

Abstract
In all herpesviruses, the space between the capsid shell and the lipid envelope is occupied by the unique tegument layer composed of proteins that, in addition to structural roles, play many other roles in the viral replication. UL37 is a highly conserved tegument protein that has activities ranging from virion morphogenesis to directional capsid trafficking to manipulation of the host innate immune response and binds multiple partners. The N-terminal half of UL37 (UL37N) has a compact bean-shaped α-helical structure that contains a surface region essential for neuroinvasion. However, no biochemical or structural information is currently available for the C-terminal half of UL37 (UL37C) that mediates most of its interactions with multiple binding partners. Here, we show that the C-terminal half of UL37 from pseudorabies virus UL37C is a conformationally flexible monomer composed of an elongated folded core and an unstructured C-terminal tail. This elongated structure, along with that of its binding partner UL36, explains the nature of filamentous tegument structures bridging the capsid and the envelope. We propose that the dynamic nature of UL37 underlies its ability to perform diverse roles during viral replication.
Keywords: herpesvirus, viral protein, multifunctional protein, small-angle X-ray scattering (SAXS), structural model, conformational flexibility, multi-angle light scattering, tegument, Thermofluor
Introduction
Herpesviridae are a large family of viruses that infect a broad range of hosts from mollusks to birds to humans. All herpesviruses are composed of a capsid containing the dsDNA genome surrounded by a layer of proteins called the tegument, which is enclosed by the glycoprotein-studded lipid envelope. The tegument layer is stabilized by extensive protein/protein interactions and is largely structurally maintained even once the viral membrane is removed (1). The tegument includes 23 distinct virally encoded proteins, in addition to several host proteins (2). The tegument is further subdivided into the inner and outer tegument. The inner tegument is directly associated with the capsid, and protein copy numbers are tightly controlled. The outer tegument is less ordered and associates with the viral envelope instead of the capsid (2).
In addition to linking the capsid to the lipid envelope (3), tegument proteins play additional roles during viral infection (2). The inner tegument protein UL36, at ∼330 kDa, the largest tegument protein (4), is involved in capsid transport (5, 6), release of viral DNA (7), and virion maturation (6, 8, 9) and contains a deubiquitinating domain (10). Other tegument proteins are involved in regulation of viral and host gene expression. For example, the outer tegument protein UL48, also known as α-transactivator, activates transcription of immediate-early viral genes (11). Another outer tegument protein, UL41, is an endonuclease that suppresses host protein expression by degrading mRNA and is commonly termed vhs (viral host shutoff) (12).
Likewise, the inner tegument protein UL37 plays multiple roles in the viral replication cycle. Along with UL36 (13–15), which is directly bound to the viral capsid (16), UL37 initiates tegument accumulation on the capsid during virion assembly and forms filamentous structures that may serve as a scaffold for the outer tegument proteins (17, 18). In HSV-1, 3 UL37 promotes formation of the enveloped viral particles, termed secondary envelopment, by binding viral envelope proteins gK and UL20, thereby linking capsid and envelope (3). Beyond structure and assembly, UL37 homologs from HSV-1 and PRV have been shown to facilitate efficient capsid trafficking during both viral entry and egress. During entry into epithelial cells, HSV-1 and PRV viruses lacking UL37 are delayed in trafficking, with about 50% of capsids failing to reach the nucleus (13). During egress, in the absence of UL37, HSV-1 and PRV capsids do not efficiently traffic from the nucleus to the sites of secondary envelopment (vesicles derived from Golgi (19) or endosomes (20)), and “naked” capsids accumulate within the cytoplasm (21, 22). In HSV-1, UL37 has been shown to promote capsid transport on microtubules during egress in epithelial cells through its interaction with host trafficking protein dystonin/BPAG1a (23). UL37 plays an even more prominent role in capsid trafficking in neurons during infection with HSV-1 and PRV. UL37-null viruses are defective in retrograde movement from axon termini to the nucleus and avirulent in the mouse model of infection, demonstrating the importance of efficient capsid trafficking in viral replication (24). Finally, HSV-1 UL37 manipulates the innate immune response by binding immune modulators TNF receptor–associated factor 6 (TRAF6) (25) and retinoic acid-inducible gene I (RIG-I) (26). Binding of UL37 to TRAF6 leads to activation of transcription factor NF-κB, which subsequently localizes to the nucleus and turns on expression of immune response genes, such as interleukin-8 (IL-8), presumably to increase transcription of early viral genes (25). UL37 also deamidates RIG-I, thus preventing its activation by viral dsRNA, which decreases the overall interferon production and stunts the antiviral response (26).
The structural characteristics underlying multiple distinct activities of UL37 are unknown. We previously reported the crystal structures of the N-terminal half of UL37 (UL37N) from PRV and HSV-1 (27, 28) that revealed the bean-shaped structure of UL37N and helped identify a conserved surface region essential for directional capsid trafficking during neuroinvasion (24). However, no structural information is yet available for the C-terminal domain of UL37 (UL37C) that mediates the majority of known interactions of UL37. Here, we show that UL37C forms an elongated, conformationally flexible species with a folded core and an unstructured C-tail. We hypothesize that, in addition to providing a large surface with many potential binding sites for its multiple binding partners, UL37C can adopt several distinct conformations, each of which mediates a specific subset of UL37 activities. We propose that this conformational flexibility contributes to the multifunctionality of UL37.
Results
Expression and purification of PRV UL37
During expression in Escherichia coli, most of PRV UL37FL is cleaved, which generates the stable N-terminal half but no detectable C-terminal fragment (27) (Fig. 1A). Affinity tagging both the N and C termini of UL37FL allowed for its purification but did not improve the yield. Therefore, the following constructs of PRV UL37C were cloned, expressed, and purified instead: UL37C(478–919)-StII, UL37C(478–884), UL37C(499–884), and UL37C(618–919)-StII (Fig. 1B). Although the last resolved residue of the crystal structure of PRV UL37N is Leu-479, only Trp-477 was unambiguously assigned to UL37N due to extensive interactions within the core. Therefore, conservatively, UL37C starts at residue 478, and UL37C(478–919)-StII represents the largest UL37C construct. UL37C(478–884) lacks a large portion of the unstructured C-terminal tail, whereas UL37C(499–884) additionally lacks the unstructured N-terminal region (Fig. S1). Finally, UL37C(618–919)-StII was designed based on a stable UL37 degradation product observed during expression of HSV-1 UL37 in mammalian cells. 4
Figure 1.

PRV UL37 and HSV-1 UL37N constructs and properties. A, schematic of PRV UL37 WT with domain boundaries labeled based on crystal structure determination of PRV UL37N (PDB code 4K70). B, schematics of PRV UL37FL (green/blue, Gene ID: 2952491), PRV UL37N (green), HSV1-UL37N (orange, Gene ID: 2703358), and PRV UL37C (blue). Linker resulting from cleavage of the His6-SUMO tag with PreScission protease (pink) and linker preceding Strep-II tag (orange) is shown. C, theoretical molecular mass (MM) for all constructs as calculated from amino acid sequence; presence (✓) or absence (×) of crystal formation; MM as calculated from SEC (SEC MM), SEC-SAXS (SEC-SAXS MM), or SEC-MALS (SEC-MALS MM); and melting temperature as determined from the Thermofluor assay (Thermofluor TM).
