolayers were generated by co-dissolving lipids (40 mol % 18:0–22:6 PC, 30.0 mol % 16:0–20:4 PE, 20 mol % (18:2)4CL, 5 mol % DOPI, 3 mol % DOPS, 2 mol % Chol) in chloroform (10 μg/μl). For select lipid monolayer experiments in which CL's content and acyl chain composition was altered, (18:2)4CL concentration was decreased 50% by mass or replaced with MLCL. Native mitochondrial monolayers were initially generated by aliquoting native mitochondrial phospholipids (0.25 μmol/25 μl) extracted from murine (C57BL6/J) cardiac tissue. Lipid monolayers were constructed by spotting either 9.0 nmol (biomimetic mitochondrial monolayers) or 6.0 nmol of total lipid (native mitochondrial monolayers) on a subphase of 10 mm sodium phosphate buffer (pH 7.4). Biomimetic and native mitochondrial lipid monolayers were analyzed in the presence and absence of cytochrome c (cyt c; 3.8–17 μm final concentration) by gently injecting protein underneath the subphase immediately after spotting lipid. Excess chloroform was allowed to evaporate for 10 min prior to monolayer compression. Pressure-area isotherms were analyzed at physiologically relevant surface pressures (i.e. 30 mN/m) as previously described (26, 68). Raw isotherms were used to calculate the surface elasticity modulus (Cs−1),
| (Eq. 1) |
A represents the mean molecular area of the lipid mixture of interest at the indicated surface pressure (π) (75). The Gibbs-free energy of mixing (ΔGmix) was calculated by the following relationship to quantify lipid-lipid mixing,
| (Eq. 2) |
ΔGex represents the excess Gibbs-free energy of mixing and ΔGideal represents the ideal Gibbs-free energy of mixing, which were calculated by the following expressions,
| (Eq. 3) |
An and Xn are the mean molecular area and mol fraction of each component, respectively, at a given surface pressure (π) (76). ΔGideal was determined by,
| (Eq. 4) |
R represents the ideal gas constant and T is temperature in Kelvin (76). All lipid mixtures were acquired multiple times on differing days to ensure reproducibility.
Overexpression and purification of recombinant α-synuclein
Electrocompetent E. coli BL21 STAR (dE3) cells were transformed with expression plasmid pReceiver-B31 (GeneCopeia, Rockville, MD) encoding for the N-terminally His-tagged α-synuclein. Protein was expressed in 20-liter batch cultures of LB media, supplemented with 200 μg/ml of ampicillin for plasmid maintenance, and inoculated with a 1-liter overnight culture of transformed E. coli. Large-scale fermentations were carried out in a 30-liter carboy equipped with an air inlet hose connected to, and fitted with, a gas diffusion stone for proper aeration and agitation of the media during growth and induction. Batch cultures were grown at 37 °C to an A600 = 0.9–1.2. The culture was then chilled on ice for 20 min prior the addition of isopropyl 1-thio-β-d-galactopyranoside (final concentration 1 mm). Protein expression was induced at 16 °C for ∼20–24 h. Cells were harvested via centrifugation, yielding 100–200 × g of cell paste. Cell paste was flash frozen in liquid N2 and stored at −80 °C until needed.
