

Abstract
The neuropeptide relaxin-3 and its receptor relaxin family peptide receptor-3 (RXFP3) play key roles in modulating behavior such as memory and learning, food intake, and reward seeking. A linear relaxin-3 antagonist (R3 B1-22R) based on a modified and truncated relaxin-3 B-chain was recently developed. R3 B1-22R is unstructured in solution; thus, the binding conformation and determinants of receptor binding are unclear. Here, we have designed, chemically synthesized, and pharmacologically characterized more than 60 analogues of R3 B1-22R to develop an extensive understanding of its structure–activity relationships. We show that the key driver for affinity is the nonnative C-terminal Arg23. Additional contributors to binding include amino acid residues that are important also for relaxin-3 binding, including Arg12, Ile15, and Ile19. Intriguingly, amino acid residues that are not exposed in native relaxin-3, including Phe14 and Ala17, also interact with RXFP3. We show that R3 B1-22R has a propensity to form a helical structure, and modifications that support a helical conformation are functionally well-tolerated, whereas helix breakers such as proline residues disrupt binding. These data suggest that the peptide adopts a helical conformation, like relaxin-3, upon binding to RXFP3, but that its smaller size allows it to penetrate deeper into the orthosteric binding site, creating more extensive contacts with the receptor.
Keywords: relaxin-3, G protein-coupled receptor (GPCR), neuropeptide, peptide conformation, peptide interaction, antagonist, R3 B1-22R, RXFP3, alanine scan, peptide stapling
Introduction
Relaxin-3 is a highly conserved novel neuropeptide (1) that has been shown to modulate food intake (2, 3), stress (4, 5), arousal, memory and learning, and addiction in animal models (6, 7). These functions are consistent with relaxin-3 innervation pathways and sites of expression of its endogenous receptor, relaxin family peptide receptor-3 (RXFP3). 3 Relaxin-3 has been shown to be highly expressed in the nucleus incertus in the rodent brain (1, 8). The relaxin-3–expressing neurons co-localize with the corticotropin-releasing factor type 1 receptor, and relaxin-3 expression has been shown to be up-regulated in stressed animals (4). Projection of relaxin-3 fibers to the paraventricular nucleus of hypothalamus and the bed nucleus strial terminalis, both regions with high RXFP3 expression, are thought to drive increased food intake and modulate pathways involved in addiction/reward seeking, respectively (3, 6). Furthermore, projections of relaxin-3 neurons to the septohippocampus pathway modulate arousal and spatial memory (7).
Relaxin-3 is a member of the insulin/relaxin superfamily of peptide hormones. Although insulin and the insulin-like growth factors signal through tyrosine kinase receptors, the relaxin peptides, including relaxins 1–3, and insulin-like peptides 3–6 (INSL3–6) signal through RXFPs, which are G protein–coupled receptors (9). Currently, four RXFPs have been identified. Relaxin-2 acts through RXFP1 (10), INSL3 through RXFP2 (11), and INSL5 through RXFP4 (12). Relaxin-3 is, in addition to its cognate receptor, RXFP3, also able to activate both RXFP1 and RXFP4 (13–15).
Relaxin-3, like the other members of the insulin/relaxin superfamily, consists of two peptide chains cross-braced by one intrachain and two interchain disulfide bonds (Fig. 1) (16). Multiple studies have investigated structure–activity relationships of relaxins to develop better analogues (17). For relaxin-3, amino acid residues Arg8, Arg12, Ile15, Arg16, Ile19, and Phe20 in the helical region of the relaxin-3 B-chain have been identified as important for binding to RXFP3. The same amino acid residues, except Arg12, are also essential for binding to RXFP4 (18). For activation of RXFP3 and RXFP4, Arg26 and Trp27 of the B-chain tail are critical (18). Interestingly, removal of N-terminal amino acid residues in the A-chain significantly reduces the binding affinity of relaxin-3 to RXFP1, whereas the affinity and potency for RXFP3 are unaffected (19, 20). Replacement of the relaxin-3 A-chain with the INSL5 A-chain results in increased specificity for RXFP3, by reducing the interaction with RXFP1 (21, 22). The ability of relaxin-3 to retain the interaction with RXFP3 even after changes to the A-chain strongly suggests that the A-chain functions only as a structural support, ensuring the correct conformation of the relaxin-3 B-chain (20). Indeed, it has now been shown that introducing a helical “staple” that supports the native conformation is sufficient to create a high-affinity agonist (23, 24). Receptor mutagenesis studies have shed some light on the amino acid residues in the receptor that are involved in the relaxin-3/RXFP3 interaction. Arg12, Arg16, and Arg26 of the relaxin-3 B-chain form electrostatic interactions with Glu244, Asp145, and Glu141 of RXFP3, respectively (25, 26). The flexible C-terminal of relaxin-3 has been suggested to undergo a conformational change to allow Trp27 to interact with Trp138 buried deep in the transmembrane region of RXFP3 (27).
Figure 1.
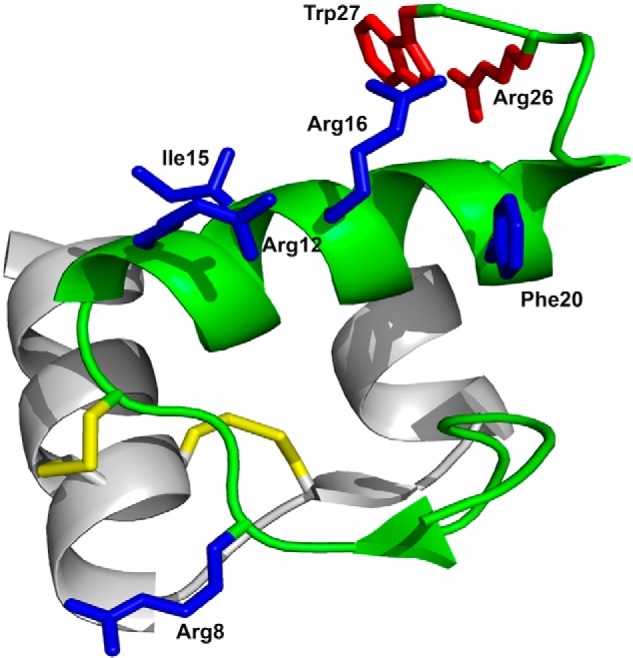
Structure and receptor-interacting amino acid residues of relaxin-3. The A-chain, shown in gray, consists of two antiparallel helices connected by a β-strand. The B-chain, shown in green, comprises a helical segment spanning from Gly11 to Cys22. Disulfides are shown in yellow. Side chains important for binding to RXFP3 and for activating RXFP3 are shown in blue and red, respectively. The R3 B1-22R antagonist retains only the B-chain with the two cysteines at positions 10 and 22 replaced with Ser and the C-terminal five amino acid residues 23–27 replaced with a nonnative Arg23.