UL37C is an elongated monomer
Upon size-exclusion chromatography (SEC), all PRV UL37C constructs migrated as apparent dimers or trimers (Fig. 2A). However, SEC-coupled multi-angle light scattering (SEC-MALS) estimated molecular masses of PRV UL37C(478–919)-StII and UL37C(478–884) to be 50 and 46 kDa, which is close to their respective theoretical molecular masses of 48 and 43 kDa. Thus, PRV UL37C is a monomer in solution (Fig. 2, B and C). To rule out concentration-dependent oligomerization, SEC-MALS of UL37C(478–884) was performed at a range of concentrations: 1, 3, and 5 mg/ml. At all three concentrations, UL37C(478–884) was a monomer with an estimated molecular mass of 46 kDa (data not shown). To confirm the lack of dimerization, the PRV UL37C constructs were incubated with bis(sulfosuccinimidyl)suberate (BS3), a homobifunctional cross-linking reagent that reacts with primary amines, including the N-terminal amine and lysine side chains. Monomeric PRV UL37N (27) was used as a negative control. No high-molecular-mass species were observed for any construct in the presence of the cross-linker, consistent with a lack of oligomerization (Fig. 2D). The largest UL37C construct, UL37C(478–919)-StII, migrated at ∼55 kDa on SDS-PAGE (Fig. 2D), higher than its theoretical molecular mass of 48 kDa, yet migrated at ∼48 kDa after incubation with BS3. By contrast, UL37C(478–884) migrated at ∼43 kDa with or without cross-linker (Fig. 2D). The increased mobility of UL37C(478–919)-StII is indicative of intramolecular cross-linking, which prevents complete unfolding under SDS-PAGE conditions. An apparent lack of intramolecular cross-linking in UL37C(478–884), which lacks the second half of the unstructured C-terminal tail, suggests that BS3 reacts with a lysine side chain near the C terminus.
Figure 2.

Biochemical and biophysical characterization of PRV UL37C. A, PRV UL37C(478–919)-StII (royal blue), PRV UL37C(478–884) (medium blue), and PRV UL37C(618–919)-StII (pale blue) with standard proteins (arrows) analyzed by SEC. Standardization using elution volumes, molecular masses of the standard proteins, and column specifics yielded a standard curve for molecular mass estimations for PRV UL37C. B, SEC-MALS of PRV UL37C(478–919)-StII; C, PRV UL37C(478–884) in solution. The signal from the 90° scattering detector is shown as a dashed line, and the signal from the UV absorbance is shown in color. Experimental molecular masses are plotted as black dots against the right y axis, as calculated across the protein elution peak. Theoretical molecular masses corresponding to those of a monomer are indicated as horizontal dashed gray lines. D, BS3 chemical cross-linking of PRV UL37N (54 kDa), PRV UL37C(478–919)-StII (48 kDa), and PRV UL37C(478–884) (43 kDa). Gels were split to hide unrelated lanes, but contrast settings and molecular weight ladder remain consistent between gel sections. E, CD of PRV UL37C(478–919)-StII. Helical content from UL37C was estimated by the K2D2 program (http://k2d2.ogic.ca/) using the spectrum over the range from 190 to 240 nm. Helical content from UL37N was reported previously (27).
UL37C has a proteolytically resistant α-helical core
UL37C has high α-helical content, according to its CD spectra (Fig. 2E), but is predicted to have an unstructured C-terminal tail (Fig. S1). We used limited proteolysis to map the boundaries of the folded core of UL37C. Digests of UL37C(478–919)-StII with either chymotrypsin or trypsin resulted in a single major proteolytic product migrating at ∼40 kDa, with the trypsin-generated product being slightly larger (Fig. 3, A and B). N-terminal sequencing of both 40-kDa proteolytic products showed that both start with the GPGS sequence, generated by cleavage of the N-terminal His6-SUMO tag by PreScission protease during protein purification (Fig. 1B), implying that trypsin and chymotrypsin cleavage leave the N terminus intact. Neither degradation product retained the C-terminal StII tag, as determined by streptavidin–horseradish peroxidase Western blotting (data not shown). Thus, both proteases cleave UL37C near the C terminus. Mass spectrometry analysis of the chymotrypsin digest showed a predominant peak at 41.8 kDa, consistent with cleavage after residue Leu-866 (Fig. 3C and Fig. S2) (theoretical mass of 41.6 kDa). Trypsin generated a product ∼2 kDa larger, which is consistent with cleavage after residue Arg-884 (theoretical mass 43.5 kDa) (Fig. 3C). According to the secondary structure prediction, both proteolytic sites are located within the unstructured C-terminal tail (Fig. 3C).
Figure 3.

Limited proteolysis of PRV UL37C(478–919)-StII. A and B, limited proteolysis of PRV UL37C(478–919)-StII using either chymotrypsin (A) or trypsin (B) with decreasing amounts of protease. Solid arrows, original protein; open arrows, digestion products. C, secondary structure prediction for proteolytically sensitive C-terminal tail as calculated by PsiPred (http://bioinf.cs.ucl.ac.uk/psipred) (49). Blue cylinders, predicted helices. Cleavage sites for chymotrypsin and trypsin are indicated in blue and purple, respectively, based on MALDI-TOF data (Fig. S2). Linker (orange) and Strep-II tag (gray) are included.
UL37C has a low melting temperature
To assess the overall stability of UL37C, we used the Thermofluor, or differential scanning fluorimetry, assay (29), which estimates the melting temperature (Tm) by measuring binding of a hydrophobic dye to hydrophobic regions exposed during unfolding. Thermal melting was done in 24 different buffer and salt conditions to identify those that could potentially stabilize the protein. The highest Tm for UL37C(478–919)-StII and UL37C(478–884) was 49 and 45 °C, respectively (Fig. 1C, Fig. S3, and Table S1). By contrast, the highest Tm for PRV UL37N is 61 °C (Fig. 1C, Fig. S3, and Table S1). The highest Tm for any construct fell within a pH range of 7.0–7.5 and increased with the addition of 150 mm NaCl and 5–10% glycerol (Table S1). Regardless of buffer conditions, the lowest Tm for PRV UL37N was still higher than any Tm for PRV UL37C. The relatively low melting temperature of UL37C could be due to a weak hydrophobic core (30).
UL37C is an elongated, conformationally flexible molecule
We have been unable to crystallize any PRV UL37C constructs despite extensive efforts. To obtain structural information on UL37C, we employed small-angle X-ray scattering coupled with SEC (SEC-SAXS), which is used to characterize conformational state and flexibility of macromolecules in solution. SAXS profiles were obtained for three constructs of PRV UL37C and for HSV-1 UL37N, a compact, conformationally rigid protein that served as a control (Fig. 4 and Fig. 5A). PRV UL37C(478–919)-StII and HSV-1 UL37N had only one intensity peak, whereas UL37C(478–884) and UL37C(618–919)-StII each had two peaks. The later peaks correspond to a smaller radius of gyration (Rg) around 5–10 Å, potentially due to degradation products (Fig. 4, B and C). The Rg profile across the primary peak for all samples slopes downward, but frames chosen for analyses had Rg values within ±1 Å of each other. Parameters calculated from SAXS data are summarized in Table 1.
Figure 4.

SEC-SAXS curves. Plots show intensity (colored lines) and Rg (colored dots) as a function of time for the SEC-SAXS run. Buffer frames in the dashed boxes were subtracted from the selected and averaged data frames in the solid boxes for PRV UL37C(478–919)-StII (A), PRV UL37C(478–884) (B), PRV UL37C(618–919)-StII (C), and HSV-1 UL37N (D).
Figure 5.

SAXS analysis of PRV UL37C and HSV-1 UL37N. A, scattering profiles are shown after averaging and subtraction of solvent scattering from solid and dashed boxes, respectively, in Fig. 4. I(q) versus q are shown as log-linear plots. B, Guinier plots (including linear fits) at the low-angle region (qRg < 1.3). Data points in gray were excluded from the Guinier region. C, distance distribution functions shown for when forced to zero (inset) and not forced to zero (primary panel). Faded lines, curve after the determined Dmax. D, dimensionless Kratky plots were calculated from scattering curves in A. Data for PRV UL37C(478–919)-StII and HSV-1 UL37N have been deposited into the SASBDB under codes SASDDQ9 and SASDDP9, respectively.
Table 1.
