

Abstract
IFNϵ and IFNκ are interferons that induce microbial immunity at mucosal surfaces and in the skin. They are members of the type-I interferon (IFN) family, which consists of 16 different IFNs, that all signal through the common IFNAR1/IFNAR2 receptor complex. Although IFNϵ and IFNκ have unique expression and functional properties, their biophysical properties have not been extensively studied. In this report, we describe the expression, purification, and characterization of recombinant human IFNϵ and IFNκ. In cellular assays, IFNϵ and IFNκ exhibit ∼1000-fold lower potency than IFNα2 and IFNω. The reduced potency of IFNϵ and IFNκ are consistent with their weak affinity for the IFNAR2 receptor chain. Despite reduced IFNAR2-binding affinities, IFNϵ and IFNκ exhibit affinities for the IFNAR1 chain that are similar to other IFN subtypes. As observed for cellular IFNAR2 receptor, the poxvirus antagonist, B18R, also exhibits reduced affinity for IFNϵ and IFNκ, relative to the other IFNs. Taken together, our data suggest IFNϵ and IFNκ are specialized IFNs that have evolved to weakly bind to the IFNAR2 chain, which allows innate protection of the mucosa and skin and limits neutralization of IFNϵ and IFNκ biological activities by viral IFN antagonists.
Keywords: interferon, receptor structure-function, recombinant protein expression, viral protein, protein complex, protein-protein interaction, structure-function, B18R, IFNAR, IFNϵ, IFNκ
Introduction
IFNϵ and IFNκ are part of the human type-I interferon (IFN) 2 family that consists of 16 different cytokines whose signaling properties are critical for the control and elimination of microbial pathogens (1–5). IFNs activate innate immunity through the induction of IFN-stimulated genes (ISGs) that exhibit antiviral activity (2, 6, 7). IFNs also promote adaptive immunity through up-regulation of major histocompatibility complex and the induction of chemokines (8–11). They also control cell growth and apoptosis (12, 13), B-cell lineage commitment (14), and induction of T regulatory cells (15, 16). Due to the importance of IFNs, medical applications of IFNs have been developed including the treatment of viral infections (17), cancer (18), and multiple sclerosis (19).
The biological activities of all 16 IFNs are initiated upon binding to the cell-surface receptors IFNAR1 and IFNAR2. IFN-IFNAR interactions activate JAK1 and TYK2 kinases and the transcription factors STAT1 and STAT2 (20–23). JAK/STAT signaling, and additional kinases and transcription factors (24), ultimately induce IFN gene expression programs that protect the host from virus, bacteria, and even fungi (5, 7, 25, 26). Due to the critical role IFNs play in protecting the host from infection, many pathogens produce proteins that block IFN activity at multiple steps in the IFN signaling pathway. For example, Dengue, West Nile, and Zika viruses disrupt IFN-mediated STAT2 signaling (27, 28). In contrast, poxviruses encode IFN-binding proteins that neutralize IFN activity by binding to secreted IFNs, which prevents them from engaging cell-surface IFNAR1 and IFNAR2 (29). The IFN-binding proteins, B18R and B19R, were identified in vaccinia virus strains Western Reserve and Copenhagen, respectively (30, 31). B18R/B19R encode the same secreted ∼65-kDa glycoprotein that promiscuously binds to all of the type-I IFNs (32). Deletion of B18R from vaccinia virus resulted in an attenuated virus, emphasizing the importance of the type-I IFNs in controlling vaccinia virus infection (30, 31).
IFNϵ and IFNκ are unique from the other IFNs based on their amino acid sequences and limited expression in mucosa and skin. IFNϵ and IFNκ share 35% sequence identity with one another and the 14 other IFNs. The additional 14 IFNs consist of 12 IFNα subtypes, IFNω, and IFNβ. The IFNαs share 77–95% sequence identity with one another. IFNβ exhibits 35% sequence identify with all other IFNs, whereas IFNω shares the highest sequence identity (60%) with both IFNϵ/κ/β and the IFNαs. Based on sequence and structural comparisons, all IFNs exhibit an α-helical fold consisting of five helices, which are labeled from the N terminus as helix A, B, C, D, E, and F (33–35). A unique feature of IFNϵ is that it encodes an 18-amino acid C-terminal tail following helix F, whereas IFNκ has a 13-amino acid peptide insertion between helices D and E.
In addition to these novel sequence features, IFNϵ and IFNκ are expressed predominantly in the female reproductive track (FRT) and keratinocytes, respectively (26, 36). In fact, analysis of the IFNϵ gene identified putative progesterone-binding sites in the promoter, suggesting hormones regulate IFNϵ expression (37). Subsequent studies confirmed IFNϵ expression is induced by hormones but not by viral infection (e.g. TLRs) like other IFNs (26). Consistent with IFNϵ's expression in the reproductive tract, IFNϵ is able to induce important restriction factors that prevent HIV-1 infection (38, 39). In fact, HIV-1 negative female sex workers express high levels of IFNϵ in their cervical tissues (40). IFNκ is constitutively expressed in keratinocytes, which are found in skin and in the mucosa of the FRT. Thus, IFNκ and IFNϵ are both found in the FRT, but IFNκ is inducible by virus and dsRNA, whereas IFNϵ is not (36). Although it appears that IFNϵ appears to play an important role in immunity against HIV-1, IFNκ expression is rapidly reduced in keratinocytes that are infected with human papilloma virus strains that induce cervical cancer (41). These data strongly argue that IFNϵ and IFNκ are essential components to the host response against pathogens in the FRT, whereas the specific role of IFNκ, produced by keratinocytes in the skin, remains to be determined.
Despite a critical role of IFNϵ and IFNκ signaling in mucosa and skin, their interactions with the IFNARs and their functional activities have not been extensively characterized. To address this issue, we have expressed human IFNϵ and IFNκ for comparative biophysical and functional studies with other IFN family members (often called IFN subtypes). Studies with purified IFNϵ and IFNκ reveal they induce ISFG3-mediated gene expression that is ∼1000-fold weaker than IFNα2 or IFNω. The weaker potency of IFNϵ and IFNκ is consistent with their reduced affinities for the IFNAR2 receptor chain. However, IFNϵ and IFNκ exhibit IFNAR1 binding affinity that is similar to IFNα2 and IFNω.
Because poxviruses cause disseminated infections of the skin and mucosa and can block type-I IFN signaling, we evaluated the ability of B18R to neutralize IFNϵ and IFNκ cellular activity. Subsequent kinetic binding studies determined IFNϵ, IFNκ, as well as IFNα1, exhibit reduced binding to B18R, relative to the other IFNs. Sequence and structural models of IFNϵ/κ-IFNAR2 and IFNϵ/κ-B18R complexes identified residues responsible for the disrupted IFNAR2 and B18R binding phenotypes. Our data suggests IFNϵ and IFNκ have evolved to exhibit reduced IFNAR2 binding affinity and biological potency optimized for tissue-specific expression and escape from viral type-I IFN antagonists.
Results
Expression and characterization of IFNϵ and IFNκ proteins
Expression plasmids encoding human IFNϵ and IFNκ protein sequences, which also encode C-terminal histidine tags, were synthesized using optimized codons. Expression studies were performed in Escherichia coli where IFNϵ and IFNκ formed insoluble inclusion bodies. The guanidine-solubilized IFNs were refolded by rapid dilution into a refolding buffer. The refolded IFNs were purified using a 2-step strategy consisting of nickel affinity and cation exchange chromatography. Endotoxin levels for IFNϵ and IFNκ were less than 1 EU/μg, which is within the range of values observed in IFNα preparations obtained from commercial sources (42). SDS-PAGE gel analysis of the final purified IFNϵ and IFNκ protein preparations is shown in Fig. 1. The molecular weight of IFNϵ on SDS-PAGE gels matched its theoretical molecular weight of 23,249. However, IFNκ ran larger than expected (∼25,000 kDa) in SDS-PAGE gels, suggesting the peptide could be a frameshift product (43) or exhibit aberrant gel migration. To resolve these possibilities, MS was performed on the samples, which demonstrated the molecular masses were consistent with full-length IFNϵ and IFNκ proteins without the N-terminal initiating methionine residues (Table 1).
Figure 1.
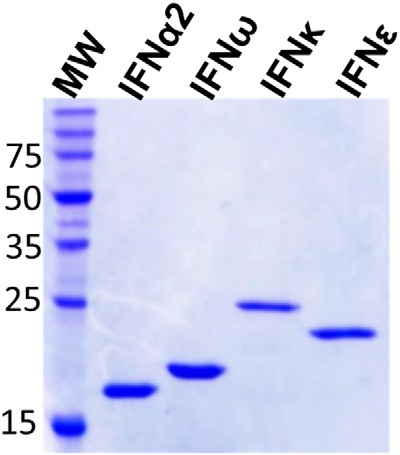
Final SDS-PAGE gel of purified IFNϵ, IFNκ, and two control IFNs, IFNα2a and IFNω.