All purification steps were performed at 4 °C. 5–10 ml of nickel-nitrilotriacetic acid resin (ThermoFisher Scientific, Waltham, MA) was equilibrated with wash buffer (20 mm NaPO4, pH 7.5, 300 mm NaCl). Cell paste (20 g) was thawed, while stirring, in 300 ml of lysis buffer (20 mm NaPO4, pH 8.0, 300 mm NaCl, 1 mm phenylmethylsulfonyl fluoride, and 100 μg/ml of lysozyme) for 30 min. Cells were disrupted via sonication and the lysate was clarified by centrifugation (20 min). Clarified lysate was incubated with equilibrated resin (batch binding) for 2 h while rotating. After supernatant decantation, batch-bound resin was loaded into a glass column and washed with 100 ml (1.5 ml/min) of wash buffer (20 mm NaPO4, pH 7.5, 300 mm NaCl, 10 mm imidazole). α-Synuclein was eluted using a linear gradient of 150 ml of 10–750 mm imidazole at a flow rate of 1.5 ml/min. SDS-PAGE (5% stacking, 12% resolving) was used to identify those fractions containing α-synuclein. Fractions were pooled and dialyzed overnight (2 × 2 liters) against the storage buffer containing 20 mm MOPS (pH 7.2), 150 mm NaCl, and 1 mm DTT. After dialysis, monomeric α-synuclein was separated from higher molecular weight α-synuclein aggregates using centrifugal spin concentrators (30 kDa MWCO). The resulting flow-through contained the purified protein that was further concentrated (1.5–3 mg/ml) with a 3-kDa MWCO membrane. Protein concentration was determined using a BCA assay (ThermoFisher Scientific). Purified protein was aliquoted, flash frozen using liquid N2, and stored at −80 °C.
Overexpression and purification of human mtCK
The human sarcomeric mtCK localized in skeletal and heart muscle was expressed in bacteria as a tag-free protein and was purified in a two-dimensional HPLC protocol as previously described (54). Purified protein was aliquoted and stored at −20 °C.
UV-visible spectroscopy and oxidation of cytochrome c
UV-visible spectra of cytochrome c were recorded with a Shimadzu 1800 spectrophotometer fitted with a thermostated cell holder. Stock solutions of proteins, lipids, and buffers/substrates were made fresh prior to use. The 3-ml reactions contained HEPES (30 mm, pH 7.4), catalase (10 μg/ml), glucose (50 μm), glucose oxidase (50 μg/ml), and cytochrome c (5 μm). Initial UV-visible spectra were recorded (200–800 nm) and ascorbate (final concentration 10 μm) was added to completely reduce the cytochrome c. Spectra were recorded every minute for 5 min after the addition of ascorbate to ensure all cytochrome c was reduced. 2–3 μl of 1 mg/ml of solubilized lipid (DOPC, (18:2)4 CL, and MLCL) was subsequently titrated into the reaction mixture, and after a 3-min equilibration period, spectra were recorded. After all titrations were complete, K3[FeCN6] (55 μm, final concentration) was added, and after a 3-min equilibration period, the final spectra of fully oxidized cytochrome c were recorded.
Statistics
Data were analyzed with GraphPad Prism version 7.0. Data were ensured to display normalized distributions. Statistical analyses were conducted using unpaired two-tailed t-tests or one-way analysis of variance followed by a post hoc Bonferroni test. p values less than 0.05 were considered significant. Results are plotted as mean ± S.D. from multiple independent experiments.
Author contributions
E. R. P., U. S., R. A. C., A. J. C., D. A. B., and S. R. S. conceptualization; E. R. P., E. M. S., A. F., S. D., T. N. Z., and A. D. data curation; E. R. P., T. N. Z., and S. R. S. formal analysis; E. R. P. validation; E. R. P., E. M. S., A. F., S. D., T. N. Z., A. D., and S. R. S. investigation; E. R. P., E. M. S., A. F., S. D., T. N. Z., and A. D. methodology; E. R. P. writing-original draft; E. R. P., T. N. Z., U. S., R. A. C., A. J. C., D. A. B., and S. R. S. writing-review and editing; T. N. Z., U. S., R. A. C., and D. A. B. resources; D. A. B. and S. R. S. funding acquisition; S. R. S. project administration.
Supplementary Material
This work was supported by National Institutes of Health Grant R01HL123647 (to D. B. and S. R. S.) and internal funds from the University of North Carolina, Chapel Hill (to S. R. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S3.