Removal of the Arg26–Trp27 activation domain of relaxin-3 results in an antagonist peptide (18). Intriguingly, as a result of the recombinant production strategy, a nonnative Arg was left as an artifact in place of the C-terminal five amino acid residues during these studies, and this addition appeared to benefit affinity (18). Given that the interaction with RXFP3 is only dependent on the B-chain, this modification was further explored in single-chain variants, allowing the development of the antagonist R3 B1-22R (28). The R3 B1-22R variant has binding affinity comparable with that of native relaxin-3 for RXFP3, despite R3 B1-22R being unstructured in solution (28). R3 B1-22R may adopt a defined conformation upon binding to RXFP3; however, its smaller size and flexibility will allow it to optimize interactions in a substantially different way than the larger and constrained relaxin-3. R3 B1-22R has been an important tool for studying the relaxin-3 system and shown that antagonizing RXFP3 reduces food intake in mice and alcohol seeking in rat addiction models (2, 6, 29). Therefore, an investigation of the structure–activity relationship of this peptide is a critical step to further develop it as a drug lead.
In this study, we have explored the structure–activity relationships of R3 B1-22R through alanine scanning and followed up changes in pharmacology with additional substitutional approaches. The binding conformation was also explored by introducing helix-breaking amino acid residues, such as prolines, or helix-promoting features, such as side-chain “staples,” to further understand the binding conformation of R3 B1-22R. We show that some features that are important in relaxin-3 are also important in the antagonist, but that additional amino acid residues also contribute to receptor binding. These findings provide new mechanistic insights into the activity of R3 B1-22R and highlight regions that can be explored for further improving the activity of this peptide.
Results
Alanine scan of R3 B1-22R identifies amino acids involved in the RXFP3 interaction
Using an alanine scan strategy, a series of peptide variants, each carrying a single Ala substitution, was synthesized using standard Fmoc solid-phase peptide synthesis. The influence of the amino acid side-chain substitutions in R3 B1-22R were assessed through a competition binding assays using a europium-labeled R3 B1-22R tracer (30). Truncation of the N-terminal part has previously been shown to be relatively well-tolerated; thus, we focused on residue 5 onward (28). Strikingly, removal of the majority of side chains resulted in a reduction of the binding affinity of the mutant peptides to RXFP3, compared with R3 B1-22R (Table 1 and Fig. 2A). Substitution of amino acid residues before the helical region to Ala (Y5A, G6A, V7A, R8A, L9A, and S10A) showed a modest but significant reduction in binding affinity by 3–6-fold. A slightly larger reduction in affinity, up to 15-fold, was observed in the mutants R12A, F14A, I15A R16A, V18A, I19A, T21A, and S22A in what constitutes the helical region in the native relaxin-3 structure. Intriguingly, although these included Arg8, Arg12, Ile15, Arg16, and Ile19, which all have been shown to be important for relaxin-3 binding (18), amino acid residues that are not surfaced-exposed in relaxin-3 due to the interaction with the A-chain, such as Phe14 and Val18 also appear to contribute to binding of the antagonist. In contrast, Phe20, which is a key residue for affinity in relaxin-3, can be replaced with an Ala without loss of affinity in R3 B1-22R. Position 17 is an Ala in the native sequence. Changing this to a polar Asn showed a significant 50-fold reduction in binding affinity compared with native R3 B1-22R. Ala17 is also buried in relaxin-3, and its replacement should be well-tolerated if a similar binding position was adopted. A >300-fold reduction in binding affinity, essentially abolishing the interaction, was observed when the nonnative Arg23 was mutated to Ala; this residue by far is the biggest contributor to affinity.
Table 1.
Effects of point modifications on binding affinity for RXFP3

† All peptides were synthesized with an amidated C-terminus. Orn, ornithine; Har, homoarginine; Cit, citrulline; Agb, norarginine.
a p < 0.001 versus R3 B1-22R.
b p < 0.01 versus R3 B1-22R.
c p < 0.05 versus R3 B1-22R.
Figure 2.

Competition binding of R3 B1-22R variants at RXFP3. The effect of representative R3 B1-22R substitutions including Ala substitutions (A), Arg variants (B), helix breakers (Pro) or helix promotors (Aib) (C), and side-chain and backbone cyclization (D) on the ability to compete for binding with europium-labeled R3 B1-22R is shown. Data are presented as mean ± S.E. (error bars) from a minimum of three independent experiments.
Only a small subset of amino acid residues contribute specific interactions when binding to RXFP3
Alanine is the smallest chiral amino acid residue. To investigate how specific the interactions contributed by the native side chains were, we tested whether simply providing some additional “bulk” was sufficient to restore some of the loss seen in the Ala variants. In this series, the nonprotein-encoded amino acid aminobutyric acid (Abu) was introduced, and again the affinity for RXFP3 was tested. Strikingly, improvements of binding affinity to a level not significantly different from those of the native peptide were seen for positions Arg8, Phe14, Arg16, Val18, and Thr21 (Table 1). Substitution of the native Ala17 for Abu was also well-tolerated; however, for Arg12, Ile15, and Ile19, no improvements were observed. At these three positions, the particular side-chain features thus appear to be required for the interaction, whereas for others, including the positive charges of Arg8 and Arg16, they are dispensable. These amino acid residues contact the receptor but can be replaced by other types, allowing different types of interactions.
Arg23 at the R3 B1-22R C terminus cannot be altered
The alanine scan highlighted the extraordinary importance of Arg23 for ensuring high-affinity binding to RXFP3. Therefore, we set out to investigate whether any type of subtle change to this Arg could be tolerated, or even favored, by introducing nonprotein-encoded Arg variants (Fig. 3). Citrulline lacks a positive charge, whereas lysine and ornithine retain a positive charge but in the form of a smaller amine rather than the large guanidinium group. These modifications were all found to be detrimental for binding, reducing affinity by at least 70-fold. Homoarginine and norarginine both retain the native guanidinium group, but its position is altered as the side chain is extended or shortened by one carbon, respectively. For these, the effect was less dramatic, yet the binding affinity was still reduced >20-fold. (Table 1 and Fig. 2B). Thus, both the nature and relative position of the Arg guanidinium group are optimal and critical for affinity. All analogues in this study were produced as C-terminal amides, as a free acid at Arg23 has been previously shown to be unfavorable and result in a ∼10-fold drop in affinity (28).
Figure 3.

Chemical structure of arginine and its variants. Nonproteinogenic amino acids were used to substitute arginine at position 23 to investigate the optimal features for binding to RXFP3.
Can an Arg at position 23 in a full-length B-chain improve affinity of an agonist?