Table 1.
Mass spectrometry of IFNϵ and IFNκ
| Treatment | Observed mass | Expected massa | Deltab | Interpretation | |
|---|---|---|---|---|---|
| IFNϵ | None | 23,082 | 23,118 | 36 | |
| IA | 23,145 | 23,139 | 6 | + 1 IA | |
| IA + DTT | 23,256 | 23,253 | 3 | + 3 IA | |
| IFNκ | None | 23,201 | 23,220 | 19 | |
| IA | 23,259 | 23,258 | 1 | + 1 IA | |
| IA + DTT | 23,496 | 23,486 | 10 | + 5 IA |
a Calculated mass without N-terminal methionine.
b Mass difference between expected and observed masses.
The IFNϵ and IFNκ amino acid sequences include three and five cysteine residues, respectively. Based on prior structural and biophysical analysis of other IFNs (42, 44), IFNϵ and IFNκ are predicted to contain one free cysteine and form one and two disulfide bonds, respectively, in their folded forms. To confirm disulfide bond formation had occurred during the refolding process, IFNϵ and IFNκ were treated with iodoacetamide (IA) in the presence, or absence, of the reducing agent dithiothreitol (DTT, Table 1). IA selectively binds to free cysteines, resulting in an increase in mass of 57 Da. Upon IA treatment in the absence DTT, the mass of IFNϵ and IFNκ increased by 57 Da, which is consistent with one free cysteine in the folded proteins (Table 1). However, in the presence of DTT, the mass of IFNϵ and IFNκ increased by three IA and five IA mass units, respectively. This is consistent with the protection of two (IFNϵ) and four (IFNκ) cysteines, due to disulfide bond formation, in the folded proteins (Table 1). Thus, the MS data are consistent with the predicted IFNϵ and IFNκ disulfide bonding patterns.
IFNϵ and IFNκ have disrupted IFNAR2-binding properties
Three preparations of IFNϵ and IFNκ were characterized for their ability to bind to soluble IFNARs (Fig. 2). The IFNs were injected over Biacore chips coupled with IFNAR1-FC, IFNAR2-FC, or an IFNAR1/IFNAR2-FC heterodimer, as previously described (45). Binding is reported as receptor occupancy (RO) for each IFN. For comparison with other IFNs, binding studies were also performed with IFN subtypes, IFNα2 and IFNω.
Figure 2.

Receptor binding and biological activity of IFNϵ and IFNκ preparations. A, receptor binding of IFNϵ and IFNκ, as well as IFNα2 and IFNω, measured as percent RO. RO corresponds to the SPR RU observed upon IFN injection over an IFNAR Biacore chip surface, divided by the theoretical RUmax of the surface, multiplied by 100. B, dose-response curves for IFNϵ, IFNκ, IFNα2, and IFNω-mediated activation of the IFI6 gene reporter in HL116 cells. The color code for the dose-response curves is the same as the receptor binding analysis in A.
IFNα2 and IFNω bound to IFNAR2 with RO values of 53 and 69%, respectively (Fig. 2). In contrast, IFNϵ and IFNκ bound very poorly to IFNAR2, exhibiting IFNAR2 occupancies of 3 and 5%, respectively. Despite poor IFNAR2-binding properties, IFNϵ bound to IFNAR1 (RO = 26%) better than IFNα2 (RO = 19%), whereas IFNκ-IFNAR1 RO values were lower than IFNα2 (RO = 11%). In addition to binding to the single IFNARs, we evaluated IFN binding to an IFNAR1/IFNAR2-FC heterodimer, which positions IFNAR1 and IFNAR2 close to one another in space through the FC heterodimer (45). IFNϵ and IFNκ binding to IFNAR1/IFNAR2-FC remained low, relative to IFNα2 and IFNω. However, IFNκ binding, in particular, was significantly enhanced (RO = 36%) relative to the IFNAR2 and IFNAR1 single receptor experiments.
IFNϵ and IFNκ weakly induce ISGF3-mediated gene expression in reporter cells
The biological activity of IFNϵ and IFNκ were compared against IFNω and IFNα2 using a reporter cell line (HL116), which contains a firefly luciferase gene downstream of the IFI6 promoter. Dose-response curves were generated from at least six independent measurements, to derive half-maximal effective concentrations (EC50, Fig. 2B, Table 2). Consistent with the receptor binding data, IFNϵ (60 nm ± 26 nm) and IFNκ (22 ± 16 nm) exhibited ∼1000-fold lower EC50 values, compared with IFNα2 (11 ± 3 pm) or IFNω (6 ± 1 pm). The dose-response curves were repeated using a different reporter cell line that contains the ISG54 promoter, with similar results.
Table 2.
Surface plasmon resonance-derived binding constants and ISFG3 assay EC50 values
—, not determined.
| ka | kd | KD | ||
|---|---|---|---|---|
| m−1 s−1 | s−1 | m | ||
| IFNα2 | IFNAR1 | — | — | 2.3 (± 0.5) × 10−6 |
| EC50 = 11 pm (±3 pm) | IFNAR2 | 5.9 (± 0.4) × 106 | 0.031 (± 0.002) | 5.3 (± 0.2) × 10−9 |
| IFNAR1/2 | 8 (± 1) × 106 | 4.2 (± 0.4) × 10−4 | 54 (± 3) × 10−12 | |
| IFNϵ | IFNAR1 | — | — | 2.2 (± 0.3) × 10−6 |
| EC50 = 60 nm (±26 nm) | IFNAR2 | 4 (± 3) × 105 | 0.01 (± 0.01) | 70 (± 16) × 10−9 |
| IFNAR1/2 | 3.3 (± 0.8) × 105 | 9 (± 1) × 10−4 | 3.5 (± 0.8) × 10−9 | |
| IFNω | IFNAR1 | — | — | 0.5 (± 0.1) × 10−6 |
| EC50 = 6 pm (±1 pm) | IFNAR2 | 7 (± 2) × 107 | 0.01 (± 0.07) | 237 (± 37) × 10−12 |
| IFNAR1/2 | 3 (± 2) × 108 | 4 (± 2) × 10−3 | 25 (± 8) × 10−12 | |
| IFNκ | IFNAR1 | — | — | 0.34 (± 0.13) × 10−6 |
| EC50 = 22 nm (±16 nm) | IFNAR2 | 1.2 (± 0.2) × 105 | 2.1 (± 0.3) × 10−3 | 21 (± 4) × 10−9 |
| IFNAR1/2 | 4 (± 2) × 105 | 8 (± 1) × 10−4 | 2 (± 1) × 10−9 | |
| IFNκ-M2 | IFNAR1/2 | 7 (± 2) × 105 | 3 (± 1) × 10−4 | 0.4 (± 0.3) × 10−9 |
| EC50 = 4 nm (±2 nm) | ||||
| IFNα1 | IFNAR1 | — | — | 0.4 × 10−6 |
| EC50 = 258 pm (±73 pm) | IFNAR2 | 5 (± 0.1) × 105 | 0.18 (± 0.02) | 353 (± 83) × 10−9 |
| IFNAR1/2 | 1.8 (± 0.2) × 106 | 4 (± 2) × 10−4 | 222 (± 92) × 10−12 | |
| IFNα1-M1 | IFNAR1 | — | — | 0.2 × 10−6 |
| EC50 = 26 pm (±10 pm) | IFNAR2 | 2.4 (± 0.1) × 106 | 0.06 (± 0.01) | 26.0 (± 0.4) × 10−9 |
| IFNAR1/2 | 5.0 (± 0.1) × 106 | 3.5 (± 0.5) × 10−4 | 70 (± 9) × 10−12 | |
| IFNϵ | B18R | 1.5 × 105 | 3.1 × 10−5 | 2.10 × 10−10 |
| IFNα1 | B18R | 5.5 × 105 | 2.3 × 10−5 | 4.2 × 10−11 |
| IFNα1-M1 | B18R | 2.2 × 106 | 2.3 × 10−5 | 1.1 × 10−11 |
| IFNα4 | B18R | 5.8 × 106 | 1.5 × 10−5 | 2.6 × 10−12 |
| IFNα14 | B18R | 1.1 × 107 | 2.1 × 10−5 | 1.9 × 10−12 |
Distinct neutralization of IFNϵ and IFNκ biological activity
To further understand how IFNϵ and IFNκ engage cell-surface IFNARs, IFNϵ- and IFNκ-induced gene expression was monitored in reporter cells treated with a series of reagents that block IFN biological activity (Fig. 3). For comparative purposes, IFNα2a and IFNω were also included in the analysis. To validate that reporter activity was due to IFNϵ and IFNκ receptor binding, the pan-anti-IFNα neutralizing antibody (IFNα NAb) was added to the assay. Consistent with its specificity profile, the IFNα NAb efficiently blocked IFNα2 reporter activity, but not the activity of IFNω, IFNϵ, or IFNκ. To neutralize the activity of all four IFNs, the IFNs were incubated with the poxvirus antagonist B18R, which has been reported to block the activity of all IFN subtypes (31, 32). As expected, B18R efficiently neutralized IFNα2 and IFNω reporter activity. However, IFNϵ and IFNκ activity was only blocked at the higher concentration of B18R tested. Thus, IFNϵ and IFNκ are less sensitive to neutralization by B18R than IFNα2 and IFNω.
