

Abstract
Class I ribonucleotide reductase (RNR) consists of a catalytic subunit (NrdA) and a radical-generating subunit (NrdB) that together catalyze reduction of ribonucleotides to their corresponding deoxyribonucleotides. NrdB from the firmicute Facklamia ignava is a unique fusion protein with N-terminal add-ons of a glutaredoxin (Grx) domain followed by an ATP-binding domain, the ATP cone. Grx, usually encoded separately from the RNR operon, is a known RNR reductant. We show that the fused Grx domain functions as an efficient reductant of the F. ignava class I RNR via the common dithiol mechanism and, interestingly, also via a monothiol mechanism, although less efficiently. To our knowledge, a Grx that uses both of these two reaction mechanisms has not previously been observed with a native substrate. The ATP cone is in most RNRs an N-terminal domain of the catalytic subunit. It is an allosteric on/off switch promoting ribonucleotide reduction in the presence of ATP and inhibiting RNR activity in the presence of dATP. We found that dATP bound to the ATP cone of F. ignava NrdB promotes formation of tetramers that cannot form active complexes with NrdA. The ATP cone bound two dATP molecules but only one ATP molecule. F. ignava NrdB contains the recently identified radical-generating cofactor MnIII/MnIV. We show that NrdA from F. ignava can form a catalytically competent RNR with the MnIII/MnIV-containing NrdB from the flavobacterium Leeuwenhoekiella blandensis. In conclusion, F. ignava NrdB is fused with a Grx functioning as an RNR reductant and an ATP cone serving as an on/off switch.
Keywords: ribonucleotide reductase, allosteric regulation, oxidation-reduction (redox), radical, manganese, ATP-cone, dATP inhibition, dithiol–monothiol, glutaredoxin, tetramers
Introduction
Ribonucleotide reductase (RNR) 2 is an essential enzyme that catalyzes the synthesis of the DNA building blocks (dNTPs) by reduction of the four ribonucleotides. RNR plays a key role in DNA synthesis and DNA repair and consequently attracts biomedical interest as a potential target for antibacterial substances and for anticancer therapies. Currently, the RNR enzyme family comprises three different RNR classes and several subclasses. The three classes have a common reaction mechanism that builds on radical chemistry but differ in the way they initiate the radical mechanism (1–5). The class I RNRs consist of a larger catalytic component (NrdA) and a smaller radical-generating metal-containing component (NrdB) in which the dinuclear metal site differs between subclasses. Currently, class I RNRs have been subclassified based on radical cofactor type (subclasses Ia, Ib, Ic, Id, and Ie) or evolutionary history (subclasses NrdA/B followed by a small letter plus subclass NrdE/F) (1, 6). Metal content does not always follow phylogeny because two unrelated Mn2 subclasses exist, where one subclass contains a tyrosyl radical in the vicinity of a MnIII/MnIII center (Ib, NrdE/F), and another recently identified subclass (Id, NrdAi/Bi) contains a mixed valent MnIII/MnIV metal center that harbors the unpaired electron (7–9). In eukaryotic RNRs and several evolutionarily unrelated bacterial class I subclasses, the NrdB component contains a stable tyrosyl radical in the vicinity of a diferric metal center (Ia). In another bacterial subclass (Ic), a mutational change in the radical-carrying tyrosine to phenylalanine is accompanied by a mixed valent MnIV/FeIII metal center (10, 11). Recently, a metal independent subclass (Ie) with an intrinsically modified dopa radical cofactor was discovered (12).
All class I RNRs contain a C-terminal redox-active cysteine pair in NrdA that functions as a reductant of a cysteine pair in the active site that is oxidized during catalysis. Physiological regeneration of active NrdA is performed by members of the redoxin family, with NADPH as ultimate electron source (13–15). Three types of redoxin have been found to reduce the C-terminal cysteines in class I RNRs: (i) thioredoxin that receives the electrons from NADPH via thioredoxin reductase, (ii) glutaredoxin (Grx) that receives the electrons from NADPH via GSH reductase and GSH, and (iii) NrdH-redoxin that also receives the electrons from NADPH via thioredoxin reductase even though NrdH is more similar to Grx than to thioredoxin. Whereas the nrdA and nrdB genes are mostly encoded close to each other in bacteria, the trx and grx genes are usually found elsewhere in the genome. Only the nrdH gene is predominantly encoded in the vicinity of the corresponding RNR genes, which, for historical reasons, in this particular subclass are called nrdE (encoding the catalytic subunit) and nrdF (encoding the radical-generating subunit).
We discovered an intriguing fusion of a grx gene to the nrdB gene in the bacterium Facklamia ignava, resulting in an ORF encoding a fusion protein. The F. ignava NrdB fusion protein consists of an N-terminal Grx domain followed by an ATP-cone domain and then the radical-generating subunit. An N-terminal grx fusion to the nrdB gene in Francisella tularensis was noticed by us previously (16). The redoxin domain in both of these fusions are most similar to the grxC domain family (COG0695). Whereas the γ-proteobacterium F. tularensis is a well-studied human pathogen causing tularemia (17), the Firmicutes genus Facklamia was first described in 1997 and has since been identified in samples from a wide range of animals and as a human pathogen (18–20).
RNR has been described as a textbook example of allosteric regulation in enzymes and employs two different allosteric mechanisms to regulate the synthesis of dNTPs (21, 22). One common mechanism regulates the balance between the four dNTPs in a sophisticated feedback control at the specificity site (s-site). Additional allosteric regulation is provided by the overall activity site (a-site), which works as a general on/off switch and constitutes a separate domain called the ATP cone. In short, the enzyme is active when ATP is bound and when dATP is bound, the enzyme is turned off. We have recently shown that the ATP cone can be horizontally transferred between different RNRs and even to different subunits of the holoenzyme (8, 23). In an overwhelming number of cases, the ATP cone is an N-terminal domain of the catalytic subunit of RNR (23). F. ignava RNR instead carries an ATP cone in its NrdB protein, between the N-terminal Grx domain and the radical-generating domain. We have recently reported a similar N-terminal ATP-cone fusion to NrdB in Leeuwenhoekiella blandensis (8). Both of these fusion proteins belong to the NrdBi subclass, which harbors a few additional ATP-cone::NrdB fusions.
In this study we have used the F. ignava RNR to study two major questions: does the fused Grx domain function as a reductant for the holoenzyme, and does the fused ATP cone function as a general on/off switch? To investigate these questions, we used a series of biochemical assays to show that the Grx domain is indeed an efficient reductant of F. ignava RNR and that the fused ATP-cone domain is a functional allosteric domain.
Results
Glutaredoxin fusions to RNR components
The 496-residue F. ignava (Firmicutes) NrdB fusion protein consists of an N-terminal Grx domain (residues 4–61, with the characteristic cysteine pair at residues 12 and 15) followed by an ATP-cone domain (residues 84–169) and thereafter the NrdB proper. The nrdA gene is located 46 nucleotides downstream of the nrdB gene, and the two genes conceivably form an operon (Fig. 1). The F. ignava NrdB is a member of the NrdBi phylogenetic subclass (http://rnrdb.pfitmap.org), 3 like all other NrdBs in which we have detected N-terminal ATP cones (8).
Figure 1.

The F. ignava nrdAB operon and some other class I RNR operons with grx fusions. Transcriptional and translational directions are from left to right. The F. tularensis operon occur in all Francisella spp., and the displayed vibrio-phage operon is found in most vibrio phages (Table S1).
Spurred by the discovery of the Grx fusion to F. ignava NrdBi, we performed a search of the RefSeq database for combinations of RNR proteins and Grx domains. Grx fusions were found in all RNR components (NrdA, NrdB, NrdD, and NrdJ), in some cases together with an ATP cone (Fig. 1 and Table S1). Grx fused to NrdB were detected in all Francisella spp. and Allofrancisella guangzhouensis (both γ-proteobacteria; subclass NrdBk) and in 24 viruses (NrdBe, NrdBg, and NrdBk) (Fig. 1 and Table S1). In addition, a grx::nrdE fusion was found in Streptococcus pneumoniae, a grx::nrdD fusion in Lachnospiraceae bacterium TWA4, a grx::nrdJ fusion in Labrenzia aggregata, and grx::nrdA fusions in two viruses (Table S1).