SAXS structural parameters
| UL37C(478–919)-StII | UL37C(478–884) | HSV-1 UL37N | |
|---|---|---|---|
| Guinier analysis | |||
| I(0) (cm−1)a | 0.017 ± 0.00007 | 0.0081 ± 0.00007 | 0.02786 ± 0.00008 |
| Rg (Å)b | 41.97 ± 0.95 | 38.86 ± 0.51 | 33.34 ± 0.14 |
| qmin (Å−1)c | 0.00855 | 0.0108 | 0.00912 |
| qmaxRgd | 1.29208 | 1.30719 | 1.29265 |
| Coefficient of correlation, R2e | 0.9909 | 0.9695 | 0.9968 |
| P(r) analysis (forced to 0/not forced)f | |||
| I(0) (cm−1) | 0.017 ± 0.00008/0.019 ± 0.0004 | 0.0078 ± 0.00004/0.0085 ± 0.0002 | 0.02785 ± 0.00005 |
| Rg (Å) | 43.00 ± 0.17/50.2 ± 0.30 | 39.34 ± 0.16/47.63 ± 0.38 | 33.81 ± 0.065 |
| Dmax (Å)g | 140/250 | 125/250 | 120 |
| q range (Å−1)h | 0.007–0.2808/0.007–0.2808 | 0.007–0.2808/0.007–0.2808 | 0.007–0.2808 |
| χ2 (total estimate from GNOM)i,j | 1.169 (0.80)/1.087 (0.68) | 1.112 (0.80)/1.055 (0.66) | 1.106 (0.88) |
| DAMAVER | |||
| Symmetry,k anisotropy assumptionsl | P1, none | P1, none | P1, none |
| Models averagedm (omitted)n | 10 (0) | 10 (0) | 10 (0) |
| Mean NSDo (S.D.) | 0.619 (0.014) | 0.660 (0.017) | 0.644 (0.014) |
| χ2 rangep | 1.186–1.197 | 1.172–1.175 | 1.013–1.015 |
| Resolutionq (from SASRES (48)) (Å) | 41 ± 3 | 42 ± 3 | 38 ± 3 |
a I(0) = scattering intensity at zero angle.
b Radius of gyration.
c qmin = lowest q value used in the Guinier analysis.
d Maximum q value used in the Guinier analysis times Rg.
e Linear fit of the Guinier plot.
f P(r) forced to 0 at Dmax in DATGNOM/P(r) not forced to 0.
g Maximum dimension calculated from P(r).
h q values used in P(r) plot.
i χ2 fit of P(r) to data.
j Total quality estimate from GNOM.
k Symmetry used for DAMMIN input.
l Prolate, oblate, or none for DAMMIN input.
m Number of DAMMIN models averaged using DAMAVER.
n Number of DAMMIN models omitted from DAMAVER if NSD > mean + 2 × S.D.
o Mean NSD for each model used in DAMAVER average.
p Range of χ2 values for input DAMMIN models.
q Resolution of final filtered average from DAMAVER.
For PRV UL37C(478–919)-StII, UL37C(478–884), and HSV-1 UL37N, SAXS-derived molecular masses (31) were consistent with the theoretical values (Fig. 1C), and Guinier plots were linear, which indicated good data quality and no significant protein aggregation (Fig. 5B). By contrast, the SAXS-derived molecular mass of PRV UL37C(618–919)-StII was 85 kDa, which is more than twice its theoretical molecular mass of 34 kDa (Fig. 1C). In the Guinier plot, the slight upturn at the lowest q values and steeper slope indicated aggregation for this construct (Fig. 5B). In addition, the Rg increased before flattening out within the intensity peak instead of decreasing, as seen with the other three traces (Fig. 4). Such highly variable Rg indicates poor data quality and an unreliable Rg calculation, and therefore the UL37C(618–919)-StII data were not analyzed further.
SAXS data show that whereas both PRV UL37C(478–919)-StII and UL37C(478–884) constructs are at least 10 kDa smaller than HSV-1 UL37N, they have larger Rg and maximum dimensions (Dmax) (Table 1). Globular proteins have symmetrical bell-shaped distance distribution curves. The distance distribution curve of HSV-1 UL37N is slightly asymmetric, which is consistent with a bean-shaped structure observed in the crystals (Fig. 5C). In contrast, the obviously asymmetrical distance distribution curves that trail off at large distances for PRV UL37C(478–919)-StII and UL37C(478–884) (Fig. 5C) are characteristic of elongated and/or flexible structures.
The default settings in the program DATGNOM (used to evaluate the distance distribution) force the distance distribution function to 0 at the determined Dmax. Using these default settings, the Dmax for PRV UL37C(478–884) and UL37C(478–919)-StII was estimated as ∼125 and ∼140 Å, respectively (Fig. 5C, inset). However, when not forced to 0 at Dmax, the distance distribution function for UL37C has a pronounced “tail” that runs asymptotically to 0, well beyond 200 Å (Fig. 5C). Such behavior of the distribution function can be attributed to either aggregation or flexibility. Given that all other quality checks indicated that UL37C samples were not aggregated, based on the shape of the distribution function, we conclude that PRV UL37C is flexible. Whereas Dmax is difficult to define for a flexible protein, ∼125 and ∼140 Å represent the major transitions toward zero (i.e. transitions from average electron density of the protein to average electron density of the solvent), suggesting that they represent the Dmax values of the most common conformations adopted by each of the two UL37C constructs, respectively. In contrast, the long tail is representative of more extended conformations present in lower frequencies. Structural parameters for the distance distribution analysis are reported both for the scenario when the distance distribution curve is forced to zero at Dmax and when it is not (Table 1).
To estimate conformational flexibility, we used the dimensionless Kratky plots. HSV-1 UL37N has a symmetrical bell-shaped curve that reaches its peak at qRg = √3 and then returns to zero, which is characteristic of globular, ordered proteins (Fig. 5D and Fig. S4). In contrast, intrinsically disordered proteins have Kratky plots that continue to increase after qRg = √3. Both PRV UL37C(478–919)-StII and UL37C(478–884) had mixed profiles, with a small decrease after qRg = √3, indicating a balance between order and disorder, which is characteristic of a conformationally flexible yet mostly folded protein (Fig. 5D and Fig. S4). Taken together, the SAXS data suggest that UL37C is an elongated, conformationally flexible protein, in contrast to the slightly elongated, rigid UL37N.
The flexibility of the UL37C constructs was further analyzed using ensemble optimization modeling (EOM), which describes the size distribution of species in a heterologous mixture (32, 33). EOM version 2.0 uses the Rflex metric to quantify the differences between flexible and rigid systems, where Rflex = 0% for a fully rigid system and Rflex = 100% for a fully flexible system (33). The Rflex of a random pool, which represents 10,000 possible conformations, is typically between 85 and 90%. For a rigid system, Rflex would be expected to decrease significantly after ensemble optimization modeling. However, the Rflex for either UL37C(478–919)-StII or UL37C(478–884) decreased only 2–3%, which is characteristic of a highly flexible system (Fig. 6, A and B).
Figure 6.

SAXS modeling of PRV UL37C and HSV-1 UL37N. A and B, EOM of UL37C(478–919)-StII (A) and UL37C(478–884) (B). Each graph shows the Dmax distribution for a pool of 10,000 randomly generated structures and three sets of ensembles that best fit the data. Rflex, which represents flexibility of the ensemble, is shown for the pool, and Rflex for the best fitting curve is shown in parentheses. The structures of the best fit ensemble for PRV UL37C(478–919)-StII (C) and PRV UL37C(478–884) (D) are shown below. These models suggest possible behavior of the flexible protein in solution but do not represent the only solution. E, ab initio bead modeling of PRV UL37C(478–919)-StII. F, ab initio bead modeling of PRV UL37C(478–884). G, ab initio bead modeling of HSV-1 UL37N shown compared with the crystal structure of HSV-1 UL37N (PDB code 5VYL).
EOM generates probability density functions, representing the likelihood that the protein is found in a specific conformation and the relative ratios of those conformations. Multiple ensembles for both constructs show that the majority of the protein is found in a compact conformation, centered around a Dmax of ∼150 and 130 Å for the longer and the shorter construct, respectively (Fig. 6, A and B). The ensembles also indicate the presence of extended conformations for both constructs, with Dmax of 200–250 Å. No discrete extended conformation was found across all ensembles (Fig. 6, A and B), indicating that the extended conformation is highly flexible. Representative models of the compact and extended conformations for each construct (Fig. 6, C and D) show possible conformations but do not represent the only solution.
