

Abstract
Tissue plasminogen activator (tPA) thrombolysis continues to be the gold standard therapy for ischemic stroke. Due to the time-limited treatment window, within 4.5 h of stroke onset, and a variety of potentially deadly complications related to delayed administration, particularly hemorrhagic transformation (HT), clinical use of tPA is limited. Combination therapies with other interventions, drug or nondrug, have been hypothesized as a logical approach to enhancing tPA effectiveness. Here, we discuss various potential pharmacological and nondrug treatments to minimize adverse effects, primarily HT, associated with delayed tPA administration. Pharmacological interventions include many that support the integrity of the blood–brain barrier (i.e., atorvastatin, batimastat, candesartan, cilostazol, fasudil, and minocycline), promote vascularization and preserve cerebrovasculature (i.e., coumarin derivative IMM-H004 and granulocyte-colony stimulating factor), employing other mechanisms of action (i.e., oxygen transporters and ascorbic acid). Nondrug treatments are comprised of stem cell transplantation and gas therapies with multi-faceted approaches. Combination therapy with tPA and the aforementioned treatments demonstrated promise for mitigating the adverse complications associated with delayed tPA treatment and rescuing stroke-induced behavioral deficits. Therefore, the conjunctive therapy method is a novel therapeutic approach that can attempt to minimize the limitations of tPA treatment and possibly increase the therapeutic window for ischemic stroke treatment.
Keywords: Blood–brain barrier, hemorrhage, matrix metalloproteinase, stem cell, tissue plasminogen activator
Current Status of Stroke
Stroke continues to be one of the most detrimental diseases in America, in addition to its persistent threat to millions around the world.[1] Many therapeutic treatments for this disease demonstrate narrow efficacy for rescuing neurological function. In addition, the only Food and Drug Administration (FDA)-approved drug for stroke, tissue plasminogen activator (tPA), is limited by a time-constrained treatment window which is >4.5 h from stroke onset. If tPA is administered outside of this window adverse effects, such as hemorrhagic transformation (HT), have been documented.[2] Due to these restraints, tPA only benefits 3% of ischemic stroke patients.[3,4,5] As the repertoire of effective treatments is limited, preclinical, and clinical studies for novel stroke treatments have begun.
A variety of drugs which range from enhancing neurogenesis to alternative thrombolytic agents has been assessed and demonstrated poor efficacy.[6,7,8] As tPA continues to be the gold standard treatment for ischemic stroke, aims to widen the treatment window and minimize adverse side-effects are of significant interest.[9] Identifying treatments to augment tPA administration are equally important to discovering novel treatments for ischemic stroke.[9] The lengthening of the tPA treatment window is two-fold, minimizing the significant adverse effects of delayed tPA treatment and expanding the window of neuroplasticity, increasing the chance for improved functional outcomes poststroke.
Thrombolytics and Ischemic Stroke
Damage to the blood–brain barrier (BBB), microvessels, along with the dangerous nonthrombolytic properties of tPA has been highlighted as the likely cause for the adverse effects of delayed tPA, particularly HT.[8,10,11,12,13] Treatments that encourage resistance to the aforementioned events, minimizing the BBB disturbance and inducing vascularization, are potential therapies that could be given in conjunction with tPA to curtail adverse effects. Furthermore, therapies which attack on multiple fronts are favorable when addressing a complex disease such as stroke and delayed tPA complications.[9,14] In the subsequent sections, we will examine pharmacological and nondrug therapies which have been evaluated to reduce delayed tPA complications, chiefly HT. We emphasize treatments that have been assessed in animal models, in which delayed tPA treatment is defined at >4.5 h following the onset of stroke. If available, the behavior of these therapeutic agents in clinical settings is also examined. These conjunctive therapies, their suggested mechanism of action and effects are displayed in Figure 1 and outlined in Table 1.
Figure 1.
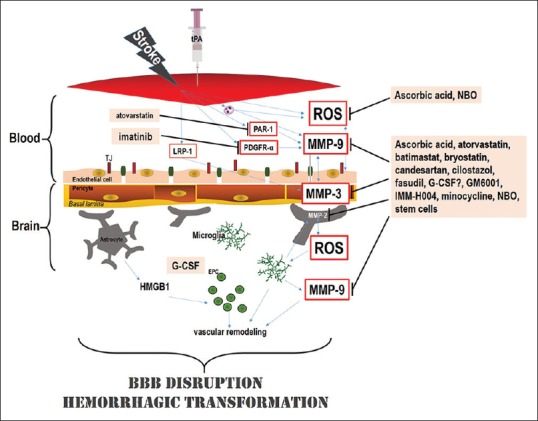
Putative mechanism(s) of action of conjunctive treatments to enhance thrombolytic therapy for acute ischemic stroke. Acute stroke potentially injures endothelial cells which lead to the formation of reactive oxygen species and pro-inflammatory cytokines. Moreover, delayed tissue plasminogen activator therapy could also damage the neurovascular unit and increase blood–brain barrier (BBB) leakage, neurovascular cell death, and hemorrhagic transformation. The hemorrhagic transformation that ensues after delayed Tissue plasminogen activator therapy has also been ascribed to enhanced reperfusion and the influence of tissue plasminogen activator on metalloproteinase activity and other signaling pathways, such as LRP, PAR1, and PDGRF-α signaling. In preclinical stroke modes, adjunctive treatment of ascorbic acid, and NBO has been shown to counteract delayed Tissue plasminogen activator -induced complications through dampening of ROS production and BBB breakdown. Administering atorvastatin, minocycline, cilostazol, GM6001, fasudil, candesartan, bryostatin, and IMM-H004 also reduced delayed tissue plasminogen activator-induced hemorrhagic transformation by preserving the BBB through their effects on MMPs and tight junction proteins. Granulocyte-colony stimulating factor and IMM-H004 have also been demonstrated to reduce the hemorrhagic transformation by enhancing neurovascularization and preserving the integrity of the BBB. Imatinib administration also reduced hemorrhagic transformation by acting through the PDGRF-α receptor, while atorvastatin decreased hemorrhagic transformation by inhibiting PAR1. Stem cells have also been shown to affect multiple targets including BBB and MMPs. EPC: Endothelial progenitor cell; G-CSF: Granulocyte-colony stimulating factor; HMGB1: High-mobility-group-box-1; ROS: Reactive oxygen species; LRP: Lipoprotein receptor-related protein; PAR1: Protease-activated receptor; PDGFR-α: Platelet-derived growth factor α-receptor; NBO: Normobaric oxygen
Table 1.
