

Abstract
Endothelial activation is a hallmark of the high-glucose (HG)-induced retinal inflammation associated with diabetic retinopathy (DR). However, precisely how HG induces retinal endothelial activation is not fully understood. We hypothesized that HG-induced up-regulation of lysyl oxidase (LOX), a collagen-cross-linking enzyme, in retinal capillary endothelial cells (ECs) enhances subendothelial basement membrane (BM) stiffness, which, in turn, promotes retinal EC activation. Diabetic C57BL/6 mice exhibiting a 70 and 50% increase in retinal intercellular adhesion molecule (ICAM)-1 expression and leukocyte accumulation, respectively, demonstrated a 2-fold increase in the levels of BM collagen IV and LOX, key determinants of capillary BM stiffness. Using atomic force microscopy, we confirmed that HG significantly enhances LOX-dependent subendothelial matrix stiffness in vitro, which correlated with an ∼2.5-fold increase in endothelial ICAM-1 expression, a 4-fold greater monocyte–EC adhesion, and an ∼2-fold alteration in endothelial NO (decrease) and NF-κB activation (increase). Inhibition of LOX-dependent subendothelial matrix stiffening alone suppressed HG-induced retinal EC activation. Finally, using synthetic matrices of tunable stiffness, we demonstrated that subendothelial matrix stiffening is necessary and sufficient to promote EC activation. These findings implicate BM stiffening as a critical determinant of HG-induced retinal EC activation and provide a rationale for examining BM stiffness and underlying mechanotransduction pathways as therapeutic targets for diabetic retinopathy.—Yang, X., Scott, H. A., Monickaraj, F., Xu, J., Ardekani, S., Nitta, C. F., Cabrera, A., McGuire, P. G., Mohideen, U., Das, A., Ghosh, K. Basement membrane stiffening promotes retinal endothelial activation associated with diabetes.
Keywords: lysyl oxidase, matrix stiffness, nitric oxide, inflammation, diabetic retinopathy
Diabetic retinopathy (DR) is an important microvascular complication of diabetes and the leading cause of blindness in the working-age population (1). Growing evidence indicates that DR is a multifactorial condition that is strongly regulated by retinal inflammation (2, 3). A critical early step in diabetic retinal inflammation is the activation of retinal capillary endothelial cells (ECs) (3, 4). Activated retinal ECs up-regulate cell adhesion molecules such as intercellular adhesion molecule (ICAM)-1 to promote leukocyte–EC adhesion (leukostasis). The adherent leukocytes subsequently release proinflammatory and vascular permeability factors that together disrupt the blood–retinal barrier, leading to an increase in vascular permeability that culminates in macular edema (3, 5). Blocking endothelial ICAM-1 alone prevents diabetic retinal leukostasis and vascular hyperpermeability in animal models of DR (4), thereby implicating retinal EC activation as a major determinant of DR pathogenesis.
Studies aimed at understanding diabetic retinal EC activation have focused primarily on the role of abnormal epigenetic, metabolic, and inflammatory factors (3, 6). However, a recent study reported that diabetic rat retinal ECs express increased levels of lysyl oxidase (LOX) (7), a copper-dependent enzyme that cross-links collagen and enhances extracellular matrix stiffness (8). Notably, LOX overexpression and the associated increase in extracellular matrix stiffness have been shown to promote endothelial dysfunction, resulting in lung vascular hyperpermeability (9). Whether LOX up-regulation in diabetic retinal ECs leads to subendothelial basement membrane (BM) stiffening and EC activation, however, remains unknown.
It is important to understand the role of subendothelial BM stiffening in diabetic retinal EC activation and inflammation, as it can modulate the efficacy of existing and new anti-inflammatory DR therapies that commonly target soluble factors and their signaling pathways. This modulation occurs because changes in extracellular matrix stiffness are known to regulate intracellular biochemistry independently and thereby to mediate cell response to soluble cues via alteration in cytoskeletal tension and cell–matrix adhesion strength (10). This conversion of matrix stiffness–dependent mechanical cues into intracellular biochemical signaling (termed mechanotransduction) plays an important role in regulating tissue function and disease. For instance, abnormal subendothelial matrix stiffness and associated alterations in intracellular mechanical signaling have been strongly implicated in vascular malformations (11), loss of capillary ECs (12), pulmonary edema (9), and atherosclerosis (13). Thus, we hypothesized that LOX up-regulation in diabetic retinas promotes increased cross-linking and stiffening of retinal subendothelial BM, which, in turn, actively contributes to the retinal inflammation in DR.
Using atomic force microscopy, we showed that high glucose (HG), the major risk factor for DR, causes a significant increase in the stiffness of retinal EC-secreted matrix in vitro, which correlates with increased subendothelial collagen IV deposition, LOX up-regulation, NF-κB–dependent retinal EC activation, and monocyte–EC adhesion. These findings are consistent with in vivo observations showing that diabetic mouse retinal inflammation correlates with higher levels of LOX and BM collagen IV. Finally, by pharmacological inhibition of LOX and development of synthetic matrices, we showed that HG-induced subendothelial matrix stiffening is necessary and sufficient for diabetic retinal EC activation.
MATERIALS AND METHODS
Animal model
All animal studies were performed according to the Association for Research in Vision and Ophthalmology (ARVO) Statement for the Use of Animals in Ophthalmic and Vision Research and were in compliance with the Institutional Animal Care and Use Committee protocols approved by University of California, Riverside, and the University of New Mexico. Adult (8-wk-old) male C57BL6/J mice (Jackson Laboratory, Bar Harbor, ME, USA) were injected with streptozotocin (60 mg/kg body weight; Sigma-Aldrich, St. Louis, MO, USA) in 10 mM citrate buffer (pH 4.5) daily for 5 consecutive days. Animals with fasting blood glucose greater than 300 mg/dl were considered diabetic and were studied at 4 mo duration of diabetes. Age-matched mice receiving no streptozotocin injection were used as the nondiabetic control.