Because many Firmicutes lack GSH and instead produce another low molecular weight reductant called bacillithiol (24), we also searched the F. ignava genome for the presence of GSH biosynthesis and bacillithiol biosynthesis genes (gshA, gshB, gshF, bshA, bshB1, bshB2, and bshC). F. ignava and all other Facklamia spp. except one, encode the bifunctional gshF gene that is primarily found in Firmicutes (25). The Facklamia gshF has extensive similarity primarily to the gshA gene (Table S2). The GSH reductase gene gor was only found in Facklamia sourekii. The closest ortholog in F. ignava is a mercury(II) reductase and a dihydrolipoyl dehydrogenase, both with ∼50% similarity to F. sourekii gor. There were no genes corresponding to the bacillithiol biosynthesis genes in any Facklamia spp., apart from a glycosyl transferase gene with some similarity to bshA. Our results show that F. ignava and other Facklamia spp. have the capacity to synthesize GSH.
Redox activity of the NrdB-fused glutaredoxin
Using a series of cysteine-to-serine mutant proteins, we delineated the reaction mechanism of the fused Grx domain. Grx proteins usually reduce RNRs via a dithiol mechanism, but e.g. a human Grx has been reported to work via a glutathionylation mechanism (26–28). To test the capacity of the Grx domain in F. ignava NrdB to perform a dithiol reduction, we constructed two mutant proteins with a serine instead of cysteine in one or the other of the two redox-active residues in the Grx domain (C12S and C15S) and the corresponding double mutant (C12S/C15S).
In a first set of experiments the mutants were compared with the WT protein in a redox cycle with the artificial substrate 2-hydroxyethyl disulfide (HED). As evident from fig. 2A, the WT and C15S mutant proteins reduced the HED substrate, whereas the C12S mutant and the double mutant did not. The Km for HED was 0.6 ± 0.09 mm for the WT protein and 1.3 ± 0.24 mm for the C15S protein, and the Vmax was ∼2-fold higher for the WT compared with C15S at saturating HED (Fig. 2B). In a GSH titration experiment with constant HED, the Km for GSH was 3 ± 0.9 mm for the C15S mutant protein, and the rate was 44 μm/min (Fig. 2C), corresponding to a redoxin kcat of 7.3 s−1. Activity in the absence of one cysteine demonstrates that the Grx domain in F. ignava NrdB can work via a monothiol mechanism utilizing Cys-12 as the redox-active cysteine in presence of HED. The behavior of the WT protein in the GSH titration experiment (Fig. 2C) cannot be explained by a pure dithiol reaction mechanism. One possible explanation is that a monothiol mechanism may interfere at higher GSH concentrations.
Figure 2.

HED reduction capacity of F. ignava NrdB. Glutaredoxin activity was measured as NADPH consumption in presence of 0.1 μm protein. A, WT and mutant proteins in the presence of 0.75 mm HED and 4 mm GSH. Assays were performed in triplicate with standard deviations shown. B, HED titration of WT and C15S proteins in the presence of 4 mm GSH. C, GSH titration of WT and C15S proteins in presence of 0.75 mm HED.
In a second set of experiments, we compared the ability of the WT and mutant Grx domains to function as reductants in RNR assays. High specific activity (kcat 1.4 ± 0.06 s−1) with an apparent Km for GSH of 1.2 ± 0.2 mm was only obtained with the WT protein (Fig. 3). Of the mutant proteins, C12S and the C12S/C15S were deficient in ribonucleotide reduction with both 4 and 10 mm GSH, whereas their specific activity was on par with the WT enzyme when the Grx domain was bypassed using DTT as reductant (Fig. 3C, inset). Interestingly, the C15S mutant promoted a low but significant GSH-dependent ribonucleotide reductase activity, as measured both as consumption of NADPH (Fig. 3A) and as formation of dCDP (Fig. 3C), but it was not possible to reach a Vmax for the RNR activity of the C15S protein even at 20 mm GSH (Fig. 3B and data not shown). The GSH concentration of Facklamia spp. is not known, but GSH concentrations in studied bacteria range between 0.1 and 10 mm, with Firmicutes generally on the high side (25, 29). Conceivably, the Grx fused to F. ignava NrdB is most efficiently promoting turnover of the F. ignava RNR via a dithiol mechanism, and at 10 mm GSH concentration, the C15S mutant protein can promote ∼4-fold less efficient ribonucleotide reduction via a monothiol mechanism involving Cys-12.
Figure 3.
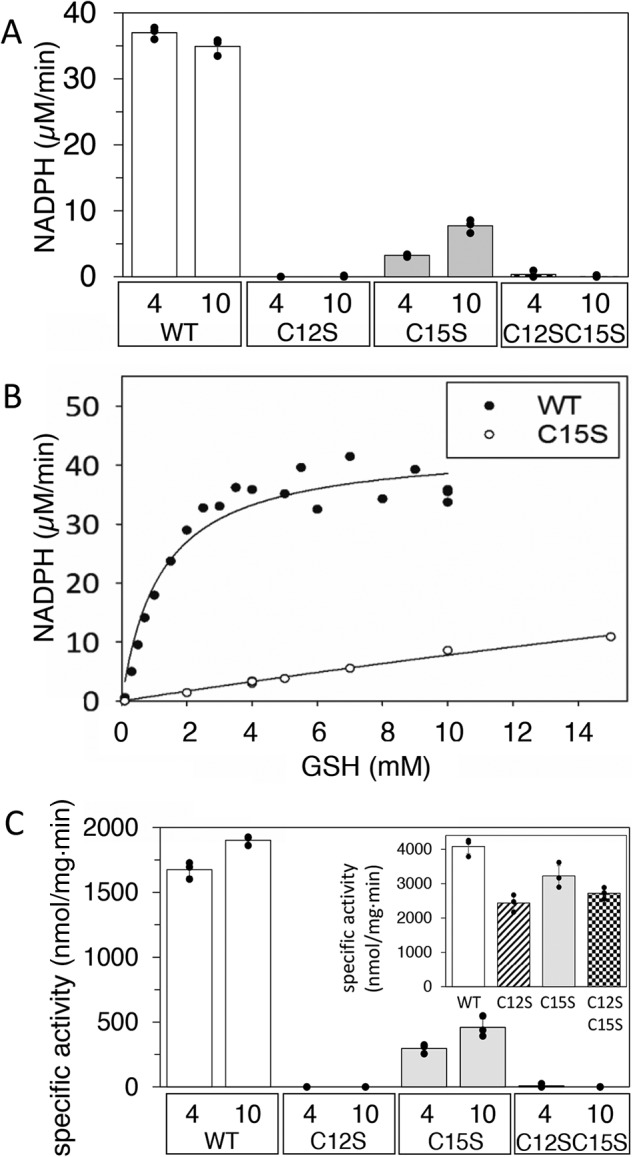
GSH-dependent RNR activity of F. ignava NrdB WT and mutant proteins. CDP was used as substrate, and 3 mm ATP was used as effector. A, RNR activity measured as NADPH consumption in presence of 0.5 μm NrdB. B, GSH-dependent NADPH consumption of the WT (●) and the C15S (○) NrdB. C, GSH-dependent specific activity measured as dCDP formation. Inset, DTT-dependent (10 mm) specific activity measured as dCDP formation. GSH concentrations (4 and 10 mm) are indicated in A and C. Assays in A and C were performed in triplicate with standard deviations shown.
Substrate specificity regulation of F. ignava RNR via the s-site
Using a four-substrate activity assay in the presence of saturating concentrations of the substrate specificity site (s-site) effectors ATP, dTTP, or dGTP, we found that F. ignava RNR has a similar specificity regulation pattern to most characterized RNRs (3). ATP stimulated the reduction of CDP and UDP, whereas dTTP stimulated the reduction of GDP, and dGTP stimulated the reduction of ADP and GDP (Fig. 4). There was also a low activity of predominantly CDP reduction in the absence of allosteric effectors. Using mixtures of allosteric effectors, we observed that dTTP-induced GDP reduction increased in the presence of ATP (Fig. 5A), as is commonly seen in RNRs (3).
Figure 4.

Substrate specificity of F. ignava class I RNR. Enzyme assays were performed in mixtures with 0.5 mm of each of the four substrates (ADP, CDP, GDP, and UDP) and a saturating concentration of one effector nucleotide at a time. Assays were performed in triplicate with standard deviations shown.
Figure 5.

Inhibition and activation of WT and mutant enzyme activity by dATP and ATP. A and B, ATP titration (A) and dATP titration (B) of enzyme loaded with 2 mm dTTP and GDP as substrate. Specific activities of NrdB proteins were measured with a 10-fold excess of NrdA. WT NrdB (●) had a starting activity of 830 ± 120 nmol/min·mg in the absence of added ATP and reached a Vmax of 1850 ± 200 nmol/min·mg (kcat = 1.8 s−1) in the presence of ATP, whereas NrdBΔ169 (○) had a specific activity of 3400 ± 500 nmol/min·mg (kcat = 2.2 s−1) in the absence of ATP that was not affected by addition of ATP or dATP.