To complement EOM analysis, we turned to ab initio bead modeling, which can generate an envelope shape to match the experimental data (Fig. 5A). Ten ab initio models were generated independently and averaged for PRV UL37C(478–919)-StII, UL37C(478–884), and HSV-1 UL37N. The ab initio bead model of HSV-1 UL37N closely resembles the bean-shaped crystal structure (Fig. 6G), whereas the ab initio bead models of UL37C(478–919)-StII and UL37C(478–884) show extended, S-shaped envelopes (Fig. 6, E and F). In a flexible system, however, the ab initio model represents an average of all possible conformations. Therefore, the ab initio bead models of PRV UL37C(478–919)-StII and UL37C(478–884) should not be construed as accurate structures. Nevertheless, the Dmax values of the models fall at the larger end of the compact populations identified in EOM (Fig. 6, A and B), supporting the observation that PRV UL37C is predominantly found in this conformation.
UL37FL is composed of two independently folded halves that do not interact
The low yield of purified PRV UL37FL ruled out SAXS measurements yet permitted thermostability analysis using the Thermofluor method. UL37FL showed two distinct melting transitions, at 46 and 60 °C (Fig. 1C, Fig. S3D, and Table S1). These temperatures are close to the individual melting temperatures of UL37C and UL37N, 49 and 61 °C, respectively (Fig. 1C and Fig. S3, and Table S1). The melting profile of PRV UL37FL suggests that UL37N and UL37C have individual hydrophobic cores with distinct thermal stabilities. Individually expressed PRV UL37N and PRV UL37C(478–919)-StII do not form a stable complex under the conditions of the SEC experiment (Fig. 7) and are not cross-linked by BS3 (Fig. 2D). We conclude that UL37N and UL37C do not interact and, within UL37FL, exist as independent domains connected by a hinge or linker.
Figure 7.
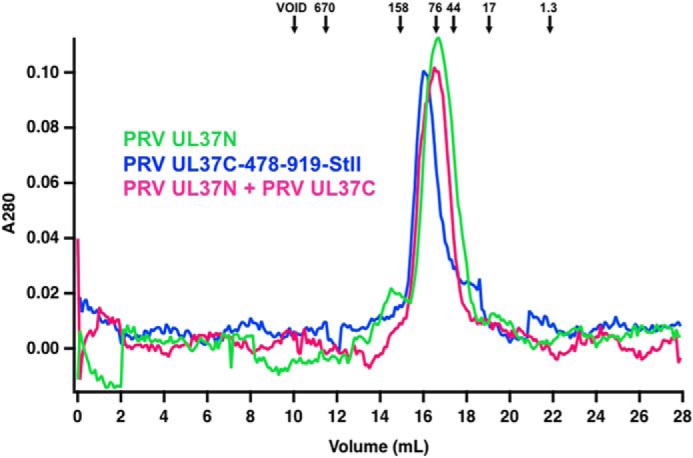
SEC of PRV UL37N and PRV UL37C(478–919)-StII. Standard proteins (arrows), PRV UL37N, PRV UL37C(478–919)-StII, and PRV UL37N+UL37C were analyzed by size-exclusion chromatography. Standardization using elution times, molecular weights of the standard proteins, and column specifics yielded a standard curve for molecular weight estimations for UL37N and UL37C.
Discussion
UL37 is a conserved inner tegument protein that plays multiple diverse roles in viral replication. In HSV-1 alone, UL37 interacts with viral proteins UL36 (34) and gK (3) and host proteins dystonin (23), TRAF6 (25), and RIG-I (26). Previously, we determined the crystal structures of UL37N from HSV-1 and PRV. Given that most of these interactions map to the C-terminal half of UL37, there is a considerable interest in determining the structure of UL37C. Here, we characterized biochemical properties of PRV UL37C and determined its molecular envelope by SAXS. We found that whereas UL37N has a compact bean-shaped structure, PRV UL37C is an elongated molecule. Due to this nonglobular shape, PRV UL37C constructs migrate as apparent dimers or trimers by SEC, which depends not only on molecular mass but also shape of the protein. Nonetheless, SEC-SAXS, MALS, and cross-linking clearly show that PRV UL37C constructs are monomeric in solution. Thus, PRV UL37C is an elongated monomer. Surprisingly, PRV UL37C has a relatively low melting temperature, 45–49 °C, in contrast to the melting temperature of 61 °C for UL37N. A low melting temperature could reflect a weak hydrophobic core (30), potentially due to its elongated shape and the unstructured C terminus. Proteins with intrinsically disordered regions typically have fewer hydrophobic residues and, thus, weaker hydrophobic cores (35). The differences in order between UL37N and UL37C can also be seen in a plot indicating which residues in a protein sequence are folded versus unfolded (Fig. 8A and Fig. S1) (36). The residues predicted to be unfolded in PRV and HSV-1 UL37 are found almost exclusively within the C-terminal half of the protein (Fig. S1) (36, 37). The specific residues important for UL37 interactions with gK/UL20, UL36, RIG-I, and BPAG map to ordered regions (Fig. 8A and Fig. S1), suggesting that although the binding sites may be rigid, the surrounding disordered regions could provide the flexibility needed to modulate the accessibility of the binding site. In this way, UL37 could control its choice of binding partners in different cellular locations or during different stages of the viral replication cycle.
Figure 8.

Predicted disorder and model of UL37. Shown is predicted flexibility of PRV (A) and HSV-1 UL37FL (B) as calculated by FoldUnfold (http://bioinfo.protres.ru/ogu/) (36). The gray dotted line marked 20.4 is the threshold for the parameter that determines if a residue is folded or unfolded; anything above 20.4 is considered folded and anything below is unfolded. Folded residues are indicated in blue, and unfolded residues are indicated in red. Residues in light blue are treated as folded despite lying below the 20.4 threshold. Binding regions within HSV-1 UL37 are shown for gK/UL20 (light pink), UL36 (cyan), BPAG (purple), RIG-I (green), and TRAF6 (pink). C, model of PRV UL37FL including crystal structure of UL37N (green; PDB code 4K70) and possible conformations of UL37C (blue) based on EOM structures (Fig. 6, C and D).
UL37C has a high α-helical content, consistent with secondary structure predictions, and a large proteolytically stable core connected to an unstructured C-terminal tail (C-tail). BS3 treatment of UL37C(478–919)-StII yielded faster-migrating species indicative of intramolecular cross-linking. The BS3 cross-linker is 11.4 Å long and can generate intramolecular cross-links due to flexibility in both the side chains and carbon backbone (38). The UL37C(478–919)-StII construct has four lysine residues: three within the UL37C itself (Lys-574, Lys-765, and Lys-919) and one within the StII tag. Lack of intramolecular cross-linking in UL37C(478–884) implies that either Lys-919 within the unstructured C-tail or the lysine within StII is cross-linked with either one of the other two lysines or the N-terminal amine, resulting in the observed faster-migrating species in UL37C(478–919)-StII. This suggests that the unstructured C-tail contacts the folded core of UL37C.
In the absence of a crystal structure, SAXS was used to determine the shape of UL37C and to assess its conformational flexibility. The distance distribution function for both UL37C(478–919)-StII and UL37C(478–884) indicates that UL37C is an elongated protein, in agreement with the SEC results, whereas the Kratky plots suggest that it is conformationally flexible, but not intrinsically disordered. The downward sloping Rg profiles within SEC intensity peaks are probably representative of the inherent flexibility of UL37C, where the more extended conformations are on one side of the peak and more compact conformations are on the other (Fig. 4). Further, the extensive tailing in the distance distribution functions for PRV UL37C constructs, when not forced to 0, supports our observations that UL37C is flexible. Whereas defining an accurate Dmax for a flexible protein is difficult, ∼125 and ∼140 Å represent the Dmax of the conformations most frequently adopted by each of the two UL37C constructs, respectively. These values are consistent with ensemble modeling of both UL37C constructs, which demonstrate a preference for the most compact conformations between 130 and 150 Å in length. EOM also shows that the remaining population is found in various extended conformations, but at lower frequencies, which contribute to the asymptotic tail in the distance distribution function. The unstructured C-tail of UL37C likely contributes significantly to this conformational flexibility by adopting various orientations. However, the construct UL37–478-884, which lacks about half of the C-tail, remains conformationally flexible, suggesting that the core is likely not rigid either. Spectrin repeats provide a useful model for the conformational flexibility of the UL37C core. Spectrin repeats are composed of α-helical bundles that assemble into filaments. SAXS experiments have shown that whereas individual spectrin repeats are rigid, constructs containing several repeats yielded Kratky plots similar to UL37C, suggesting that rigid units can be flexible in relationship to each other without being intrinsically disordered (39). We hypothesize that the inherent flexibility of UL37C precluded its crystallization.
