

Abstract
Because castration-resistant prostate cancer (CRPC) does not respond to androgen deprivation therapy and has a very poor prognosis, it is critical to identify a prognostic indicator for predicting high-risk patients who will develop CRPC. Here, we report a dataset of whole genomes from four pairs of primary prostate cancer (PC) and CRPC samples. The analysis of the paired PC and CRPC samples in the whole-genome data showed that the average number of somatic mutations per patients was 7,927 in CRPC tissues compared with primary PC tissues (range, 1,691 to 21,705). Our whole-genome sequencing data of primary PC and CRPC may be useful for understanding the genomic changes and molecular mechanisms that occur during the progression from PC to CRPC.
Keywords: castration-resistant prostate cancer, DNA variants, whole-genome sequencing
Introduction
Prostate cancer (PC) is the most common malignancy in males [1]. It is known that about 20% of PC patients experience disease progression and distant metastasis [2, 3]. The therapeutic options for patients with aggressive PC include prostatectomy, radiation therapy, and androgen deprivation therapy (ADT) [4]. Although ADT therapy induces short 2–3-year remissions, unfortunately, most PCs eventually progress into castration-resistant prostate cancer (CRPC) [3], which does not respond to ADT therapy and shows poor clinical behavior. Therefore, it is crucial to understand the molecular characteristics and identify robust biomarkers that are associated with the development of CRPC from primary PC.
High-throughput next-generation sequencing technologies have gradually uncovered the molecular characteristics of PC, along with CRPC [5–8]. However, many genomic studies on CRPC have been conducted on metastasized CRPC that has been discovered at distant organs. These studies of metastatic sites do not reflect the precise molecular characteristics of CRPC, because these sites are not the primary PC site and because metastatic sites have completely different microenvironments from the primary site [9]. Here, we generated a dataset of whole genomes from four pairs of primary PC and CRPC samples from the same patient (i.e., a total of eight paired samples from four PC patients). In this report, the genomic status of samples with primary PC and CRPC was explored, and different variants between these two distinct phenotypes were identified by comparing the genomes of primary PC and its paired CRPC.
Methods
Tissues samples
Four pairs of primary PC and CRPC tissues were obtained from Chungbuk National University Hospital (Korea) with informed consent and approval of the Internal Review Board at Chungbuk National University. To obtain a consistent variant profile that was associated with the development of CRPC, primary CRPCs were obtained from homogenous biopsy sites, and none of our PC samples was from distant metastatic sites. Detailed clinical characteristics of the four pairs of primary PC and CRPC tissues are described in Supplementary Table 1.
Whole-genome sequencing library construction and sequencing
Genomic DNA was isolated using the DNeasy Blood and Tissue kit (Qiagen, Carlsbad, CA, USA), and the sequencing library was constructed using the Illumina TruSeq DNA Library Prep Kit (San Diego, CA, USA). Next, paired-end sequencing was performed on an Illumina HiSeq X Ten sequencing instrument, yielding ~ 150-bp short sequencing reads.
Data analysis
The sequenced reads were aligned to human reference genome 19 using Burrows Wheelers Aligner [10], and duplicate reads were removed using Picard (Broad Institute). Then, the remaining reads were calibrated and realigned using the Genome Analysis Toolkit [11]. The realigned Binary Alignment Map files were analyzed using Strelka [12] to detect somatic single-nucleotide variants and insertions/ deletions. For all programs, the default parameter settings were applied.
Results and Discussion
Quality and quantity of the sequencing data
The whole-genome sequencing (WGS) data, including the mapping rate, genome coverage, scores of the mapping quality, and duplicate reads, are summarized in Table 1. Briefly, the mapping rate and scores of the mapping quality in the four pairs of primary PC and CRPC samples were higher than 95% and 53%, respectively. In addition, the average genome coverage of our samples was over 30× (between 31.81× and 53.54×). Although coverage of several hundred times is required for detecting low-level mutations in next-generation sequence data [13], WGS with 30× sequence coverage is appropriate for comprehensive identification of tumor-specific somatic mutations [14]. These results suggest that the quality and quantity of our sequencing data are adequate for mutational analysis during the progression from PC to CRPC.