Drug and nondrug conjunctive treatments to lengthen thrombolytic therapy for acute ischemic stroke
| Agent | Species and stroke model | tPA dose, mode and time of treatment | Timing of outcome evaluation | Result | Reference |
|---|---|---|---|---|---|
| Ascorbic acid (500 mg, p.o.) 5 h poststroke | Male rats MCA cauterization | 1 mg/kg, i.v., 5 h poststroke | 48 h poststroke | Decreased infarct volume, brain edema, and brain permeability Decreased MMP-9 Improved sensorimotor functions |
[15] |
| Atorvastatin (first dose: 20 mg/kg 4 h after stroke, second dose: 20 mg/kg at 24 h after the first dose, s.c.) | Male Wistar rats; embolic | 10 mg/kg, i.v., 6 h poststroke | 7 h 30 h poststroke |
Reduced HT, infarct volume Improved neurological functions Increased thrombolysis and vascular patency Reduced expression of ICAM-1, PAR-1, Collagen IV Increased MMP-9 |
[16] |
| Batimastat (MMP inhibitor; 50 mg/kg; i.p., 3 and 6 h after stroke) | Male spontaneously hypertensive rats; embolic | 10 mg/kg, i.v., 6 h poststroke | 24 h poststroke | Decreased HT, infarct volume and mortality Improved neurological functions |
[17] |
| Bryostatin (PKC modulator; 2.5 mg/kg., i.v., alongside tPA) | Female SD rats, 18-20 months old; embolic | 5 mg/kg, i.v., 6 h poststroke | 24 h poststroke | Decreased HT Decreased MMP-9 Infarct volume, MMP-2, PKCα, PKCδ not changed Increased PKCɛ |
[18] |
| Candesartan (AT1R blocker; 1 mg/kg, i.v., 3 h after stroke) | Male Wistar rats (330-350 g); embolic | 10 mg/kg., i.v., 6 h poststroke | 24 h poststroke | Decreased HT Decreased MMP-3, NF-κB, TNF-α, p-eNOS Infarct volume, MMP-9, MMP-2 not changed |
[19] |
| Cilostazol (PDEIII-inhibitor; 10 mg/kg, i.p., before tPA) | 10 mg/kg., i.v., 6 h poststroke, before reperfusion | Male ddY (22-26 g) 4 weeks old; intraluminal filament/reperfusion | 18 h postreperfusion 7 d poststroke | Decreased HT and infarct volume Decreased MMP-9 Increased claudin 5 Improved locomotor behavior |
[20] |
| Agent | Species and stroke model | tPA dose, mode and time of treatment | Timing of outcome evaluation | Result | Reference |
| DDFPe nanodroplets 0.3 ml/kg, i.v. 1 h after stroke, and 5 additional doses at 90 min intervals | 0.9 mg/kg tPA | New Zealand male or female rabbits; 3.4 to 4.7 kg/between; embolic 9 h after last DDFPe dose | 24 h poststroke | Decreased infarct volume Improved neurological functions |
[21] |
| Fasudil (ROCK inhibitor; 3 mg/kg, i.p., before tPA) | Male SD rats (250-330 g); intraluminal filament/reperfusion | 10 mg/kg., i.v., 6 h poststroke, after reperfusion | 18 h postreperfusion 7 d post stroke | Decreased HT Decreased MMP-9 Infarct volume not changed Improved locomotor behavior |
[22] |
| G-CSF (300 µg/kg, i.v., alongside tpa) | Male SD rats, (200-250 g) 9-10 wk old; intraluminal filament/reperfusion | 10 mg/kg., i.v., poststroke, before reperfusion | 24 h post-drug treatment | Decreased HT Infarct volume not changed Improved neurological functions Increased Ang-2, CD34, eNOS, VEGFR2, vWF |
[23] |
| GM6001 (MMP inhibitor; 100 mg/kg, i.p., alongside tPA) | Male ddY mice (22-30 g) 4 weeks old; intraluminal filament/reperfusion | 10 mg/kg., i.v., 6 h poststroke, after reperfusion | 48 h poststroke/reperfusion | Decreased HT Decreased MMP-9 Claudin not changed Enhanced occludin, ZO-1 |
[24] |
| Imatinib (PDGFR-α antagonist | C57BL/6J mice, 10 weeks old, photothrombotic induction of MCAO | 10 mg/kg, i.v., 5 h after stroke | 24 h poststroke | Decreased HT | [25] |
| Agent dosage, mode, and time of treatment | Species and stroke model | tPA dose, mode, and time of treatment | Timing of outcome evaluation | Result | Reference |
| IMM-H004 (Coumarin derivative; 6 mg/kg, i.v., alongside tPA) | Male SD rats (300-320 g); embolic Male SD rats (260-280 g); intraluminal filament/reperfusion |
10 mg/kg, i.v., poststroke | 18 h poststroke 24 h poststroke 1,2,3 d poststroke 24 h poststroke 1-7 d poststroke 24 h poststroke/reperfusion 7 d poststroke/reperfusion | Decreased HT Decreased infarct volume Improved neurological functions Decreased HT and infarct volume Improved neurological functions Decreased pro-MMP-9 and Akt, occludin Increased Ang-1, CD31, CD31+Ki67, Tie2 |
[26] |
| Minocycline (antibiotic; 3 mg/kg, i.v., 4 h after stroke) | Male SHR; embolic | 10 mg/kg., i.v., 6 h poststroke | 24 h poststroke | Decreased HT, infarct volume Decreased MMP-9 (plasma) |
[27] |
| Neural stem cells (1 day poststroke) + minocycline | Aged mice Intraluminal filament model | 10 mg/kg, i.v., 6 h poststroke | 48 h posstroke | Improved neurological functions Reduced mortality | [28] |
| Normobaric oxygen (100% O2) | Male Sprague-Dawley rats (290-320 g) suture occlusion, and reperfusion |
10 mg/kg, i.v., 5 and 7 h poststroke, 15 min before reperfusion | 24 h poststroke | Decreased HT, infarct volume, brain edema, neurological deficits, mortality Reduced BBB disruption, MMP-9 Increased occluding, claudin-5 | [29] |
I.v.: Intravenous, TPA: Tissue plasminogen activator, MMP: Matrix metalloproteinase, HT: Hemorrhagic transformation, ICAM-1: Intercellular adhesion molecule-1, PKC: Protein kinase C, TNF- α: Tumor necrosis factor-α, NF-κB: Nuclear factor kappa-B, VEGFR: Vascular endothelial growth factor, BBB: Blood-brain barrier, DDFPe: Dodecafluoropentane emulsion, SHR: Spontaneously hypertensive rat, PAR-1: Protease-activated receptor 1
Drug-Based Therapeutics for Ischemic Stroke Treatment
The ability to attenuate HT resulting from delayed tPA administration could be achieved by minimizing the disruption of the BBB, which has also been shown to improve the overall effectiveness of tPA treatment.[11] Due to the role, metalloproteinases (MMPs) play in the disturbance of the BBB, therapies focusing on several MMPs has been investigated.[8,10,11,12,13] Furthermore, conserving endothelial tight junction proteins (TJP) have also been evaluated, as TJPs are the basic subunit of the BBB structure.[30,31,32] Pharmacological agents shown to display therapeutic effects through conservation of the BBB is atorvastatin, batimastat, bryostatin, candesartan, cilostazol, fasudil, and minocycline. The vascular disturbance is a vital aspect of intracerebral hemorrhage, causing BBB leakage.[33] Improving neovascularization and angiogenesis are both viable options, beyond rescuing BBB integrity, to counter the adverse effects of delayed tPA HT.[34] The pharmacological agents studied to minimize the adverse effects of delayed tPA administration though preserving cerebrovasculature and augmenting vascularization contain coumarin derivative IMM-H004 and granulocyte-colony stimulating factor (G-CSF). Taking into account, the function of free radicals in the adverse effects of delayed tPA administration, the beneficial properties of antioxidants have also been examined[15] Furthermore, promising preclinical studies show that oxygen transporters have the potential to lengthen the treatment window of tPA.[21]