Given the significance of Arg23 in R3 B1-22R, we wanted to explore whether introducing the same modification in a single-chain variant retaining the activation domain would be beneficial for creating an agonist. The variant R3 B1-22RGSRW was designed to retain the relative spacing between the helical domain and the C-terminal Arg26–Trp27 activation domain, whereas in R3 B1-22RGGSRW, the full five-residue C-terminal tail was added after Arg23. Notably, both variants showed poor binding with a pKi of ∼5.5; thus, the inclusion of Arg23 made no improvement over the linear B-chain (Fig. S1 and Table S1), confirming that the interaction of Arg23 is not compatible with the endogenous receptor binding mode of native relaxin-3. A third variant that included a single Ala residue extension to the R3 B1-22R antagonist (R3 B1-22RA) was also produced. Again, this variant showed significantly lower affinity than R3 B1-22R, highlighting that the chain cannot be C-terminally extended for Arg23 to be able to optimally engage RXFP3.
Modifications supporting a helical conformation of R3 B1-22R are well-tolerated, but ones that disrupt it are not
Next, we turned to investigating the binding conformation of the flexible R3 B1-22R. Retro-inverso variants, in which the sequence order and chirality of each amino acid has been inverted, have been shown to be able to recreate the positioning of side chains in peptide loops, resulting in native-like binding surfaces (31). This strategy is, however, not compatible with recreating elements of secondary structure, such as helices. Consistent with the need to refold into a helix, the retro-inverso variant of R3 B6–22R showed no binding to RXFP3 (Table 2). To further investigate whether the ability to form a helix upon binding to RXFP3 is required for R3 B1-22R, we incorporated Pro residues in the sequence. Pro residues are helix breakers due to the constrained conformation and lack of hydrogen bonding potential. Pro substitution at Leu9 and Glu13, before the helix and at the N-terminal part of the helix, resulted in a modest drop in affinity (Table 2 and Fig. 2C). In contrast, disrupting the C-terminal portion of the helix by the modification A17P or F20P caused a total loss of binding.
Table 2.
Effects of helical supportive or disruptive modifications on binding affinity for RXFP3

† All peptides were synthesized with an amidated C-terminus.
a p < 0.001 versus R3 B1-22R.
b p < 0.001 versus R3 B6–22R.
c p < 0.01 versus R3 B1-22R.
Instead, substitutions promoting a helical structure were introduced. Aminoisobutyric acid (Aib) has, as a result of the substitution of its α proton with a methyl group, a conformationally restricted backbone that favors the dihedral angles adopted in helices (32). Single-residue substitutions with Aib at positions Ala17, Val18, and Thr21 in the C-terminal part of the helix were all well-tolerated, with neither variant showing impaired binding to RXFP3 relative to R3 B1-22R. Using a shorter version of the antagonist template R3 B6–22R, which has affinity comparable with R3 B1-22R (Table 2), we investigated whether incorporation of multiple Aib residues could drive helix formation and improve binding beyond the affinity of the native sequence. However, incorporating three Aib residues resulted in a reduction rather than improvement in binding affinity by ∼5-fold (Aib20, Aib21, and Aib22). Binding affinity dropped a further 2.5 times when the positions Phe14, Val18, and Thr21 were replaced with Aib. This drop is probably due to the cumulative effect of removing side chains, each of which makes a small contribution to binding. Other helix-promoting strategies were also explored, including introducing side chain “staples” in the form of lactam bonds between helical positions i and i + 4. The sequence modifications to install Glu–Lys pairs at positions 16–20 and 18–22 were both found to completely disrupt binding, but notably some affinity was restored when the side chains were linked to stabilize a helical conformation. The substitution A17K, as previously noted, resulted in a significant drop in affinity; however, linking it to Glu13 via a lactam negated some of this effect. Finally, introducing a Lys-Asp pair at positions 13–17 also reduced affinity, but linking them to support a helical structure restored this drop to a closer to native affinity (Table 2 and Fig. 2D).
In addition to the short-range helical staples, we explored the effect of more global cyclization restraints. In the NMR structure of native relaxin-3, the N-terminal tail loops around and lies parallel to the helical segment, an arrangement that could be supported by a covalent link between these regions in the antagonist. Cyclizing R3 B1-22R via a lactam bond from the N-terminal amino group to the side chain carboxyl group of Glu13 caused a reduction in pKi from 7.69 to 6.78. Introducing the same linkage in the shorter antagonist version R3 B6–22R also reduced affinity, but to a lesser extent. To introduce a different anchor point, the native Glu13 was mutated to Gln13 and Thr21 to Glu21. Cyclizing the full-length peptide from the N terminus to Glu21 in this variant was surprisingly well-tolerated, despite this position being close to the key binding site of Arg23. However, cyclization of the shorter R3 B6–22R variant from the N terminus to Glu21 resulted in complete loss of binding. In this variant, the N-terminal tail is too short to be able to wrap around to the C-terminal end of the helix without disrupting it.
Finally, we explored the inherent ability of R3 B1-22R to adopt a helical conformation in solution by studying the effect of the addition of trifluoroethanol (TFE). TFE is well-known to support the formation of helical structure; thus, we prepared a sample containing 70% water, 30% TFE and recorded two-dimensional solution NMR spectroscopy data. The data were assigned using sequential assignment methods, and the secondary shifts, which are highly sensitive indicators of secondary structure, were determined by subtracting random coil chemical shifts from the observed chemical shifts. The comparison of Hα secondary shifts of R3 B1-22R in water and TFE, as well as the B-chain in relaxin-3, are presented in Fig. 4. Remarkably, not only was the helical conformation of relaxin-3 restored, as the negative secondary shifts in the region 13–21 closely match the ones observed in the native peptide, but the positive secondary shifts resulting from the extended conformation in the region 8–11 were also observed in the linear peptide in TFE. Thus, R3 B1-22R readily adopts a native-like conformation even in the absence of the A-chain when placed in a more hydrophobic environment.
Figure 4.

Secondary Hα shifts (experimental shifts − random coil shifts) for R3 B1-22R in H2O and 30% TFE solvents. For comparison, the secondary Hα shifts of the B-chain in native relaxin-3 are included. The secondary shifts, and consequently the secondary structure, in TFE closely mimics the structure in native relaxin-3.
Discussion
Studies on relaxin-3 and related peptides have long been hampered by the complex and expensive synthesis of their two chain structure. The development of the single-chain relaxin-3 antagonist R3 B1-22R was the first example of a variant that retained native-like binding affinity in a single peptide chain (28). It has since been shown that linear variants of relaxin-2 can also be achieved, possessing potent anti-fibrotic activity through targeting RXFP1 (33). These peptides have been a game changer, as they allow large-scale production and extensive in vivo studies into rodent physiology and behavior. Data showing that modulation of the relaxin-3/RXFP3 system controls important behaviors highlight R3 B1-22R as a potential therapeutic lead. However, further improvements of this peptide are required, as unstructured amino acid sequences are readily degraded by proteases, and current studies have relied on intracerebroventricular administration. For the peptide to be a viable candidate for further preclinical studies, it must be modified to be able to pass the blood–brain barrier to engage its neuronal target after systemic administration. Although much is known about how relaxin-3 engages RXFP3, there is little information about how R3 B1-22R interacts with the receptor, both in terms of contributions from individual amino acid residues and in terms of its binding conformation. In this study, we investigated the changes to R3 B1-22R binding interactions that arise from amino acid substitutions to further understand the binding mode of the antagonist at RXFP3. These new insights set the scene for the development of next-generation analogues.