Overall activity of F. ignava RNR is regulated via the NrdB-linked ATP-cone
We performed a series of activity assays with CDP as substrate to elucidate the potential roles of ATP and dATP in activating and inhibiting the enzyme. The presence of ATP activated the enzyme, whereas dATP showed a dual effect: activating the enzyme at low concentrations and inhibiting enzyme activity at 3 μm and higher (Fig. S1). The kinetics are complex, because ATP and dATP can bind both to the s-site in NrdA, as well as the ATP cone in NrdB. To analyze the effects of ATP or dATP at the ATP cone of NrdB, the specificity site of NrdA was saturated with dTTP, and GDP was used as substrate, giving a starting specific activity (normalized to 100% in Fig. 5) even in the absence of added ATP. In WT NrdB KL for ATP-dependent activation was 47 ± 12 μm (Fig. 5A), and Ki for dATP-dependent inhibition was 1.3 ± 0.23 μm (Fig. 5B). The activity of the deletion mutant that lacks both the Grx domain and the ATP cone (NrdBΔ169) was not affected by addition of either ATP or dATP (Fig. 5, A and B). The initial kcat of the NrdBΔ169 in the presence of dTTP-loaded NrdA was 2.2 s−1, i.e. almost three times higher than that of full-length NrdB (0.8 s−1). However, ATP addition increased the activity of WT NrdB to 1.8 s−1 (Fig. 5A), i.e. on par with the NrdBΔ169 mutant and other RNR enzymes. Titration with dADP inhibited the WT enzyme activity (Fig. S2), although less strongly than dATP did.
dATP binding to NrdB induces formation of higher oligomeric complexes
To elucidate the mechanism of allosteric overall activity regulation governed by the NrdB-linked ATP-cone, oligomer-distribution experiments were performed by gas-phase electrophoretic macromolecule analysis (GEMMA). GEMMA analysis showed that the NrdB subunit (β) was in a dimer–tetramer equilibrium and that the tetramer formation was stimulated by dATP and suppressed by ATP (Fig. 6A). If the ATP cone is removed as in NrdBΔ169, the protein lost the ability to form tetramers, indicating that the process depends on the ATP cone (Fig. 6B). In the Grx deletion mutant, the ability to form tetramers was decreased but not lost completely (Fig. 6B). The NrdA subunit (α) was in a monomer–dimer equilibrium favoring dimers, especially in the presence of dATP where the monomers were below the detection limit (Fig. 6C). When both proteins were mixed together with dATP, an additional peak corresponding to an α2β4 complex appeared and to a minor extent also an α4β4 complex (Fig. 6D). In the absence of allosteric effectors or in the presence of ATP, α2β2 complexes were formed instead. The subunit compositions of the 234-, 344-, and 470-kDa peaks were determined by comparing the results with each subunit alone. NrdB tetramer formation was very inefficient in the absence of effectors or in the presence of ATP, indicating that the 234-kDa peak only to a minor extent can be explained by NrdB tetramers and mostly contains α2β2 complexes, resulting from the interaction of NrdA and NrdB, the major two species formed in the absence of effectors. In the presence of dATP, the two major species NrdA dimers and NrdB tetramers interacted to form the α2β4 complex and to some extent also an α4β4 complex if an additional NrdA dimer binds.
Figure 6.

GEMMA analysis of the F. ignava ribonucleotide reductase. A, analysis of 0.05 mg/ml NrdB (0.9 μm) in the absence or presence of 50 μm dATP or 100 μm ATP. B, similar experiments as in A but with NrdB mutant proteins lacking the ATP cone (NrdBΔ169) or the Grx domain (NrdBΔGrx) analyzed with and without 50 μm dATP. C, analysis of 0.05 mg/ml NrdA protein in the absence or presence of 50 μm dATP. D, experiments with NrdA–NrdB mixtures containing 0.025 mg/ml of each protein and no effector, 50 μm dATP, or 100 μm ATP.
To complement the GEMMA analyses of oligomer formation, we performed analytical size-exclusion chromatography (Fig. 7) using higher protein concentrations and physiologically reasonable concentrations of effectors (3 mm ATP and 0.1 mm dATP) (30, 31). The SEC experiments confirmed the GEMMA results. The NrdA protein was predominantly a dimer, and the dimeric form was further enhanced by binding of dATP and ATP to the s-site (Fig. 7A). The NrdB subunit doubled in mass in the presence of dATP compared with when ATP was present, supporting the conclusion from GEMMA that it is a dimer with ATP and a tetramer with dATP (Fig. 7B). In SEC, both the dimer and tetramer had larger masses than expected, indicating that the shape of the protein is not perfectly globular. Without effector, the NrdB protein seemed to be a dimer that gradually went through a transition to a larger species at higher protein concentration (Fig. 7B). This is in agreement with the GEMMA results that there is a dimer–tetramer equilibrium with the majority of the protein being dimeric (Fig. 6A). When the NrdA and NrdB proteins were mixed, they formed an α2β2 complex both with ATP and without effector, and a larger species with dATP. There was a gradual movement to a larger species when the protein concentration was increased up to a mass indicating an α4β4 complex.
Figure 7.
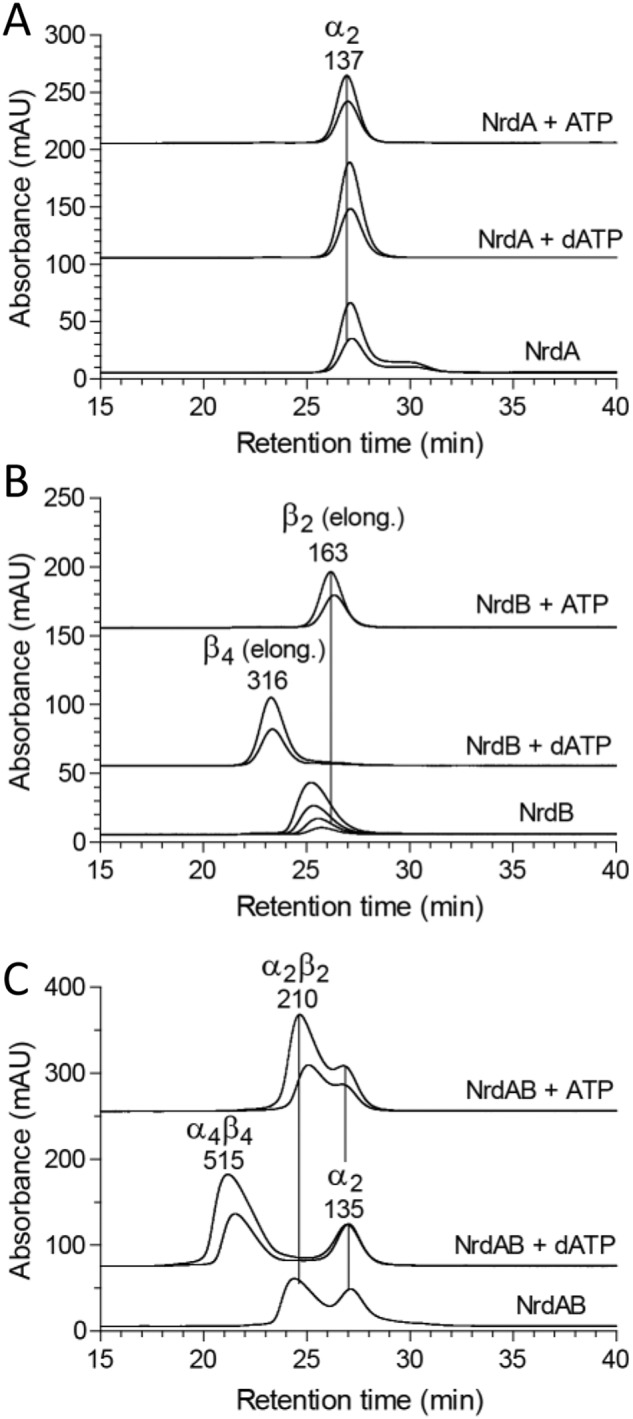
Size-exclusion chromatography analysis of F. ignava RNR components in the presence of nucleotides. A, 5 and 10 μm of the NrdA subunit was analyzed in the presence of 3 mm ATP or 100 μm dATP or without effector. B, corresponding analysis of the NrdB subunit. In this case the experiment without effector was performed at 1.25, 2.5, 5, and 10 μm protein. The position of the peaks indicate a larger size than expected, which is typical for elongated proteins, and the interpretation above the peaks is based on a comparison with the GEMMA results. C, analysis of the combination of both subunits. Each subunit was used at 10 and 20 μm concentration except in the experiment without effector, where only 10 μm was used.