Retinal whole-mount immunofluorescence
Enucleated eyes from diabetic and control mice were fixed in 4% paraformaldehyde (PFA; Electron Microscopy Sciences, Fort Washington, PA, USA) for 4 h before retinal isolation. Next, isolated retinas were permeabilized in 1% Triton X-100 and labeled with rabbit anti-collagen IV antibody (cat. no. ab6586; Abcam, Cambridge, MA, USA) followed by fluorescence-labeled anti-rabbit IgG (cat. no. DI-1488; Vector Laboratories, Burlingame, CA, USA). Retinal capillaries were counterstained with isolectin GS-IB4 (cat. no. I21412; Thermo Fisher–Life Technologies, Carlsbad, CA, USA) and imaged with an SP5 confocal microscope (Leica Microsystems, Buffalo Grove, IL, USA). To confirm the specificity of anti-collagen IV immunostaining, some retinal whole mounts were labeled with secondary antibody alone.
Cell culture and glucose treatment
Human retinal ECs, monkey chorioretinal ECs (RF/6A), and human monocytes (U937) were purchased from Cell Systems (Kirkland, WA, USA) and ATCC (Manassas, VA, USA), respectively. Human retinal ECs were grown in MCDB131 medium (Mediatech, Manassas, VA, USA) supplemented with 10% fetal bovine serum (FBS; Thermo Fisher–Life Technologies), 2 mM l-glutamine (Thermo Fisher–Life Technologies), 0.03 mg/ml Endothelial Cell Growth Supplement (Sigma-Aldrich), 1× antimycotic/antibiotic mixture (Thermo Fisher–Life Technologies), 10 ng/ml human epidermal growth factor (Millipore, Billerica, MA, USA), 1 μg/ml hydrocortisone, and 0.09 mg/ml heparin (Sigma-Aldrich). Monkey chorioretinal ECs were grown in Eagle’s minimum essential medium (ATCC) supplemented with 10% FBS and 1× antimycotic/antibiotic mixture. U937 monocytes were grown in RPMI-1640 medium (GE Healthcare, Pittsburgh, PA, USA) supplemented with 10% FBS, 2 mM l-glutamine, 1.5 mg/ml sodium bicarbonate (Thermo Fisher–Life Technologies), 1× antimycotic/antibiotic mixture, 1 mM sodium pyruvate (Thermo Fisher–Life Technologies), and 4.5 mg/ml glucose (Sigma-Aldrich). Human CD14+ peripheral blood monocytes (PBMs) were freshly isolated by centrifuging peripheral blood (Zenbio, Durham, NC, USA) in Ficoll-Paque (GE Healthcare Life Sciences) to obtain a buffy coat, which was subsequently spun down, decanted, resuspended in PBS buffer [containing 1% bovine serum albumin (BSA) and 2 mM EDTA], at 250 × 106 cells/ml, and mixed with 20 μl CD14+ magnetic microbeads (Miltenyi Biotec, Auburn, CA, USA) per 10 × 106 cells for 20 min at 4°C. Finally, the cell/bead suspension was spun down, decanted, suspended in PBS buffer, and subjected to magnet-activated cell sorting.
For in vitro studies, EC monolayers were cultured in regular growth medium containing normal glucose (NG; 5.5 mM) or HG (30 mM) ± LOX inhibitor β-aminopropionitrile (BAPN; C3H6N2·0.5C4H4O4; 0.1 mM; ≥98% purity; Sigma-Aldrich) and supplemented with ascorbic acid (200 μg/ml; Sigma-Aldrich) to facilitate BM deposition. BAPN is a specific and irreversible inhibitor of LOX enzymatic activity (7, 14, 15). After 9 d, culture medium was replaced with low-serum (2.5% FBS) medium (with all other components at original concentration), and monolayers were allowed to grow overnight before they were used in specific assays.
Western blot analysis
To detect LOX, ICAM-1, and CD11b expression in vivo, mouse retinas were perfused, isolated, and homogenized in ice-cold RIPA lysis buffer (containing protease and phosphatase inhibitors). To detect LOX expression in cultured ECs, cells were grown in medium containing NG or HG ± BAPN for 10 d, followed by lysis in RIPA lysis buffer. Supernatants from centrifuged retinal and EC culture lysates were subjected to Western blot analysis using nitrocellulose membrane, which was probed with antibodies against LOX (cat. no. NB110-59729; Novus Biologicals, San Diego, CA, USA), CD11b (cat. no. NB110-89474; Novus Biologicals), and ICAM-1 (cat. no. SC-1511; Santa Cruz Biotechnology, Santa Cruz, CA, USA), followed by appropriate horseradish peroxidase–conjugated secondary antibodies (cat. no. DI-1488; Vector Laboratories). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH; cat. no. G9545; Sigma-Aldrich) or β-tubulin (cat. no. ab6046; Abcam) was used as a loading control. Western blot protein bands were visualized with a camera-based imaging system (Biospectrum AC Imaging System; UVP, Upland, CA, USA) that automatically adjusts exposure time to avoid pixel saturation. ImageJ (National Institutes of Health, Bethesda, MD, USA) was used to perform densitometric analysis, and the measurements (within the 10–250 pixel intensity range) were normalized with respect to the corresponding loading controls.