The reduction of glutathione and ascorbic acid (AA) levels with increased free radical formation following ischemic stroke, provides evidence for the possible benefits of an AA supplement to ameliorate outcomes after ischemic stroke. AA, or vitamin C, may act by protecting endothelial function from ischemic oxidative injury in diabetes and prevent the development of free radicals in the brain parenchyma, potentially reducing the adverse side-effects of delayed tPA administration.[15] In rats subjected to permanent middle cerebral artery occlusion (MCAO) and treated with low-dose tPA (1 mg/kg, intravenous [IV]) and oral vitamin C (500 mg/kg) 5 h poststroke, infarct volume, and edema were attenuated at 48 h poststroke, when compared to rats only administered low-dose tPA.[15] MMP-9 production is stimulated by oxidative stress, which induces BBB disruption following ischemic reperfusion. The amplified MMP-9 levels and BBB damage were attenuated following Vitamin C treatment with tPA.[15] Therefore, Vitamin C administration with tPA reduces some of the adverse effects of delayed tPA treatment and demonstrates neuroprotection, signifying its possible role as a conjunctive therapy that may lengthen the tPA treatment window. Although vitamin C has demonstrated an ability to reduce stroke volume, the influence on HT has yet to be investigated.
The pleiotropic (BBB-protecting, antithrombotic, and anti-inflammatory) properties of statins have made them appealing conjunctive therapies to minimize adverse effects of delayed tPA administration, through the lengthening of the therapeutic window.[35] In rats, when atorvastatin was administered 4 h following embolic stroke embolus size at the origin of the middle cerebral artery was reduced with enhanced microvascular patency and reduced infarct volume after being treated with tPA at 6 h poststroke. In addition, the adjunctive therapies did not raise the prevalence of HT. The tPA-induced upregulation of MMP-9, intercellular adhesion molecule-1, and protease-activated receptor-1 were all reduced by atorvastatin. Moreover, atorvastatin decreased cerebral, fibrin, neutrophil, and microvascular platelet deposits. It has been hypothesized that atorvastatin-mediated neuroprotective effects are responsible for the reduction of adverse effects resulting from delayed tPA treatment.[35] It has been proposed that the effectiveness of atorvastatin as a thrombolytic agent boosts cerebrovascular patency and stability.
Treatment with batimastat, a broad spectrum MMP inhibitor (50 mg/kg, intraperitoneal [IP]), in spontaneously hypertensive rats which were subjected to embolic stroke demonstrated a significant reduction of volume associated with cerebral hemorrhage following delayed tPA treatment.[16] However, despite the diminished hemorrhage volume, no significant rescue of neurological function was observed poststroke in batimastat-treated animals. The study also did not investigate specific MMPs and mechanisms involved in the reduction of hemorrhage volume. Further studies examining time- and dose-dependent results are necessary to conclude the optimal treatment schedule for administering batimastat with tPA in future stroke models.
Protein kinase C (PKC) modulator bryostatin (2.5 mg/kg, IV) was given 2 h following MCAO to evaluate its efficacy to attenuate delayed tPA (5 mg/kg, IV)-potentiated cerebral hemorrhage, edema, and mortality at 24 h poststroke in rats.[17] Bryostatin reduced ischemic cerebral injury in aged female rats.[18] When administered in conjunction with delayed tPA, a reduction of HT and BBB damage was observed, along with a downregulation of MMP-9 expression and upregulation of PKCε.[17] The upregulation of PKCε has been proposed to limit damage to TJPs within the BBB and therefore attenuate HT. On the other hand, the downregulation in MMP-9 is potentially responsible for functional outcome improvements poststroke.[17] PKCε interplay of MMP-9 through a regulatory pathway is also suggested to play a vital role in the beneficial properties of bryostatin in minimizing delayed tPA-induced BBB damage and HT.[17]
Early administration of candesartan (1 mg/kg), which blocks angiotensin II type 1 receptors and protects from ischemic stroke damage, 3 h poststroke reduces brain hemorrhaging and leads to neurological improvements in animals subjected to embolic stroke and administered tPA (10 mg/kg, IV) at 6 h after stroke onset.[36] These treatments in conjunction upregulated and downregulated MMP-9 and MMP-3 levels, respectively. Intracranial hemorrhaging after delayed tPA was reduced in the MMP-3-null groups but not in MMP-9-null mice when compared to wild-type controls.[19] Taking this into account, it was suggested that the upregulation of MMP-9 alone cannot elevate the risk of HT in embolic stroke. However, the therapies in conjunction reduced nuclear factor kappa-B (NF-kB) expression, which has been known to regulate MMP-3 expression in endothelial cells following tPA therapy, tumor necrosis factor-α (TNF-α) was also diminished following NF-kB activation. After being administered candesartan, levels of endothelial nitric oxide synthase, an enzyme required for vascular function and homeostasis, were increased.[37]
Combination therapy of cilostazol (10 mg/kg, IV), used to treating intermittent claudication, with tPA (10 mg/kg, IV) 6 h after stroke reperfusion has demonstrated a reduction of cerebral edema, HT, morbidity, mortality, and a rescue of neurological function in mice at 18 h and 7 days following reperfusion.[32,38] Cilostazol has been effective in preventing upregulation of MMP-9 after delayed tPA treatment, as well as limiting downregulation of claudin 5, a vital molecule for the formation of tight junctions in microvascular endothelial cells.[20,32] In vitro, cilostazol blocked damage of pericytes and endothelial cells from delayed tPA treatment through its ability to modulate cyclic adenosine monophosphate (cAMP) activity.[32] However, the long-term beneficial neuroprotective effects of cilostazol have yet to be elucidated following stroke.
Dodecafluoropentane emulsion (DDFPe) facilitates oxygen-transportation through a perfluorocarbon, demonstrated to exhibit neuroprotective effects in rabbits subjected to ischemic stroke.[21] DDFPe (0.3 mL/kg, IV) administration following embolic stroke was shown to rescue neurological function and reduce stroke volume 24 h poststroke, in conjunction with tPA (0.9 mg/kg) given 9 h poststroke.[21] The ability to increase oxygen transport without the necessity of red blood cells is suggested to be the mechanism of action attributed to the neurological protective effects of DDFPe.[21] The effect of DDFPe on HT following delayed tPA has yet to be investigated.