Table 1.
Quality and quantity of the sequencing data
| Sample ID | Total No. of reads | Mapped reads, n/% | Duplicate reads, n/% | Genome coverage (mean) | Mapping quality |
|---|---|---|---|---|---|
| P1_PC | 848,047,506 | 808,804,115/95.37 | 68,671,081/8.10 | 37.89 | 54.01 |
| P1_CRPC | 948,133,472 | 906,103,176/95.57 | 261,605,851/27.59 | 42.86 | 54.05 |
| P2_PC | 850,014,794 | 812,799,767/95.62 | 132,111,659/15.54 | 38.24 | 54.10 |
| P2_CRPC | 1,119,621,752 | 1,074,841,222/96.00 | 178,460,836/15.94 | 50.85 | 54.32 |
| P3_PC | 873,087,626 | 831,226,978/95.21 | 219,433,437/25.13 | 39.29 | 54.05 |
| P3_CRPC | 1,217,525,626 | 1,164,525,389/95.65 | 182,382,654/14.98 | 53.54 | 53.79 |
| P4_PC | 740,468,590 | 703,382,210/94.99 | 138,235,516/18.67 | 31.81 | 53.21 |
| P4_CRPC | 915,523,140 | 875,358,078/95.40 | 215,503,940/23.49 | 41.39 | 54.03 |
Mutation patterns identified from CRPC compared with PC
The average number of somatic mutations per patients was 7,927 in CRPC tissues compared with primary PC tissues (range, 1691 to 21,705). In particular, patient P2 had hypermutations (n = 21,705), whereas patient P1 had a low mutation frequency (n = 1,691) (Fig. 1A). To observe the mutation signatures in the development of CRPC from primary PC, we examined the spectrum of base substitutions. This analysis revealed an unusually high proportion of C:G > T:A and A:T>G:C transversions (Fig. 1B), similar to a previous study [15]. Next, the mutated sites were annotated as non-synonymous, synonymous, stop, and gain mutations. The number of mutations affecting protein-coding genes was 9, 321, 22, and 10 for the four patients (Table 2), and we observed recurrent mutations in the ANKRD20A4, ANDRK38B, AQP7, GGT1, and TAS2R31 genes. Detailed information for the non-synonymous and recurrent mutations is summarized in Supplementary Table 2. Further study will be needed to examine whether the mutated genes are associated with the development of CRPC from primary PC.
Fig. 1.
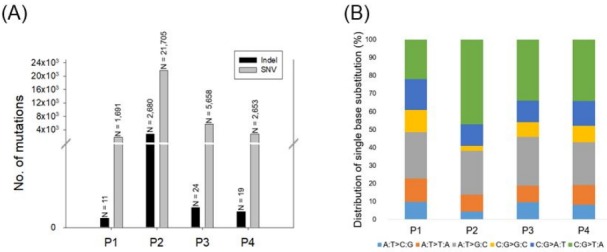
Number of mutations and distribution of mutation type. (A) Somatic mutations were detected using the Strelka package with default parameter settings. (B) Relative distribution of single-base substitutions by type in each of the four paired castration-resistant prostate cancer patients. SNV, single nucleotide variant.
Table 2.
Summary of mutation in exonic regions
| Sample ID | Synonymous mutations | Non-synonymous mutations | Stop or gain | Mutated genes |
|---|---|---|---|---|
| P1 | 3 | 6 | 0 | 9 |
| P2 | 104 | 226 | 10 | 321 |
| P3 | 11 | 13 | 0 | 22 |
| P4 | 3 | 8 | 1 | 10 |
In conclusion, PC is a heterogeneous disease and has various steps in its disease progression, including CRPC, the poorest prognostic status during the progression of PC. Understanding the molecular characteristics of the development of CRPC will help identify high-risk PC patients and develop novel therapeutic strategies to block the progression of CRPC. We generated a set of WGS data, consisting of eight PC samples containing four pairs of primary PC and CRPC samples from the same patient, because genetic mutations have the greatest potential to play a role in the progression of PC and CRPC and the therapeutic management of CRPC [16, 17]. By comparing primary PC and its paired CRPC, many somatic mutations that were significantly associated with the development of CRPC were identified, including TP53 and KMT2C, which are known to be involved in the progression of PC [16, 17]. We hope that our whole-genome sequence data of the four paired PC and CRPC tissues will be utilized by many researchers to understand the progression of PC and the resistance to androgen deprivation therapy.