EC-secreted BM
Decellularized (cell-free) subendothelial BM was obtained by using a technique that we and others have reported (12, 16). In brief, ECs in NG or HG ± BAPN medium were grown on glass coverslips coated with glutaraldehyde cross-linked gelatin for 10 d, followed by decellularization of the EC monolayers with a mild detergent composed of 20 mM ammonium hydroxide and 0.5% Triton X-100 (both from Sigma-Aldrich) and fixation of the resultant BM with 1% PFA. Decellularized BMs were labeled with rabbit anti-collagen IV (cat. no. ab6586; Abcam) and fluorescein-conjugated anti-rabbit secondary antibody (cat. no. DI-1488; Vector Laboratories) and imaged with a Leica SP5 confocal microscope. BM thickness (n = 3 per condition) was analyzed from x-z plane images with ImageJ.
Measurement of subendothelial matrix stiffness
The stiffness of unfixed decellularized subendothelial matrices obtained from NG- or HG ± BAPN-treated EC cultures was measured with a biologic-grade AFM (Veeco Instruments, Plainview, NY, USA) operated in tapping mode with an ∼40 nm silicon nitride tip attached to a 140 μm long microcantilever (MLCT; Bruker Corp., Billerica, MA, USA) with a bending–spring constant of 0.1 N/m. The maximum indentation force applied to the sample was ∼8 nN. Measurements were made in force–curve mode, and the cantilever deflection was measured through the photodiode difference signal (Sdef, in volts). For each region, ∼25 force curves were measured, and only the linear region of the force curves was considered for matrix stiffness analysis. Sample stiffness is calculated as ksample = F/ zs, where F is the applied force and zs is the sample deformation. Average stiffness was obtained from indentations in at least 9 different regions across the subendothelial matrix samples.
Real-time PCR
Total RNA was isolated from cells (Direct-zol RNA MiniPrep; Zymo Research, Irvine, CA, USA), converted to cDNA with High Capacity cDNA Reverse Transcription (Thermo Fisher–Applied Biosystems), and amplified with the appropriate TaqMan assay for ICAM-1 (cat. no. 4331182; Thermo Fisher–Applied Biosystems) on the ABI 7500 Fast system (Thermo Fisher–Applied Biosystems). Relative mRNA levels were determined by the comparative cycle threshold method with normalization to GAPDH.
Flow cytometry
ECs were detached and labeled with rabbit anti–ICAM-1 antibody (SC-107; Santa Cruz Biotechnology) followed by fluorescence-labeled anti-mouse IgG (cat. no. DI-2488; Vector Laboratories). Next, ECs were fixed with 1% PFA, detected by a Cell Lab Quanta SC flow cytometer (Beckman Coulter, Brea, CA, USA), and analyzed by FlowJo (Treestar, Inc., Ashland, OR, USA).
Synthetic matrix fabrication
Thin (∼100 μm thick) elastic synthetic matrices of various stiffnesses were prepared by mixing polyacrylamide and bis-acrylamide at different mass ratios (17, 18). Polyacrylamide gels have been extensively used to study extracellular matrix mechanosensing, because they can be synthesized over a broad range of stiffness through simple variation in bis-acrylamide (cross-linker) concentration, in addition to permitting a chemically activated surface for covalent conjugation of any cell-binding matrix protein. Synthetic matrices were prepared at 1000 Pa, which mimics the average stiffness of normal subendothelial matrix (unpublished results), as well as 2000 and 4000 Pa to mimic progressive matrix stiffening. To promote EC spreading, synthetic matrix surfaces were activated by using sulfo-Sanpah (Thermo Scientific–Pierce, Rockford, IL, USA) before covalently conjugating human plasma fibronectin (BD Biosciences) at ∼3 μg/cm2 for 2 h at 37°C. Retinal ECs grown in NG or HG ± BAPN conditions for 10 d were detached and replated on fibronectin-coated synthetic matrices in low-serum medium and incubated overnight before use in assays.
Monocyte–EC adhesion
ECs were cultured in NG or HG ± BAPN medium for 10 d before addition of fluorescence-labeled human PBMs or U937 cells (125,000 cells/cm2) for 30 min at 37°C. After they were rinsed with PBS, adherent monocytes were fixed with 1% PFA, imaged with an Eclipse Ti microscope fitted with a DS-Qi1Mc camera (Nikon, Tokyo, Japan), and counted using ImageJ (≥10 images per condition). To confirm the effect of subendothelial matrix stiffness on monocyte–EC adhesion, NG- and HG-treated ECs were grown on synthetic matrices of normal (1000 Pa) or higher (2000 and 4000 Pa) stiffness in low-serum medium and subjected to the monocyte–EC adhesion assay described above.
NF-κB activation
ECs grown in NG or HG ± BAPN medium for 10 d were detached and replated (in low-serum medium containing NG or HG ± BAPN) on decellularized subendothelial matrices obtained from parallel NG or HG ± BAPN cultures. At 6 h after plating, the cells were fixed with 1% PFA, permeabilized, blocked with 2% BSA, and labeled with rabbit anti–NF-κB p65 antibody (cat. no. SC-109; Santa Cruz Biotechnology) followed by fluorescence-labeled anti-rabbit IgG (cat. no. DI-1488; Vector Laboratories). Cells were counterstained with DAPI to label nuclei and imaged with an Eclipse Ti microscope (Nikon). NF-κB activation was determined by quantifying the fraction of cells (expressed as a percentage of the total) that exhibit significant nuclear NF-κB p65 staining (n ≥ 20 cells per condition), as previously reported (19). To confirm the effect of HG-induced subendothelial matrix stiffening on NF-κB activation, NG-treated ECs were plated (in low-serum medium) on the corresponding decellularized matrices for 6 h before assessment of NF-κB activation. ECs treated with TNF-α (25 ng/ml; 5 h), an inflammatory cytokine that induces NF-κB activation (20), as the positive control, whereas antibody specificity was confirmed by labeling some samples with secondary antibody alone.