The results from all point modifications introduced are summarized in Fig. 5. As expected, and consistent with relaxin-3 structure–activity data (18), the Ala scan showed that Arg8, Arg12, Ile15, Arg16, and Ile19 contribute to RXFP3 binding. Notably, these contributions are significantly less pronounced than in relaxin-3, where many modifications at these sites resulted in a drop of affinity of 100-fold or more. Instead, a larger number of amino acid residues appear to make smaller contributions to binding in R3 B1-22R as a reduction of side-chain functionality through replacement with a small Ala residue at the majority of positions leading to a decrease in binding. These changes include positions that are involved in the interaction with the A-chain in native relaxin-3. Interestingly, Ala substitution at Phe20 did not show any change in binding affinity in the single-chain antagonist (18). Taken together, these findings highlight significant differences in the binding of R3 B1-22R compared with relaxin-3 at RXFP3.
Figure 5.

Summary of the SAR data from point substitutions in R3 B1-22R. Substitutions resulting in a drop of binding p > 0.01 versus R3 B1-22R are shown in yellow circles, and substitutions resulting in a drop in affinity p > 0.001 versus R3 B1-22R are shown in red circles. No changes to Arg23 were tolerated.
Given that many amino acid residues made small contributions, including ones that have evolved to maintain structural integrity in the two-chain relaxin-3, as opposed to maintaining an optimal receptor interaction, we wanted to further investigate the type of interactions present (i.e. are they specific in terms of requiring a particular residue type, or can different types of side chains provide similar level of binding contribution?). To analyze this, we made a series of mutants incorporating an Abu residue rather than Ala, increasing the side-chain “bulk” by one carbon to allow additional receptor contacts. Indeed, we found that for both Phe14 and Val18, amino acid residues that would not be expected to be optimized for binding given their structural role in relaxin-3, incorporation of an Abu instead of Ala was sufficient to restore a native-like affinity. In contrast, for Arg12, Ile15, and Ile19, which are part of the native binding site, introducing an Abu made no improvement over an Ala. Intriguingly, at positions Arg8 and Arg16, which are critical for relaxin-3 binding, an improved affinity was observed when Abu was introduced instead of Ala. This suggests that the positive charge is no longer essential for the interaction, and the added peptide flexibility allows local adaptation of interactions in the antagonist. An Abu residue was tolerated at position Ala17, in contrast to the larger polar residue Asn, which resulted in a large drop in affinity. The latter likely induces clashes in the binding position of the antagonist. Overall, it appears that the main contributions of interactions from the helical region are hydrophobic in nature.
In contrast to the modest contributions from the helical amino acid residues, the C-terminal Arg23 is vital for the interaction with RXPF3, and an Ala substitution resulted in a >300-fold loss in binding affinity. Although Arg23 is not part of the original relaxin-3 sequence, it was serendipitously realized in a two-chain C-terminally truncated antagonist that an extra Arg residue at this position increased affinity (18). Complete removal of the Arg in the two-chain antagonist resulted in significantly reduced binding to RXFP3 (34), but the effect is even more dramatic in the single-chain antagonist, resulting in a complete loss of binding (28). Here, we investigated whether the affinity can be further increased in the single-chain antagonist by using subtle variants of Arg. From Table 1, it was clear that the guanidinium group is essential for binding because replacement with an amine or amide functional group showed at least a 70-fold reduction in binding to RXFP3. Citrulline substitution resulted in the loss of measurable affinity. Surprisingly, even a change in side-chain length by one carbon (using norarginine or homoarginine) is sufficient to significantly disrupt the binding to the receptor. This is a strong indication that the spatial relationship between the key Arg-binding site and the binding site of other groups are optimally targeted by R3 B1-22R, and no alteration to Arg at position 23 is tolerated. Given the significance of the Arg23 interactions, we tried to incorporate this residue in single-chain variants that also included the activation domain of relaxin-3. However, in neither variant was the Arg able to increase affinity above the one already observed for the relaxin-3 B-chain. These data highlight that the binding mode of the antagonist is not compatible with the interactions required for receptor activation.
NMR studies have shown that the antagonist is disordered in solution (28). It was suggested that a native conformation of R3 B1-22R would form when the peptide binds to RXFP3 (28). Here, we used NMR spectroscopy to analyze the influence of changing the solvent conditions on the structure of R3 B1-22R. The addition of TFE induced a structure with striking resemblance to the one seen in native relaxin-3, confirming that it is a preferred conformation even in the absence of covalent fixation to the A-chain via disulfide bonds. In relaxin-3, the helix ends at Cys22; it is, however, possible that in the active conformation of R3 B1-22R, it extends all the way to include Arg23. If so, Arg23 may further contribute to activity by stabilizing this conformation through an electrostatic interaction between its positive charge and the negative end of the helical dipole. Such a conformation might also explain why a C-terminal amide is favored, as the negative charge of a free C terminus would interact unfavorably with the dipole and destabilize the helix.
Given the clear difference in binding contributions from individual amino acids between relaxin-3 and R3 B1-22R, we still wanted to further investigate the binding conformation of R3 B1-22R using sequence modifications. Proline residues are known to break helical structures and favoring turns because of their constrained structure and lack of hydrogen bonding potential due to the missing amide proton. We therefore introduced Pro residues at different positions to see what effect conformational restraints would have on binding. Substitution at Leu9 or Glu13 was relatively well-tolerated; however, binding was completely abolished with a Pro substitution at Ala17 or Phe20. Leu9 is located N-terminally to the helical segment and Glu13 in the N-terminal first helical turn; thus, these changes may not majorly influence the overall conformation. In contrast, Ala17 and Phe20 are located toward the C-terminal end of the helix, closer to the key binding residue Arg23. At position 20, the native Phe and an Ala substitution are equally well-tolerated; thus, the Pro is unlikely to remove an interaction or introduce a clash. The effect is instead highly likely to be due its structural effect, preventing the formation of the correct helical conformation for the interaction with RXFP3.