- CL
- cardiolipin
- GUV
- giant unilamellar vesicle
- GMV
- giant mitochondrial vesicle
- MLCL
- monolyso-cardiolipin
- NAO
- nonyl acridine orange
- cyt c
- cytochrome c
- α-syn
- α-synuclein
- mtCK
- mitochondrial creatine kinase
- (18:1)4CL
- 1,1′,2,2′-tetra-(9Z-octadecenoyl) cardiolipin
- DOPC
- 1,2-di-(9Z-octadecenoyl)-sn-glycero-3-phosphocholine
- 18:0-22:6 PC
- 1-stearoyl-2-docosahexaenoyl-sn-glycero-3-phosphocholine
- 16:0–20:4 PE
- 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphoethanolamine
- DOPI
- 1,2-dioleoyl-sn-glycero-3-phospho-(1′-myo-inositol)
- DOPS
- 1,2-dioleoyl-sn-glycero-3-phospho-l-serine
- (18:2)4CL
- bovine heart cardiolipin
- MIM
- mitochondrial isolation medium
- DRM
- detergent-resistant membrane
- DSM
- detergent-soluble membrane
- Chol
- cholesterol
- mN
- millinewton.
References
- 1. Ren M., Phoon C. K. L., and Schlame M. (2014) Metabolism and function of mitochondrial cardiolipin. Prog. Lipid Res. 55, 1–16 10.1194/jlr.E045732, 10.1016/j.plipres.2014.04.001 [DOI] [PubMed] [Google Scholar]
- 2. Paradies G., Paradies V., De Benedictis V., Ruggiero F. M., and Petrosillo G. (2014) Functional role of cardiolipin in mitochondrial bioenergetics. Biochim. Biophys. Acta 1837, 408–417 10.1016/j.bbabio.2013.10.006 [DOI] [PubMed] [Google Scholar]
- 3. Claypool S. M., and Koehler C. M. (2012) The complexity of cardiolipin in health and disease. Trends Biochem. Sci. 37, 32–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schlame M., and Ren M. (2009) The role of cardiolipin in the structural organization of mitochondrial membranes. Biochim. Biophys. Acta 1788, 2080–2083 10.1016/j.bbamem.2009.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mileykovskaya E., Zhang M., and Dowhan W. (2005) Cardiolipin in energy transducing membranes. Biochemistry 70, 154–158 [DOI] [PubMed] [Google Scholar]
- 6. Houtkooper R. H., and Vaz F. M. (2008) Cardiolipin, the heart of mitochondrial metabolism. Cell. Mol. Life Sci. 65, 2493–2506 10.1007/s00018-008-8030-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Osman C., Voelker D. R., and Langer T. (2011) Making heads or tails of phospholipids in mitochondria. J. Cell Biol. 192, 7 10.1083/jcb.201006159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Khalifat N., Puff N., Bonneau S., Fournier J., and Angelova M. I. (2008) Membrane deformation under local pH gradient: mimicking mitochondrial cristae dynamics. Biophys. J. 95, 4924–4933 10.1529/biophysj.108.136077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rampelt H., Zerbes R. M., van der Laan M., and Pfanner N. (2017) Role of the mitochondrial contact site and cristae organizing system in membrane architecture and dynamics. Biochim. Biophys. Acta 1864, 737–746 10.1016/j.bbamcr.2016.05.020 [DOI] [PubMed] [Google Scholar]
- 10. Schug Z. T., Frezza C., Galbraith L. C. A., and Gottlieb E. (2012) The music of lipids: how lipid composition orchestrates cellular behaviour. Acta Oncol. 51, 301–310 10.3109/0284186X.2011.643823 [DOI] [PubMed] [Google Scholar]
- 11. Mileykovskaya E., and Dowhan W. (2014) Cardiolipin-dependent formation of mitochondrial respiratory supercomplexes. Chem. Phys. Lipids. 179, 42–48 10.1016/j.chemphyslip.2013.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ikon N., and Ryan R. O. (2017) Cardiolipin and mitochondrial cristae organization. Biochim. Biophys. Acta 1859, 1156–1163 10.1016/j.bbamem.2017.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Klingenberg M. (2009) Cardiolipin and mitochondrial carriers. Biochim. Biophys. Acta 1788, 2048–2058 10.1016/j.bbamem.2009.06.007 [DOI] [PubMed] [Google Scholar]