Intracellular NO production
Retinal ECs grown in NG or HG ± BAPN medium for 10 d were detached and replated on the corresponding decellularized subendothelial matrices in low-serum medium containing NG or HG ± BAPN. After overnight incubation, the cells were loaded with the fluorescent NO-sensitive dye DAF-FM diacetate (2 µM; Thermo Fisher–Life Technologies), as per the manufacturer’s protocol. After excess dye was rinsed off and the cells were recovered in NG or HG ± BAPN medium, the medium was replaced with Krebs-Henseleit buffer. Dye-loaded cells were imaged with the Nikon Eclipse Ti microscope, and intracellular net fluorescence intensity was quantified with ImageJ in the aforementioned manner (n ≥ 20 cells per condition). To confirm the effect of subendothelial matrix stiffness on intracellular NO, NG- or HG-treated ECs were grown overnight on synthetic matrices of normal (1000 Pa) or high (4000 Pa) stiffness in low-serum medium and subjected to intracellular NO measurement.
LOX activity assay
Retinal ECs were cultured in NG or HG ± BAPN medium for 9 d, followed by overnight starvation in NG or HG ± BAPN-containing phenol red-free medium. LOX activity was determined by the conventional Amplex Red fluorescence assay (21, 22). In brief, culture supernatants were incubated (at 1:1 ratio) with a reaction buffer (37°C; 30 min) containing 10 µM Amplex Red Reagent (Thermo Fisher–Life Technologies), and the LOX activity was determined by measuring emitted fluorescence (540/590 nm), resulting from hydrogen peroxide produced by active LOX, with a fluorescence spectrophotometer (n ≥ 8) and comparing it with a standard curve generated from serially diluted hydrogen peroxide solution.
Statistics
All data were obtained from multiple replicates and are expressed as means ± sem. For studies involving 3 experimental conditions, statistical significance was determined by ANOVA (Instat; GraphPad, San Diego, CA, USA), followed by Tukey’s post hoc analysis. For studies involving two experimental conditions, statistical significance was determined by unpaired Student’s t test (Instat; GraphPad), followed by Welch correction, as well as Pearson’s correlation analysis (SPSS Statistics; IBM, Inc., Armonk, NY, USA). Results were considered significant if P < 0.05.
RESULTS
Retinal endothelial activation is a chief characteristic of diabetes
Activated ECs express ICAM-1, a key EC adhesion molecule that promotes adhesion of circulating leukocytes. Predictably, we found that retinal ECs in diabetic mice exhibit ∼70% higher (P < 0.01) ICAM-1 expression than nondiabetic controls (Fig. 1A). This significant increase in retinal ICAM-1 expression correlated with a markedly (∼50%; P < 0.05) greater accumulation of CD11b+ monocytes/macrophages in diabetic retinas than in nondiabetic controls.
Figure 1.
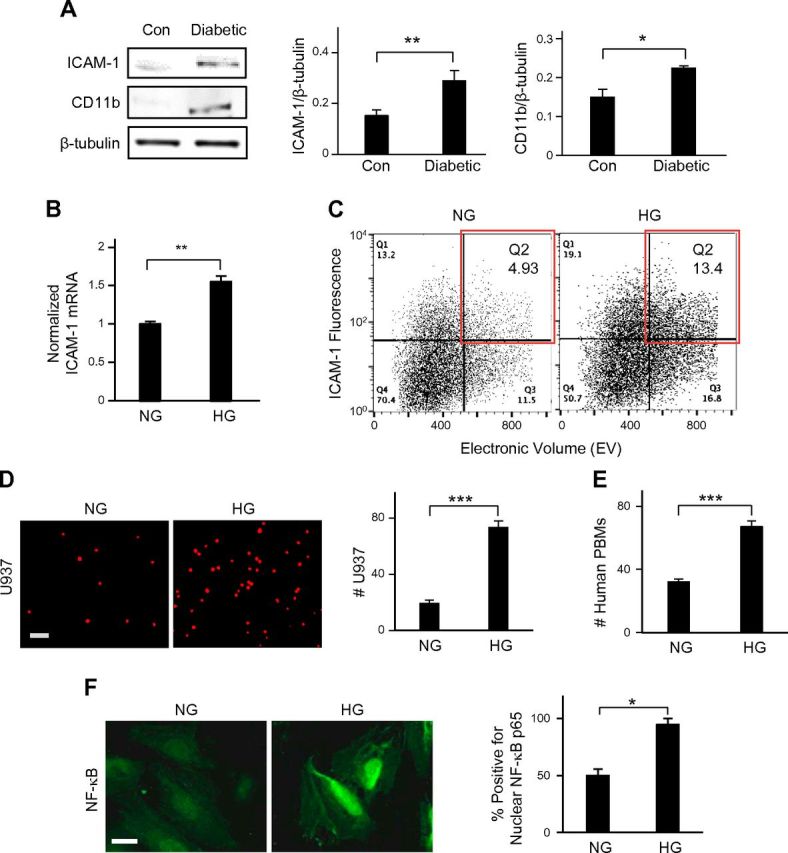
Retinal endothelial activation is a chief characteristic of diabetes. A) Representative Western blot bands and their densitometric analyses (bar graphs) together revealed that the levels of ICAM-1 in ECs and CD11b in monocytes/macrophages were significantly higher in diabetic mouse retinas vs. control retinas (n = 5). *P < 0.05; **P < 0.01. ICAM-1 and CD11b levels were normalized with respect to the corresponding levels of β-tubulin (loading control). B) In real-time RT-PCR analysis, HG-treated retinal EC cultures exhibited a 60% increase in ICAM-1 mRNA expression vs. NG-treated cells. **P < 0.01. ICAM-1 mRNA levels were normalized with respect to NG levels. C) Surface expression of endothelial ICAM-1 was determined by flow cytometry. Quantitative dot plot analysis of representative fluorescence vs. size (electronic volume) indicated that HG treatment produced an ∼2.7-fold increase in the number of high ICAM-1–expressing ECs (red box) than was produced by NG treatment. D and E) Fluorescence-labeled U937 cells or freshly isolated human PBMs were added to the EC monolayer for 30 min. Representative fluorescent images of adherent U937 cells or human PBMs and subsequent cell count indicated a significant (2.1- to 3.5-fold) increase in monocyte–EC adhesion in HG conditions. Data are averages ± sem (per square millimeter) from multiple images of monocyte–EC cocultures (n ≥ 10). Scale bar, 100 μm. ***P < 0.001. F) Representative fluorescent images of NG- and HG-treated ECs labeled with anti–NF-κB p65 and quantification of percent total cells (bar graph; n ≥ 20 cells) with NF-κB p65 nuclear translocation revealed significantly greater incidence of NF-κB activation in HG- vs. NG-treated ECs. Significance was determined by unpaired Student’s t test, followed by Welch correction, and confirmed by Pearson’s correlation analysis. *P < 0.05. Scale bar, 20 µm.