Fasudil, a Rho kinase inhibitor, has been utilized as a treatment for cerebral vasospasms following a subarachnoid hemorrhage.[39] In combined therapy with tPA (10 mg/kg, IV) 6 h poststroke, Fasudil (3 mg/kg, IP) has been demonstrated to reduce HT at 18 h postreperfusion in mice subjected to a 6-h transient MCAO.[30] It also a significant reduction in mortality and increase in locomotor function 7 days after reperfusion. However, there were no evident neuroprotective effects of fasudil when compared to the tPA treatment only group and controls.[30] In vitro investigations observed the fasudil protected human brain microvascular endothelial cells (HBMECs) from delayed tPA-induced injury through the downregulation of MMP-9.[30] Further studies focusing on dose-dependent response would help elucidate the optimal doses of fasudil to use in conjunction with tPA and investigating the long-term neuroprotective effects in stroke are important for further consideration of this treatment.
G-CSF is an FDA-approved treatment to enhance survival of patients following exposure to myelosuppressive levels of radiation, by regulating the cell cycle of hematopoietic stem cells and progenitor cells.[22] Combination therapy of G-CSF (300 μg/kg, IV) with tPA (10 mg/kg, IV) 6 h poststroke in an MCAO model has been shown to decrease the incidence of HT.[40] In addition, elevated levels of angiogenesis marker Ang-2, vasculogenesis marker vWF, phosphorylated-eNOS, endothelial progenitor cell markers cluster of differentiation 34+ and vascular endothelial growth factor (VEGF) receptor-2 were found in the ischemic hemispheres in rats receiving the combination therapy compared to tPA alone. Rescue of neurological deficits was also observed in the G-CSF group at 24 h post-drug administration. It has been suggested that G-CSF reduced the incidence of tPA-proliferated HT and improved neurological function poststroke through the various angiogenic and endothelial factors associated with G-CSF.[40] A recent clinical study elucidated that although growth factors (GFs) VEGF, Ang-1, and G-CSF improved recanalization; only Ang-1 increased HT.[23] High serum levels of G-CSF were found to be associated with enhanced functional outcomes following 90 days posttreatment, highlighting its ability to be utilized as a countermeasure for delayed tPA-induced complications.[23]
GM6001 (100 mg/kg, IP) functions by attaching to active sites on MMPs and prohibiting the conversion of pro-MMPs to active MMPs which degrade the matrix.[41] GM6001 treatment, in conjunction with tPA (10 mg/kg, IV) 6 h poststroke, in mice after being subjected to MCAO lead to significantly reduced elevation of brain hemoglobin, associated with delayed tPA-HT.[42] In vitro studies demonstrated GM6001 facilitates stroke recovery via minimizing delayed tPA damage to endothelial cells and reducing transendothelial electrical resistance. GM6001 treatment also reduced MMP-9 upregulation, degradation of occludin and ZO-1 at 42 h postreperfusion.[42] These results were associated with greater survival rate and a rescue of locomotor function at 7 days’ poststroke.[42] GM6001 suppresses TNF-α converting enzyme expression, and the resulting upregulation of TNF-α is associated with HT, the interplay between these molecules needs to be examined further for a more comprehensive understanding.[24]
Imatinib is an FDA-approved treatment for chronic myelogenous leukemia, along with other cancers, which functions as a platelet-derived GF α-receptor inhibitor. When imatinib (200 mg/kg, oral) was administered 1 h after stroke onset before delayed tPA treatment 5 h after the ischemic event, instances of HT were significantly reduced.[43] The combination treatments led to reduced BBB permeability and lesion volume. Due to the limited time window to administer imatinib, 1 h, it presents similar concerns as tPA and further studies should investigate its affects at later time points after stroke onset.[43]
IMM-H004 (10 mg/kg, IV), an organic heterocyclic compound, was given 6 h after stroke onset along with tPA (10 mg/kg, IV) to rats which were subjected to embolic stroke.[25] IMM-H004 was associated with reduced hemorrhage, infarct volume, and brain swelling, along with fewer incidents of tPA-induced HT.[44] Decreased levels of MMP-9 and MMP-2, enhancing co-localization of astrocytes with MMP-2 and IgG leakage, and upregulation of occludin were suggested mechanisms of action to describe the beneficial properties of IMM-H004. In addition, IMM-H004 was shown to improve vascularization 7 days poststroke, leading to enhanced cerebral blood flow through the stability of vascular endothelial cells. Other in vitro studies have shown that IMM-H004 elevates levels of ATP, protein kinase A (PKA), and PI3K-dependent activation of Akt in HBMECs and PC12 cells, pointing to the role PI3K/Akt and cAMP/PKA signaling pathways.[44] Suggesting IMM-H004 may reduce delayed tPA HT by promoting neurovascularization and promoting BBB integrity.[44]
Minocycline (3 mg/kg, IV), currently utilized for acne vulgaris, given 4 h after stroke onset with delayed tPA (10 mg/kg, IV) at 6 h has demonstrated diminished infarct volume and reduced HT at 24 h following embolic stroke.[26] Minocycline functions as an MMP inhibitor, reducing plasma MMP-9 levels which correspond to increased infarct volume and HT.[26,27] However, cerebral levels of MMP-9 were not determined and therefore can be further studied to elucidate the potential correlation between brain MMP-9 levels and ischemic volume and hemorrhaging.[26] In a clinical trial to assess the safety and efficacy of minocycline in conjunction with tPA, 60% of patients were administered a loading dose of minocycline in a 6-h window followed up by maintenance dosing for 3 days, which resulted in no intracerebral hemorrhaging.[45] When tPA was given with minocycline, patients showed reduced levels of plasma MMP-9.[46] Various other clinical trials with varying populations have been initiated and pending results.[47]
Nondrug Therapeutics for Ischemic Stroke Treatment
Lengthening the treatment window for tPA administration may be attainable through nondrug therapies, in addition to pharmacological approaches.[9] Multi-faceted abilities of stem cells suggest their potential as combination therapies to alleviate adverse effects of delayed tPA treatment.[9,48] Gas therapy has also been studied for its potential as a conjunctive therapy to attenuate the various complications associated with delayed tPA treatment.[49] Additional nondrug therapies are well-defined methods, such as brain imaging and endovascular procedures, which have been shown to assist in visualization of stroke pathology and prolong the treatment window for tPA therapy in ischemic stroke.[9,50,51,52]
As noted above, minocycline has been demonstrated to decrease the incidence of HT related to delayed tPA administration.[16] Intracranial transplantation of neural stem cells (hNSCs) has also been shown to support BBB integrity following ischemic stroke.[53] In vivo studies have demonstrated the reduced mortality related to delayed tPA treatment when administered as a co-treatment with tPA 6 h poststroke in aged mice.[54] In addition, significant mitigation of adverse effects associated with delayed tPA treatment was observed when mice were administered minocycline and intracranially transplanted hNSCs 24 h poststroke.[54,55] This combination therapy suggests tPA, minocycline with stem cell transplantation could attenuate delayed tPA adverse effects as well as induce neuroplasticity following stroke.