Figure 3.

Neutralization of IFN biological activity, by antibodies, soluble IFNARs, and the viral antagonist B18R. IFNs and antagonists were incubated with reporter cells for 18 h, followed by measurement of ISG54 reporter activity. All measurements are normalized relative to the IFN control, which is the average of 4 measurements corresponding to IFN alone and IFN + three isotype antibody controls.
IFNϵ- and IFNκ-mediated bioactivity was also studied in the presence of NAbs against cell-surface IFNAR1 and IFNAR2. The IFNAR1 NAb was equally effective in blocking IFNα2, IFNϵ, and IFNκ activity, but was much less effective in blocking IFNω activity. The anti-IFNAR2 NAb exhibited a different blocking profile than the IFNAR1 NAb. The IFNAR2 NAb most efficiently blocked IFNα2 activity at both high and low concentrations tested. Neutralization of IFNϵ and IFNω activity was intermediate, relative to IFNα2 neutralization, whereas IFNκ activity was largely insensitive to IFNAR2 NAb inhibition. These data suggest neutralization by the IFNAR2 NAb is sensitive to differences between the IFN subtypes that go beyond receptor affinity. Soluble IFNAR1-FC and IFNAR2-FC proteins were also used to block IFNϵ and IFNκ activity. As expected based on its low affinity, IFNAR1-FC was unable to block the activity of any of the IFNs. In contrast, soluble IFNAR2-FC blocked IFNα2 and IFNω activity, but was unable to block IFNϵ or IFNκ activity. Thus, IFNAR1-FC and IFNAR2-FC-mediated neutralization was consistent with IFN-IFNAR receptor affinities.
Surface plasmon resonance of IFNϵ and IFNκ receptor binding
IFNϵ/κ-IFNAR interactions were studied using surface plasmon resonance (SPR) (Fig. 4, Table 2). SPR analysis demonstrated IFNϵ (KD = 2.2 μm) and IFNα2a (KD = 2.3 μm), as well as IFNκ (KD = 0.3 μm) and IFNω (KD = 0.5 μm), share similar IFNAR1 affinities. Despite similar IFNAR1 affinities, IFNϵ's affinity for IFNAR2 (KD = 70 nm) is 14-fold lower than IFNα2. Analysis of the rate constants for the interactions reveals the IFNϵ/IFNAR2 association rate constant (ka) is 10-fold lower than for IFNα2/IFNAR2. Despite a slightly higher affinity (IFNκ/IFNAR2 KD = 21 nm) IFNκ also exhibits a very slow ka value (1 × 105 m−1 s−1) relative to IFNα2a (5.9 × 106 m−1 s−1) or IFNω (7 × 107 m−1 s−1). Thus, IFNϵ and IFNκ both exhibit poor IFNAR2 affinities due to reduced association rate constants.
Figure 4.

SPR analysis of IFNϵ, IFNκ, IFNα2, and IFNω binding to IFNAR1-FC, IFNAR2-FC, and the IFNAR1/IFNAR2-FC heterodimer. SPR sensorgrams for each IFN-IFNAR interaction are shown in black. The calculated sensorgrams, derived from fitting the data to a 1:1 binding model, are shown in black (IFNα2), red (IFNκ), blue (IFNϵ), and green (IFNω). Kinetic and equilibrium constants derived from the data are shown in Table 2.
IFNϵ and IFNκ binding to the IFNAR1/IFNAR2-FC was also determined (Table 2). The IFNAR1/IFNAR2-FC provides a biochemical mimic of the cell-surface IFNAR1/IFNAR2 heterodimer and the resulting affinities are due to IFN binding to both IFNAR1 and IFNAR2 (45). As a control, IFNα2 bound to the IFNAR1/IFNAR2-FC with a KD of 54 pm. Notably, this affinity constant is on the same order of magnitude of IFNα2's EC50 (11 pm) in the ISFG3 reporter assays (Fig. 2B, Table 2). Similarly, IFNω exhibits a KD of 25 pm for IFNAR1/IFNAR2-FC and an EC50 value of 6 pm in the reporter assay. In contrast to the picomolar affinities observed for IFNα2 and IFNω, IFNϵ and IFNκ exhibit nanomolar KD values for the IFNAR1/IFNAR2-FC (IFNϵ KD = 3.5 × 10−9 and IFNκ KD = 2 × 10−9). These affinity values are consistent with the reduced activity of IFNϵ and IFNκ in the reporter assays. The results obtained with the IFNAR1/IFNAR2-FC also show a 24–66-fold reduction in the ka values of the interactions. Thus, IFNϵ and IFNκ bind with reduced affinity to IFNAR2, relative to other IFN subtypes, such as IFNα2 and IFNω.
Distinct binding of IFNϵ and IFNκ to the poxvirus antagonist B18R
IFNϵ and IFNκ exhibit unique B18R neutralization profiles, relative to IFNα2 and IFNω (Fig. 3). To study this further, IFNϵ/B18R and IFNκ/B18R interactions were compared with the other 14 IFNs using SPR (Fig. 5). The 16 IFNs were injected at two concentrations (high, 50 nm; low, 5 nm), over a Biacore chip coupled with B18R from poxvirus strain Copenhagen (Fig. 5). Consistent with the reduced ability of B18R to neutralize IFNϵ and IFNκ activity in the reporter assay (Fig. 3), B18R bound 80 and 64% of IFNϵ and IFNκ, respectively, when injected at the high concentration. However, the percentage dropped to 14% for IFNϵ and 7% for IFNκ when the IFNs were injected at the low concentration (Fig. 5B). Interestingly, the B18R binding profile of IFNα1 mimicked IFNϵ and IFNκ yielding 52% binding at high concentration and 11% at the low concentration.
Figure 5.

IFNϵ, IFNκ, and IFNα1 exhibit reduced binding to the viral IFN antagonist B18R. Type-I IFNs, at 50 (A) or 5 (B) nm concentrations, were injected over the Copenhagen strain of B18R (B19R-FC) captured on a Biacore chip. Binding is reported as fraction RU bound, which corresponds to the SPR RU observed upon IFN injection over the B19R-FC surface, divided by the theoretical RUmax of the surface. The type-III IFNλ proteins (IL29, IL28A, IL28B), which do not bind to B18R, were used as negative controls. Based on the IFN-B18R binding profiles in A and B, kinetic analyses were performed for selected strong B18R binders (IFNα14 and IFNα4) and weak B18R binders (IFNϵ and IFNα1). The experimental (black) and calculated (colored) sensorgrams for strong and weak binders are shown. The rate constants derived from the sensorgrams are found in Table 2.
Based on the results of the binding screen (Fig. 5, A and B), four IFNs that span the strong B18R binder group (IFNα14 and IFNα4), and the weak binders (IFNα1 and IFNϵ), were subjected to further kinetic analysis (Fig. 5). Elucidating their binding parameters revealed the strong and weak binders were distinguished by their association rate constants (ka, Table 2). Specifically, the poor binders had small ka values (ka = 1.5 × 105 to 5.5 × 105 m−1 s−1), whereas the ka values of the strong binders were 10- to 73-fold faster (ka = 5.8 × 106 to 1 × 107 m−1 s−1). Importantly, all four IFNs tested exhibited very slow (t½ ∼ 10 h) dissociation rates that differ from one another by no more than 2-fold. Thus, as observed for IFNAR2 interactions, the B18R neutralization profiles of IFNϵ and IFNκ correlate with reduced association rates, which reduce the ability of B18R to bind and neutralize their biological activity (Fig. 3). These observations suggest IFNϵ, IFNκ, and even IFNα1, may have a selective advantage during poxvirus infections by partially escaping B18R-mediated IFN neutralization.