Rather than disrupting the conformation, we investigated whether instead introducing modifications promoting a helical structure would be beneficial for binding. The achiral Aib residue contains an extra methyl group at the Cα carbon. The increased steric hindrance restricts the number of backbone Φ and Ψ angle combinations that are favored and has been shown to promote helicity in peptides (32). Introduction of a single Aib residue at positions Ala17, Val18, and Thr21 at the critical C-terminal end of the helix was well-tolerated but did not improve binding. Multiple Aib residues were thus introduced to further strengthen the helical character. Rather than improving binding, this led to a modest reduction in binding. We speculate that this is likely a result of removal of multiple side chains that make some contribution to the binding, but the possibility that removal of too much of the freedom of the backbone is negatively influencing the favored backbone conformation cannot be ruled out.
Because Aib did not improve the binding interaction between R3 B1-22R and RXFP3, lactam bridge “staples” were introduced. The constraints were introduced to mimic one turn of an α-helix (i, i + 4). Similar stapling motif has been used for helix induction of a single-chain relaxin-3 agonist using lactam and hydrocarbon staples (23) and also in the related peptide INSL3 (35). Although the affinity and activity of single-chain relaxin-3 agonists were successfully improved with helical stapling compared with linear B-chain analogs, no improvement was seen for the relaxin-3 antagonist. Notably, it was the installment of the point modifications that would allow the stapling that resulted in the drop in affinity. The formation of the lactam bridges did improve affinity for RXFP3 compared with the linear peptide variants, but not enough to rescue binding to a level comparable with the native single-chain antagonist. The positions of the lactams were chosen to face away from the key binding motif; thus, if the antagonist was bound in a similar position to native relaxin-3, it would not interfere with the interaction. Indeed, this was the case for the stapled B-chain agonist, which would be required to bind in a native-like fashion to induce the structural changes required for receptor activation (23). However, consistent with the point modification data, the unfavorable results from the stapling of the antagonist highlight a difference in binding position that brings both sides of the helix into contact with the receptor. The Lys–Asp lactam at position 13–17 was the one most tolerated for RXFP3 binding. The Lys–Asp lactam has been found to be one of the most promising strategies to induce α-helicity using a pentapeptide screen (36). The 13–17 position is furthest away from the C-terminal binding site, and it was also found to be the one most suited for modification in the agonist variants (23).
Rather than trying to induce α-helicity, we tried alternative cyclizations. Cyclization has been widely used to stabilize peptides to improve protection against protease degradation, including in bioactive drug leads such as conotoxins (37). The relaxin-3 N-terminal region turns back along the helix in native relaxin-3; thus, we envisaged that linking the N terminus to a side chain could provide some conformational support. However, none of the head-to-side-chain cyclizations improved binding affinity for RXFP3. For the shorter R3 B6–22R, cyclization from the N terminus to position 21 via an introduced Glu residue resulted in complete loss of binding. This linkage likely compromised the ability to form the required helical conformation.
RXFP3 mutational studies primarily targeting acidic amino acid residues that are likely binding partners to the several important Arg residues in relaxin-3 have provided some insights into how native relaxin-3 binds to RXFP3 with high affinity (14, 21). The peptide uses Arg12 and Arg16 in the helical domain (18) to form electrostatic interactions with Asp145 and Glu244 in RXFP3, located in extracellular loops 1 and 2, respectively (26). The neighboring Ile15, Ile19, and Phe20 in relaxin-3 likely provide complementary hydrophobic contacts, creating an extensive continuous binding surface (Fig. 6). The terminal Arg26–Trp27 residues are known to be critical for activation and target a second binding site deeper in the binding pocket of RXFP3. Glu141 at the top of TM2 has been identified as the binding partner for Arg26 and Trp138 has been identified as the binding partner of Trp27 (14, 21). We have shown here that the antagonist R3 B1-22R likely adopts a helical conformation similar to relaxin-3 upon binding to RXFP3. It also utilizes amino acid residues in this helical region for interactions with RXFP3. In a companion article (41), we have explored the roles of features in RXFP3 in ligand recognition. The data from this study confirm that the binding sites of Arg12, Ile15, Arg16, and Ile19 are likely the same as for native relaxin-3. Notably, the antagonist relies far less on individual contributions from these positions, but instead, it is also able to target a second binding site in RXFP3, using features on the other face of the helix. We thus propose that the smaller size in the absence of an A-chain allows a different positioning of the helix, probably deeper in the binding pocket, which promotes this interaction. This arrangement will allow Arg23 to insert deep into the pocket, forming a perfect arrangement of interactions that are the key drivers of high affinity (Fig. 6). In the companion paper (41), this interaction is shown to involve both a cation-π interaction with Trp138 and a salt bridge with Glu141 in RXFP3.
Figure 6.

Differences in binding mode of R3 B1-22R and relaxin-3. Structural modifications in the form of staples, Aib substitutions, and Pro substitutions strongly suggest that R3 B1-22R adopts a native-like helical conformation when interacting with RXFP3. Important amino acid residues that are buried and involved in intramolecular interactions in relaxin-3 do contribute to the RXFP3 interactions in R3 B1-22R. We propose that the positioning of the helix differs in R3 B1-22R, creating an additional interaction surface. Positions that showed no tolerance to substitution are shown in red, positions with some tolerance are shown in yellow, and positions where substitutions were widely accepted are shown in green. Positions in relaxin-3 that are not exposed were not probed for binding.
Conclusion
In this study, we have reported extensive structure–activity data for the R3 B1-22R RXFP3 antagonist. This peptide has been shown to effectively reduce food intake and alcohol seeking in rodents, thus representing an important lead for treatment of obesity and addiction. Antagonist peptides are frequently developed from naturally occurring agonist ligands, and it was initially envisaged that the binding mode of R3 B1-22R would mimic the one of the native peptide. All evidence suggest that the antagonist adopts a helical conformation, but that it binds in a different configuration. These new insights suggest new avenues for the development of next-generation antagonists. It is unlikely that stapling strategies will be beneficial for this peptide, given that its positioning in RXFP3 means that they are likely to introduce clashes with the receptor. Instead, efforts could be directed at modifying side chains, in particular for amino acid residues on the face of the helix that is buried in relaxin-3, as these represent a new nonnative interaction that to date has not been optimized by nature or by medicinal chemistry approaches. This study also highlights the need for the development of individual SAR data for agonists and antagonists when targeting peptide G protein-coupled receptors.