Binding of nucleotides to F. ignava NrdB was investigated using isothermal titration calorimetry (ITC). Binding curves for dATP and ATP to NrdB at 20 °C were consistent with a single set of binding sites (Fig. 8). In dATP titrations, the fitted apparent N value was significantly above 1 (n = 1.4 ± 0.1), suggesting that the protein binds two dATP molecules per ATP cone provided our preparation contains ∼70% active protein. Fit of ATP titrations, performed with the same protein preparation and at the same day resulted in n = 0.55 ± 0.02, suggesting binding of only one ATP per ATP cone. Kd for the different nucleotides (Fig. 8E) indicated a 20-fold lower affinity for ATP compared with dATP. Thermodynamic parameters (Fig. 8E) indicated that the interactions are predominantly enthalpy-driven, with negative ΔH values of −80 and −60 kJ/mol for dATP and ATP, respectively. As observed earlier for L. blandensis NrdB (8) dADP also binds to the ATP cone of F. ignava NrdB with a Kd of 5.8 μm at 25 °C, i.e. considerably weaker than the Kd for dATP (Fig. S3).
Figure 8.

Representative ITC thermograms obtained by titration of ligands into NrdB. A, titration of dATP to NrdB at 20 °C. B, titration of dATP to NrdB at 10 °C. C, titration of ATP to NrdB at 20 °C. D, titration of ATP to NrdB at 10° °C. Isothermal calorimetric enthalpy changes are shown. E, thermodynamic parameters of ligand binding to NrdB. Binding isotherms were fitted using a one-set-of-sites binding model. All titrations were performed at 20 and 10 °C as described under “Experimental procedures.”
We performed an additional set of ITC experiments at 10 °C, which resulted in lower Kd values (0.4 μm for dATP and 6.8 μm for ATP) but otherwise similar conclusions. Fitted stoichiometries were 1.5 ± 0.1 for dATP and 0.57 ± 0.01 for ATP in agreement with the 20 °C results and underscoring our interpretation that the F. ignava NrdB protein binds two molecules of dATP and one molecule of ATP.
Type of radical cofactor in the F. ignava NrdB protein
To elucidate the nature of the radical cofactor in the F. ignava NrdB protein, we employed EPR spectroscopy. X-band EPR spectra recorded on samples of NrdBΔ169 expressed in the presence of excess Mn2+ and purified via affinity chromatography revealed an intense multiline signal with a signal width of 125–130 mT (Fig. 9). The signal varied in a uniform fashion in the interval 5–15 K and can thus be attributed to a single paramagnetic species (Fig. 9, compare 5-, 10-, and 15-K spectra). Increasing the temperature further resulted in a complete disappearance of the signal at 30 K, with no new signal appearing. The shape, width, and temperature dependence of the signal is in good agreement with an anti-ferromagnetically coupled MnIII/MnIV complex, where the complex line shape is a result of an S = ½ system where the unpaired electron is interacting with two I = 5/2 manganese centers. In a biological context, similar MnIII/MnIV species have been observed in the case of superoxidized manganese catalase and as a short-lived intermediate during the assembly of the MnIII2–Y• cofactor in NrdF (32, 33). The presence of such an intense multiline signal in our purified samples suggests that this high-valent species is stable at least in the time scale of hours.
Figure 9.
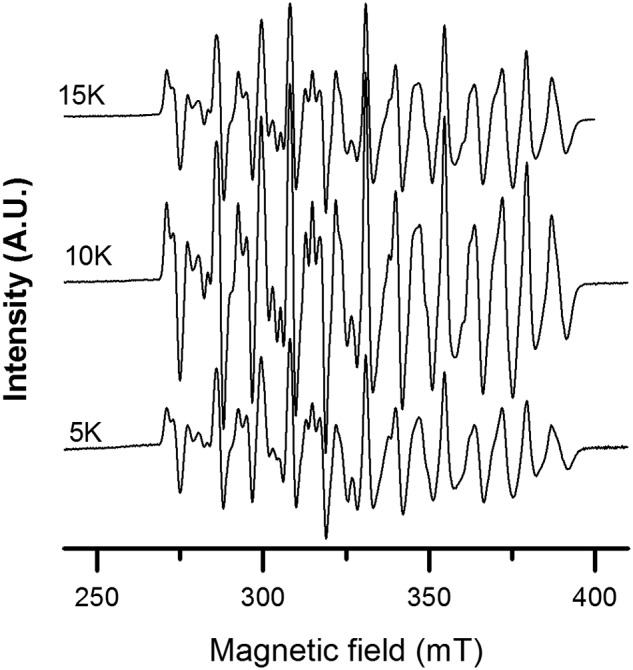
X-band EPR spectra of F. ignava NrdBΔ169 recorded at 5–15 K. Spectra were recorded on samples with 1.13 mm protein. Instrument settings were as follows: microwave frequency, 9.28 GHz; modulation amplitude, 10 G; modulation frequency, 100 kHz; microwave power, 1 milliwatt; and temperature, 5–15 K.
RNR activity in mixtures of F. ignava and L. blandensis NrdA and NrdB proteins
The NrdB core of the firmicute F. ignava from residue 170 and onwards has extensive similarity (61% sequence identity; Fig. S4) to the core of the NrdB protein from the flavobacterium L. blandensis. They both harbor a mixed valent MnIII//MnIV center with capacity to initiate the radical-based enzyme reaction (this study and Ref. 8). Both the F. ignava NrdB and the L. blandensis NrdB also harbor an ATP-cone domain that functions as an on/off switch for the activity of its RNR holoenzyme by forming tetrameric NrdB structures in presence of dATP to which the NrdA protein is prevented from binding in a productive fashion (above and in Ref. 8). However, the ATP cones of F. ignava and L. blandensis NrdB proteins are more different (28% sequence identity; Fig. S4) and align extensively only over their C-terminal sequences, which in the L. blandensis structure has been shown to interact primarily with one of the two bound dATP molecules (8). The similarity between the two corresponding NrdA proteins is extensive (61% sequence identities; Fig. S5). Based on these similarities, we designed a set of experiments to test whether RNR enzyme activity can be achieved in heterologous mixtures of F. ignava and L. blandensis NrdA and NrdB proteins and whether the unique Grx domain would disturb a heterologous interaction. Heterologous mixtures of class I RNR subunits have primarily been tested for distantly related enzymes, e.g. class I RNR subunits from Escherichia coli and bacteriophage T4 with negative results (34). On the other hand, several thioredoxins are known to cross-react with heterologous RNRs, whereas Grxs usually do not (35).
Fig. 10 shows that the heterologous F. ignava NrdA/L. blandensis NrdB holoenzyme was active and regulated by ATP and dATP via the ATP cone linked to L. blandensis NrdB, whereas the heterologous L. blandensis NrdA/F. ignava NrdB holoenzyme was inactive. The same was true for heterologous mixtures with F. ignava NrdBΔGrx, as well as for F. ignava NrdBΔ169 (Fig. 10A). KL for ATP was ≈300 μm, and Ki for dATP was ≈70 μm for the ATP cone of L. blandensis NrdB in the heterologous mixture (Fig. 10B), i.e. more than 3 times higher than the KL(ATP) of 96 μm and the Ki(dATP) of 20 μm for the L. blandensis holoenzyme (8). The Vmax obtained in the heterologous holoenzyme was 250 nmol/min·mg, corresponding to a kcat of ∼0.2 s−1, approximately a fourth of the activity of the L. blandensis holoenzyme (Fig. 10A).
Figure 10.

Enzyme activity in heterologous mixtures of NrdA and NrdB protein from F. ignava and L. blandensis. A, GDP reduction in presence of dTTP as effector. Assays were performed in triplicate with standard deviations shown. B, ATP (●) and dATP (○) titrations of F. ignava NrdA plus L. blandensis NrdB in presence of 2 mm dTTP and with GDP as substrate. ATP and dATP titrations in assays with CDP as substrate are shown in Fig. S6.
Discussion
We have shown that the multidomain radical-generating component of the F. ignava class I RNR contains a gene fusion of an N-terminal Grx that is fully functional as a reductant of the RNR holoenzyme and an ATP-cone that serves as a general on/off-switch of the enzyme. We also identified fusions of Grx-domains with NrdB proteins in Francisella spp., A. guangzhouensis, and several viruses (Fig. 1 and Table S1), but none of the other cases were in the NrdBi subclass that the F. ignava protein belongs to. This strongly suggests that the F. ignava grx::nrdBi fusion was a separate evolutionary event, not related to the grx::nrdB fusions discovered in other organisms and viruses. On the contrary, the presence in F. ignava of a fusion between an ATP-cone domain and NrdBi appears to be the result of horizontal gene transfer because the majority of ATP cones fused with nrdBi genes occurs in flavobacteria (8). It thus appears most parsimonious to suggest that the ATP cone::NrdBi fusion gene was first transferred to F. ignava and that the gene was subsequently fused with the grx gene in the F. ignava genome.