A molecular model to explain IFNϵ/κ binding to IFNAR2 and B18R
SPR analysis revealed IFNϵ, IFNκ, as well as IFNα1, have reduced association rate constants for B18R and IFNAR2, relative to the other type-I IFNs. To identify regions of IFNϵ, IFNκ, and IFNα1 that could explain their unique binding properties, we aligned the amino acid sequences of all of the IFNs. This analysis revealed IFNκ is the only IFN that has replaced the conserved IFNAR2-binding residue, Arg-56, with an asparagine (Fig. 6E). Structural and biophysical studies confirm Arg-56 makes extensive interactions with IFNAR2 and an R56A mutant drastically disrupts the IFN-IFNAR2 interaction (46, 47). These data suggest the change of Arg-56 to an asparagine in IFNκ could be sufficient to explain its reduced affinity for IFNAR2.
Figure 6.
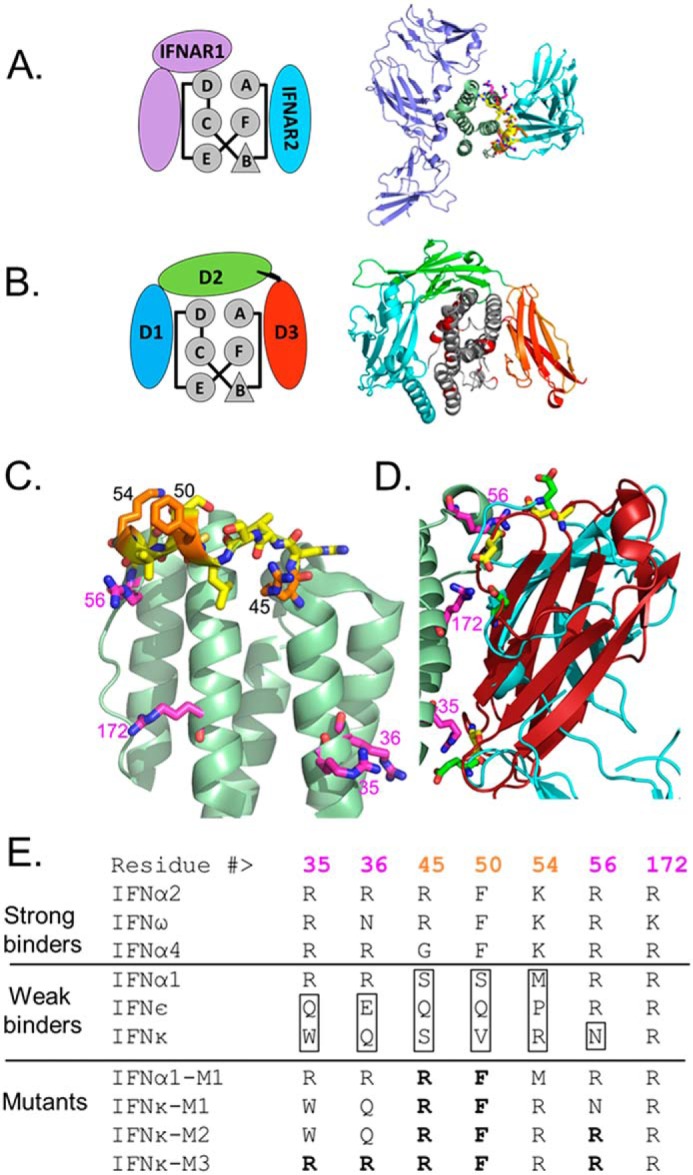
Molecular model of unique IFNϵ/κ and IFNα1 residues that modulate IFNAR2 and B18R binding. A and B, schematic models and structures of the (A) IFNα2/IFNAR1/IFNAR2 complex (PDB 3SE3) and the (B) IFNα2/B18R complex (IFNα2 from PDB 3S9D, and B18R derived from the C12R structure, PDB 3OQ3). C, ribbon diagram of the IFNα2 backbone with key residues that regulate IFNAR2 and B18R binding affinity, as discussed in the text, shown in yellow, orange, and magenta. D, superposition of IFNs in IFN/IFNAR2 and IFN/B18R structures with B18R D3 domain in red and the IFNAR2 D1 domain cyan. Three negatively charged residues conserved in IFNAR2 (green) and B18R (yellow) are positioned by IFNα arginines: Arg-35, Arg-56, and Arg=172 (magenta). E, amino acids within the AB loop region of IFNs that distinguish strong and weak IFNAR2 and B18R binders. Amino acid substitutions made to produce IFNα1 and IFNκ mutants are shown in bold.
To define the impact of the IFNκ Arg to Asn change on B18R binding, we generated a three-dimensional model of the human IFNα2/B18R complex based on the structure of the murine ectromelia virus type-I IFN antagonist, C12R (PDB 3OQ3). The B18R model is highly reliable because C12R shares 89% sequence identity with B18R (Fig. 6). Our analysis focused on the C-terminal domain of B18R (D3 domain 91% sequence identity with C12R), which forms contacts with the IFNAR2-binding site of the IFNs (Fig. 6B). Consistent with our hypothesis, Arg-56 in the IFNα2/B18R complex forms a salt bridge with glutamate 297 (C12R numbering) that would be significantly disrupted by replacing it with an asparagine as found in IFNκ. Because electrostatic interactions drive association rate constants, we looked for additional salt bridge interactions that are conserved in the high affinity IFNα2/IFNAR2 and IFNα2/B18R complexes. This analysis identified two structurally conserved salt bridge interactions, formed with IFNα2 residues Arg-35 and Arg-36 (to IFNAR2 Glu-190 and B18R Asp-260) and Arg-172 (to IFNAR2 Glu-77 and B18R Asp-276). Arg-172, located in helix F, is conserved in all 16 IFNs. In contrast, Arg-35 and Arg-36 are not conserved in either IFNϵ (Gln-35/Glu-36) or IFNκ (Trp-35/Gln-36), suggesting these residues contribute to the reduced IFNAR2 and B18R association rates observed for IFNϵ and IFNκ.
This analysis suggests that IFNϵ and IFNκ have reduced affinities for IFNAR2 and B18R because they have replaced Arg-35, Arg-36, and Arg-56 (IFNκ) with alternative residues that are suboptimal for IFNAR2 and B18R binding. Although this provides a possible explanation for disruption of IFNϵ/κ binding, all three arginine residues are conserved in the sequence of IFNα1. Thus, other residues must be responsible for the reduced affinity of IFNα1 for IFNAR2 and B18R. Further sequence analysis identified three residues that are unique to IFNα1, relative to the high affinity B18R binders (Fig. 6). The three residues in the high affinity IFNs (Arg-45, Phe-50, and Lys-54) are replaced with Ser-45, Ser-50, and Met-54 in IFNα1. Interestingly, these residues are all located adjacent to Arg-56 in the AB loop of IFNα1. This suggests IFNα1 alters IFNAR2 affinity by indirectly changing the conformation of Arg-56, and/or stability of the entire AB loop, rather than replacing Arg-56 with an Asn, as observed for IFNκ. Notably, the same three residues are also different in the sequences of IFNϵ and IFNκ, relative to the high affinity IFNs (Fig. 6). This suggests IFNα1, IFNϵ, and IFNκ modulate IFNAR2 and B18R affinity through salt bridge/hydrogen bond interactions that particularly manipulate Arg-56-mediated interactions.
Analysis of IFNα1 and IFNκ mutants
To test the structural model, mutants of IFNα1 and IFNκ were made that replaced putative “poor binding” residues with residues found in high affinity IFNs (Fig. 6). First, an IFNα1 double mutant (S45R/S50F) was evaluated (IFNα1-M1). Consistent with the modeling study, IFNα1-M1 exhibited 10-fold higher activity (26 ± 10 pm) in the ISFG3 reporter assay than IFNα1 (Fig. 7A, Table 2). IFNα1-M1 also exhibited 14-fold higher affinity for IFNAR2, 2-fold high affinity for IFNAR1, and 3-fold higher affinity for the IFNAR1/2 heterodimer, relative to IFNα1 (Table 2). IFNα1-M1 also exhibited 4-fold higher affinity for B18R than IFNα1, which was almost entirely due to a faster association rate constant (ka) for the IFNα1-M1/B18R interaction (Fig. 7C, Table 2).
Figure 7.