Experimental procedures
Synthesis of relaxin-3 mutants
All R3 B1-22R variants were synthesized using Fmoc-based solid-phase peptide synthesis. The peptides were made at 0.125-mmol scale on Pal-PEG-PS or Tentagel-XV-RAM resins, resulting in a favored C-terminal amide (28). Excess Fmoc-protected amino acids (4 eq), 0.5 m HBTU (4 eq), and 1 m DIPEA (4 eq) were used to couple each amino acid, and Fmoc deprotection was carried out using 20% piperidine in N,N-dimethylformamide. For lactam bridge–containing peptides, Fmoc-Asp(Opis)-OH, Fmoc-Glu(Opis)-OH, and Fmoc-Lys(Mtt)-OH (CS Bio, Shanghai, China) were incorporated and side-chain deprotected on resin using 1% TFA in dichloromethane. Side-chain lactam bond cyclization was done on resin through treatment with HBTU (4 eq) and DIPEA (4 eq) under microwave heating, when required. Upon completion of synthesis, the peptides were cleaved from resin using a mixture of TFA/TIPS/DODT/H2O (92.5:2.5:2.5:2.5), precipitated with diethyl ether, and lyophilized. All peptides were purified by RP-HPLC using C-18 columns and characterized using electrospray ionization-MS (API 2000, Ab Sciex). Analytical RP-HPLC using a C18 column with a flow rate of 1 ml/min and a 1% linear gradient was used to confirm the high purity of the synthesized peptides. Theoretical and experimental masses and pI values for all peptides are presented in Tables S2 and S3.
Cell culture
CHO-K1 cells stably transfected with RXFP3 (38) were maintained in Dulbecco's modified Eagle's medium/Ham's F-12 medium supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, 1% l-glutamine, and 400 ng/ml G418 for RXFP3-expressing cell selection.
Competition binding assay
Determination of binding affinity was carried out as described previously (30). Briefly, CHO-K1 cells stably transfected with RXFP3 were seeded into 96-well plates and incubated with 5 nm europium-DTPA-R3 B1-22R and increasing concentrations of R3 B1-22R antagonist analogues (0.001 nm to 10 μm). Fluorescence measurements of the europium-labeled tracer were recorded with 340-nm excitation and 614-nm emission wavelengths. The assays were conducted with a minimum of three independent experiments. Data are presented as mean ± S.E. pKi values were determined using one-site fit Ki and a Kd value of 26 nm. Statistical analyses were conducted using one-way analysis of variance with uncorrected Fisher's least significant difference in GraphPad Prism version 7.
NMR spectroscopy
A sample for NMR spectroscopy was prepared containing 0.2 mg of R3 B1-22R in 0.5 ml of 60% H2O, 10% D2O, 30% TFE. NMR data, including two-dimensional TOCSY and NOESY, were recorded at 298 K and 700 MHz on a Bruker Avance II spectrometer equipped with a cryoprobe. The data were processed using Topspin and analyzed in CARA (39). The data were referenced to 4,4-dimethyl-4-silapentane-1-sulfonic acid at 0.0 ppm, and secondary shifts were determined using random coil shifts reported by Wishart et al. (40).
Author contributions
L. M. H.-K., H. S. L., and M. V. J. data curation; L. M. H.-K., H. S. L., K. J. R., and R. A. B. formal analysis; L. M. H.-K., H. S. L., M. V. J., A. S., V. R., and M. A. H. investigation; L. M. H.-K., H. S. L., A. S., and R. A. B. methodology; L. M. H.-K., H. S. L., and K. J. R. writing-original draft; L. M. H.-K., H. S. L., R. A. B., and K. J. R. writing-review and editing; A. S. and K. J. R. supervision; R. A. B. and K. J. R. conceptualization; R. A. B. and K. J. R. resources; R. A. B. and K. J. R. funding acquisition; R. A. B. validation; K. J. R. project administration.
Supplementary Material
Acknowledgments
We thank Tania Ferraro and Sharon Layfield for technical assistance.
This work was supported by the National Health and Medical Research Council (NHMRC), Australia, through Project Grants 1063425 and 1066369 (to R. A. D. B. and K. J. R.). Research at the Florey was supported by the Victorian Government Operational Infrastructure Support Program. L. M. H. K., K. J. R., and R. A. D. B. are inventors on Australian Patent 2010904046 and United States patent application 13/821726, Modified Relaxin B Chain Peptides.
This article contains Fig. S1 and Tables S1–S3.
- RXFP
- relaxin family peptide receptor
- Fmoc
- N-(9-fluorenyl)methoxycarbonyl
- Abu
- aminobutyric acid
- Aib
- aminoisobutyric acid
- TFE
- trifluoroethanol
- HBTU
- 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- DIPEA
- N,N-diisopropylethylamine
- TIPS
- triisopropylsilane
- DODT
- 3,6-dioxa-1,8-octane-dithiol.
References
- 1. Bathgate R. A., Samuel C. S., Burazin T. C., Layfield S., Claasz A. A., Reytomas I. G., Dawson N. F., Zhao C., Bond C., Summers R. J., Parry L. J., Wade J. D., and Tregear G. W. (2002) Human relaxin gene 3 (H3) and the equivalent mouse relaxin (M3) gene: novel members of the relaxin peptide family. J. Biol. Chem. 277, 1148–1157 10.1074/jbc.M107882200 [DOI] [PubMed] [Google Scholar]
- 2. Smith C. M., Chua B. E., Zhang C., Walker A. W., Haidar M., Hawkes D., Shabanpoor F., Hossain M. A., Wade J. D., Rosengren K. J., and Gundlach A. L. (2014) Central injection of relaxin-3 receptor (RXFP3) antagonist peptides reduces motivated food seeking and consumption in C57BL/6J mice. Behav. Brain Res. 268, 117–126 10.1016/j.bbr.2014.03.037 [DOI] [PubMed] [Google Scholar]
- 3. McGowan B. M., Stanley S. A., Smith K. L., White N. E., Connolly M. M., Thompson E. L., Gardiner J. V., Murphy K. G., Ghatei M. A., and Bloom S. R. (2005) Central relaxin-3 administration causes hyperphagia in male Wistar rats. Endocrinology 146, 3295–3300 10.1210/en.2004-1532 [DOI] [PubMed] [Google Scholar]
- 4. Tanaka M., Iijima N., Miyamoto Y., Fukusumi S., Itoh Y., Ozawa H., and Ibata Y. (2005) Neurons expressing relaxin 3/INSL 7 in the nucleus incertus respond to stress. Eur. J. Neurosci. 21, 1659–1670 10.1111/j.1460-9568.2005.03980.x [DOI] [PubMed] [Google Scholar]