We did not observe any interaction between the individually expressed UL37N and UL37C from PRV by either SEC or chemical cross-linking. We hypothesize that within full-length UL37, these domains either have limited interactions or do not interact at all. This hypothesis is supported by the biphasic melting profile of full-length PRV UL37, where transitions occur at the temperatures corresponding to melting temperatures of individually expressed PRV UL37N and UL37C. UL37N and UL37C are thus independently folded domains connected by a linker or a hinge within UL37. Fig. 8B shows a model of PRV UL37, composed of a stable, rigid UL37N and a conformationally flexible UL37C that resembles an arm or hook (Fig. 8B). We propose that the dynamic nature of UL37C may contribute to the multifunctionality of UL37, whereby distinct conformations could engage different binding partners and perform different roles during viral replication. Flexibility of disordered regions has been proposed as a significant factor in the ability of intrinsically disordered proteins to bind to two or more partners (40). Such a concept could help to explain the ability of UL37 to perform diverse roles during replication.
Viral proteomes have been shown to contain a greater percentage of intrinsic disorder than their human host proteome (41). Moreover, the intrinsically disordered regions of proteins have been shown to significantly contribute to the overall pathogenicity or oncogenicity of viruses (41). One well-characterized example is the E6 protein from human papillomavirus. Previous analysis showed that E6 from human papillomavirus genotypes that have higher oncogenic potential (high risk) contain a greater percentage of predicted intrinsically disordered residues when compared with low-risk genotypes (42). Whereas intrinsically disordered viral proteins implicated in pathogenesis have been identified in several viral families, there are no current examples from herpesviruses. Structural information on tegument proteins is scarce. By demonstrating the inherent flexibility within a highly conserved tegument protein, our work expands our knowledge of tegument proteins and will inform future studies of this complex virion layer.
Previously, it has been proposed that UL36 and UL37 form long filamentous structures extending away from the capsid vertices within the virion (17, 18). A recombinant construct corresponding to the central third of UL36 forms long fibers in vitro (43). Here, we showed that UL37C forms an extended, conformationally flexible structure. Given that UL37N has a compact, slightly elongated structure, we hypothesize that the extended, flexible UL37C contributes to the filamentous structures observed in the virion. Future characterization of UL37 structure and interactions will lead to a more complete understanding of the complex tegument protein UL37.
Experimental procedures
Cloning
The PRV UL37 gene with a C-terminal StII tag, codon-optimized for E. coli expression, was synthesized by GeneArt. The PRV UL37(1–919)-StII fragment was excised from the GeneArt plasmid using BamHI and HindIII and subcloned into either pJP4 or pGEX-6P1. The pJP4 plasmid contains a His6-SUMO-PreScission tag in frame with the BamHI restriction site of the multiple-cloning site in a pET24b vector. The pGEX-6P1 plasmid contains a GSH S-transferase (GST)-PreScission tag in frame with the BamHI restriction site of the multiple-cloning site. The resulting plasmids encode PRV UL37 1–919-StII with either a cleavable N-terminal His6-SUMO tag (pJP14) or an N-terminal GST tag (pAK26). DNA encoding UL37C(478–919)-StII was amplified by PCR from the codon-optimized PRV UL37 gene using the primers JP46 (CTAGGGATCCGGTCTGCGTGCAGATGGTGCC, BamHI site underlined) and AK7 (CGCAAGCTTCATTTTTCAAACTG, HindIII site underlined) and subcloned into pJP4 vector using the BamHI and HindIII restriction sites to yield plasmid pAK1. DNA encoding UL37C(478–884) was amplified by PCR from the codon-optimized PRV UL37 gene using the primers JP46 and AK23 (AAAAAACTAGAAGCTTCTAACGCAGAACTTCCAGATTAAC, HindIII site underlined) and subcloned into pJP4 using the BamHI and HindIII restriction sites to yield plasmid pAK13. DNA encoding UL37C(499–884) was amplified by PCR from the codon-optimized PRV UL37 gene using the primers JP79 (CTAGGGATCCGATCTGGCAGCCGCAGCCGAT, BamHI site underlined) and AK23 and subcloned into pJP4 using the BamHI and HindIII restriction sites to yield plasmid pAK3. DNA encoding UL37C(618–919)-StII was amplified by PCR from the codon-optimized PRV UL37 gene using the primers AK118 (CTAAAGGATCCGCTCCGGCACCTCCGA, BamHI site underlined) and AK7 and subcloned into pJP4 using the BamHI and HindIII restriction sites to yield plasmid pAK19. Construction of plasmid pJP23 encoding PRV UL37N with an N-terminal His6-SUMO tag was described previously (27). Construction of plasmid pAK11 encoding HSV-1 UL37N with an N-terminal His6-SUMO tag was described previously (28).
Recombinant protein expression and purification
All constructs except pAK26 were expressed in low background strain (LoBSTr) E. coli (a gift from Thomas Schwartz, Massachusetts Institute of Technology). Freshly transformed cells were incubated at 37 °C overnight in 5 ml of LB starter culture supplemented with 50 μg/ml kanamycin and 34 μg/ml chloramphenicol. The starter culture was diluted into 1 liter of LB supplemented with 50 μg/ml kanamycin and 34 μg/ml chloramphenicol and grown at 37 °C until the A600 reached 0.8–1.0. At this point, the temperature was shifted to 16 °C, and the cells were induced with 0.5 mm isopropyl-β-d-thiogalactopyranoside for 16–20 h. Cells were harvested by centrifugation at 5,000 × g for 40 min, resuspended in 40 ml of buffer A (100 mm HEPES, pH 7.5, 150 mm NaCl, 5% glycerol, 0.1 mm tris(2-carboxyethyl)phosphine (TCEP)) for pAK1, pAK3, pAK13 or buffer B (20 mm HEPES, pH 7.5, 50 mm NaCl, 5% glycerol, 0.1 mm TCEP) for pJP14 or buffer C (100 mm PIPES, pH 7.5, 150 mm NaCl, 5% glycerol, 0.1 mm TCEP) for pAK19 and lysed using a Microfluidizer. All lysis buffers were supplemented with 15 mm imidazole to reduce nonspecific binding during the subsequent metal-affinity capture step. The insoluble fraction was removed by centrifugation at 13,000 × g for 30 min at 4 °C. Soluble lysate was loaded onto a 5-ml nickel-Sepharose 6B FF column (GE Healthcare). The column was subsequently washed with buffer A, B, or C, depending on the construct, containing increasing amounts of imidazole from 15 to 100 mm. Protein was eluted in buffer A, B, or C containing 300 mm imidazole. The protein concentration was determined from the absorbance at 280 nm using a calculated extinction coefficient. Purified recombinant GST-tagged HRV3C (PreScission) protease was added to the protein solution at a 1:30 protease/protein ratio, and the protein was cleaved overnight at 4 °C to remove the His6-SUMO tag. Cleaved constructs were further purified by SEC using a Superdex 200 column in buffer A, B, or C (GE Healthcare).
pAK26 was purified as described above, with some modifications. pAK26 was expressed in LB supplemented with 100 μg/ml ampicillin and 34 μg/ml chloramphenicol. Cells were harvested and lysed in buffer C. The soluble fraction was captured on GSH-Sepharose 4B (GE Healthcare), eluted with 10 mm reduced GSH in buffer C, and further purified by SEC. pJP23 and pAK11 were expressed and purified as described previously (27, 28). Protein purity was assessed by SDS-PAGE and GelCode Blue staining (Thermo Fisher Scientific).
Thermofluor assay
Protein was diluted into 1× TBS to a final concentration of 0.15 mg/ml. The fluorescent dye SYPRO Orange (Invitrogen) was added at a 1:1,000 dilution. Ten μl of the protein-dye solution was pipetted into each well of a 96-well PCR microplate. Next, 10 μl of buffer (from a custom-made screen containing buffers at pH 4.5–10.5 and sodium chloride concentrations ranging from 0 to 500 mm, 24 conditions in total) was added to wells containing the protein-dye solution. The plate was sealed and centrifuged for 1 min at 500 × g and 25 °C. Samples were analyzed on a Roche Applied Science LightCycler 480 quantitative PCR machine using an excitation wavelength of 465 nm and detection of emission at 610 nm. The emission signal was analyzed from 25 to 95 °C at a continuous acquisition rate of 3 measurements/°C. Data were analyzed using the ThermoQ software program.