- 14. Mileykovskaya E., and Dowhan W. (2009) Cardiolipin membrane domains in prokaryotes and eukaryotes. Biochim. Biophys. Acta 1788, 2084–2091 10.1016/j.bbamem.2009.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Brown D. A., Sabbah H. N., and Shaikh S. R. (2013) Mitochondrial inner membrane lipids and proteins as targets for decreasing cardiac ischemia/reperfusion injury. Pharmacol. Ther. 140, 258–266 10.1016/j.pharmthera.2013.07.005 [DOI] [PubMed] [Google Scholar]
- 16. Renner L. D., and Weibel D. B. (2011) Cardiolipin microdomains localize to negatively curved regions of Escherichia coli membranes. Proc. Natl. Acad. Sci. U.S.A. 108, 6264–6269 10.1073/pnas.1015757108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sorice M., Manganelli V., Matarrese P., Tinari A., Misasi R., Malorni W., and Garofalo T. (2009) Cardiolipin-enriched raft-like microdomains are essential activating platforms for apoptotic signals on mitochondria. FEBS Lett. 583, 2447–2450 10.1016/j.febslet.2009.07.018 [DOI] [PubMed] [Google Scholar]
- 18. Epand R. F., Tokarska-Schlattner M., Schlattner U., Wallimann T., and Epand R. M. (2007) Cardiolipin clusters and membrane domain formation induced by mitochondrial proteins. J. Mol. Biol. 365, 968–980 10.1016/j.jmb.2006.10.028 [DOI] [PubMed] [Google Scholar]
- 19. Schug Z. T., and Gottlieb E. (2009) Cardiolipin acts as a mitochondrial signalling platform to launch apoptosis. Biochim. Biophys. Acta 1788, 2022–2031 10.1016/j.bbamem.2009.05.004 [DOI] [PubMed] [Google Scholar]
- 20. Dudek J. (2017) Role of cardiolipin in mitochondrial signaling pathways. Front. Cell Dev. Biol. 5, 90 10.3389/fcell.2017.00090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schlattner U., Tokarska-Schlattner M., Rousseau D., Boissan M., Mannella C., Epand R., and Lacombe M. (2014) Mitochondrial cardiolipin/phospholipid trafficking: the role of membrane contact site complexes and lipid transfer proteins. Chem. Phys. Lipids 179, 32–41 10.1016/j.chemphyslip.2013.12.008 [DOI] [PubMed] [Google Scholar]
- 22. Stepanyants N., Macdonald P. J., Francy C. A., Mears J. A., Qi X., and Ramachandran R. (2015) Cardiolipin's propensity for phase transition and its reorganization by dynamin-related protein 1 form a basis for mitochondrial membrane fission. Mol. Biol. Cell 26, 3104–3116 10.1091/mbc.e15-06-0330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Maniti O., Lecompte M. F., Marcillat O., Desbat B., Buchet R., Vial C., and Granjon T. (2009) Mitochondrial creatine kinase binding to phospholipid monolayers induces cardiolipin segregation. Biophys. J. 96, 2428–2438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Beales P. A., Bergstrom C. L., Geerts N., Groves J. T., and Vanderlick T. K. (2011) Single vesicle observations of the cardiolipin-cytochrome c interaction: induction of membrane morphology changes. Langmuir 27, 6107–6115 10.1021/la104924c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bergstrom C. L., Beales P. A., Lv Y., Vanderlick T. K., and Groves J. T. (2013) Cytochrome c causes pore formation in cardiolipin-containing membranes. Proc. Natl. Acad. Sci. U.S.A 110, 6269–6274 10.1073/pnas.1303819110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pennington E. R., Fix A., Sullivan E. M., Brown D. A., Kennedy A., and Shaikh S. R. (2017) Distinct membrane properties are differentially influenced by cardiolipin content and acyl chain composition in biomimetic membranes. Biochim. Biophys. Acta 1859, 257–267 10.1016/j.bbamem.2016.