Bioactivity and B18R-binding properties of IFNα1 and IFNκ mutants. A and B, representative dose-response curves from ISFG3 reporter cell assays performed using IFNα1/IFNα1-M1 (A) and IFNκ/IFNκ-M1/IFNκ-M2/IFNk-M3 (B). The EC50 values for all mutants are shown in Table 2. SPR sensorgrams for IFNα1-M1 binding to B18R are shown (C). Experimental (black) and calculated (blue) sensorgrams, used to derive kinetic parameters in Table 2, are shown. Kinetic parameters for IFNκ and IFNκ-M2/B18R interactions could not be uniquely determined from the sensorgrams. To compare their binding properties, 5 nm concentrations of the IFNs were injected over the B18R-FC surface (D). Binding is reported as fraction RU bound as described in the legend to Fig. 5.
The activity profiles of three IFNκ mutants (IFNκ-M1, -M2, and -M3, Fig. 6) confirmed the importance of the N56R substitution for increasing IFNκ biological potency (Fig. 7B, Table 2). However, the mutants also demonstrated that the conformation/biophysical properties of the IFNκ N-terminal region (e.g. helix A, AB loop, and helix B) are distinct from other IFNs. For example, making the S45R/V50F double mutant in IFNκ (IFNκ-M1), the same mutations that significantly increased the bioactivity and receptor binding affinity of IFNα1, resulted in decreased bioactivity (Fig. 7B). In contrast, the IFNκ-M2 triple mutant (S45R/V50F/N56R) increased ISGF3 bioactivity (6-fold) and IFNAR1/2 binding affinity (5-fold), relative to IFNκ. This demonstrates IFNκ is not optimized to induce maximal bioactivity, or to bind with the highest affinity possible to the IFNAR receptors. Although accurate rate constants could not be derived from IFNκ/B18R or IFNκ-M2/B18R sensorgrams, B18R bound more IFNκ-M2 (53%) than IFNκ (7%) when equal concentrations (5 nm) of the IFNs were injected over B18R surfaces (Fig. 7D).
We also tested if additional amino acid changes (W35R and Q36R) in IFNκM2 (e.g. IFNκ-M3) would further increase IFNκ bioactivity. However, IFNκ-M3 exhibited the poorest bioactivity of the three IFNκ mutants tested (Fig. 7B), suggesting these mutations may significantly disrupt the structure of IFNκ's IFNAR-binding sites.
Discussion
Genes encoding IFNϵ and IFNκ protein sequences were identified over a decade ago (36, 37), yet only recently has their importance in host defense, especially in the FRT, come to light (26, 38, 41). Although animal studies are progressing, studies that directly characterize the purified human proteins are lacking. At least one reason for the paucity of information about IFNϵ and IFNκ is they cannot be expressed using protocols suitable for other IFNs (42, 47). In addition, as functional studies about IFNϵ and IFNκ are beginning to be reported, the proteins used in the assays often have not been biochemically evaluated. In addition, high quality antibodies for robust detection of IFNϵ and IFNκ by flow cytometry, ELISA, or other methods, are not available (48). To address these issues, we have identified protocols for producing purified, and functionally active, IFNϵ and IFNκ in E. coli. We focused on optimizing the E. coli expression/refolding system because continuing functional, biophysical, and structural studies require significant amounts of protein.
A major question we sought to address in this report is how IFNϵ and IFNκ bind to the IFNAR1 and IFNAR2 receptors, compared with previously characterized IFNs. These analyses demonstrated both IFNϵ and IFNκ have greatly reduced affinity for the IFNAR2 chain, yet retain IFNAR1 affinities similar to other IFNs. In addition to evaluating binding to the single IFNAR chains, we also characterized IFNϵ/κ binding to an IFNAR1/IFNAR2-FC, which provides a soluble mimic of the cell-surface receptor heterodimer. All three analyses support the conclusion that the reduced potency of IFNϵ and IFNκ is due to reduced receptor binding, predominantly IFNAR2. We followed up these studies by blocking IFNϵ/IFNκ functional activity with a variety of antagonists against the cell-surface receptors, soluble receptors, or using the poxvirus antagonist B18R. The main finding of this analysis was IFNϵ and IFNκ exhibited a different neutralization profile against B18R, compared with IFNα2 or IFNω. To explore this in greater detail, the interaction of all 16 IFNs with B18R were evaluated by SPR. As suggested in the cell-based neutralization studies, B18R bound weakly to IFNϵ and IFNκ. Surprisingly, IFNα1 also exhibited a B18R binding profile that matched IFNϵ and IFNκ. The common biophysical property between IFNϵ, IFNκ, and IFNα1 is that they all interact with the IFNAR2 chain with much lower affinity than IFNα2, IFNω (this study), or other IFNs (49–51). Rate constant analysis revealed the three low affinity IFNs, IFNϵ, IFNκ, and IFNα1, all share reduced association rates for IFNAR2, but different dissociation rates. In particular, IFNα1 dissociates very fast (kd = 0.18 s−1) from IFNAR2, whereas IFNϵ dissociates from IFNAR2 similar to IFNα2 and IFNω (kd = 0.01 s−1), and IFNκ dissociates very slowly (kd = 0.002 s−1) from IFNAR2, as observed for IFNβ (51).
The amino acid sequences of IFNϵ, IFNκ, and IFNα1 share ∼30% identity with one another. Thus, global analysis of the sequences do not explain how these three IFNs bind weakly to IFNAR2 and B18R, relative to other IFNs. However, analysis of the sequences that form the IFNAR2-binding site, which were identified from the IFN/IFNAR1/IFNAR2 crystal structure (46), and a structural model of B18R derived from the ectromelia virus C12R structure (PDB 3OQ3), revealed a series of conserved contacts in both interfaces. Overall, the modeling and mutagenesis data suggest the three low affinity IFNs have evolved a strategy to modulate IFNAR2 and B18R binding that is beneficial in protecting individuals from pathogen challenge. These data support a unified molecular mechanism of manipulating the AB loop to reduce the potency of IFNϵ, IFNκ, and IFNα1, relative to the other IFNs. These molecular changes may also allow IFNϵ, IFNκ, and IFNα1 to partially escape neutralization by poxvirus. In the case of IFNκ, it is interesting that cancer-inducing strains of human papilloma virus have found an alternative strategy, inhibition of IFNκ expression, to evade IFNκ antiviral activity (41).
The biochemical properties of murine IFNϵ (murIFNϵ) were recently characterized (52). Interestingly, murine and human IFNϵ share 59% sequence identity, which is higher than human IFNϵ shares with any other human type-I IFN. The similarity of the murine and human IFNϵ sequences, at least partially, explains why murIFNϵ is active on human cells (52). As reported in our study for human IFNϵ, murIFNϵ also exhibits higher affinity for the murine IFNAR1 chain than for the murine IFNAR2 chain. However, in the context of the entire murine type-I IFN family, other murine IFN-IFNAR interactions appear to be very different from what is observed for the human IFNs. For example, the KD for murine IFNα1/IFNAR2 interaction is 2.2 nm (52), whereas our analysis of the human IFNα1-IFNAR2 interaction results in a KD of 353 nm, which is similar to the value reported by Jaks et al. (49). Interestingly, the high affinity of murine IFNα1 for IFNAR2 is consistent with murine IFNα1 exhibiting the same residue combinations identified for the high affinity (strong binding) human IFNs (Fig. 6E). Thus, there does not appear to be a direct correspondence in binding or function between human IFNα1 and murine IFNα1. However, both humans and mice have evolved IFNs that exhibit very different affinities for the IFNAR1 and IFNAR2 chains.
There is some evidence to suggest weak IFN-IFNAR2 interactions, as observed for IFNϵ, IFNκ, and IFNα1, may result in distinct signaling properties of the IFNs, beyond simply a weaker response. In an extreme case, cells from IFNAR2 knockout mice, when treated with IFNβ, could still induce inflammatory signals through the IFNAR1 chain (53). However, murine IFNβ exhibits very high affinity for the IFNAR1 chain (49), suggesting a possible mechanism for IFNβ-mediated activation of IFNAR1 that is not conserved by IFNϵ, IFNκ, or IFNα1. Thus, if these IFNs induce IFNAR1-specific signals, it must occur by a distinct mechanism relative to a previously described mechanism for murine IFNβ. Although differences in signaling outputs by IFNϵ and IFNκ require further studies, it is clear that human IFNϵ can inhibit HIV-1 at several steps in its replication cycle (38, 39). Furthermore, studies in mice demonstrate IFNϵ controls Chlamydia and herpes simplex virus 2 (26). These data suggest that, just as IFNα2 was formulated as an anti-hepatitis C therapy, the unique biochemical properties of IFNϵ and IFNκ may be useful to protect women from a variety of pathogens that colonize surfaces of the reproductive tract (5).