Acknowledgments
This research was supported by a National Research Foundation of Korea (NRF) grant (NRF-2014M3C9A3068 554 and NRF-2017MBA9B5060884), funded by the Korean government (MEST), and a grant from the KRIBB Research Initiative Program.
Footnotes
Availability: The whole-genome data are available in the Korean Bioinformation Center (KOBIC) biodata (http://biodata.kr/) public database under accession numbers KBRS20180406_0000001–KBRS20180406_0000008.
Authors’ contribution
Conceptualization: SYK
Sample and data curation: SJY, WJK, WTK, PJ, HWK
WGS data generation: JHK, PJ
Data analysis: JLP, SKK
Writing – original draft: JLP, SKK
Writing – review & editing: SYK
Supplementary Materials
Supplementary data including two tables can be found with this article online at https://doi.org/10.5808/GI.2018.16.3.71.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 2.Maximum androgen blockade in advanced prostate cancer: an overview of 22 randomised trials with 3283 deaths in 5710 patients. Prostate Cancer Trialists’ Collaborative Group. Lancet. 1995;346:265–269. [PubMed] [Google Scholar]
- 3.Yap TA, Smith AD, Ferraldeschi R, Al-Lazikani B, Workman P, de Bono JS. Drug discovery in advanced prostate cancer: translating biology into therapy. Nat Rev Drug Discov. 2016;15:699–718. doi: 10.1038/nrd.2016.120. [DOI] [PubMed] [Google Scholar]
- 4.Droz JP, Aapro M, Balducci L, Boyle H, Van den Broeck T, Cathcart P, et al. Management of prostate cancer in older patients: updated recommendations of a working group of the International Society of Geriatric Oncology. Lancet Oncol. 2014;15:e404–e414. doi: 10.1016/S1470-2045(14)70018-X. [DOI] [PubMed] [Google Scholar]
- 5.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215–1228. doi: 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu J, Yu J, Mani RS, Cao Q, Brenner CJ, Cao X, et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell. 2010;17:443–454. doi: 10.1016/j.ccr.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park ES, Kim SJ, Kim SW, Yoon SL, Leem SH, Kim SB, et al. Cross-species hybridization of microarrays for studying tumor transcriptome of brain metastasis. Proc Natl Acad Sci U S A. 2011;108:17456–17461. doi: 10.1073/pnas.1114210108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28:1811–1817. doi: 10.1093/bioinformatics/bts271. [DOI] [PubMed] [Google Scholar]
- 13.Li M, Stoneking M. A new approach for detecting low-level mutations in next-generation sequence data. Genome Biol. 2012;13:R34. doi: 10.1186/gb-2012-13-5-r34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alioto TS, Buchhalter I, Derdak S, Hutter B, Eldridge MD, Hovig E, et al. A comprehensive assessment of somatic mutation detection in cancer using whole-genome sequencing. Nat Commun. 2015;6:10001. doi: 10.1038/ncomms10001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burns MB, Temiz NA, Harris RS. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat Genet. 2013;45:977–983. doi: 10.1038/ng.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rubin MA, Demichelis F. The genomics of prostate cancer: a historic perspective. Cold Spring Harb Perspect Med. 2018 Apr 30; doi: 10.1101/cshperspect.a034942. [Epub]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hovelson DH, Tomlins SA. The role of next-generation sequencing in castration-resistant prostate cancer treatment. Cancer J. 2016;22:357–361. doi: 10.1097/PPO.0000000000000217. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.