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chicco A. J., and Sparagna G. C. (2007) Role of cardiolipin alterations in mitochondrial dysfunction and disease. Am. J. Physiol. Cell Physiol. 292, C33–C44 10.1152/ajpcell.00243.2006 [DOI] [PubMed] [Google Scholar]
- 28. Shen Z., Ye C., McCain K., and Greenberg M. L. (2015) The role of cardiolipin in cardiovascular health. BioMed Res. Int. 2015, 891707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Paradies G., Paradies V., Ruggiero F. M., and Petrosillo G. (2014) Cardiolipin and mitochondrial function in health and disease. Antioxid. Redox Signal. 20, 1925–1953 10.1089/ars.2013.5280 [DOI] [PubMed] [Google Scholar]
- 30. Ye C., Shen Z., and Greenberg M. L. (2016) Cardiolipin remodeling: a regulatory hub for modulating cardiolipin metabolism and function. J. Bioenerg. Biomembr. 48, 113–123 10.1007/s10863-014-9591-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sullivan E. M., Fix A., Crouch M. J., Sparagna G. C., Zeczycki T. N., Brown D. A., and Shaikh S. R. (2017) Murine diet-induced obesity remodels cardiac and liver mitochondrial phospholipid acyl chains with differential effects on respiratory enzyme activity. J. Nutr. Biochem. 45, 94–103 10.1016/j.jnutbio.2017.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sparagna G. C., Chicco A. J., Murphy R. C., Bristow M. R., Johnson C. A., Rees M. L., Maxey M. L., McCune S. A., and Moore R. L. (2007) Loss of cardiac tetralinoleoyl cardiolipin in human and experimental heart failure. J. Lipid Res. 48, 1559–1570 10.1194/jlr.M600551-JLR200 [DOI] [PubMed] [Google Scholar]
- 33. Sparagna G. C., and Lesnefsky E. J. (2009) Cardiolipin remodeling in the heart. J. Cardiovasc. Pharmacol. 53, 290–301 10.1097/FJC.0b013e31819b5461 [DOI] [PubMed] [Google Scholar]
- 34. Shi Y. (2010) Emerging roles of cardiolipin remodeling in mitochondrial dysfunction associated with diabetes, obesity, and cardiovascular diseases. J. Biomed. Res. 24, 6–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. He Q., and Han X. (2014) Cardiolipin remodeling in diabetic heart. Chem. Phys. Lipids 179, 75–81 10.1016/j.chemphyslip.2013.10.007 [DOI] [PubMed] [Google Scholar]
- 36. Han X., Yang J., Yang K., Zhao Z., Abendschein D. R., and Gross R. W. (2007) Alterations in myocardial cardiolipin content and composition occur at the very earliest stages of diabetes: a shotgun lipidomics study. Biochemistry 46, 6417–6428 10.1021/bi7004015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Han X., Yang J., Cheng H., Yang K., Abendschein D. R., and Gross R. W. (2005) Shotgun lipidomics identifies cardiolipin depletion in diabetic myocardium linking altered substrate utilization with mitochondrial dysfunction. Biochemistry 44, 16684–16694 10.1021/bi051908a [DOI] [PubMed] [Google Scholar]
- 38. Schlame M., and Ren M. (2006) Barth syndrome, a human disorder of cardiolipin metabolism. FEBS Lett. 580, 5450–5455 10.1016/j.febslet.2006.07.022 [DOI] [PubMed] [Google Scholar]