Crystal screening
PRV UL37C constructs were screened at various concentrations in nine screens (in-house Harrison Lab Grid Screen, Classics Suite (Qiagen), Protein Complex Suite (Qiagen), Index (Hampton Research), Peg/Ion (Hampton Research), SaltRx (Hampton Research), Top 96 (Anatrace), and Wizard 1–4 (Rigaku)) using a 96-well sitting-drop vapor diffusion format with drops containing 0.2 μl of crystallization solution and 0.2 μl of protein dispensed by a Crystal Phoenix liquid handling robot (Art Robbins). Plates were stored at room temperature and were evaluated using a stereo microscope daily for a week and throughout subsequent months.
Circular dichroism
Secondary structure content of UL37(478–919)StII was estimated using CD. Purified UL37(478–919)StII was exchanged into CD buffer (20 mm Tris, pH 8.0, 150 mm sodium chloride, 5% glycerol, 0.5 mm TCEP) and diluted to 0.2 mg/ml. The far-UV CD spectrum from 190 to 250 nm was measured at room temperature on a JASCO model 810 spectropolarimeter using a 50-nm/s scan speed, 1-nm band pass value, and 1-s response time. For each sample, three scans were collected and data were averaged, a buffer blank spectrum was subtracted, and the sample was processed for noise elimination. Measurements are presented in units of δϵ. The spectra were analyzed using Dichroweb (http://dichroweb.cryst.bbk.ac.uk/html/home.shtml) 5 (50).
Multi-angle light scattering
The oligomeric state of UL37C(478–884) was assessed by SEC-MALS using a Wyatt Dawn Heleos II multi-angle light scattering detector and Optilab TrEX refractive index monitor with an Agilent isocratic HPLC system. The construct was purified as described above and prepared at three different concentrations (1, 3, and 5 mg/ml) in SEC-MALS buffer (100 mm HEPES, pH 7.5, 150 mm sodium chloride, 5% glycerol, 0.1 mm TCEP). Separation steps were performed in SEC-MALS buffer with a Tosoh G4SWxl column at a flow rate of 0.5 ml/min. The oligomeric state of UL37C(478–919)-StII was assessed by SEC-MALS as described above, except using an Akta isocratic HPLC system with a GE Superose 6 10/300 column at a flow rate of 0.3 ml/min. Data analysis for both constructs was performed with the Astra software package version 6.0.5.3 (Wyatt).
Cross-linking
BS3 (Thermo Fisher Scientific) was added to 100 pmol of purified UL37(478–884)StII at a 0:1, 25:1, 50:1, 250:1, and 500:1 cross-linker/protein molar ratio. Samples were incubated in amber tubes in the dark at room temperature for 30 min, at which point 1 m Tris, pH 8.0, was added to a final concentration of 50 mm and incubated for 15 min to stop the reaction. Purified HSV-1 UL37N was used as a negative control. Half of the reaction was analyzed by SDS-PAGE and Coomassie staining.
Limited proteolysis, MS, and N-terminal sequencing
3 μg of purified UL37C(478–919)-StII was incubated for 1 h at room temperature with either tosylphenylalanyl chloromethyl ketone–treated trypsin or 1-chloro-3-tosylamido-7-amino-2-heptanone–treated chymotrypsin (Sigma) in buffer A at 1:25, 1:50, 1:100, 1:200, 1:500, 1:1,000 or 1:2,000 protease/protein mass ratio. To stop the reaction, samples were boiled for 5 min at 95 °C with 1× loading buffer (2% SDS, 4% glycerol, 40 mm Tris, pH 6.8, 2% β-mercaptoethanol) and analyzed with SDS-PAGE.
For MS, 200 μg of purified UL37C(478–919)-StII was digested with chymotrypsin at a 1:200 protease/protein mass ratio as described above. After 1 h, the reaction was stopped by adding phenylmethanesulfonyl fluoride to a final concentration of 10 mm. The mixture was analyzed by MALDI-TOF on a Voyager DE-PRO instrument using a dihydroxybenzoic acid matrix at the Tufts University Core Facility.
For N-terminal sequencing, 75 μg of purified UL37C(478–919)-StII was digested with either chymotrypsin at a 1:200 or trypsin at a 1:100 protease/protein mass ratio, separated on a 4–15% SDS-PAGE, and transferred to polyvinylidene fluoride membrane in 50 mm CAPS, pH 10.5, supplemented with 10% methanol using a semi-dry transfer apparatus. The membrane was stained in 40% methanol with 0.05% Coomassie Blue R-250, followed by destaining, first in 50% methanol and then water. Pieces cut from the dried membrane were subjected to N-terminal sequencing by Edman degradation at the Tufts University Core Facility.
SEC-SAXS
SEC-SAXS experiments were performed at the BioSAXS facility at beamline G1 at the Cornell High Energy Synchrotron Source (Ithaca, NY). UL37C(478–919)-StII at ∼6 mg/ml, UL37C(478–884) at 3 mg/ml, and HSV-1 UL37N at 6 mg/ml were precleared of aggregates by centrifugation at 16,000 × g for 5 min at 4 °C. 100-μl aliquots were injected onto a Superdex 200 Increase 5/150 column (GE Healthcare) in buffer A at 0.3 ml/min at 4 °C and fed directly into a proprietary SEC-SAXS flow cell with a glass window. Samples were irradiated with a 9.9496-keV (1.246122-Å) beam with 5.7 × 1011-photon/s flux and diameter of 250 μm × 250 μm, and 2-s images were recorded through the duration of the run on a Pilatus 100K-S detector (Dectris) at a sample-to-detector distance of 1531.09 mm, resulting in a q range of 0.006 Å−1 < q < 0.8 Å−1, where scattering vector q = 4πsin(θ)/λ.
Data were processed using the RAW software package (https://sourceforge.net/projects/bioxtasraw/)5 (44) by averaging frames with a consistent Rg value (±1 Å) and subtracting averaged buffer frames. Specific frames were chosen based on which region gave the highest quality DATGNOM solution, which sometimes required exclusion of frames even with consistent Rg values. Subtracted curves were further analyzed in RAW and using programs in the ATSAS software package (https://www.embl-hamburg.de/biosaxs/software.html)5 (51), including DATRG, DATGNOM, DAMMIN (45), DAMAVER (46), and EOM version 2.0 (33). DATRG was used to evaluate uncertainty in Rg estimates. DATGNOM was used to generate and automatically evaluate the pair distance distribution function (P(r)) from the subtracted experimental data before manual adjustment of the automatically assigned Dmax value to improve P(r) shape. DAMMIN was used to generate ab initio shape determination by simulated annealing using a single-phase dummy atom. DAMAVER was used to align 10 models generated from DAMMIN (using P(r) functions not forced to 0, with a Dmax of 250 Å), select the most typical one, and build an averaged model. In EOM, a pool of 10,000 random native-like structures representing most of the structural space available to the sequence of UL37C were generated. Theoretical SAXS curves were generated for random combinations of these models, and the ensemble was optimized with a genetic algorithm to best fit the experimental data. No symmetry information or structured domains were provided. The initial pool and the final ensemble were given flexibility metrics relating directly to the amount of information entropy in the system. Rg and Dmax values calculated from the species in each group were represented in histograms. Some low-angle data points in the Guinier region were excluded from the other three approximations, indicated by the gray dots in the Guinier plot (Fig. 4B). Data for UL37C(478–919)-StII, UL37C(478–884), and HSV-1 UL37N have been deposited into the SASBDB under codes SASDDQ9, SASDEL4, and SASDDP9, respectively.
Author contributions
A. L. K. and E. E. H. conceptualization; A. L. K. resources; A. L. K. and E. E. H. formal analysis; A. L. K. validation; A. L. K. investigation; A. L. K. visualization; A. L. K. and E. E. H. methodology; A. L. K. and E. E. H. writing-original draft; A. L. K. and E. E. H. writing-review and editing; E. E. H. supervision; E. E. H. funding acquisition; E. E. H. project administration.