Experimental procedures
Protein expression and refolding
DNA sequences encoding the mature IFNϵ and IFNκ protein sequences were synthesized with optimized codons for expression in E. coli (ATUM). The codon-optimized cDNAs were subcloned into the PET21b plasmid. All mutagenesis was performed using the QuikChange site-directed mutagenesis kit (Stratagene). The plasmids were transformed into E. coli BL21-CodonPlus (DE3)-RIPL cells (Agilent) for expression. Cultures were grown at 37 °C for ∼3.5 h to A600 values of 0.6–0.8, before induction with 1 mm isopropyl 1-thio-β-d-galactopyranoside. After induction, cultures were grown for an additional 3 h at 37 °C before they were harvested by centrifugation. Protein expression of IFNα1 was as previously described (42).
Expression of IFNϵ and IFNκ resulted in the formation of insoluble inclusion bodies. The inclusion bodies were solubilized in 6 m guanidine HCl and full-length denatured IFNϵ and IFNκ were purified by nickel affinity chromatography (Takara). The denatured proteins were subsequently refolded by a rapid 1:10 dilution into a refolding buffer consisting of 0.1 m Tris-HCl, pH 8, 50 mm NaCl, 2.5 mm EDTA, 0.2 mm oxidized GSH, 2 mm reduced GSH, and 0.8 m arginine. The refolding mixture was incubated for 18 h at 10 °C and then dialyzed into 20 mm Tris, pH 7.5, and 20 mm NaCl. Refolded IFNϵ and IFNκ were subsequently purified using SP cation exchange chromatography (GE Healthcare), which resulted in highly purified IFNϵ and IFNκ preparations. Residual endotoxin was removed from the samples using the High Capacity Endotoxin Removal Spin Columns (Pierce). Endotoxin levels were determined using a Limulus Amoebocyte Lysate Endotoxin Quantitation Kit (Pierce).
Mass spectrometry
Mass analyses were performed using MALDI-TOF MS. Briefly, samples were analyzed in the positive mode on a Voyager Elite mass spectrometer with delayed extraction technology (PerSeptive Biosystems, Framingham MA). The acceleration voltage was set at 25 kV and 100 laser shots were summed. Experiments were performed using sinapinic acid (Sigma) at 5 mg/ml dissolved in acetonitrile, 0.1% TFA (1:1) as the matrix. The mass spectrometer was calibrated using apomyoglobin (Sigma). Samples were diluted 1:10 with matrix, and 1 μl was pipetted onto a stainless steel 96-spot plate for analysis. The data were smoothed and processed using the Data Explorer Software (AB Sciex).
IFI6 reporter assay
HT1080 cells, containing a luciferase reporter downstream of the IFN inducible IFI6 promoter (HL116 cells), were used to characterize IFN activity. HL116 cells (4 × 104 cells) were plated in white opaque 96-well plates and incubated overnight at 37 °C. IFN were added to the cells the next morning and incubated for 5 h at 37 °C. Following the 5-h incubation, the plates were moved to room temperature for 10 min, followed by the addition of 50 μl of luciferase assay reagent (Steady-Glo, Promega) to each well. Luminescence was measured on a Biotek Synergy 2 plate reader and the dose-response curves were analyzed using PRISM with a three-parameter fit (GraphPad Inc.).
ISG54 reporter assay
HEK-Blue IFN-α/β cells (Invivogen) were used to characterize IFN-mediated stimulation of the ISG54 promoter. Activation of the ISG54 promoter results in dose-dependent secretion of embryonic alkaline phosphatase (SEAP). For the assay, plates were seeded with 50,000 cells/well and incubated overnight. The following day, the cells were stimulated with IFNs (IFNα2 and IFNω from PBL Assay Science) in a total volume of 100 μl and incubated for 18 h. Following the 18-h incubation, SEAP levels are quantified by mixing 40 μl of cell supernatant with 160 μl of quanti-blue substrate (Invivogen), followed by an additional 5-min incubation. The plates were then read using a Spectramax plate reader at 650 nm.
Neutralization assays
Neutralization assays were performed using the ISG54 reporter assay with HEK-Blue IFNα/β cells. Reagents used in the assays included anti-IFNα (MMHA-2, PBL Assay Science), anti-IFNAR1 (ab10739, Abcam), and anti-IFNAR2 (MMHAR-2, PBL Assay Science) neutralizing antibodies, soluble receptors IFNAR1-FC (245-AB-050, R&D Systems) and IFNAR2-FC (4015-AB-050, R&D Systems), and viral type-I antagonist B18R (34-8185-85, eBioscience). The neutralization reagents were added to cells at low (3.5 nm) and high (67 nm) concentrations with each IFN. After 18 h, SEAP levels were determined. The data are reported as the mean ± S.D. of three experiments using Prism.
Surface plasmon resonance
SPR experiments were performed using a Biacore T200 (GE Healthcare) at 25 °C using a running buffer consisting of 10 mm HEPES, pH 7.4, 150 mm NaCl, 0.0125% P20, and 125 μg/ml of bovine serum albumin (BSA). Monomeric IFNAR1-FChk, IFNAR2-FChk, and the IFNAR1/IFNAR2-FChk heterodimer proteins (as described in Ref. 45) were captured onto CM-5 sensor chips using an anti-murine FC antibody (Ab, GE Healthcare). All SPR experiments were performed in duplicate and double referenced (e.g. sensorgram data were subtracted from a control surface and from a buffer blank injection) as previously described (54). The control surface for all experiments consisted of the capture Ab. Approximately 100–500 response units (RU) of the IFNAR-FChks were captured onto the chip surfaces. Fresh IFNAR-FChks were coupled to the surfaces for each injection. The surfaces were regenerated between injections with a 3-min injection of 10 mm glycine, pH 1.7. The buffer flow rate for all studies was 50 μl/min. Sensorgrams were globally fit to a 1:1 binding model using Biacore T-200 evaluation software version 1.0.
Receptor occupancies were calculated from the formula (RUobs/RUmax × 100) where RUobs is the RU value observed at the end of 90-s injections of the IFNs over each IFNAR-FC. RUmax values were derived by multiplying the number of RUs of each IFNAR-FC coupled to the chip surface by the ratio of IFN and IFNAR-FC molecular weights (Mr) used in the experiment (e.g. RUmax = RUs of IFNAR2-FC coupled × (IFN MW/IFNAR2-FC MW)). IFN-B18R interactions were characterized by capturing B18R-FC (R&D Systems) on CM-5 sensor chips using the anti-murine FC Ab. IFN subtypes, expressed as described in Ref. 42, were injected over the B18R-FC surface. IFN-B18R-FC interaction screening was performed at IFN concentrations of 50 and 5 nm using a flow rate of 40 μl/min. For kinetic analyses, IFNs were injected over the B18R-FC surface for 2 min and dissociation was monitored for 20 min. The resulting sensorgrams were globally fit to a 1:1 binding model using Biacore T-200 evaluation software version 1.0.
Author contributions
B. D. H. and M. C. formal analysis; B. D. H. and M. R. W. validation; B. D. H., J. S., and M. C. investigation; B. D. H. and M. R. W. methodology; B. D. H., J. S., M. C., J. L. J., and M. R. W. writing-review and editing; M. C., J. L. J., and M. R. W. conceptualization; M. C., J. L. J., and M. R. W. resources; M. C., J. L. J., and M. R. W. supervision; M. R. W. funding acquisition; M. R. W. writing-original draft; M. R. W. project administration.
Acknowledgments
We thank Ashlesha Deshpande for helpful discussions on protein purification. We thank Gilles Uzé for HL116 cells.
This work was supported by a Novel Research Grant from the Alliance for Lupus Research and a Research Grant from Janssen Research & Development, LLC. The authors declare that they have no conflicts of interest with the contents of this article.
- IFN
- interferon
- ISG
- IFN-stimulated gene
- FRT
- female reproductive track
- IA
- iodoacetamide
- RO
- receptor occupancy
- NAb
- neutralizing antibody
- SPR
- surface plasmon resonance
- SEAP
- secretion of embryonic alkaline phosphatase
- RU
- response units
- PDB
- Protein Data Bank.