- 39. Xu Y., Phoon C. K., Berno B., D'Souza K., Hoedt E., Zhang G., Neubert T. A., Epand R. M., Ren M., and Schlame M. (2016) Loss of protein association causes cardiolipin degradation in Barth syndrome. Nat. Chem. Biol. 12, 641–647 10.1038/nchembio.2113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xu Y., Sutachan J. J., Plesken H., Kelley R. I., and Schlame M. (2005) Characterization of lymphoblast mitochondria from patients with Barth syndrome. Lab. Invest. 85, 823–830 10.1038/labinvest.3700274 [DOI] [PubMed] [Google Scholar]
- 41. Schlame M., Towbin J. A., Heerdt P. M., Jehle R., DiMauro S., and Blanck T. J. (2002) Deficiency of tetralinoleoyl-cardiolipin in Barth syndrome. Ann. Neurol. 51, 634–637 10.1002/ana.10176 [DOI] [PubMed] [Google Scholar]
- 42. Schlame M., Kelley R. I., Feigenbaum A., Towbin J. A., Heerdt P. M., Schieble T., Wanders R. J., DiMauro S., and Blanck T. J. (2003) Phospholipid abnormalities in children with Barth syndrome. J. Am. Coll. Cardiol. 42, 1994–1999 10.1016/j.jacc.2003.06.015 [DOI] [PubMed] [Google Scholar]
- 43. Juhasz J., Davis J. H., and Sharom F. J. (2010) Fluorescent probe partitioning in giant unilamellar vesicles of “lipid raft” mixtures. Biochem. J. 430, 415 10.1042/BJ20100516 [DOI] [PubMed] [Google Scholar]
- 44. Klymchenko A. S., and Kreder R. (2014) Fluorescent probes for lipid rafts: from model membranes to living cells. Chem. Biol. 21, 97–113 10.1016/j.chembiol.2013.11.009 [DOI] [PubMed] [Google Scholar]
- 45. Garcia Fernandez M. I., Ceccarelli D., and Muscatello U. (2004) Use of the fluorescent dye 10-N-nonyl acridine orange in quantitative and location assays of cardiolipin: a study on different experimental models. Anal. Biochem. 328, 174–180 10.1016/j.ab.2004.01.020 [DOI] [PubMed] [Google Scholar]
- 46. Mileykovskaya E., Dowhan W., Birke R. L., Zheng D., Lutterodt L., and Haines T. H. (2001) Cardiolipin binds nonyl acridine orange by aggregating the dye at exposed hydrophobic domains on bilayer surfaces. FEBS Lett. 507, 187–190 10.1016/S0014-5793(01)02948-9 [DOI] [PubMed] [Google Scholar]
- 47. Schlame M. (2013) Cardiolipin remodeling and the function of tafazzin. Biochim. Biophys. Acta 1831, 582–588 10.1016/j.bbalip.2012.11.007 [DOI] [PubMed] [Google Scholar]
- 48. Paradies G., Paradies V., Ruggiero F. M., and Petrosillo G. (2015) Cardiolipin alterations and mitochondrial dysfunction in heart ischemia/reperfusion injury. Clin. Lipidol. 10, 415–429 10.2217/clp.15.31 [DOI] [Google Scholar]
- 49. Baile M. G., Sathappa M., Lu Y. W., Pryce E., Whited K., McCaffery J. M., Han X., Alder N. N., and Claypool S. M. (2014) Unremodeled and remodeled cardiolipin are functionally indistinguishable in yeast. J. Biol. Chem. 289, 1768–1778 10.1074/jbc.M113.525733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Grevengoed T. J., Martin S. A., Katunga L., Cooper D. E., Anderson E. J., Murphy R. C., and Coleman R. A. (2015) Acyl-CoA synthetase 1 deficiency alters cardiolipin species and impairs mitochondrial function. J. Lipid Res. 56, 1572–1582 10.1194/jlr.M059717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schlattner U., and Wallimann T. (2000) Octamers of mitochondrial creatine kinase isoenzymes differ in stability and membrane binding. J. Biol. Chem. 275, 17314–17320 10.1074/jbc.M001919200 [DOI] [PubMed] [Google Scholar]