- 5. Banerjee A., Shen P. J., Ma S., Bathgate R. A., and Gundlach A. L. (2010) Swim stress excitation of nucleus incertus and rapid induction of relaxin-3 expression via CRF1 activation. Neuropharmacology 58, 145–155 10.1016/j.neuropharm.2009.06.019 [DOI] [PubMed] [Google Scholar]
- 6. Ryan P. J., Kastman H. E., Krstew E. V., Rosengren K. J., Hossain M. A., Churilov L., Wade J. D., Gundlach A. L., and Lawrence A. J. (2013) Relaxin-3/RXFP3 system regulates alcohol-seeking. Proc. Natl. Acad Sci. U.S.A. 110, 20789–20794 10.1073/pnas.1317807110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ma S., Olucha-Bordonau F. E., Hossain M. A., Lin F., Kuei C., Liu C., Wade J. D., Sutton S. W., Nuñez A., and Gundlach A. L. (2009) Modulation of hippocampal theta oscillations and spatial memory by relaxin-3 neurons of the nucleus incertus. Learn. Mem. 16, 730–742 10.1101/lm.1438109 [DOI] [PubMed] [Google Scholar]
- 8. Burazin T. C., Bathgate R. A., Macris M., Layfield S., Gundlach A. L., and Tregear G. W. (2002) Restricted, but abundant, expression of the novel rat gene-3 (R3) relaxin in the dorsal tegmental region of brain. J. Neurochem. 82, 1553–1557 10.1046/j.1471-4159.2002.01114.x [DOI] [PubMed] [Google Scholar]
- 9. Shabanpoor F., Separovic F., and Wade J. D. (2009) The human insulin superfamily of polypeptide hormones. Vitam. Horm. 80, 1–31 10.1016/S0083-6729(08)00601-8 [DOI] [PubMed] [Google Scholar]
- 10. Hsu S. Y., Nakabayashi K., Nishi S., Kumagai J., Kudo M., Sherwood O. D., and Hsueh A. J. (2002) Activation of orphan receptors by the hormone relaxin. Science 295, 671–674 10.1126/science.1065654 [DOI] [PubMed] [Google Scholar]
- 11. Kumagai J., Hsu S. Y., Matsumi H., Roh J. S., Fu P., Wade J. D., Bathgate R. A., and Hsueh A. J. (2002) INSL3/Leydig insulin-like peptide activates the LGR8 receptor important in testis descent. J. Biol. Chem. 277, 31283–31286 10.1074/jbc.C200398200 [DOI] [PubMed] [Google Scholar]
- 12. Liu C., Kuei C., Sutton S., Chen J., Bonaventure P., Wu J., Nepomuceno D., Kamme F., Tran D. T., Zhu J., Wilkinson T., Bathgate R., Eriste E., Sillard R., and Lovenberg T. W. (2005) INSL5 is a high affinity specific agonist for GPCR142 (GPR100). J. Biol. Chem. 280, 292–300 10.1074/jbc.M409916200 [DOI] [PubMed] [Google Scholar]
- 13. Liu C., Chen J., Sutton S., Roland B., Kuei C., Farmer N., Sillard R., and Lovenberg T. W. (2003) Identification of relaxin-3/INSL7 as a ligand for GPCR142. J. Biol. Chem. 278, 50765–50770 10.1074/jbc.M308996200 [DOI] [PubMed] [Google Scholar]
- 14. Liu C., Eriste E., Sutton S., Chen J., Roland B., Kuei C., Farmer N., Jörnvall H., Sillard R., and Lovenberg T. W. (2003) Identification of relaxin-3/INSL7 as an endogenous ligand for the orphan G-protein-coupled receptor GPCR135. J. Biol. Chem. 278, 50754–50764 10.1074/jbc.M308995200 [DOI] [PubMed] [Google Scholar]
- 15. Sudo S., Kumagai J., Nishi S., Layfield S., Ferraro T., Bathgate R. A., and Hsueh A. J. (2003) H3 relaxin is a specific ligand for LGR7 and activates the receptor by interacting with both the ectodomain and the exoloop 2. J. Biol. Chem. 278, 7855–7862 10.1074/jbc.M212457200 [DOI] [PubMed] [Google Scholar]
- 16. Rosengren K. J., Lin F., Bathgate R. A., Tregear G. W., Daly N. L., Wade J. D., and Craik D. J. (2006) Solution structure and novel insights into the determinants of the receptor specificity of human relaxin-3. J. Biol. Chem. 281, 5845–5851 10.1074/jbc.M511210200 [DOI] [PubMed] [Google Scholar]
- 17. Patil N. A., Rosengren K. J., Separovic F., Wade J. D., Bathgate R. A. D., and Hossain M. A. (2017) Relaxin family peptides: structure-activity relationship studies. Br. J. Pharmacol. 174, 950–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kuei C., Sutton S., Bonaventure P., Pudiak C., Shelton J., Zhu J., Nepomuceno D., Wu J., Chen J., Kamme F., Seierstad M., Hack M. D., Bathgate R. A., Hossain M. A., Wade J. D., Atack J., Lovenberg T. W., and Liu C. (2007) R3(BΔ23–27)R/I5 chimeric peptide, a selective antagonist for GPCR135 and GPCR142 over relaxin receptor LGR7: in vitro and in vivo characterization. J. Biol. Chem. 282, 25425–25435 10.1074/jbc.M701416200 [DOI] [PubMed] [Google Scholar]
- 19. Hossain M. A., Rosengren K. J., Haugaard-Jönsson L. M., Zhang S., Layfield S., Ferraro T., Daly N. L., Tregear G. W., Wade J. D., and Bathgate R. A. (2008) The A-chain of human relaxin family peptides has distinct roles in the binding and activation of the different relaxin family peptide receptors. J. Biol. Chem. 283, 17287–17297 10.1074/jbc.M801911200 [DOI] [PubMed] [Google Scholar]
- 20. Shabanpoor F., Akhter Hossain M., Ryan P. J., Belgi A., Layfield S., Kocan M., Zhang S., Samuel C. S., Gundlach A. L., Bathgate R. A., Separovic F., and Wade J. D. (2012) Minimization of human relaxin-3 leading to high-affinity analogues with increased selectivity for relaxin-family peptide 3 receptor (RXFP3) over RXFP1. J. Med. Chem. 55, 1671–1681 10.1021/jm201505p [DOI] [PubMed] [Google Scholar]
- 21. Liu C., Chen J., Kuei C., Sutton S., Nepomuceno D., Bonaventure P., and Lovenberg T. W. (2005) Relaxin-3/insulin-like peptide 5 chimeric peptide, a selective ligand for G protein-coupled receptor (GPCR)135 and GPCR142 over leucine-rich repeat-containing G protein-coupled receptor 7. Mol. Pharmacol. 67, 231–240 10.1124/mol.104.006700 [DOI] [PubMed] [Google Scholar]
- 22. Haugaard-Jönsson L. M., Hossain M. A., Daly N. L., Bathgate R. A., Wade J. D., Craik D. J., and Rosengren K. J. (2008) Structure of the R3/I5 chimeric relaxin peptide, a selective GPCR135 and GPCR142 agonist. J. Biol. Chem. 283, 23811–23818 10.1074/jbc.M800489200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hojo K., Hossain M. A., Tailhades J., Shabanpoor F., Wong L. L., Ong-Pålsson E. E., Kastman H. E., Ma S., Gundlach A. L., Rosengren K. J., Wade J. D., and Bathgate R. A. (2016) Development of a single-chain peptide agonist of the relaxin-3 receptor using hydrocarbon stapling. J. Med. Chem. 59, 7445–7456 10.1021/acs.jmedchem.6b00265 [DOI] [PubMed] [Google Scholar]