Supplementary Material
Acknowledgments
We thank Xuanzong Guo for purifying PreScission protease and Abraham Sonenshein for the use of the Roche LightCycler 480 quantitative PCR machine. We thank MacCHESS BioSAXS staff for help with SAXS data collection and analysis, especially Jesse Hopkins for helpful discussions. We also thank Peter Cherepanov (Francis Crick Institute) for the gift of the GST-PreScission protease expression plasmid and Thomas Schwartz (Massachusetts Institute of Technology) for the gift of LoBSTr cells. Multi-angle light scattering experiments were performed at the Center for Macromolecular Interactions in the Department of Biological Chemistry and Molecular Pharmacology at Harvard Medical School. All software was installed and maintained by SBGrid (47). This work is based upon research conducted at the Cornell High Energy Synchrotron Source (CHESS), which is supported by the National Science Foundation and NIGMS/National Institutes of Health under NSF award DMR-0936384, using the Macromolecular Diffraction at CHESS (MacCHESS) facility, which is supported by NIGMS/National Institutes of Health Grant GM-103485.
This work was supported by National Institutes of Health Grant R01 AI056346 (to E. E. H.), the Investigators in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund (to E. E. H.), and a Faculty Scholar grant from the Howard Hughes Medical Institute (to E. E. H.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Table S1 and Figs. S1–S4.
Small-angle scattering data have been deposited into the SASBDB under codes SASDDQ9, SASDEL4, and SASDDP9, respectively.
P. Feng, unpublished observations.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- HSV-1
- herpes simplex virus-1
- PRV
- pseudorabies virus
- UL37C
- C terminus of UL37
- UL37N
- N terminus of UL37
- SEC
- size-exclusion chromatography
- MALS
- multi-angle light scattering
- SAXS
- small angle X-ray scattering
- Rg
- radius of gyration
- Dmax
- maximum dimension
- EOM
- ensemble optimization modeling
- BS3
- bis(sulfosuccinimidyl)suberate
- SUMO
- small ubiquitin-like modifier
- C-tail
- C-terminal tail
- GST
- glutathione S-transferase
- LB
- lysogeny broth
- TCEP
- tris(2-carboxyethyl)phosphine
- CAPS
- 3-(cyclohexylamino)propanesulfonic acid
- PDB
- Protein Data Bank
- NSD
- normalized spatial discrepancy.
References
- 1. McLauchlan J., and Rixon F. J. (1992) Characterization of enveloped tegument structures (L particles) produced by alphaherpesviruses: integrity of the tegument does not depend on the presence of capsid or envelope. J. Gen. Virol. 73, 269–276 10.1099/0022-1317-73-2-269 [DOI] [PubMed] [Google Scholar]
- 2. Owen D. J., Crump C. M., and Graham S. C. (2015) Tegument assembly and secondary envelopment of alphaherpesviruses. Viruses 7, 5084–5114 10.3390/v7092861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jambunathan N., Chouljenko D., Desai P., Charles A. S., Subramanian R., Chouljenko V. N., and Kousoulas K. G. (2014) Herpes simplex virus 1 protein UL37 interacts with viral glycoprotein gK and membrane protein UL20 and functions in cytoplasmic virion envelopment. J. Virol. 88, 5927–5935 10.1128/JVI.00278-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kelly B. J., Fraefel C., Cunningham A. L., and Diefenbach R. J. (2009) Functional roles of the tegument proteins of herpes simplex virus type 1. Virus Res. 145, 173–186 10.1016/j.virusres.2009.07.007 [DOI] [PubMed] [Google Scholar]
- 5. Luxton G. W., Lee J. I., Haverlock-Moyns S., Schober J. M., and Smith G. A. (2006) The pseudorabies virus VP1/2 tegument protein is required for intracellular capsid transport. J. Virol. 80, 201–209 10.1128/JVI.80.1.201-209.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schipke J., Pohlmann A., Diestel R., Binz A., Rudolph K., Nagel C. H., Bauerfeind R., and Sodeik B. (2012) The C terminus of the large tegument protein pUL36 contains multiple capsid binding sites that function differently during assembly and cell entry of herpes simplex virus. J. Virol. 86, 3682–3700 10.1128/JVI.06432-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Abaitua F., Hollinshead M., Bolstad M., Crump C. M., and O'Hare P. (2012) A nuclear localization signal in herpesvirus protein VP1–2 is essential for infection via capsid routing to the nuclear pore. J. Virol. 86, 8998–9014 10.1128/JVI.01209-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fuchs W., Klupp B. G., Granzow H., and Mettenleiter T. C. (2004) Essential function of the pseudorabies virus UL36 gene product is independent of its interaction with the UL37 protein. J. Virol. 78, 11879–11889 10.1128/JVI.78.21.11879-11889.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Desai P. J. (2000) A null mutation in the UL36 gene of herpes simplex virus type 1 results in accumulation of unenveloped DNA-filled capsids in the cytoplasm of infected cells. J. Virol. 74, 11608–11618 10.1128/JVI.74.24.11608-11618.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kattenhorn L. M., Korbel G. A., Kessler B. M., Spooner E., and Ploegh H. L. (2005) A deubiquitinating enzyme encoded by HSV-1 belongs to a family of cysteine proteases that is conserved across the family Herpesviridae. Mol. Cell 19, 547–557 10.1016/j.molcel.2005.07.003 [DOI] [PubMed] [Google Scholar]
- 11. Campbell M. E., Palfreyman J. W., and Preston C. M. (1984) Identification of herpes simplex virus DNA sequences which encode a trans-acting polypeptide responsible for stimulation of immediate early transcription. J. Mol. Biol. 180, 1–19 10.1016/0022-2836(84)90427-3 [DOI] [PubMed] [Google Scholar]
- 12. Smiley J. R. (2004) Herpes simplex virus virion host shutoff protein: immune evasion mediated by a viral RNase? J. Virol. 78, 1063–1068 10.1128/JVI.78.3.1063-1068.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Leege T., Granzow H., Fuchs W., Klupp B. G., and Mettenleiter T. C. (2009) Phenotypic similarities and differences between UL37-deleted pseudorabies virus and herpes simplex virus type 1. J. Gen. Virol. 90, 1560–1568 10.1099/vir.0.010322-0 [DOI] [PubMed] [Google Scholar]
- 14. Vittone V., Diefenbach E., Triffett D., Douglas M. W., Cunningham A. L., and Diefenbach R. J. (2005) Determination of interactions between tegument proteins of herpes simplex virus type 1. J. Virol. 79, 9566–9571 10.1128/JVI.79.15.9566-9571.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Klupp B. G., Fuchs W., Granzow H., Nixdorf R., and Mettenleiter T. C. (2002) Pseudorabies virus UL36 tegument protein physically interacts with the UL37 protein. J. Virol. 76, 3065–3071 10.1128/JVI.76.6.3065-3071.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dai X., and Zhou Z. H. (2018) Structure of the herpes simplex virus 1 capsid with associated tegument protein complexes. Science 360, eaao7298 10.1126/science.aao7298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Grünewald K., Desai P., Winkler D. C., Heymann J. B., Belnap D. M., Baumeister W., and Steven A. C. (2003) Three-dimensional structure of herpes simplex virus from cryo-electron tomography. Science 302, 1396–1398 10.1126/science.1090284 [DOI] [PubMed] [Google Scholar]