Grx was first described as a physiological reductant for RNR in E. coli (26) and has since also been observed to be involved in sulfate assimilation, detoxification, and development and proliferation, primarily in eukaryotic cells (27, 36). Similarly to other redoxins, the active site of dithiol Grxs consists of a cysteine pair separated by two residues (predominantly -CPYC-) (27). The corresponding sequence in F. ignava Grx::NrdB is -CPWC- (Fig. S4). Grxs differ from other redoxins in that they form mixed disulfides with GSH and also promote glutathionylation/deglutathionylation reactions, which may lead to reduction of protein disulfides (36). E. coli Grx has been shown to use the dithiol mechanism in its reduction of E. coli RNR, whereas a human Grx was interpreted to reduce mammalian RNR via a glutathionylation mechanism (28). However, recent theoretical studies, as well as thorough experimental studies on Grx-dependent reduction of protein disulfides with heterologous components from eukaryotic and bacterial origins, show that the monothiol–dithiol mechanisms occur in parallel and that GSH concentration and dominance of specific steps in the mechanism determine the preferred path taken (37–39). In this study we show that the Grx::NrdB fusion protein of F. ignava can reduce its class I RNR holoenzyme via a dithiol mechanism and that the C15S mutant in the Grx active site can reduce RNR less efficiently via a monothiol mechanism (Fig. 11). To our knowledge this is the first demonstration of parallel dithiol–monothiol reduction mechanisms in a native system between Grx and its oxidized substrate from the same species.
Figure 11.

Schematic mechanisms for dithiol and monothiol reduction of F. ignava RNR. The solid arrows show the dithiol mechanism used by the WT Grx::NrdB reducing NrdA, and the dashed arrows show the step taken by the C15S Grx::NrdB (mutant protein indicated by the OH group) reducing NrdA. The dashed arrow step may also be used by the WT Grx::NrdB at high GSH levels, whereas the C15S Grx::NrdB can only use the monothiol mechanism. Adapted after Ref. 39.
Over and above the fused Grx domain, the remarkable F. ignava NrdB protein exhibits two other unusual characters: a fused ATP cone and a mixed valent MnIII/MnIV metal site. Both of these features were recently described in L. blandensis NrdB and in several NrdBi proteins in Flavobacteriales (8). The ATP cone in F. ignava NrdB binds two dATP molecules like the cone in L. blandensis NrdB, but their amino acid sequences across the ATP cones are surprisingly dissimilar in the N-terminal half (Fig. S4). This may relate to our finding that the cone in F. ignava binds only one ATP molecule, whereas the cone in L. blandensis binds two ATP molecules. The ATP-loaded active F. ignava holoenzyme is α2β2, whereas the dATP-inhibited complexes are β4 for NrdB and α2β4 plus α4β4 for the holoenzyme. All of these complexes were also observed in the L. blandensis RNR (8).
The mixed valent MnIII/MnIV metal site in F. ignava NrdB has a distinct EPR signal in the temperature range of 5–15 K, with no other manganese-related EPR signals at 30 K and no trace of a tyrosyl radical. A similar high valent manganese dimer was recently found to be present in NrdB from L. blandensis (8). Later, Boal and co-workers (9) also reported a similar multiline signal in Flavobacterium johnsoniae class I RNR. However, in both of these latter cases, the multiline feature represented only a fraction of the total metal content. Conversely, in our F. ignava NrdB samples presented here, the MnIII/MnIV signal is clearly the dominant metal species. These observations underscore the catalytic relevance of the MnIII/MnIV site and support the notion that the NrdBi proteins represent a new subclass of class I RNRs, denoted subclass Id (6, 8, 9).
The similarities between F. ignava and L. blandensis NrdB proteins is further manifested by the enzyme activity observed in a heterologous mixture of F. ignava NrdA and L. blandensis NrdB, which is almost a third of that in the L. blandensis holoenzyme. Conversely, heterologous mixtures of L. blandensis NrdA and F. ignava NrdB lack activity even in the absence of the Grx domain or for the NrdBΔ169 protein that lacks both the Grx domain and the ATP cone. The F. ignava NrdB core may have undergone significant structural changes to accept the fusion of the Grx domain, which may also pertain to the divergent N-terminal of the ATP-cone sequence. Future studies will be directed to clarify this point.
All in all, we have shown that the unique NrdB protein in F. ignava carries an N-terminal Grx domain with capacity to act as a physiological reductant of its corresponding holoenzyme via a dithiol mechanism and less efficiently via a monothiol mechanism in the C15S mutant variant. The ATP-cone domain, which is fused between the Grx domain and the NrdB core, functions as an allosteric on/off switch, promoting an enzymatically active α2β2 complex in presence of ATP and enzymatically inactive α2β4 and α4β4 complexes in the presence of dATP. The radical cofactor in F. ignava NrdB is a mixed valent dinuclear MnIII/MnIV site, which forms in the absence of an NrdI activase and lacks a tyrosyl radical. F. ignava NrdB is an enthralling illustration of how RNR subclasses continuously evolve via gain and loss of accessory domains and RNR-related proteins.
Experimental procedures
Bioinformatics
The RefSeq database (40) was downloaded March 16, 2018, and searched with the Pfam (41) profiles for Grx (PF00462) and the ATP cone (PF03477) plus profiles developed in-house for RNR proteins (http://rnrdb.pfitmap.org)3 using the HMMER software version 3.1b2 (42). For RNR proteins, only hits covering at least 90% of the length of the profile were kept. For the Grx and ATP-cone profiles, only hits with a higher bitscore than the Pfam-specified gathering scores (21.50 in both cases) were kept.
Cloning
DNA fragments encoding NrdAi (WP_006702002) and NrdBi (EKB53615/WP_006702003) were amplified by PCR from F. ignava CCUG 37419 genomic DNA, obtained from the Culture Collection at the University of Gothenburg, using specific primers: NrdA, FiR1_For 5′-tctcCATATGACCGCACAATTAAAGAATC-3′, and FiR1_Rev 5′-cagaGGATCCTTAAGCTTCACAAGCTAAGC-3′; NrdB: FiR2_For 5′-tctaCATATGACTCAAGTACAAGTTTATAG-3′ and FiR2_REV 5′-cagaGGATCCTTAGAATAGGTCGTCGGC-3′.
The PCR products were purified, cleaved with NdeI and BamHI restriction enzymes, and inserted into a pET-28a(+) expression vector (Novagen, Madison, WI). The obtained constructs pET-nrdA and pET-nrdB contained an N-terminal His6 tag and a thrombin cleavage site. To construct the truncated NrdB mutant lacking the Grx domain (residues 1–78), a new forward primer FiR2ΔGrx_For 5′-tctaCATATGAGCAAAATCCCGCAACAC-3′ was used with FiR2_REV to yield pET-nrdBΔGrx. To construct the truncated NrdB mutant lacking both the Grx and the ATP-cone domains (residues 1–169), a new forward primer FiR2Δ169_For 5′-tctaCATATGGCGCGTCAACGTGATATA-3′ was used with FiR2_REV to yield pET-nrdBΔ169. To obtain NrdB bearing point mutations of individual cysteine residues to serines at the Grx active site, constructs pET-nrdB_C12S, pET-nrdB_C15S, and pET-nrdB_C12SC15S containing nucleotide mismatches T34A, G44C, and T34A/G44C, respectively, were ordered from GenScript.
Protein expression
Overnight cultures of E. coli BL21(DE3)/pET28a(+) bearing pET-nrdA, pET-nrdB, pET-nrdBΔGrx, pET-nrdBΔ169, pET-nrdB_C12S, pET-nrdB_C15S, or pET-nrdB_C12SC15S were diluted to an absorbance at 600 nm of 0.1 in LB (Luria-Bertani) liquid medium, containing kanamycin (50 μg/ml) and shaken vigorously at 37 °C. At an absorbance of A600 = 0.8 isopropyl-β-d-thiogalactopyranoside (Sigma) was added to a final concentration of 0.5 mm; the cultures expressing NrdB were further supplemented with MnSO4 (final concentration, 0.5 mm) during the induction. The cells were grown overnight at 30 °C and harvested by centrifugation.
Protein purification
The cell pellet was resuspended in lysis buffer: 50 mm Tris-HCl, pH 7.6, containing 300 mm NaCl, 10% glycerol, 2 mm DTT, 10 mm imidazole, 1 mm phenylmethylsulfonyl fluoride. The cells were disrupted by high pressure homogenization, and the lysate was centrifuged at 18,000 × g for 45 min at 4 °C. The recombinant His-tagged protein was first isolated by metal-chelate affinity chromatography using ÄKTA prime system (GE Healthcare): the supernatant was loaded on a HisTrap FF nickel-Sepharose column (GE Healthcare), equilibrated with lysis buffer (without phenylmethylsulfonyl fluoride), washed thoroughly with buffer, and eluted with buffer containing 500 mm imidazole.