- 24. Jayakody T., Marwari S., Lakshminarayanan R., Tan F. C., Johannes C. W., Dymock B. W., Poulsen A., Herr D. R., and Dawe G. S. (2016) Hydrocarbon stapled B chain analogues of relaxin-3 retain biological activity. Peptides 84, 44–57 10.1016/j.peptides.2016.08.001 [DOI] [PubMed] [Google Scholar]
- 25. Zhang W. J., Wang X. Y., Guo Y. Q., Luo X., Gao X. J., Shao X. X., Liu Y. L., Xu Z. G., and Guo Z. Y. (2014) The highly conserved negatively charged Glu141 and Asp145 of the G-protein-coupled receptor RXFP3 interact with the highly conserved positively charged arginine residues of relaxin-3. Amino Acids 46, 1393–1402 10.1007/s00726-014-1705-3 [DOI] [PubMed] [Google Scholar]
- 26. Bathgate R. A., Oh M. H., Ling W. J., Kaas Q., Hossain M. A., Gooley P. R., and Rosengren K. J. (2013) Elucidation of relaxin-3 binding interactions in the extracellular loops of RXFP3. Front. Endocrinol. (Lausanne) 4, 13 10.3389/fendo.2013.00013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hu M. J., Shao X. X., Wang J. H., Wei D., Liu Y. L., Xu Z. G., and Guo Z. Y. (2016) Identification of hydrophobic interactions between relaxin-3 and its receptor RXFP3: implication for a conformational change in the B-chain C-terminus during receptor binding. Amino Acids 48, 2227–2236 10.1007/s00726-016-2260-x [DOI] [PubMed] [Google Scholar]
- 28. Haugaard-Kedström L. M., Shabanpoor F., Hossain M. A., Clark R. J., Ryan P. J., Craik D. J., Gundlach A. L., Wade J. D., Bathgate R. A. D., and Rosengren K. J. (2011) Design, synthesis, and characterization of a single-chain peptide antagonist for the relaxin-3 receptor RXFP3. J. Am. Chem. Soc. 133, 4965–4974 10.1021/ja110567j [DOI] [PubMed] [Google Scholar]
- 29. Calvez J., de Ávila C., Matte L. O., Guèvremont G., Gundlach A. L., and Timofeeva E. (2016) Role of relaxin-3/RXFP3 system in stress-induced binge-like eating in female rats. Neuropharmacology 102, 207–215 10.1016/j.neuropharm.2015.11.014 [DOI] [PubMed] [Google Scholar]
- 30. Haugaard-Kedström L. M., Wong L. L., Bathgate R. A., and Rosengren K. J. (2015) Synthesis and pharmacological characterization of a europium-labelled single-chain antagonist for binding studies of the relaxin-3 receptor RXFP3. Amino Acids 47, 1267–1271 10.1007/s00726-015-1961-x [DOI] [PubMed] [Google Scholar]
- 31. Fischer P. M. (2003) The design, synthesis and application of stereochemical and directional peptide isomers: a critical review. Curr. Protein Pept. Sci. 4, 339–356 10.2174/1389203033487054 [DOI] [PubMed] [Google Scholar]
- 32. Mahalakshmi R., and Balaram P. (2006) Non-protein amino acids in the design of secondary structure scaffolds. Methods Mol. Biol. 340, 71–94 [DOI] [PubMed] [Google Scholar]
- 33. Hossain M. A., Kocan M., Yao S. T., Royce S. G., Nair V. B., Siwek C., Patil N. A., Harrison I. P., Rosengren K. J., Selemidis S., Summers R. J., Wade J. D., Bathgate R. A. D., and Samuel C. S. (2016) A single-chain derivative of the relaxin hormone is a functionally selective agonist of the G protein-coupled receptor, RXFP1. Chem. Sci. 7, 3805–3819 10.1039/C5SC04754D [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hossain M. A., Bathgate R. A., Rosengren K. J., Shabanpoor F., Zhang S., Lin F., Tregear G. W., and Wade J. D. (2009) The structural and functional role of the B-chain C-terminal arginine in the relaxin-3 peptide antagonist, R3(BΔ23–27)R/I5. Chem. Biol. Drug Des. 73, 46–52 10.1111/j.1747-0285.2008.00756.x [DOI] [PubMed] [Google Scholar]
- 35. Shabanpoor F., Hughes R. A., Zhang S., Bathgate R. A., Layfield S., Hossain M. A., Tregear G. W., Separovic F., and Wade J. D. (2010) Effect of helix-promoting strategies on the biological activity of novel analogues of the B-chain of INSL3. Amino Acids 38, 121–131 10.1007/s00726-008-0219-2 [DOI] [PubMed] [Google Scholar]
- 36. de Araujo A. D., Hoang H. N., Kok W. M., Diness F., Gupta P., Hill T. A., Driver R. W., Price D. A., Liras S., and Fairlie D. P. (2014) Comparative α-helicity of cyclic pentapeptides in water. Angew. Chem. Int. Ed. Engl. 53, 6965–6969 10.1002/anie.201310245 [DOI] [PubMed] [Google Scholar]
- 37. Clark R. J., Fischer H., Dempster L., Daly N. L., Rosengren K. J., Nevin S. T., Meunier F. A., Adams D. J., and Craik D. J. (2005) Engineering stable peptide toxins by means of backbone cyclization: stabilization of the α-conotoxin MII. Proc. Natl. Acad. Sci. U.S.A. 102, 13767–13772 10.1073/pnas.0504613102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Van der Westhuizen E. T., Sexton P. M., Bathgate R. A., and Summers R. J. (2005) Responses of GPCR135 to human gene 3 (H3) relaxin in CHO-K1 cells determined by microphysiometry. Ann. N.Y. Acad. Sci. 1041, 332–337 10.1196/annals.1282.053 [DOI] [PubMed] [Google Scholar]
- 39. Keller R. L. J. (2004) The Computer Aided Resonance Assignment Tutorial, Cantina Verlag, Goldau, Switzerland [Google Scholar]
- 40. Wishart D. S., Bigam C. G., Holm A., Hodges R. S., and Sykes B. D. (1995) 1H, 13C and 15N random coil NMR chemical shifts of the common amino acids. I. Investigations of nearest-neighbor effects. J. Biomol. NMR 5, 67–81 10.1007/BF00227471 [DOI] [PubMed] [Google Scholar]
- 41. Wong L. L. L., Scott D. J., Hossain M. A., Kaas Q., Rosengren K. J., and Bathgate R. A. D. (2018) Distinct but overlapping binding sites of agonist and antagonist at the relaxin family peptide 3 (RXFP3) receptor. J. Biol. Chem. 293, 15777–15789 10.1074/jbc.RA118.002645 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.