Consistent with our in vivo observations, HG treatment caused significantly greater activation of cultured human retinal ECs, as judged by the ∼1.6-fold higher (P < 0.01) ICAM-1 mRNA expression in HG-treated ECs than in the NG-treated counterparts (Fig. 1B). This increase in ICAM-1 mRNA levels in HG conditions correlated with an ∼2.7-fold higher number of HG-treated ECs than NG-treated cells expressing high surface ICAM-1 (Fig. 1C). Further, consistent with a significant increase in monocyte accumulation in diabetic retinas, HG-treated ECs exhibited ∼4-fold greater binding (P < 0.001) of fluorescence-labeled U937 human monocytic cells than did NG-treated ECs (Fig. 1D). Human PBMs freshly isolated from whole blood also exhibited preferential binding to HG-treated ECs (Fig. 1E), thereby indicating that U937 cells mimic human circulating monocytes with regard to their ability to differentially adhere to normal vs. diabetic retinal ECs.
To further understand the mechanism underlying higher endothelial ICAM-1 expression on HG-treated stiffer BM, we looked at the activation of NF-κB, the major transcriptional factor that up-regulates various inflammatory genes, including ICAM-1 (20). Immunostaining for NF-κB p65 and subsequent quantitative analysis revealed an ∼2-fold greater (P < 0.05) incidence of its nuclear translocation, an indicator of NF-κB activation (19) in HG-treated cells than in NG-treated cells (Fig. 1F; Supplemental Fig. 1).
Diabetes is associated with a significant increase in retinal subendothelial matrix cross-linking and stiffening
To test our hypothesis that BM stiffening actively contributes to retinal EC activation associated with DR, we examined retinas of diabetic mice for levels of LOX, a collagen cross-linking enzyme, and BM collagen IV, which, together, confer stiffness to capillary BM. LOX expression was found to be significantly higher (1.6-fold; P < 0.05) in diabetic retinas than in nondiabetic controls (Fig. 2A). Further, whole-mount retinal immunofluorescent staining revealed that diabetic retinal capillaries also exhibit markedly greater BM collagen IV deposition than their nondiabetic counterparts (Fig. 2B; Supplemental Fig. 2), probably because of greater LOX-mediated collagen cross-linking in diabetic retinas.
Figure 2.

Diabetes is associated with a significant increase in retinal subendothelial matrix cross-linking and stiffening. A) Representative Western blot bands and their densitometric analyses (bar graph) together revealed that LOX expression was significantly higher in diabetic mouse retinas when compared with control retinas. LOX levels were normalized with respect to the corresponding levels of β-tubulin (loading control). *P < 0.05. B) Representative fluorescent images (n ≥ 15) of nondiabetic control and diabetic mouse retinas immunostained with anti-collagen IV indicated that diabetes led to a marked increase in collagen IV deposition in retinal capillary BM. Scale bar, 50 μm. C) Representative Western blot bands and their densitometric analyses (bar graph) together revealed that HG-treated retinal ECs expressed 2-fold higher levels of LOX than NG-treated cells. LOX levels were normalized with respect to the corresponding levels of GAPDH (loading control). ***P < 0.001. D) Decellularized matrices obtained from NG- and HG-treated human retinal EC cultures were immunostained with anti-collagen IV. Representative top (x-y plane) and cross-sectional (x-z plane) views of the collagen IV network showed that, compared to NG-treated ECs, those treated with HG deposited a markedly denser and thicker matrix. Matrix thickness was quantified from multiple (n = 3) samples and plotted as averages ± sem (bar graph). ***P < 0.001. Scale bar, 10 μm. E) Stiffness of EC-secreted matrix was measured by an AFM fitted with an ∼40 nm pyramidal silicon nitride tip (schematic). Quantitative analysis of multiple (n ≥ 25) force-indentation measurements (bar graph) revealed a 2-fold increase in subendothelial matrix stiffness in HG vs. NG conditions. Significance was determined by unpaired Student’s t test, followed by Welch correction, and confirmed by Pearson’s correlation analysis. ***P < 0.001.