NrdB_C12S, NrdB_C15S, NrdB_C12SC15S, and the WT NrdB used for measuring the redox activity of the NrdB fused Grx were then desalted on a Sephadex G-25 PD10 column (GE Healthcare) equilibrated with buffer containing 50 mm Tris-HCl, pH 7.6, 300 mm NaCl, 10% glycerol, and 1 mm DTT; frozen in liquid nitrogen; and stored at −80 °C until used.
For NrdA, NrdB, NrdBΔGrx, and NrdBΔ169, further purification was accomplished by FPLC on a 125-ml column packed with HiLoad 16/600 Superdex 200-pg column (GE Healthcare) using ÄKTA prime system, equilibrated with buffer containing 50 mm Tris-HCl, pH 7.6, 300 mm NaCl, 10% glycerol, and 2 mm DTT. Eluted protein was frozen until used.
NrdA was further applied to hydrophobic interaction chromatography using the HiLoad 16/60 phenyl-Sepharose column (GE Healthcare) in 50 mm Tris-HCl, pH 7.6, 2 mm DTT, 0.75 m (NH4)2SO4; washed extensively (15 column volumes) with the same buffer; and eluted with buffer without ammonium sulfate. The protein was resuspended in excess of buffer containing 50 mm Tris-HCl, pH 7.6, 300 mm NaCl, 10% glycerol, 2 mm DTT; concentrated; and frozen until used. The hydrophobic interaction chromatography removed residual nucleotide contamination from NrdA. L. blandensis NrdA and NrdB were expressed and purified as previously described (8).
Protein concentrations were determined by measuring the UV absorbance at 280 nm based on protein theoretical extinction coefficients 99,700 m−1 cm−1 for NrdA, 72,770 m−1 cm−1 for NrdB (and cysteine to serine mutants), 54,320 m−1 cm−1 for NrdBΔGrx, and 51,340 m−1 cm−1 for NrdBΔ169. Protein purity was evaluated by SDS–PAGE (12%) stained with Coomassie Brilliant Blue. Proteins were concentrated using Amicon Ultra-15 centrifugal filter units (Millipore), frozen in liquid nitrogen, and stored at −80 °C until used. For EPR measurements, NrdBΔ169 was purified using affinity chromatography as described above but transferred to EPR tubes and flash-frozen in liquid nitrogen in EPR tubes immediately upon elution.
RNR activity measurements
RNR activity assays were performed at room temperature in 50 mm Tris-HCl, pH 8, in volumes of 50 μl. Reaction conditions giving maximal activity were determined experimentally. In a standard reaction the constituents were 10 mm DTT, 40 or 20 mm Mg(CH3CO2)2 (when NrdA of F. ignava or L. blandensis was used, respectively), 10 mm KCl, 0.8 mm CDP, and various concentrations of allosteric effectors ATP or dATP. Mixtures of 0.1–1 μm of NrdB, 0.07 μm NrdBΔ169, 0.5 μm NrdB_C12S, NrdB_C15S, or NrdB_C12SC15S and a 10-fold excess of NrdA were used. In specific experiments some components were varied as indicated in the text.
In experiments aimed to determine the redoxin activity of the NrdB-fused Grx, DTT was omitted. Instead, 4 or 10 mm reduced GSH, 11 μg ml−1 GSH reductase (from yeast; Sigma) and 1 mm NADPH were added to the reaction mixtures. CDP (0.8 mm) was used as substrate, and ATP (3 mm) was used as effector. Protein concentration of 0.5 μm for WT NrdB, NrdB_C12S, NrdB_C15S, or NrdB_C12SC15S were used in combination with 5 μm NrdA.
When dTTP (2 mm) was used as an s-site effector, 0.8 mm GDP was used as substrate. In the four-substrate assays, the substrates CDP, ADP, GDP, and UDP were simultaneously present in the mixture at concentrations of 0.5 mm each with 2 mm of one of the effectors (ATP, dTTP, or dGTP). The substrate mixture was added last to start the reactions.
Enzyme reactions were incubated for 2–30 min at room temperature and then stopped by the addition of methanol. Substrate conversion was analyzed by HPLC using a Waters Symmetry C18 column (150 × 4.6 mm, 3.5-μm pore size) equilibrated with buffer A. Samples of 25–100 μl were injected and eluted at 0.4 ml/min at 10 °C with a linear gradient of 0–30% buffer B over 40 min for the separation of CDP and dCDP or 0–100% buffer B over 45 min for the separation of GDP and dGDP (buffer A: 10% methanol in 50 mm potassium phosphate buffer, pH 7.0, supplemented with 10 mm tributylammonium hydroxide; buffer B: 30% methanol in 50 mm potassium phosphate buffer, pH 7.0, supplemented with 10 mm tributylammonium hydroxide). Compound identification was achieved by comparison with injected standards. Relative quantification was obtained by peak height measurements in the chromatogram (UV absorbance at 271 or 254 nm) in relation to standards. Specific activities are given as nmol product formed per min and mg of protein.
From a direct plot of activity versus concentration of effector, the KL values for binding of effectors to the s-site and the a-site were calculated in SigmaPlot using the following equation.
| (Eq. 1) |
Ki for noncompetitive dATP inhibition at NrdB was calculated in Sigmaplot using the following equation.
| (Eq. 2) |
Photometric activity assays
Photometric assays for NrdB-fused Grx based on the artificial electron acceptor HED were performed as described in earlier studies (43, 44). The standard Grx assay contained 50 mm Tris, pH 8.0, 0.1 mg/ml BSA, 11 μg ml−1 GSH reductase (from Saccharomyces cerevisiae), 4 mm GSH, 0.75 mm HED, and 0.4 mm NADPH. The above ingredients were mixed and incubated for 3 min, after which the reaction was started by the addition of 0.1 μm WT or mutant Grx (NrdB fused). The reference cuvette contained all ingredients, except Grx. A340 was recorded for 3 min at room temperature using a Lambda 35 UV-visible spectrophotometer (PerkinElmer Life Science). Linear decrease in A340 was used to calculate moles of NADPH consumed using its extinction coefficient of 6220 m−1 cm−1.
The combined redoxin/RNR assays contained 0.5 μm NrdB, 5 μm NrdA, the indicated amount of GSH, 11 μg ml−1 GSH reductase, 0.25 mm NADPH, 10 mm Mg(CH3CO2)2, and 3 mm ATP. The reaction was started by the addition of 0.8 mm CDP. The reaction was monitored by the change of A340 using a Cary 60 UV-visible spectrophotometer (Agilent Technologies). In the calculation of specific activity, 1 mol of consumed NADPH equals formation of 1 mol of dCDP.
GEMMA analysis
In GEMMA, biomolecules are electrosprayed into gas phase and neutralized to singly charged particles, and the gas-phase electrophoretic mobility is measured with a differential mobility analyzer. The mobility of an analyzed particle is proportional to its diameter, which therefore allows for quantitative analysis of the different particle sizes contained in a sample (45). The GEMMA instrumental setup and general procedures were as described previously (46). NrdA, NrdB, NrdBΔGrx, and NrdBΔ169 proteins were equilibrated by Sephadex G-25 chromatography into a buffer containing 100 mm NH4CH3CO2, pH 7.8, and 2 mm DTT. Prior to GEMMA analysis, the protein samples were diluted to a concentration of 0.05 mg/ml in a buffer containing 20 mm NH4CH3CO2, pH 7.8, 1 mm DTT, 0.005% (v/v) Tween 20, nucleotides (when indicated), and Mg(CH3CO2)2 (equimolar to the total nucleotide concentration), incubated for 5 min at room temperature, centrifuged, and applied to the GEMMA instrument. The runs were conducted at low flow rate, resulting in 1.4–2 p.s.i. pressure. The GEMMA system contained the following components: 3480 electrospray aerosol generator, 3080 electrostatic classifier, 3085 differential mobility analyzer, and 3025A ultrafine condensation particle counter (TSI Corp., Shoreview, MN).
Analytical SEC
The SEC experiments were performed at room temperature with a SuperdexTM 200 10/300 column (GE Healthcare) equilibrated with a mobile phase containing 50 mm KCl, 10 mm MgCl2 0.1 mm DTT, and 50 mm Tris-HCl, pH 7.6. When nucleotide-dependent protein oligomerization was studied, 3 mm ATP or 0.1 mm dATP was also included in the mobile phase. The injection loop volume was 100 μl, and the flow rate was 0.5 ml/min. The UV trace was recorded with a Jasco UV-2075 Plus detector (Jasco Inc., Easton, MD) at 290 nm to limit the absorbance from the nucleotides. The proteins were incubated in mobile phase for 5 min prior to injection onto the column.