- 52. Cheniour M., Brewer J., Bagatolli L., Marcillat O., and Granjon T. (2017) Evidence of proteolipid domain formation in an inner mitochondrial membrane mimicking model. Biochim. Biophys. Acta 1861, 969–976 10.1016/j.bbagen.2017.02.001 [DOI] [PubMed] [Google Scholar]
- 53. Shaikh S. R., Sullivan E. M., Alleman R. J., Brown D. A., and Zeczycki T. N. (2014) Increasing mitochondrial membrane phospholipid content lowers the enzymatic activity of electron transport complexes. Biochemistry 53, 5589–5591 10.1021/bi500868g [DOI] [PubMed] [Google Scholar]
- 54. Schlattner U., Eder M., Dolder M., Khuchua Z. A., Strauss A. W., and Wallimann T. (2000) Divergent enzyme kinetics and structural properties of the two human mitochondrial creatine kinase isoenzymes. Biol. Chem. 381, 1063–1070 [DOI] [PubMed] [Google Scholar]
- 55. Schuck S., Honsho M., Ekroos K., Shevchenko A., and Simons K. (2003) Resistance of cell membranes to different detergents. Proc. Natl. Acad. Sci. U.S.A. 100, 5795–5800 10.1073/pnas.0631579100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. London E., and Brown D. A. (2000) Insolubility of lipids in Triton X-100: physical origin and relationship to sphingolipid/cholesterol membrane domains (rafts). Biochim. Biophys. Acta 1508, 182–195 10.1016/S0304-4157(00)00007-1 [DOI] [PubMed] [Google Scholar]
- 57. Nijtmans L. G., Henderson N. S., and Holt I. J. (2002) Blue native electrophoresis to study mitochondrial and other protein complexes. Methods 26, 327–334 10.1016/S1046-2023(02)00038-5 [DOI] [PubMed] [Google Scholar]
- 58. Heerklotz H. (2002) Triton promotes domain formation in lipid raft mixtures. Biophys. J. 83, 2693–2701 10.1016/S0006-3495(02)75278-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. El Khoury M., Swain J., Sautrey G., Zimmermann L., Van Der Smissen P., Décout J., and Mingeot-Leclercq M. (2017) Targeting bacterial cardiolipin enriched microdomains: an antimicrobial strategy used by amphiphilic aminoglycoside antibiotics. Sci. Rep. 7, 10697 10.1038/s41598-017-10543-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nakamura K. (2013) α-Synuclein and mitochondria: partners in crime? Neurotherapeutics 10, 391–399 10.1007/s13311-013-0182-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jo E., McLaurin J., Yip C. M., St. George-Hyslop P., and Fraser P. E. (2000) α-Synuclein membrane interactions and lipid specificity. J. Biol. Chem. 275, 34328–34334 10.1074/jbc.M004345200 [DOI] [PubMed] [Google Scholar]
- 62. Schlattner U., Tokarska-Schlattner M., and Wallimann T. (2006) Mitochondrial creatine kinase in human health and disease. Biochim. Biophys. Acta 1762, 164–180 10.1016/j.bbadis.2005.09.004 [DOI] [PubMed] [Google Scholar]
- 63. Speer O., Bäck N., Buerklen T., Brdiczka D., Koretsky A., Wallimann T., and Eriksson O. (2005) Octameric mitochondrial creatine kinase induces and stabilizes contact sites between the inner and outer membrane. Biochem. J. 385, 445–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Petrosillo G., Di Venosa N., Ruggiero F. M., Pistolese M., D'Agostino D., Tiravanti E., Fiore T., and Paradies G. (2005) Mitochondrial dysfunction associated with cardiac ischemia/reperfusion can be attenuated by oxygen tension control: role of oxygen-free radicals and cardiolipin. Biochim. Biophys. Acta 1710, 78–86 10.1016/j.bbabio.2005.10.003 [DOI] [PubMed] [Google Scholar]