ECs cultured in NG and HG conditions (for 10 d) exhibited a similar trend in LOX expression and collagen IV deposition. Specifically, HG-treated cells were found to exhibit 2-fold higher (P < 0.001) LOX expression than NG-treated cells (Fig. 2C), which correlated with a similar 2-fold increase (P < 0.001) in LOX activity in HG-treated ECs (Supplemental Fig. 3). Immunofluorescent staining of decellularized retinal EC monolayers for collagen IV showed a significant increase (P < 0.001) in the density and thickness of subendothelial collagen IV matrix secreted in HG conditions (Fig. 2D), which correlates with HG-induced LOX up-regulation.
To determine whether higher levels of LOX and collagen IV observed in diabetic retinas and HG-treated EC cultures are associated with retinal subendothelial matrix stiffening, we performed AFM force indentation on matrices obtained from decellularized NG- and HG-treated EC cultures. Quantitative analysis of multiple (n ≈ 25) AFM force-indentation curves revealed that the subendothelial matrix deposited by HG-treated ECs is 2 times stiffer (P < 0.001) than that produced by NG-treated cells (Fig. 2E).
Inhibition of subendothelial matrix stiffening prevents HG-induced retinal EC activation
To determine whether HG-induced subendothelial matrix stiffening actively contributes to retinal EC activation, we suppressed LOX cross-linking in HG-treated cells by using a pharmacological LOX inhibitor, BAPN. AFM force indentations of decellularized matrices indicated that inhibition of LOX cross-linking completely prevented HG-induced subendothelial matrix stiffening (Fig. 3A). This inhibition of matrix stiffening correlated with reduced matrix deposition by HG-treated cells, as judged by immunofluorescent staining of collagen IV (Supplemental Fig. 4). More important, suppression of HG-induced subendothelial matrix stiffening by BAPN prevented the increase in U937 cell adhesion to HG-treated ECs (Fig. 3B). Notably, BAPN exerted comparable anti-inflammatory effect when added 10 d after the onset of HG treatment (Supplemental Fig. 5), thereby indicating that the proinflammatory effects of subendothelial matrix stiffening are reversible.
Figure 3.

Inhibition of HG-induced retinal subendothelial matrix stiffening prevents monocyte–EC adhesion. A) AFM force-indentation measurements of decellularized EC-secreted matrix confirmed that pharmacological inhibition of LOX activity (by BAPN) in HG-treated ECs prevented HG-induced subendothelial matrix stiffening. Data are averages ± sem from multiple (n ≥ 25) force curves. ***P < 0.001. B) Fluorescence-labeled U937 cells were added to EC monolayers for 30 min. Representative fluorescent images of adherent U937 cells and subsequent cell count indicated that the significant increase in monocyte adhesion to HG-treated ECs was prevented by suppressing LOX-dependent subendothelial matrix stiffening. Data are averages ± sem (per square millimeter) from multiple (n ≥ 10) images of monocyte–EC cocultures. Significance was determined by ANOVA, followed by Tukey’s post hoc analysis. ***P < 0.001. Scale bar, 100 μm.
The anti-inflammatory effect produced by inhibition of subendothelial matrix stiffening was associated with significant suppression of NF-κB activation (nuclear translocation) (Fig. 4A). Notably, NF-κB activation is regulated upstream by endothelial NO, a potent anti-inflammatory factor that suppresses NF-κB activation and ICAM-1-mediated leukocyte–EC adhesion (23–25). Thus, we looked to see whether BM stiffness–dependent mechanical control of NF-κB activation correlates inversely with endothelial NO production. Quantitative analyses of cells labeled with NO-sensitive dye revealed that, indeed, HG-mediated EC activation was associated with a significant (∼40%; P < 0.001) reduction in intracellular NO, which was prevented by suppression of subendothelial matrix stiffening (Fig. 4B). Taken together, these findings strongly implicate subendothelial BM stiffening as a critical determinant of diabetic retinal EC activation.
Figure 4.

Inhibition of HG-induced subendothelial matrix stiffening prevents NF-κB activation and restores endothelial NO production. A) Representative fluorescent images of ECs labeled with anti-NF-κB p65 and subsequent quantitative analysis from multiple (n ≈ 35) cells (bar graph) indicated that the significant increase in the incidence of NF-κB nuclear translocation (activation) in HG-treated ECs was effectively prevented by BAPN-mediated LOX inhibition. Scale bar, 20 μm. B) Representative fluorescent images of ECs labeled with DAF-FM diacetate, an NO-sensitive dye, and subsequent intensity measurements from multiple (n ≥ 30) cells (bar graph) revealed that the loss of NO production caused by HG treatment was prevented by BAPN-mediated inhibition of subendothelial matrix stiffening. Significance was determined by ANOVA, followed by Tukey’s post hoc analysis. ***P < 0.001. Scale bar, 100 µm.