Isothermal titration calorimetry measurements
ITC experiments were carried out on a MicroCal ITC 200 system (Malvern Instruments Ltd.) in a buffer containing 50 mm Tris, pH 7.65, 300 mm NaCl, 10% glycerol, 2 mm tris(2-carboxyethyl)phosphine, and 10 mm MgCl2. Measurements were done at 20 and 10 °C. The initial injection volume was 0.5 μl over a duration of 1 s. All subsequent injection volumes were 2–2.5 μl over 4–5 s with a spacing of 150–180 s between the injections. Data for the initial injection were not considered. For dATP binding analysis, the concentration of NrdB in the cell was 40 μm, and dATP in the syringe was 600 μm. For titration of ATP into NrdB, cell and syringe concentrations were 103 μm NrdB and 1.2 mm ATP. The data were analyzed using the built-in one set of sites model of the MicroCal PEAQ-ITC analysis software (Malvern Panalytical). Standard deviations in thermodynamic parameters, N and Kd were estimated from the fits of three different titrations.
EPR spectroscopy
Measurements were performed on a Bruker ELEXYS E500 spectrometer using an ER049X SuperX microwave bridge in a Bruker SHQ0601 cavity equipped with an Oxford Instruments continuous flow cryostat and using an ITC 503 temperature controller (Oxford Instruments). The Xepr software package (Bruker) was used for data acquisition and processing of spectra.
Author contributions
I. R. G., D. L., M. S., G. B., A. H., and B.-M. S. conceptualization; I. R. G., G. B., A. H., and B.-M. S. data curation; I. R. G., D. L., G. B., A. H., and B.-M. S. formal analysis; I. R. G., D. L., G. B., A. H., and B.-M. S. validation; I. R. G., D. L., G. B., A. H., and B.-M. S. investigation; I. R. G., D. L., G. B., A. H., and B.-M. S. visualization; I. R. G., D. L., M. S., M. C., G. B., and A. H. methodology; I. R. G., D. L., M. S., M. C., G. B., A. H., and B.-M. S. writing-original draft; I. R. G., D. L., M. S., G. B., A. H., and B.-M. S. writing-review and editing; D. L. and B.-M. S. resources; D. L. software; G. B. and B.-M. S. supervision; G. B., A. H., and B.-M. S. funding acquisition; G. B., A. H., and B.-M. S. project administration.
Supplementary Material
Acknowledgments
We thank Al Claiborne (Wake Forest University, Winston-Salem, NC) for useful advice on bacillithiol and GSH biosynthesis; Ann Magnusson and Sigrid Berglund (Uppsala University, Uppsala, Sweden) for valuable discussions on dinuclear manganese centers; Ilya Borovok (Tel Aviv University, Tel Aviv, Israel) for discussions on ATP cones; Thomas Nyman (Protein Science Facility, Karolinska Institute, Stockholm, Sweden) for help with the ITC measurements; and Malvern Panalytical for kindly sharing the MicroCal PEAQ-ITC analysis software for the analysis of ITC data.
This work was supported by Swedish Cancer Society Grant CAN 2016/670 (to B.-M. S.), Swedish Research Council Grant 2016-01920 (to B.-M. S.), a Wenner–Gren Foundations grant (to B.-M. S.), and a Carl Trygger Foundation grant (to A. H.). Work in the laboratory of G. B. is supported by Swedish Research Council Grant 621-2014-5670; Swedish Research Council for Environment, Agricultural Sciences and Spatial Planning Grant 213-2014-880; and European Research Council Grant 714102. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Tables S1 and S2 and Figs. S1–S6.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- RNR
- ribonucleotide reductase
- a-site
- allosteric overall activity site in the ATP cone
- GEMMA
- gas-phase electrophoretic macromolecule analysis
- Grx
- glutaredoxin
- HED
- 2-hydroxyethyl disulfide
- ITC
- isothermal titration calorimetry
- NrdBΔGrx
- NrdB protein lacking 69 N-terminal residues corresponding to the glutaredoxin domain
- NrdBΔ169
- NrdB protein lacking the glutaredoxin domain and the ATP-cone domain
- SEC
- size-exclusion chromatography
- s-site
- allosteric specificity site in NrdA.
References
- 1. Lundin D., Berggren G., Logan D. T., and Sjöberg B. M. (2015) The origin and evolution of ribonucleotide reduction. Life (Basel) 5, 604–636 10.3390/life5010604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Torrents E. (2014) Ribonucleotide reductases: essential enzymes for bacterial life. Front. Cell Infect. Microbiol. 4, 52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hofer A., Crona M., Logan D. T., and Sjöberg B. M. (2012) DNA building blocks: keeping control of manufacture. Crit. Rev. Biochem. Mol. Biol. 47, 50–63 10.3109/10409238.2011.630372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Reichard P. (2010) Ribonucleotide reductases: substrate specificity by allostery. Biochem. Biophys. Res. Commun. 396, 19–23 10.1016/j.bbrc.2010.02.108 [DOI] [PubMed] [Google Scholar]
- 5. Stubbe J. (2000) Ribonucleotide reductases: the link between an RNA and a DNA world? Curr. Opin. Struct. Biol. 10, 731–736 10.1016/S0959-440X(00)00153-6 [DOI] [PubMed] [Google Scholar]
- 6. Berggren G., Lundin D., and Sjöberg B.-M. (2017) Homo- and heterometallic dinuclear manganese proteins: active site assembly. In Metalloprotein Active Site Assembly (Johnson M. K., and Scott R. A., eds) John Wiley & Sons, Ltd., Chichester, UK: 10.1002/9781119951438.eibc2480 10.1002/9781119951438.eibc2480 [DOI] [Google Scholar]
- 7. Cox N., Ogata H., Stolle P., Reijerse E., Auling G., and Lubitz W. (2010) A tyrosyl-dimanganese coupled spin system is the native metalloradical cofactor of the R2F subunit of the ribonucleotide reductase of Corynebacterium ammoniagenes. J. Am. Chem. Soc. 132, 11197–11213 10.1021/ja1036995 [DOI] [PubMed] [Google Scholar]
- 8. Rozman Grinberg I., Lundin D., Hasan M., Crona M., Jonna V. R., Loderer C., Sahlin M., Markova N., Borovok I., Berggren G., Hofer A., Logan D. T., and Sjöberg B. M. (2018) Novel ATP-cone-driven allosteric regulation of ribonucleotide reductase via the radical-generating subunit. Elife 7, e31529 10.7554/eLife.31529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rose H. R., Ghosh M. K., Maggiolo A. O., Pollock C. J., Blaesi E. J., Hajj V., Wei Y., Rajakovich L. J., Chang W. C., Han Y., Hajj M., Krebs C., Silakov A., Pandelia M. E., Bollinger J. M. Jr., et al. (2018) Structural basis for superoxide activation of Flavobacterium johnsoniae class I ribonucleotide reductase and for radical initiation by Its dimanganese cofactor. Biochemistry 57, 2679–2693 10.1021/acs.biochem.8b00247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jiang W., Yun D., Saleh L., Barr E. W., Xing G., Hoffart L. M., Maslak M. A., Krebs C., and Bollinger J. M. Jr. (2007) A manganese(IV)/iron(III) cofactor in Chlamydia trachomatis ribonucleotide reductase. Science 316, 1188–1191 10.1126/science.1141179 [DOI] [PubMed] [Google Scholar]
- 11. Högbom M., Stenmark P., Voevodskaya N., McClarty G., Gräslund A., and Nordlund P. (2004) The radical site in chlamydial ribonucleotide reductase defines a new R2 subclass. Science 305, 245–248 10.1126/science.1098419 [DOI] [PubMed] [Google Scholar]
- 12. Srinivas V., Lebrette H., Lundin D., Kutin Y., Sahlin M., Lerche M., Eirich J., Branca R. M. M., Cox N., Sjöberg B. M., and Högbom M. (2018) Metal-independent ribonucleotide reduction powered by a DOPA radical in mycoplasma pathogens. bioRxiv, e348268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sengupta R., and Holmgren A. (2014) Thioredoxin and glutaredoxin-mediated redox regulation of ribonucleotide reductase. World J. Biol. Chem. 5, 68–74 10.4331/wjbc.v5.i1.68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Arnér E. S., and Holmgren A. (2000) Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 267, 6102–6109 10.1046/j.1432-1327.2000.01701.x [DOI] [PubMed] [Google Scholar]