- 65. Chen Q., and Lesnefsky E. J. (2006) Depletion of cardiolipin and cytochrome c during ischemia increases hydrogen peroxide production from the electron transport chain. Free Radic. Biol. Med. 40, 976–982 10.1016/j.freeradbiomed.2005.10.043 [DOI] [PubMed] [Google Scholar]
- 66. Lesnefsky E. J., Chen Q., Slabe T. J., Stoll M. S., Minkler P. E., Hassan M. O., Tandler B., and Hoppel C. L. (2004) Ischemia, rather than reperfusion, inhibits respiration through cytochrome oxidase in the isolated, perfused rabbit heart: role of cardiolipin. Am. J. Physiol. Heart Circ. Physiol. 287, H258–H267 10.1152/ajpheart.00348.2003 [DOI] [PubMed] [Google Scholar]
- 67. Mulligan C. M., Le C. H., deMooy A. B., Nelson C. B., and Chicco A. J. (2013) Inhibition of delta-6 desaturase reverses cardiolipin remodeling and prevents contractile dysfunction in the aged mouse heart without altering mitochondrial respiratory function. J. Gerantol. A Biol. Sci. Med. Sci. 69, 799–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sullivan E. M., Pennington E. R., Sparagna G. C., Torres M. J., Neufer P. D., Harris M., Washington J., Anderson E. J., Zeczycki T. N., Brown D. A., and Shaikh S. R. (2018) Docosahexaenoic acid lowers cardiac mitochondrial enzyme activity by replacing linoleic acid in the phospholipidome. J. Biol. Chem. 293, 466–483 10.1074/jbc.M117.812834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Liu G.-Y., Moon S. H., Jenkins C. M., Li M., Sims H. F., Guan S., and Gross R. W. (2017) The phospholipase iPLA2γ is a major mediator releasing oxidized aliphatic chains from cardiolipin, integrating mitochondrial bioenergetics and signaling. J. Biol. Chem. 292, 10672–10684 10.1074/jbc.M117.783068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Mitsopoulos P., Chang Y. H., Wai T., König T., Dunn S. D., Langer T., and Madrenas J. (2015) Stomatin-like protein 2 is required for in vivo mitochondrial respiratory chain supercomplex formation and optimal cell function. Mol. Cell. Biol. 35, 1838–1847 10.1128/MCB.00047-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bligh E. G., and Dyer W. J. (1959) A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 10.1139/y59-099 [DOI] [PubMed] [Google Scholar]
- 72. Schmid E. M., Richmond D. L., and Fletcher D. A. (2015) Reconstitution of proteins on electroformed giant unilamellar vesicles. Methods Cell Biol. 128, 319–338 10.1016/bs.mcb.2015.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Angelova M. I., Soleau S., Meleard P., Faucon F., and Bothorel P. (1992) Preparation of giant vesicles by external AC electric fields. Prog. Colloid. Polym. Sci. 89, 127–131 10.1007/BFb0116295 [DOI] [Google Scholar]
- 74. Shaikh S. R., Rockett B. D., Salameh M., and Carraway K. (2009) Docosahexaenoic acid modifies the clustering and size of lipid rafts and the lateral organization and surface expression of MHC class I of EL4 cells. J. Nutr. 139, 1632–1639 10.3945/jn.109.108720 [DOI] [PubMed] [Google Scholar]
- 75. Shaikh S. R., Dumaual A. C., Jenski L. J., and Stillwell W. (2001) Lipid phase separation in phospholipid bilayers and monolayers modeling the plasma membrane. Biochim. Biophys. Acta 1512, 317–328 10.1016/S0005-2736(01)00335-2 [DOI] [PubMed] [Google Scholar]
- 76. Nichols-Smith S., Teh S. Y., and Kuhl T. L. (2004) Thermodynamic and mechanical properties of model mitochondrial membranes. Biochim. Biophys. Acta 1663, 82–88 10.1016/j.bbamem.2004.02.002 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.