BM stiffening is necessary and sufficient for HG-induced retinal EC activation
In addition to increasing subendothelial matrix stiffness, hyperglycemia also exerts biochemical effects that lead to increased oxidative stress and inflammation (3). Thus, to determine the extent to which LOX-mediated subendothelial matrix stiffening alone causes HG-induced retinal EC activation, we plated fresh, NG-treated retinal ECs on decellularized matrices obtained from preceding NG- and HG ± BAPN-treated EC cultures and examined monocyte–EC adhesion and endothelial NO production (Fig. 5A). In these conditions where the effects of LOX is conveyed only via matrix stiffness, retinal ECs plated on HG-treated (stiffer) subendothelial matrix exhibited significantly greater U937 cell adhesion than those plated on NG- or HG + BAPN-treated matrices (Fig. 5B). These differential levels of monocyte–EC adhesion on decellularized matrices were inversely related to the levels of endothelial NO, with ECs plated on HG-treated matrix producing ∼40% lower NO (P < 0.001) than those on NG- or HG + BAPN-treated matrices (Fig. 5C). As expected, the reduction in endothelial NO on HG-treated matrix correlated with greater NF-κB activation (Supplemental Fig. 6). These findings indicate that the LOX-dependent subendothelial matrix stiffening that occurs in HG conditions is alone sufficient to cause retinal EC activation.
Figure 5.

HG-induced subendothelial matrix stiffening alone can promote retinal EC activation. A) Schematic depiction of the experimental procedure to examine the direct effect of modified BM on normal ECs. B) Fluorescence-labeled U937 cells were added (for 30 min) to NG-treated EC monolayers grown on decellularized matrices obtained from preceding NG, HG, or HG + BAPN cultures. Counting of adherent U937 cells (per square millimeter) from multiple (n ≥ 10) images of monocyte–EC cocultures indicated that HG-treated stiff matrix alone caused a significant increase in monocyte–EC adhesion, which did not occur on normalized matrix obtained from BAPN-treated cultures. ***P < 0.001. C) The lack of increase in monocyte–EC adhesion on normalized HG + BAPN-treated matrix correlated strongly with a lack of decrease in endothelial NO production on these matrices, as indicated by fluorescence intensity measurement from multiple (n ≥ 30) ECs labeled with the NO-sensitive dye DAF-FM diacetate. Significance was determined by ANOVA, followed by Tukey’s post hoc analysis. ***P < 0.001.
To unequivocally confirm the pivotal role of subendothelial matrix stiffening in retinal EC activation, NG-treated cells were grown on synthetic matrices of progressively increasing stiffness. Counting of adherent U937 cells revealed a progressive increase in monocyte–EC adhesion on stiffer synthetic matrices (Fig. 6A), which correlated inversely with endothelial NO production (Fig. 6B). Given that these significant differences in monocyte–EC adhesion and NO production are observed in NG conditions, these data corroborate the findings from decellularized matrices that subendothelial matrix stiffening is sufficient to cause retinal EC activation.
Figure 6.

Subendothelial matrix stiffening is necessary and sufficient for HG-induced retinal EC activation. A) Fresh NG-treated ECs were plated on synthetic matrices of normal (1000 Pa) or increasing (2000 and 4000 Pa) stiffness and evaluated for U937 cell adhesion. Counting of adherent U937 cells indicated that, in NG conditions, ECs grown on stiffer synthetic matrix exhibited a progressive increase in monocyte–EC adhesion. Data averages ± sem (per square millimeter) from multiple (n ≥ 10) images of monocyte–EC cocultures. Significance was determined by ANOVA, followed by Tukey’s post hoc analysis. *P < 0.05; ***P < 0.001. B) Increase in monocyte–EC adhesion on stiffer synthetic matrix correlated with a significant decrease in endothelial NO production, as indicated by fluorescence intensity measurement of multiple (n ≥ 30) ECs labeled with NO-sensitive dye DAF-FM diacetate. C and D) ECs grown in NG or HG medium for 10 d were detached and replated on synthetic matrix of normal (1000 Pa) stiffness for evaluation of U937 cell adhesion and intracellular NO production. Counting of adherent U937 cells indicated no significant difference in monocyte adhesion to NG- and HG-treated ECs grown on synthetic matrix of normal stiffness (C). This result is consistent with the similar levels of intracellular NO observed in these cells, as judged by fluorescence intensity measurement of DAF-FM diacetate–labeled ECs (D). B–D) Significance was determined by unpaired Student’s t test, followed by Welch correction, and confirmed by Pearson’s correlation analysis.
Further, when ECs grown in NG and HG medium were detached and plated on synthetic matrices of low (normal) stiffness for 24 h in corresponding NG or HG medium, both monocyte–EC adhesion and NO production were found to be comparable (Figs. 6C, D). Because 24 h is insufficient time for ECs to deposit a robust matrix, these data confirm our initial observation (Figs. 3 and 4) that subendothelial matrix stiffening is also necessary to promote HG-induced retinal EC activation.
DISCUSSION
Retinal inflammation plays a crucial role in DR pathogenesis. Past studies have demonstrated that retinal EC activation is a rate-limiting step in diabetic retinal inflammation (3, 4). Efforts aimed at understanding how retinal EC activation is regulated have revealed an important role of epigenetic, metabolic, and inflammatory factors (2, 3, 6). Our findings are the first to implicate mechanical cues in the form of subendothelial BM stiffening as an important determinant of diabetic retinal EC activation. Using cultured retinal ECs, we further showed that subendothelial matrix stiffening, caused by HG-induced LOX up-regulation, is alone sufficient to activate retinal ECs. By highlighting the critical yet previously unrecognized role of subendothelial matrix stiffening in HG-induced retinal EC activation, this study creates a new paradigm for the understanding of diabetic retinal inflammation and provides a strong rationale for detailed preclinical studies to evaluate BM stiffness normalization as a potential anti-inflammatory DR therapy, either alone or in conjunction with existing DR treatment strategies. Further, because matrix stiffening triggers mechanotransduction pathways that can independently control cell function (10), a deeper mechanistic understanding of the mechanical control of diabetic retinal EC activation may lead to the identification of new therapeutic targets for effective DR management.