Various types of stem cells have also been assessed and shown potential for attenuating the serious effects related to delayed tPA treatment. Mesenchymal stem cells (MSCs) have been demonstrated to improve functional outcomes, lower stroke volume, and HT incidence in rats following tPA treatment 1 h and 30 min after reperfusion.[28,55] The conjunctive treatment also led to diminished MMP-9 levels when compared to the tPA treatment alone.[56] The ability of MSCs to mitigate endothelial damage is suggested to be the mechanism chiefly responsible for its ability to reduce HT incidence and promote functional recovery. Bone marrow stromal cells (BMSCs) has also been demonstrated to attenuate behavioral deficits in preclinical animal models and clinical settings for stroke patients as well.[56] Liu et al. observed that following intracerebral BMSC transplantation, there was a reduction of MMP activation, resulting in diminished neurovascular damage from tPA administration 1 h and 30 min after MCAO reperfusion.[57] The mechanism highlighted for the beneficial effects of BMSCs is the secretion of neurotrophic factors of differentiated BMSCs (endothelial, glial, and neural cell types). Authors postulate the neuroprotective effects of BMSCs are primarily useful for mitigating damage sustained from tPA treatment for acute ischemic stroke. However, as MSCs, BMSCs, and other stem cells exist endogenously, a more complete understanding of the therapeutic potential of minocycline and other drugs should be assessed on endogenous and exogenous stem cells to optimize the best combination therapy.
Normobaric hyperoxia (NBO) and hyperbaric oxygen (HBO) therapies have been shown to exert neuroprotective effects when administered early following an ischemic event.[58] Studies have demonstrated that HBO can facilitate BBB integrity through the restriction of reactive oxygen species formation and MMP-9 facilitated damage of TJPs in rats subjected to stroke.[58] MMP-9 production in the afflicted microvessels was limited by early NBO induction (100% O2) of tPA-treated rats at 3, 5, and 7 h post-MCAO stroke. This also worked to minimize loss of claudin-5 and occluding associated with delayed tPA treatment, 5-and 7-h poststroke. Notably, NBO lowered incidence of HT, cerebral edema, infarct volume, and mortality for tPA-treated rats. It has been proposed that NBO could lengthen the tPA treatment window up to 7 h following stroke. Additional well-designed clinical studies are necessary to further assess the safety and efficacy of NBO and HBO as treatments to be used to mitigate adverse complication associated with tPA.[29,49,59,60,61]
Brain imaging has been utilized to evaluate patients with elevated risk of hemorrhage and poor clinical outcomes (51). This practice has assisted with treatment decisions and subsequently enhanced tPA's treatment window without sacrificing safety. Other studies have shown that endovascular procedures, such as intra-arterial thrombectomy, have enhanced clinical outcomes for stroke patients who also received IV thrombolysis. In comparison to thrombolysis alone, thrombectomy combined with thrombolysis augments functional recovery and helps to minimize mortality in patients with ischemic stroke.[51,52]
Conclusions
As we only included preclinical studies which defined delayed tPA treatment as >4.5 h poststroke onset in this review, it is important to recognize other drugs shown to mitigate HT and other adverse effects of tPA therapy administered <4.5 h after stroke in animal models.[12,13,14] Yet, as the majority of studies mentioned in this article are preclinical studies, it is imperative that conclusions be made prudently. As each study varied in examining the specific groups of animals, the effects of age and gender on poststroke outcomes, particularly HT induced by delayed tPA treatment, needs to be further studied. In addition, thorough preclinical investigations of each experimental therapy are suggested to elucidate the cause of increased HT incidence when utilizing this treatment.[62] Further preclinical studies are warranted on therapies which exhibit neuroprotection in addition to mitigating the risk of HT. Nonetheless, a meta-data analysis of 6765 patients in nine clinical trials of IV alteplase compared to controls demonstrated an increase in the incidence of HT results from various factors, including severity of the stroke.[63] Accordingly, therapies combined with tPA may facilitate neuroprotection and hasten rescue of brain tissue following stroke. As tPA is vital for reperfusion treatment, elucidating the optimal dosage and timing in respect to tPA administration is key to improving potential clinical benefits of a combined therapy. The requirements for optimal timing are rigorous and must not interfere with the known fibrinolytic activity of tPA, per the FDA.[62] Just as important as identifying new combined interventions to pair with tPA, it is equally important to understand the exact mechanism by which delayed tPA-induced HT occurs, as well as other adverse effects. In addition, determining the long-term effectiveness and safety of new combined therapies should be a priority in deciding its clinical potential. These long-term assessments should include motor and behavioral functions up to several months posttreatment in view of Stroke Treatment Academic Industry Roundtable guidelines.[64,65] If the proposed therapy includes stem cells, the Stem cell Therapeutics as an Emerging Paradigm for Stroke reference may prove beneficial in translating new interventions to the clinic.[66]
Lengthening tPA's therapeutic window through combination treatments will significantly reduce the incidence of HT and other adverse effects, leading to the improved risk-benefit ratio for thrombolytic therapy and therefore substantially increase the number of patients qualified for tPA treatment. Patients who experience “wake-up strokes,” a case where a patient awakens with stroke symptoms, also benefit from prolonged tPA treatment window as it alleviates challenges presented to acute stroke physicians.[67] The prolonged treatment window for tPA also lengthens the window of neuroplasticity which is associated with enhanced functional outcomes poststroke.