References
- 1. Samuel C. E. (2001) Antiviral actions of interferons. Clin. Microbiol. Rev. 14, 778–809, table of contents 10.1128/CMR.14.4.778-809.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ivashkiv L. B., and Donlin L. T. (2014) Regulation of type I interferon responses. Nat. Rev. Immunol. 14, 36–49 10.1038/nri3581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pestka S., Langer J. A., Zoon K. C., and Samuel C. E. (1987) Interferons and their actions. Annu. Rev. Biochem. 56, 727–777 10.1146/annurev.bi.56.070187.003455 [DOI] [PubMed] [Google Scholar]
- 4. Pfeffer L. M., Dinarello C. A., Herberman R. B., Williams B. R., Borden E. C., Bordens R., Walter M. R., Nagabhushan T. L., Trotta P. P., and Pestka S. (1998) Biological properties of recombinant α-interferons: 40th anniversary of the discovery of interferons. Cancer Res. 58, 2489–2499 [PubMed] [Google Scholar]
- 5. Borden E. C., Sen G. C., Uze G., Silverman R. H., Ransohoff R. M., Foster G. R., and Stark G. R. (2007) Interferons at age 50: past, current and future impact on biomedicine. Nat. Rev. Drug Discov. 6, 975–990 10.1038/nrd2422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Der S. D., Zhou A., Williams B. R., and Silverman R. H. (1998) Identification of genes differentially regulated by interferon α, β, or γ using oligonucleotide arrays. Proc. Natl. Acad. Sci. U.S.A. 95, 15623–15628 10.1073/pnas.95.26.15623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schoggins J. W., Wilson S. J., Panis M., Murphy M. Y., Jones C. T., Bieniasz P., and Rice C. M. (2011) A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472, 481–485 10.1038/nature09907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang C. H., Wei L., Pfeffer S. R., Du Z., Murti A., Valentine W. J., Zheng Y., and Pfeffer L. M. (2007) Identification of CXCL11 as a STAT3-dependent gene induced by IFN. J. Immunol. 178, 986–992 10.4049/jimmunol.178.2.986 [DOI] [PubMed] [Google Scholar]
- 9. Gray R. C., Kuchtey J., and Harding C. V. (2007) CpG-B ODNs potently induce low levels of IFN-αβ and induce IFN-αβ-dependent MHC-I cross-presentation in DCs as effectively as CpG-A and CpG-C ODNs. J. Leukoc. Biol. 81, 1075–1085 [DOI] [PubMed] [Google Scholar]
- 10. Loh J. E., Chang C. H., Fodor W. L., and Flavell R. A. (1992) Dissection of the interferon γ-MHC class II signal transduction pathway reveals that type I and type II interferon systems share common signalling component(s). EMBO J. 11, 1351–1363 10.1002/j.1460-2075.1992.tb05180.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhao W., Cha E. N., Lee C., Park C. Y., and Schindler C. (2007) Stat2-dependent regulation of MHC class II expression. J. Immunol. 179, 463–471 10.4049/jimmunol.179.1.463 [DOI] [PubMed] [Google Scholar]
- 12. Chawla-Sarkar M., Lindner D. J., Liu Y. F., Williams B. R., Sen G. C., Silverman R. H., and Borden E. C. (2003) Apoptosis and interferons: role of interferon-stimulated genes as mediators of apoptosis. Apoptosis 8, 237–249 10.1023/A:1023668705040 [DOI] [PubMed] [Google Scholar]
- 13. Herzer K., Hofmann T. G., Teufel A., Schimanski C. C., Moehler M., Kanzler S., Schulze-Bergkamen H., and Galle P. R. (2009) IFN-α-induced apoptosis in hepatocellular carcinoma involves promyelocytic leukemia protein and TRAIL independently of p53. Cancer Res. 69, 855–862 10.1158/0008-5472.CAN-08-2831 [DOI] [PubMed] [Google Scholar]
- 14. de Goër de Herve M. G., Durali D., Dembele B., Giuliani M., Tran T. A., Azzarone B., Eid P., Tardieu M., Delfraissy J. F., and Taoufik Y. (2011) Interferon-α triggers B cell effector 1 (Be1) commitment. PLoS ONE 6, e19366 10.1371/journal.pone.0019366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu Y., Carlsson R., Comabella M., Wang J., Kosicki M., Carrion B., Hasan M., Wu X., Montalban X., Dziegiel M. H., Sellebjerg F., Sørensen P. S., Helin K., and Issazadeh-Navikas S. (2014) FoxA1 directs the lineage and immunosuppressive properties of a novel regulatory T cell population in EAE and MS. Nat. Med. 20, 272–282 10.1038/nm.3485 [DOI] [PubMed] [Google Scholar]
- 16. Delgoffe G. M., and Vignali D. A. (2014) A Fox of a different color: FoxA1 programs a new regulatory T cell subset. Nat. Med. 20, 236–237 10.1038/nm.3493 [DOI] [PubMed] [Google Scholar]
- 17. Foster G. R. (2010) Pegylated interferons for the treatment of chronic hepatitis C: pharmacological and clinical differences between peginterferon-α-2a and peginterferon-α-2b. Drugs 70, 147–165 [DOI] [PubMed] [Google Scholar]
- 18. Kirkwood J. (2002) Cancer immunotherapy: the interferon-alpha experience. Semin. Oncol. 29, 18–26 10.1053/sonc.2002.33078 [DOI] [PubMed] [Google Scholar]
- 19. Jacobs L. D., Cookfair D. L., Rudick R. A., Herndon R. M., Richert J. R., Salazar A. M., Fischer J. S., Goodkin D. E., Granger C. V., Simon J. H., Alam J. J., Bartoszak D. M., Bourdette D. N., Braiman J., Brownscheidle C. M., et al. (1996) Intramuscular interferon β-1a for disease progression in relapsing multiple sclerosis: the multiple sclerosis collaborative research group (MSCRG). Ann. Neurol. 39, 285–294 10.1002/ana.410390304 [DOI] [PubMed] [Google Scholar]
- 20. Richter M. F., Duménil G., Uze G., Fellous M., and Pellegrini S. (1998) Specific contribution of Tyk2 JH regions to the binding and the expression of the interferon α/β receptor component IFNAR1. J. Biol. Chem. 273, 24723–24729 10.1074/jbc.273.38.24723 [DOI] [PubMed] [Google Scholar]
- 21. Colamonici O. R., Uyttendaele H., Domanski P., Yan H., and Krolewski J. J. (1994) p135tyk2, an interferon-α-activated tyrosine kinase, is physically associated with an interferon-alpha receptor. J. Biol. Chem. 269, 3518–3522 [PubMed] [Google Scholar]
- 22. Domanski P., Fish E., Nadeau O. W., Witte M., Platanias L. C., Yan H., Krolewski J., Pitha P., and Colamonici O. R. (1997) A region of the β subunit of the interferon α receptor different from box 1 interacts with Jak1 and is sufficient to activate the Jak-Stat pathway and induce an antiviral state. J. Biol. Chem. 272, 26388–26393 10.1074/jbc.272.42.26388 [DOI] [PubMed] [Google Scholar]
- 23. Stark G. R., and Darnell J. E. Jr. (2012) The JAK-STAT pathway at twenty. Immunity 36, 503–514 10.1016/j.immuni.2012.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Platanias L. C. (2005) Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 5, 375–386 10.1038/nri1604 [DOI] [PubMed] [Google Scholar]
- 25. Li T., Niu X., Zhang X., Wang S., and Liu Z. (2017) Recombinant human IFNα-2b response promotes vaginal epithelial cells defense against Candida albicans. Front. Microbiol. 8, 697 10.3389/fmicb.2017.00697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fung K. Y., Mangan N. E., Cumming H., Horvat J. C., Mayall J. R., Stifter S. A., De Weerd N., Roisman L. C., Rossjohn J., Robertson S. A., Schjenken J. E., Parker B., Gargett C. E., Nguyen H. P., Carr D. J., Hansbro P. M., and Hertzog P. J. (2013) Interferon-ϵ protects the female reproductive tract from viral and bacterial infection. Science 339, 1088–1092 10.1126/science.1233321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jones M., Davidson A., Hibbert L., Gruenwald P., Schlaak J., Ball S., Foster G. R., and Jacobs M. (2005) Dengue virus inhibits α interferon signaling by reducing STAT2 expression. J. Virol. 79, 5414–5420 10.1128/JVI.79.9.5414-5420.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Grant A., Ponia S. S., Tripathi S., Balasubramaniam V., Miorin L., Sourisseau M., Schwarz M. C., Sanchez-Seco M. P., Evans M. J., Best S. M., and Garcia-Sastre A. (2016) Zika virus targets human STAT2 to inhibit type I interferon signaling. Cell Host Microbe 19, 882–890 10.1016/j.chom.2016.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Seet B. T., Johnston J. B., Brunetti C. R., Barrett J. W., Everett H., Cameron C., Sypula J., Nazarian S. H., Lucas A., and McFadden G. (2003) Poxviruses and immune evasion. Annu. Rev. Immunol. 21, 377–423 10.1146/annurev.immunol.21.120601.141049 [DOI] [PubMed] [Google Scholar]