- 18. Laine R. F., Albecka A., van de Linde S., Rees E. J., Crump C. M., and Kaminski C. F. (2015) Structural analysis of herpes simplex virus by optical super-resolution imaging. Nat. Commun. 6, 5980 10.1038/ncomms6980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Turcotte S., Letellier J., and Lippé R. (2005) Herpes simplex virus type 1 capsids transit by the trans-Golgi network, where viral glycoproteins accumulate independently of capsid egress. J. Virol. 79, 8847–8860 10.1128/JVI.79.14.8847-8860.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hollinshead M., Johns H. L., Sayers C. L., Gonzalez-Lopez C., Smith G. L., and Elliott G. (2012) Endocytic tubules regulated by Rab GTPases 5 and 11 are used for envelopment of herpes simplex virus. EMBO J. 31, 4204–4220 10.1038/emboj.2012.262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Desai P., Sexton G. L., McCaffery J. M., and Person S. (2001) A null mutation in the gene encoding the herpes simplex virus type 1 UL37 polypeptide abrogates virus maturation. J. Virol. 75, 10259–10271 10.1128/JVI.75.21.10259-10271.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Klupp B. G., Granzow H., Mundt E., and Mettenleiter T. C. (2001) Pseudorabies virus UL37 gene product is involved in secondary envelopment. J. Virol. 75, 8927–8936 10.1128/JVI.75.19.8927-8936.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pasdeloup D., McElwee M., Beilstein F., Labetoulle M., and Rixon F. J. (2013) Herpesvirus tegument protein pUL37 interacts with dystonin/BPAG1 to promote capsid transport on microtubules during egress. J. Virol. 87, 2857–2867 10.1128/JVI.02676-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Richards A. L., Sollars P. J., Pitts J. D., Stults A. M., Heldwein E. E., Pickard G. E., and Smith G. A. (2017) The pUL37 tegument protein guides α-herpesvirus retrograde axonal transport to promote neuroinvasion. PLoS Pathog. 13, e1006741 10.1371/journal.ppat.1006741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu X., Fitzgerald K., Kurt-Jones E., Finberg R., and Knipe D. M. (2008) Herpesvirus tegument protein activates NF-κB signaling through the TRAF6 adaptor protein. Proc. Natl. Acad. Sci. U.S.A. 105, 11335–11339 10.1073/pnas.0801617105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhao J., Zeng Y., Xu S., Chen J., Shen G., Yu C., Knipe D., Yuan W., Peng J., Xu W., Zhang C., Xia Z., and Feng P. (2016) A viral deamidase targets the helicase domain of RIG-I to block RNA-induced activation. Cell Host Microbe 20, 770–784 10.1016/j.chom.2016.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pitts J. D., Klabis J., Richards A. L., Smith G. A., and Heldwein E. E. (2014) Crystal structure of the herpesvirus inner tegument protein UL37 supports its essential role in control of viral trafficking. J. Virol. 88, 5462–5473 10.1128/JVI.00163-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Koenigsberg A. L., and Heldwein E. E. (2017) Crystal structure of the N-terminal half of the traffic controller UL37 from herpes simplex virus 1. J. Virol. 91, e01244–17 10.1128/JVI.01244-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Phillips K., and de la Pena A. H. (2011) The combined use of the Thermofluor assay and ThermoQ analytical software for the determination of protein stability and buffer optimization as an aid in protein crystallization. Curr. Protoc. Mol. Biol. Chapter 10, Unit 10.28 10.1002/0471142727.mb1028s94 [DOI] [PubMed] [Google Scholar]
- 30. Franzosa E. A., Lynagh K. J., and Xia Y. (2009) Structural correlates of protein melting temperature. in Experimental Standard Conditions of Enzyme Characterizations, pp. 99–106, Beilstein-Institut, Ruedesheim/Rhein, Germany [Google Scholar]
- 31. Rambo R. P., and Tainer J. A. (2013) Accurate assessment of mass, models and resolution by small-angle scattering. Nature 496, 477–481 10.1038/nature12070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bernadó P., Mylonas E., Petoukhov M. V., Blackledge M., and Svergun D. I. (2007) Structural characterization of flexible proteins using small-angle X-ray scattering. J. Am. Chem. Soc. 129, 5656–5664 10.1021/ja069124n [DOI] [PubMed] [Google Scholar]
- 33. Tria G., Mertens H. D., Kachala M., and Svergun D. I. (2015) Advanced ensemble modelling of flexible macromolecules using X-ray solution scattering. IUCrJ 2, 207–217 10.1107/S205225251500202X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bucks M. A., Murphy M. A., O'Regan K. J., and Courtney R. J. (2011) Identification of interaction domains within the UL37 tegument protein of herpes simplex virus type 1. Virology 416, 42–53 10.1016/j.virol.2011.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Darling A. L., and Uversky V. N. (2018) Intrinsic disorder and posttranslational modifications: the darker side of the biological dark matter. Front. Genet. 9, 158 10.3389/fgene.2018.00158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Galzitskaya O. V., Garbuzynskiy S. O., and Lobanov M. Y. (2006) FoldUnfold: web server for the prediction of disordered regions in protein chain. Bioinformatics 22, 2948–2949 10.1093/bioinformatics/btl504 [DOI] [PubMed] [Google Scholar]
- 37. Prilusky J., Felder C. E., Zeev-Ben-Mordehai T., Rydberg E. H., Man O., Beckmann J. S., Silman I., and Sussman J. L. (2005) FoldIndex: a simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics 21, 3435–3438 10.1093/bioinformatics/bti537 [DOI] [PubMed] [Google Scholar]
- 38. Merkley E. D., Rysavy S., Kahraman A., Hafen R. P., Daggett V., and Adkins J. N. (2014) Distance restraints from crosslinking mass spectrometry: mining a molecular dynamics simulation database to evaluate lysine-lysine distances. Protein Sci. 23, 747–759 10.1002/pro.2458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ortega E., Manso J. A., Buey R. M., Carballido A. M., Carabias A., Sonnenberg A., and de Pereda J. M. (2016) The structure of the plakin domain of plectin reveals an extended rod-like shape. J. Biol. Chem. 291, 18643–18662 10.1074/jbc.M116.732909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hsu W. L., Oldfield C. J., Xue B., Meng J., Huang F., Romero P., Uversky V. N., and Dunker A. K. (2013) Exploring the binding diversity of intrinsically disordered proteins involved in one-to-many binding. Protein Sci. 22, 258–273 10.1002/pro.2207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xue B., Williams R. W., Oldfield C. J., Goh G. K., Dunker A. K., and Uversky V. N. (2010) Viral disorder or disordered viruses: do viral proteins possess unique features? Protein Pept. Lett. 17, 932–951 10.2174/092986610791498984 [DOI] [PubMed] [Google Scholar]
- 42. Tamarozzi E. R., and Giuliatti S. (2018) Understanding the role of intrinsic disorder of viral proteins in the oncogenicity of different types of HPV. Int. J. Mol. Sci. 19, E198 10.3390/ijms19010198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Scrima N., Lepault J., Boulard Y., Pasdeloup D., Bressanelli S., and Roche S. (2015) Insights into herpesvirus tegument organization from structural analyses of the 970 central residues of HSV-1 UL36 protein. J. Biol. Chem. 290, 8820–8833 10.1074/jbc.M114.612838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hopkins J. B., Gillilan R. E., and Skou S. (2017) BioXTAS RAW: improvements to a free open-source program for small-angle X-ray scattering data reduction and analysis. J. Appl. Crystallogr. 50, 1545–1553 10.1107/S1600576717011438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Svergun D. I. (1999) Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 76, 2879–2886 10.1016/S0006-3495(99)77443-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Volkov V. V., and Svergun D. I. (2003) Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 36, 860–864 10.1107/S0021889803000268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Morin A., Eisenbraun B., Key J., Sanschagrin P. C., Timony M. A., Ottaviano M., and Sliz P. (2013) Collaboration gets the most out of software. eLife 2, e01456 10.7554/eLife.01456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tuukkanen A. T., Kleywegt G. J., and Svergun D. I. (2016) Resolution of ab initio shapes determined from small-angle scattering. IUCrJ 3, 440–447 10.1107/S2052252516016018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. McGuffin L. J., Bryson K., and Jones D. T. (2000) The PSIPRED protein structure prediction server. Bioinformatics 16, 404–405 10.1093/bioinformatics/16.4.404 [DOI] [PubMed] [Google Scholar]
- 50. Whitmore L., and Wallace B. A. (2008) Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers 89, 392–400 [DOI] [PubMed] [Google Scholar]
- 51. Franke D., Petoukhov M. V., Konarev P. V., Panjkovich A., Tuukkanen A., Mertens H. D. T., Kikhney A. G., Hajizadeh N. R., Franklin J. M., Jeffries C. M., and Svergun D. I. (2017) ATSAS 2.8: a comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J. Appl. Crystallogr. 50, 1212–1225 10.1107/S1600576717007786 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.