Preclinical research is continuously being conducted to find novel fibrinolytics or thrombolytic agents which enhance reperfusion capacity greater than tPA.[14,68,69] Equally important is our laboratory investigations to examine other interventions to augment the only FDA-approved stroke treatment.[9]
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Koton S, Schneider AL, Rosamond WD, Shahar E, Sang Y, Gottesman RF, et al. Stroke incidence and mortality trends in US communities, 1987 to 2011. JAMA. 2014;312:259–68. doi: 10.1001/jama.2014.7692. [DOI] [PubMed] [Google Scholar]
- 2.Intracerebral hemorrhage after intravenous t-PA therapy for ischemic stroke. The NINDS t-PA stroke study group. Stroke. 1997;28:2109–18. doi: 10.1161/01.str.28.11.2109. [DOI] [PubMed] [Google Scholar]
- 3.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, et al. Heart disease and stroke statistics-2014 update: A report from the American Heart Association. Circulation. 2014;129:e28–92. doi: 10.1161/01.cir.0000441139.02102.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Graham GD. Tissue plasminogen activator for acute ischemic stroke in clinical practice: A meta-analysis of safety data. Stroke. 2003;34:2847–50. doi: 10.1161/01.STR.0000101752.23813.C3. [DOI] [PubMed] [Google Scholar]
- 5.Yip TR, Demaerschalk BM. Estimated cost savings of increased use of intravenous tissue plasminogen activator for acute ischemic stroke in Canada. Stroke. 2007;38:1952–5. doi: 10.1161/STROKEAHA.106.479477. [DOI] [PubMed] [Google Scholar]
- 6.Greenberg DA. Neurogenesis and stroke. CNS Neurol Disord Drug Targets. 2007;6:321–5. doi: 10.2174/187152707783220901. [DOI] [PubMed] [Google Scholar]
- 7.Adams H, Adams R, Del Zoppo G, Goldstein LB Stroke Council of the American Heart Association, American Stroke Association. Guidelines for the early management of patients with ischemic stroke: 2005 guidelines update a scientific statement from the stroke council of the American Heart Association/American Stroke association. Stroke. 2005;36:916–23. doi: 10.1161/01.STR.0000163257.66207.2d. [DOI] [PubMed] [Google Scholar]
- 8.Wang X, Tsuji K, Lee SR, Ning M, Furie KL, Buchan AM, et al. Mechanisms of hemorrhagic transformation after tissue plasminogen activator reperfusion therapy for ischemic stroke. Stroke. 2004;35:2726–30. doi: 10.1161/01.STR.0000143219.16695.af. [DOI] [PubMed] [Google Scholar]
- 9.Peña ID, Borlongan C, Shen G, Davis W. Strategies to extend thrombolytic time window for ischemic stroke treatment: An unmet clinical need. J Stroke. 2017;19:50–60. doi: 10.5853/jos.2016.01515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosell A, Foerch C, Murata Y, Lo EH. Mechanisms and markers for hemorrhagic transformation after stroke. Acta Neurochir Suppl. 2008;105:173–8. doi: 10.1007/978-3-211-09469-3_34. [DOI] [PubMed] [Google Scholar]
- 11.Wang W, Li M, Chen Q, Wang J. Hemorrhagic transformation after tissue plasminogen activator reperfusion therapy for ischemic stroke: Mechanisms, models, and biomarkers. Mol Neurobiol. 2015;52:1572–9. doi: 10.1007/s12035-014-8952-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jickling GC, Liu D, Stamova B, Ander BP, Zhan X, Lu A, et al. Hemorrhagic transformation after ischemic stroke in animals and humans. J Cereb Blood Flow Metab. 2014;34:185–99. doi: 10.1038/jcbfm.2013.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lapchak PA. Hemorrhagic transformation following ischemic stroke: Significance, causes, and relationship to therapy and treatment. Curr Neurol Neurosci Rep. 2002;2:38–43. doi: 10.1007/s11910-002-0051-0. [DOI] [PubMed] [Google Scholar]
- 14.Kanazawa M, Takahashi T, Nishizawa M, Shimohata T. Therapeutic strategies to attenuate hemorrhagic transformation after tissue plasminogen activator treatment for acute ischemic stroke. J Atheroscler Thromb. 2017;24:240–53. doi: 10.5551/jat.RV16006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Allahtavakoli M, Amin F, Esmaeeli-Nadimi A, Shamsizadeh A, Kazemi-Arababadi M, Kennedy D, et al. Ascorbic acid reduces the adverse effects of delayed administration of tissue plasminogen activator in a rat stroke model. Basic Clin Pharmacol Toxicol. 2015;117:335–9. doi: 10.1111/bcpt.12413. [DOI] [PubMed] [Google Scholar]
- 16.Sumii T, Lo EH. Involvement of matrix metalloproteinase in thrombolysis-associated hemorrhagic transformation after embolic focal ischemia in rats. Stroke. 2002;33:831–6. doi: 10.1161/hs0302.104542. [DOI] [PubMed] [Google Scholar]
- 17.Tan Z, Lucke-Wold BP, Logsdon AF, Turner RC, Tan C, Li X, et al. Bryostatin extends tPA time window to 6 h following middle cerebral artery occlusion in aged female rats. Eur J Pharmacol. 2015;764:404–12. doi: 10.1016/j.ejphar.2015.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tan Z, Turner RC, Leon RL, Li X, Hongpaisan J, Zheng W, et al. Bryostatin improves survival and reduces ischemic brain injury in aged rats after acute ischemic stroke. Stroke. 2013;44:3490–7. doi: 10.1161/STROKEAHA.113.002411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suzuki Y, Nagai N, Umemura K, Collen D, Lijnen HR. Stromelysin-1 (MMP-3) is critical for intracranial bleeding after t-PA treatment of stroke in mice. J Thromb Haemost. 2007;5:1732–9. doi: 10.1111/j.1538-7836.2007.02628.x. [DOI] [PubMed] [Google Scholar]
- 20.Koto T, Takubo K, Ishida S, Shinoda H, Inoue M, Tsubota K, et al. Hypoxia disrupts the barrier function of neural blood vessels through changes in the expression of claudin-5 in endothelial cells. Am J Pathol. 2007;170:1389–97. doi: 10.2353/ajpath.2007.060693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Culp WC, Brown AT, Lowery JD, Arthur MC, Roberson PK, Skinner RD, et al. Dodecafluoropentane emulsion extends window for tPA therapy in a rabbit stroke model. Mol Neurobiol. 2015;52:979–84. doi: 10.1007/s12035-015-9243-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hartung T. Anti-inflammatory effects of granulocyte colony-stimulating factor. Curr Opin Hematol. 1998;5:221–5. doi: 10.1097/00062752-199805000-00013. [DOI] [PubMed] [Google Scholar]
- 23.Sobrino T, Millán M, Castellanos M, Blanco M, Brea D, Dorado L, et al. Association of growth factors with arterial recanalization and clinical outcome in patients with ischemic stroke treated with tPA. J Thromb Haemost. 2010;8:1567–74. doi: 10.1111/j.1538-7836.2010.03897.x. [DOI] [PubMed] [Google Scholar]