- 15. Jordan A., Aslund F., Pontis E., Reichard P., and Holmgren A. (1997) Characterization of Escherichia coli NrdH: a glutaredoxin-like protein with a thioredoxin-like activity profile. J. Biol. Chem. 272, 18044–18050 10.1074/jbc.272.29.18044 [DOI] [PubMed] [Google Scholar]
- 16. Crona M. (2010) Quaternary structure and interaction approaches to allosteric regulation of class I ribonucleotide reductases, Ph.D. thesis, Stockholm University, Stockholm, Sweden [Google Scholar]
- 17. Kingry L. C., and Petersen J. M. (2014) Comparative review of Francisella tularensis and Francisella novicida. Front. Cell Infect. Microbiol. 4, 35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Collins M. D., Falsen E., Lemozy J., Akervall E., Sjödén B., and Lawson P. A. (1997) Phenotypic and phylogenetic characterization of some Globicatella-like organisms from human sources: description of Facklamia hominis gen. nov., sp. nov. Int. J. Syst. Bacteriol. 47, 880–882 10.1099/00207713-47-3-880 [DOI] [PubMed] [Google Scholar]
- 19. Collins M. D., Lawson P. A., Monasterio R., Falsen E., Sjöden B., and Facklam R. R. (1998) Facklamia ignava sp. nov., isolated from human clinical specimens. J. Clin. Microbiol. 36, 2146–2148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rahmati E., Martin V., Wong D., Sattler F., Petterson J., Ward P., Butler-Wu S. M., and She R. C. (2017) Facklamia species as an underrecognized pathogen. Open Forum Infect. Dis. 4, ofw272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mathews C. K. (2016) The most interesting enzyme in the world. Structure 24, 843–844 10.1016/j.str.2016.05.006 [DOI] [PubMed] [Google Scholar]
- 22. Mathews C. K. (2018) Still the most interesting enzyme in the world. FASEB J. 32, 4067–4069 10.1096/fj.201800790R [DOI] [PubMed] [Google Scholar]
- 23. Jonna V. R., Crona M., Rofougaran R., Lundin D., Johansson S., Brännström K., Sjöberg B. M., and Hofer A. (2015) Diversity in overall activity regulation of ribonucleotide reductase. J. Biol. Chem. 290, 17339–17348 10.1074/jbc.M115.649624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gaballa A., Newton G. L., Antelmann H., Parsonage D., Upton H., Rawat M., Claiborne A., Fahey R. C., and Helmann J. D. (2010) Biosynthesis and functions of bacillithiol, a major low-molecular-weight thiol in Bacilli. Proc. Natl. Acad. Sci. U.S.A. 107, 6482–6486 10.1073/pnas.1000928107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fahey R. C. (2013) Glutathione analogs in prokaryotes. Biochim. Biophys. Acta 1830, 3182–3198 10.1016/j.bbagen.2012.10.006 [DOI] [PubMed] [Google Scholar]
- 26. Holmgren A. (1976) Hydrogen donor system for Escherichia coli ribonucleoside-diphosphate reductase dependent upon glutathione. Proc. Natl. Acad. Sci. U.S.A. 73, 2275–2279 10.1073/pnas.73.7.2275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lillig C. H., Berndt C., and Holmgren A. (2008) Glutaredoxin systems. Biochim. Biophys. Acta 1780, 1304–1317 10.1016/j.bbagen.2008.06.003 [DOI] [PubMed] [Google Scholar]
- 28. Zahedi Avval F., and Holmgren A. (2009) Molecular mechanisms of thioredoxin and glutaredoxin as hydrogen donors for mammalian S phase ribonucleotide reductase. J. Biol. Chem. 284, 8233–8240 10.1074/jbc.M809338200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Masip L., Veeravalli K., and Georgiou G. (2006) The many faces of glutathione in bacteria. Antioxid. Redox Signal. 8, 753–762 10.1089/ars.2006.8.753 [DOI] [PubMed] [Google Scholar]
- 30. Bochner B. R., and Ames B. N. (1982) Complete analysis of cellular nucleotides by two-dimensional thin layer chromatography. J. Biol. Chem. 257, 9759–9769 [PubMed] [Google Scholar]
- 31. Buckstein M. H., He J., and Rubin H. (2008) Characterization of nucleotide pools as a function of physiological state in Escherichia coli. J. Bacteriol 190, 718–726 10.1128/JB.01020-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cotruvo J. A. Jr., Stich T. A., Britt R. D., and Stubbe J. (2013) Mechanism of assembly of the dimanganese-tyrosyl radical cofactor of class Ib ribonucleotide reductase: enzymatic generation of superoxide is required for tyrosine oxidation via a Mn(III)Mn(IV) intermediate. J. Am. Chem. Soc. 135, 4027–4039 10.1021/ja312457t [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zheng M., Khangulov S. V., Dismukes G. C., and Barynin V. V. (1994) Electronic structure of dimanganese(II,III) and dimanganese(III,IV) complexes and dimanganese catalase enzyme: a general EPR spectral simulation approach. Inorg. Chem. 33, 382–387 10.1021/ic00080a030 [DOI] [Google Scholar]
- 34. Berglund O. (1975) Ribonucleoside diphosphate reductase induced by bacteriophage T4: III. isolation and characterization of proteins B1 and B2. J. Biol. Chem. 250, 7450–7455 [PubMed] [Google Scholar]
- 35. Holmgren A. (1989) Thioredoxin and glutaredoxin systems. J. Biol. Chem. 264, 13963–13966 [PubMed] [Google Scholar]
- 36. Lillig C. H., and Berndt C. (2013) Glutaredoxins in thiol/disulfide exchange. Antioxid. Redox Signal. 18, 1654–1665 10.1089/ars.2012.5007 [DOI] [PubMed] [Google Scholar]
- 37. Mashamaite L. N., Rohwer J. M., and Pillay C. S. (2015) The glutaredoxin mono- and di-thiol mechanisms for deglutathionylation are functionally equivalent: implications for redox systems biology. Biosci. Rep. 35, e00173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Begas P., Staudacher V., and Deponte M. (2015) Systematic re-evaluation of the bis(2-hydroxyethyl)disulfide (HEDS) assay reveals an alternative mechanism and activity of glutaredoxins. Chem. Sci. 6, 3788–3796 10.1039/C5SC01051A [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ukuwela A. A., Bush A. I., Wedd A. G., and Xiao Z. (2018) Glutaredoxins employ parallel monothiol–dithiol mechanisms to catalyze thiol-disulfide exchanges with protein disulfides. Chem. Sci. 9, 1173–1183 10.1039/C7SC04416J [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Haft D. H., DiCuccio M., Badretdin A., Brover V., Chetvernin V., O'Neill K., Li W., Chitsaz F., Derbyshire M. K., Gonzales N. R., Gwadz M., Lu F., Marchler G. H., Song J. S., Thanki N., et al. (2018) RefSeq: an update on prokaryotic genome annotation and curation. Nucleic Acids Res. 46, D851–D860 10.1093/nar/gkx1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Finn R. D., Coggill P., Eberhardt R. Y., Eddy S. R., Mistry J., Mitchell A. L., Potter S. C., Punta M., Qureshi M., Sangrador-Vegas A., Salazar G. A., Tate J., and Bateman A. (2016) The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44, D279–D285 10.1093/nar/gkv1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Eddy S. R. (2011) Accelerated profile HMM searches. PLoS Comput. Biol. 7, e1002195 10.1371/journal.pcbi.1002195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Holmgren A., and Aslund F. (1995) Glutaredoxin. Methods Enzymol. 252, 283–292 10.1016/0076-6879(95)52031-7 [DOI] [PubMed] [Google Scholar]
- 44. Vlamis-Gardikas A., Aslund F., Spyrou G., Bergman T., and Holmgren A. (1997) Cloning, overexpression, and characterization of glutaredoxin 2, an atypical glutaredoxin from Escherichia coli. J. Biol. Chem. 272, 11236–11243 10.1074/jbc.272.17.11236 [DOI] [PubMed] [Google Scholar]
- 45. Kaufman S. L., Skogen J. W., Dorman F. D., Zarrin F., and Lewis K. C. (1996) Macromolecule analysis based on electrophoretic mobility in air: globular proteins. Anal. Chem. 68, 1895–1904 10.1021/ac951128f [DOI] [PubMed] [Google Scholar]
- 46. Rofougaran R., Crona M., Vodnala M., Sjöberg B.-M., and Hofer A. (2008) Oligomerization status directs overall activity regulation of the Escherichia coli class Ia ribonucleotide reductase. J. Biol. Chem. 283, 35310–35318 10.1074/jbc.M806738200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.