- 30. Symons J. A., Alcamí A., and Smith G. L. (1995) Vaccinia virus encodes a soluble type I interferon receptor of novel structure and broad species specificity. Cell 81, 551–560 10.1016/0092-8674(95)90076-4 [DOI] [PubMed] [Google Scholar]
- 31. Colamonici O. R., Domanski P., Sweitzer S. M., Larner A., and Buller R. M. (1995) Vaccinia virus B18R gene encodes a type I interferon-binding protein that blocks interferon α transmembrane signaling. J. Biol. Chem. 270, 15974–15978 10.1074/jbc.270.27.15974 [DOI] [PubMed] [Google Scholar]
- 32. Huang J., Smirnov S. V., Lewis-Antes A., Balan M., Li W., Tang S., Silke G. V., Pütz M. M., Smith G. L., and Kotenko S. V. (2007) Inhibition of type I and type III interferons by a secreted glycoprotein from Yaba-like disease virus. Proc. Natl. Acad. Sci. U.S.A. 104, 9822–9827 10.1073/pnas.0610352104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pestka S., Krause C. D., Sarkar D., Walter M. R., Shi Y., and Fisher P. B. (2004) Interleukin-10 and related cytokines and receptors. Annu. Rev. Immunol. 22, 929–979 10.1146/annurev.immunol.22.012703.104622 [DOI] [PubMed] [Google Scholar]
- 34. Pestka S., Krause C. D., and Walter M. R. (2004) Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 202, 8–32 10.1111/j.0105-2896.2004.00204.x [DOI] [PubMed] [Google Scholar]
- 35. Walter M. R. (2004) Structural analysis of IL-10 and type I interferon family members and their complexes with receptor. Adv. Protein Chem. 68, 171–223 10.1016/S0065-3233(04)68006-5 [DOI] [PubMed] [Google Scholar]
- 36. LaFleur D. W., Nardelli B., Tsareva T., Mather D., Feng P., Semenuk M., Taylor K., Buergin M., Chinchilla D., Roshke V., Chen G., Ruben S. M., Pitha P. M., Coleman T. A., and Moore P. A. (2001) Interferon-κ, a novel type I interferon expressed in human keratinocytes. J. Biol. Chem. 276, 39765–39771 10.1074/jbc.M102502200 [DOI] [PubMed] [Google Scholar]
- 37. Hardy M. P., Owczarek C. M., Jermiin L. S., Ejdebäck M., and Hertzog P. J. (2004) Characterization of the type I interferon locus and identification of novel genes. Genomics 84, 331–345 10.1016/j.ygeno.2004.03.003 [DOI] [PubMed] [Google Scholar]
- 38. Garcia-Minambres A., Eid S. G., Mangan N. E., Pade C., Lim S. S., Matthews A. Y., de Weerd N. A., Hertzog P. J., and Mak J. (2017) Interferon ϵ promotes HIV restriction at multiple steps of viral replication. Immunol. Cell Biol. 95, 478–483 10.1038/icb.2016.123 [DOI] [PubMed] [Google Scholar]
- 39. Tasker C., Subbian S., Gao P., Couret J., Levine C., Ghanny S., Soteropoulos P., Zhao X., Landau N., Lu W., and Chang T. L. (2016) IFN-ϵ protects primary macrophages against HIV infection. JCI insight 1, e88255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Abdulhaqq S. A., Zorrilla C., Kang G., Yin X., Tamayo V., Seaton K. E., Joseph J., Garced S., Tomaras G. D., Linn K. A., Foulkes A. S., Azzoni L., VerMilyea M., Coutifaris C., Kossenkov A. V., et al. (2016) HIV-1-negative female sex workers sustain high cervical IFN-ϵ, low immune activation, and low expression of HIV-1-required host genes. Mucosal Immunol. 9, 1027–1038 10.1038/mi.2015.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Reiser J., Hurst J., Voges M., Krauss P., Münch P., Iftner T., and Stubenrauch F. (2011) High-risk human papillomaviruses repress constitutive κ interferon transcription via E6 to prevent pathogen recognition receptor and antiviral-gene expression. J. Virol. 85, 11372–11380 10.1128/JVI.05279-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kuruganti S., Accavitti-Loper M. A., and Walter M. R. (2014) Production and characterization of thirteen human type-I interferon-α subtypes. Protein Expr. Purif. 103, 75–83 10.1016/j.pep.2014.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yoon S. I., and Walter M. R. (2007) Identification and characterization of a +1 frameshift observed during the expression of Epstein-Barr virus IL-10 in Escherichia coli. Protein Expr. Purif. 53, 132–137 10.1016/j.pep.2006.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Radhakrishnan R., Walter L. J., Hruza A., Reichert P., Trotta P. P., Nagabhushan T. L., and Walter M. R. (1996) Zinc mediated dimer of human interferon-α 2b revealed by X-ray crystallography. Structure 4, 1453–1463 10.1016/S0969-2126(96)00152-9 [DOI] [PubMed] [Google Scholar]
- 45. Deshpande A., Putcha B. D., Kuruganti S., and Walter M. R. (2013) Kinetic analysis of cytokine-mediated receptor assembly using engineered FC heterodimers. Protein Sci. 22, 1100–1108 10.1002/pro.2285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Thomas C., Moraga I., Levin D., Krutzik P. O., Podoplelova Y., Trejo A., Lee C., Yarden G., Vleck S. E., Glenn J. S., Nolan G. P., Piehler J., Schreiber G., and Garcia K. C. (2011) Structural linkage between ligand discrimination and receptor activation by type I interferons. Cell 146, 621–632 10.1016/j.cell.2011.06.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Piehler J., and Schreiber G. (1999) Biophysical analysis of the interaction of human ifnar2 expressed in E. coli with IFNα2. J. Mol. Biol. 289, 57–67 10.1006/jmbi.1999.2726 [DOI] [PubMed] [Google Scholar]
- 48. Couret J., Tasker C., Kim J., Sihvonen T., Fruitwala S., Quayle A. J., Lespinasse P., Heller D. S., and Chang T. L. (2017) Differential regulation of IFNα, IFNβ, and IFNϵ gene expression in human cervical epithelial cells. Cell Biosci. 7, 57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jaks E., Gavutis M., Uzé G., Martal J., and Piehler J. (2007) Differential receptor subunit affinities of type I interferons govern differential signal activation. J. Mol. Biol. 366, 525–539 10.1016/j.jmb.2006.11.053 [DOI] [PubMed] [Google Scholar]
- 50. Lavoie T. B., Kalie E., Crisafulli-Cabatu S., Abramovich R., DiGioia G., Moolchan K., Pestka S., and Schreiber G. (2011) Binding and activity of all human α interferon subtypes. Cytokine 56, 282–289 10.1016/j.cyto.2011.07.019 [DOI] [PubMed] [Google Scholar]
- 51. Jaitin D. A., Roisman L. C., Jaks E., Gavutis M., Piehler J., Van der Heyden J., Uze G., and Schreiber G. (2006) Inquiring into the differential action of interferons (IFNs): an IFN-α2 mutant with enhanced affinity to IFNAR1 is functionally similar to IFN-β. Mol. Cell. Biol. 26, 1888–1897 10.1128/MCB.26.5.1888-1897.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Stifter S. A., Matthews A. Y., Mangan N. E., Fung K. Y., Drew A., Tate M. D., Soares da Costa T. P., Hampsey D., Mayall J., Hansbro P. M., Garcia Minambres A., Eid S. G., Mak J., Scoble J., Lovrecz G., deWeerd N. A., and Hertzog P. J. (2018) Defining the distinct, intrinsic properties of the novel type I interferon, IFN. J. Biol. Chem. 293, 3168–3179 10.1074/jbc.M117.800755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Samarajiwa S. A., Mangan N. E., Hardy M. P., Najdovska M., Dubach D., Braniff S. J., Owczarek C. M., and Hertzog P. J. (2014) Soluble IFN receptor potentiates in vivo type I IFN signaling and exacerbates TLR4-mediated septic shock. J. Immunol. 192, 4425–4435 10.4049/jimmunol.1302388 [DOI] [PubMed] [Google Scholar]
- 54. Myszka D. G. (1999) Improving biosensor analysis. J. Mol. Recognit. 12, 279–284 10.1002/(SICI)1099-1352(199909/10)12:5%3C279::AID-JMR473%3E3.0.CO%3B2-3 [DOI] [PubMed] [Google Scholar]