- 24.Mayne M, Ni W, Yan HJ, Xue M, Johnston JB, Del Bigio MR, et al. Antisense oligodeoxynucleotide inhibition of tumor necrosis factor-alpha expression is neuroprotective after intracerebral hemorrhage. Stroke. 2001;32:240–8. doi: 10.1161/01.str.32.1.240. [DOI] [PubMed] [Google Scholar]
- 25.Fylaktakidou KC, Hadjipavlou-Litina DJ, Litinas KE, Nicolaides DN. Natural and synthetic coumarin derivatives with anti-inflammatory/antioxidant activities. Curr Pharm Des. 2004;10:3813–33. doi: 10.2174/1381612043382710. [DOI] [PubMed] [Google Scholar]
- 26.Murata Y, Rosell A, Scannevin RH, Rhodes KJ, Wang X, Lo EH, et al. Extension of the thrombolytic time window with minocycline in experimental stroke. Stroke. 2008;39:3372–7. doi: 10.1161/STROKEAHA.108.514026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Machado LS, Kozak A, Ergul A, Hess DC, Borlongan CV, Fagan SC, et al. Delayed minocycline inhibits ischemia-activated matrix metalloproteinases 2 and 9 after experimental stroke. BMC Neurosci. 2006;7:56. doi: 10.1186/1471-2202-7-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakazaki M, Sasaki M, Kataoka-Sasaki Y, Oka S, Namioka T, Namioka A, et al. Intravenous infusion of mesenchymal stem cells inhibits intracranial hemorrhage after recombinant tissue plasminogen activator therapy for transient middle cerebral artery occlusion in rats. J Neurosurg. 2017;127:917–26. doi: 10.3171/2016.8.JNS16240. [DOI] [PubMed] [Google Scholar]
- 29.Boussi-Gross R, Golan H, Volkov O, Bechor Y, Hoofien D, Beeri MS, et al. Improvement of memory impairments in poststroke patients by hyperbaric oxygen therapy. Neuropsychology. 2015;29:610–21. doi: 10.1037/neu0000149. [DOI] [PubMed] [Google Scholar]
- 30.Ishiguro M, Kawasaki K, Suzuki Y, Ishizuka F, Mishiro K, Egashira Y, et al. Arho kinase (ROCK) inhibitor, fasudil, prevents matrix metalloproteinase-9-related hemorrhagic transformation in mice treated with tissue plasminogen activator. Neuroscience. 2012;220:302–12. doi: 10.1016/j.neuroscience.2012.06.015. [DOI] [PubMed] [Google Scholar]
- 31.Ballabh P, Braun A, Nedergaard M. The blood-brain barrier: An overview: Structure, regulation, and clinical implications. Neurobiol Dis. 2004;16:1–13. doi: 10.1016/j.nbd.2003.12.016. [DOI] [PubMed] [Google Scholar]
- 32.Ishiguro M, Mishiro K, Fujiwara Y, Chen H, Izuta H, Tsuruma K, et al. Phosphodiesterase-III inhibitor prevents hemorrhagic transformation induced by focal cerebral ischemia in mice treated with tPA. PLoS One. 2010;5:e15178. doi: 10.1371/journal.pone.0015178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Keep RF, Zhou N, Xiang J, Andjelkovic AV, Hua Y, Xi G, et al. Vascular disruption and blood-brain barrier dysfunction in intracerebral hemorrhage. Fluids Barriers CNS. 2014;11:18. doi: 10.1186/2045-8118-11-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thiyagarajan M, Fernández JA, Lane SM, Griffin JH, Zlokovic BV. Activated protein C promotes neovascularization and neurogenesis in postischemic brain via protease-activated receptor 1. J Neurosci. 2008;28:12788–97. doi: 10.1523/JNEUROSCI.3485-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang L, Chopp M, Jia L, Cui Y, Lu M, Zhang ZG, et al. Atorvastatin extends the therapeutic window for tPA to 6 h after the onset of embolic stroke in rats. J Cereb Blood Flow Metab. 2009;29:1816–24. doi: 10.1038/jcbfm.2009.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ishrat T, Pillai B, Ergul A, Hafez S, Fagan SC. Candesartan reduces the hemorrhage associated with delayed tissue plasminogen activator treatment in rat embolic stroke. Neurochem Res. 2013;38:2668–77. doi: 10.1007/s11064-013-1185-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang Z, Huang PL, Ma J, Meng W, Ayata C, Fishman MC, et al. Enlarged infarcts in endothelial nitric oxide synthase knockout mice are attenuated by nitro-L-arginine. J Cereb Blood Flow Metab. 1996;16:981–7. doi: 10.1097/00004647-199609000-00023. [DOI] [PubMed] [Google Scholar]
- 38.Matsumoto M. Cilostazol in secondary prevention of stroke: Impact of the cilostazol stroke prevention study. Atheroscler Suppl. 2005;6:33–40. doi: 10.1016/j.atherosclerosissup.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 39.Shibuya M, Suzuki Y, Sugita K, Saito I, Sasaki T, Takakura K, et al. Effect of AT877 on cerebral vasospasm after aneurysmal subarachnoid hemorrhage. Results of a prospective placebo-controlled double-blind trial. J Neurosurg. 1992;76:571–7. doi: 10.3171/jns.1992.76.4.0571. [DOI] [PubMed] [Google Scholar]
- 40.dela Peña IC, Yoo A, Tajiri N, Acosta SA, Ji X, Kaneko Y, et al. Granulocyte colony-stimulating factor attenuates delayed tPA-induced hemorrhagic transformation in ischemic stroke rats by enhancing angiogenesis and vasculogenesis. J Cereb Blood Flow Metab. 2015;35:338–46. doi: 10.1038/jcbfm.2014.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hao JL, Nagano T, Nakamura M, Kumagai N, Mishima H, Nishida T, et al. Galardin inhibits collagen degradation by rabbit keratocytes by inhibiting the activation of pro-matrix metalloproteinases. Exp Eye Res. 1999;68:565–72. doi: 10.1006/exer.1998.0637. [DOI] [PubMed] [Google Scholar]
- 42.Mishiro K, Ishiguro M, Suzuki Y, Tsuruma K, Shimazawa M, Hara H, et al. Abroad-spectrum matrix metalloproteinase inhibitor prevents hemorrhagic complications induced by tissue plasminogen activator in mice. Neuroscience. 2012;205:39–48. doi: 10.1016/j.neuroscience.2011.12.042. [DOI] [PubMed] [Google Scholar]
- 43.Su EJ, Fredriksson L, Geyer M, Folestad E, Cale J, Andrae J, et al. Activation of PDGF-CC by tissue plasminogen activator impairs blood-brain barrier integrity during ischemic stroke. Nat Med. 2008;14:731–7. doi: 10.1038/nm1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zuo W, Chen J, Zhang S, Tang J, Liu H, Zhang D, et al. IMM-H004 prevents toxicity induced by delayed treatment of tPA in a rat model of focal cerebral ischemia involving PKA-and PI3K-dependent akt activation. Eur J Neurosci. 2014;39:2107–18. doi: 10.1111/ejn.12551. [DOI] [PubMed] [Google Scholar]
- 45.Fagan SC, Waller JL, Nichols FT, Edwards DJ, Pettigrew LC, Clark WM, et al. Minocycline to improve neurologic outcome in stroke (MINOS): A dose-finding study. Stroke. 2010;41:2283–7. doi: 10.1161/STROKEAHA.110.582601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Switzer JA, Hess DC, Ergul A, Waller JL, Machado LS, Portik-Dobos V, et al. Matrix metalloproteinase-9 in an exploratory trial of intravenous minocycline for acute ischemic stroke. Stroke. 2011;42:2633–5. doi: 10.1161/STROKEAHA.111.618215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blacker DJ, Prentice D, Alvaro A, Bates TR, Bynevelt M, Kelly A, et al. Reducing haemorrhagic transformation after thrombolysis for stroke: A strategy utilising minocycline. Stroke Res Treat. 2013;2013:362961. doi: 10.1155/2013/362961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Borlongan CV. Bone marrow stem cell mobilization in stroke: A ‘bonehead’ may be good after all! Leukemia. 2011;25:1674–86. doi: 10.1038/leu.2011.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liang J, Qi Z, Liu W, Wang P, Shi W, Dong W, et al. Normobaric hyperoxia slows blood-brain barrier damage and expands the therapeutic time window for tissue-type plasminogen activator treatment in cerebral ischemia. Stroke. 2015;46:1344–51. doi: 10.1161/STROKEAHA.114.008599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bentley P, Ganesalingam J, Carlton Jones AL, Mahady K, Epton S, Rinne P, et al. Prediction of stroke thrombolysis outcome using CT brain machine learning. Neuroimage Clin. 2014;4:635–40. doi: 10.1016/j.nicl.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Berkhemer OA, Fransen PS, Beumer D, van den Berg LA, Lingsma HF, Yoo AJ, et al. Arandomized trial of intraarterial treatment for acute ischemic stroke. N Engl J Med. 2015;372:11–20. doi: 10.1056/NEJMoa1411587. [DOI] [PubMed] [Google Scholar]
- 52.Minnerup J, Wersching H, Teuber A, Wellmann J, Eyding J, Weber R, et al. Outcome after thrombectomy and intravenous thrombolysis in patients with acute ischemic stroke: A Prospective observational study. Stroke. 2016;47:1584–92. doi: 10.1161/STROKEAHA.116.012619. [DOI] [PubMed] [Google Scholar]
- 53.Huang L, Wong S, Snyder EY, Hamblin MH, Lee JP. Human neural stem cells rapidly ameliorate symptomatic inflammation in early-stage ischemic-reperfusion cerebral injury. Stem Cell Res Ther. 2014;5:129. doi: 10.1186/scrt519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eckert AD, Hamblin M, Lee JP. Neural stem cells reduce symptomatic inflammation and mortality in aged stroke mice following delayed tpa treatment. FASEB J. 2017;31(Suppl 1):693.6–693.6. [Google Scholar]
- 55.Anderson JD, Pham MT, Contreras Z, Hoon M, Fink KD, Johansson HJ, et al. Mesenchymal stem cell-based therapy for ischemic stroke. Chin Neurosurg J. 2016;2:36. [Google Scholar]
- 56.Bang OY, Lee JS, Lee PH, Lee G. Autologous mesenchymal stem cell transplantation in stroke patients. Ann Neurol. 2005;57:874–82. doi: 10.1002/ana.20501. [DOI] [PubMed] [Google Scholar]
- 57.Liu N, Deguchi K, Yamashita T, Liu W, Ikeda Y, Abe K, et al. Intracerebral transplantation of bone marrow stromal cells ameliorates tissue plasminogen activator-induced brain damage after cerebral ischemia in mice detected by in vivo and ex vivo optical imaging. J Neurosci Res. 2012;90:2086–93. doi: 10.1002/jnr.23104. [DOI] [PubMed] [Google Scholar]
- 58.Singhal AB. A review of oxygen therapy in ischemic stroke. Neurol Res. 2007;29:173–83. doi: 10.1179/016164107X181815. [DOI] [PubMed] [Google Scholar]
- 59.Chang CF, Niu KC, Hoffer BJ, Wang Y, Borlongan CV. Hyperbaric oxygen therapy for treatment of postischemic stroke in adult rats. Exp Neurol. 2000;166:298–306. doi: 10.1006/exnr.2000.7506. [DOI] [PubMed] [Google Scholar]
- 60.Hu Q, Manaenko A, Bian H, Guo Z, Huang JL, Guo ZN, et al. Hyperbaric oxygen reduces infarction volume and hemorrhagic transformation through ATP/NAD+/Sirt1 pathway in hyperglycemic middle cerebral artery occlusion rats. Stroke. 2017;48:1655–64. doi: 10.1161/STROKEAHA.116.015753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhai WW, Sun L, Yu ZQ, Chen G. Hyperbaric oxygen therapy in experimental and clinical stroke. Med Gas Res. 2016;6:111–8. doi: 10.4103/2045-9912.184721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ehrenreich H, Weissenborn K, Prange H, Schneider D, Weimar C, Wartenberg K, et al. Recombinant human erythropoietin in the treatment of acute ischemic stroke. Stroke. 2009;40:e647–56. doi: 10.1161/STROKEAHA.109.564872. [DOI] [PubMed] [Google Scholar]
- 63.Whiteley WN, Emberson J, Lees KR, Blackwell L, Albers G, Bluhmki E, et al. Risk of intracerebral haemorrhage with alteplase after acute ischaemic stroke: A secondary analysis of an individual patient data meta-analysis. Lancet Neurol. 2016;15:925–33. doi: 10.1016/S1474-4422(16)30076-X. [DOI] [PubMed] [Google Scholar]
- 64.Albers GW, Goldstein LB, Hess DC, Wechsler LR, Furie KL, Gorelick PB, et al. Stroke treatment academic industry roundtable (STAIR) recommendations for maximizing the use of intravenous thrombolytics and expanding treatment options with intra-arterial and neuroprotective therapies. Stroke. 2011;42:2645–50. doi: 10.1161/STROKEAHA.111.618850. [DOI] [PubMed] [Google Scholar]
- 65.Lapchak PA, Zhang JH, Noble-Haeusslein LJ. RIGOR guidelines: Escalating STAIR and STEPS for effective translational research. Transl Stroke Res. 2013;4:279–85. doi: 10.1007/s12975-012-0209-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Diamandis T, Borlongan CV. One, two, three steps toward cell therapy for stroke. Stroke. 2015;46:588–91. doi: 10.1161/STROKEAHA.114.007105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rubin MN, Barrett KM. What to do with wake-up stroke. Neurohospitalist. 2015;5:161–72. doi: 10.1177/1941874415576204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Parsons M, Spratt N, Bivard A, Campbell B, Chung K, Miteff F, et al. A randomized trial of tenecteplase versus alteplase for acute ischemic stroke. N Engl J Med. 2012;366:1099–107. doi: 10.1056/NEJMoa1109842. [DOI] [PubMed] [Google Scholar]
- 69.Henninger N, Fisher M. Extending the time window for endovascular and pharmacological reperfusion. Transl Stroke Res. 2016;7:284–93. doi: 10.1007/s12975-015-0444-4. [DOI] [PubMed] [Google Scholar]