Vascular inflammation is a hallmark of diabetic complications, such as retinopathy and cardiovascular diseases. Indeed, our mouse model of DR and HG-treated retinal EC cultures demonstrated significant increases in endothelial ICAM-1 expression and monocyte–EC binding, which is consistent with the EC activation observed in diabetic aortas and arteries (26, 27). Diabetes is also associated with significant stiffening of large vessels such as arteries and aorta (28, 29), and independent studies have implicated aortic and arterial subendothelial matrix stiffening in increased vascular permeability and monocyte accumulation associated with atherosclerosis (13, 30). Thus, we asked in the present study whether diabetes also leads to an increase in retinal subendothelial matrix stiffness and, if so, whether matrix stiffening actively contributes to retinal EC activation associated with DR.
We found that EC activation and inflammation in vitro and in diabetic mouse retina correlate with higher expression and activity of the collagen cross-linking enzyme LOX, which is consistent with the LOX up-regulation reported in diabetic rat retinal capillaries (7). Given that past studies have implicated LOX up-regulation in the stiffening of lung and tumor extracellular matrix (9, 31), we asked whether LOX enhances retinal subendothelial matrix stiffness. To address this question, we used a biologic-grade AFM to perform force-indentation measurements on decellularized subendothelial matrix obtained from retinal EC cultures. AFM offers a unique, sensitive, quantitative, and reliable technique to measure micromechanical properties (e.g., cell/matrix stiffness, and intermolecular bond forces) of myriad biologic materials such as DNA, proteins, cells, and tissues (32–34). Our AFM force measurements confirmed that HG treatment significantly increases LOX-dependent retinal subendothelial matrix stiffness. These AFM measurements, which are the first to quantify retinal subendothelial matrix or BM stiffness in vitro or in vivo, formed the basis for examining the mechanical control of diabetic retinal EC activation. To this end, we showed that HG-induced retinal EC activation depends strongly on the increase in LOX-dependent subendothelial matrix stiffness. It must be noted that the subendothelial matrix deposited by retinal EC cultures is similar, but not identical, to the BM deposited in retinal capillaries, with the presence of interstitial matrix proteins (e.g., collagen I and VI) in subendothelial matrices in vitro being a major distinction (35, 36). Since the retinal capillary BM in vivo is regulated by both ECs and pericytes, coculturing these cells in vitro may lead to deposition of a matrix that more closely resembles the in vivo BM. Nonetheless, together with a past study that implicated HG-induced LOX up-regulation in retinal vascular hyperpermeability (7), our findings imply a crucial role of LOX-dependent BM stiffening in diabetic retinal inflammation.
Owing to its collagen cross-linking property, LOX up-regulation in HG-treated ECs and diabetic mouse retinas predictably correlated with increased collagen IV deposition in subendothelial matrix and BM, respectively. This observation agrees well with past reports of increased BM collagen IV accumulation in retinal vessels of diabetic rats and humans with DR (7, 37–39). Other BM components, such as fibronectin, tenascin-C, and collagen XIII, are also deposited in excessive amounts in diabetic retinal capillary BMs (38–40). Among these, fibronectin is particularly important as it interacts with both collagen IV and LOX via specific molecular domains within its N- and C-terminal domains, respectively (22, 41), and is implicated in regulating LOX activity (22). Thus, it is possible that, in addition to increasing retinal capillary BM thickness, excessive fibronectin also contributes to LOX-dependent BM stiffening and related to retinal inflammation. DR is also associated with up-regulation of plasminogen activator inhibitor (PAI)-1 in retinal ECs (42, 43). Since plasminogen activation leads to degradation of BM fibronectin (44), up-regulation of PAI-1 in DR may also, at least indirectly, contribute to BM stiffening via an increased fibronectin–LOX interaction.
In addition to increasing LOX-mediated subendothelial matrix stiffening, hyperglycemia causes other biochemical alterations, such as increased polyol and hexosamine pathway flux, glycation of plasma biomolecules, and formation of advanced glycation end-products (AGEs) that contribute to oxidative stress and inflammation (45). Thus, it may be that the inflammatory effects of HG reported herein result from dual effects of aberrant matrix stiffness–dependent mechanical and HG-dependent biochemical cues. Remarkably, however, our findings revealed that LOX-mediated subendothelial matrix stiffening alone is sufficient to promote retinal EC activation independent of the effects of HG-induced biochemical modifications. This finding is important, because it implicates BM stiffening as an independent regulator of diabetic retinal inflammation. To unequivocally confirm this, we cultured NG-treated ECs on synthetically engineered matrices made of polyacrylamide gels of increasing stiffness. Polyacrylamide gels have been widely used to examine the role of BM and matrix stiffness in endothelial permeability (30), transcriptional control of angiogenesis (46), atheroprotective effects of apolipoprotein E (13), and tumor vascular dysfunction (11). In the current study, we leveraged them to confirm that subendothelial matrix stiffening alone promotes activation of NG-treated ECs, thereby consolidating the observations made on decellularized matrices.
To determine the inflammatory pathway by which subendothelial matrix stiffening activates retinal ECs, we focused on the NO/NF-κB axis. NO is a potent endogenous anti-inflammatory factor that suppresses EC activation by inhibiting the activity of NF-κB (24, 25), a key transcriptional factor that promotes ICAM-1–dependent leukocyte-EC adhesion in DR (47–49). Indeed, diabetic vascular inflammation is associated with impaired endothelial NO (50). Further, genetic knockout of endothelial NO synthase (eNOS), the primary enzyme that produces endothelial NO, has been shown to accelerate DR pathogenesis (51). Consistent with these reports, we found that the inflammatory effects of HG-induced subendothelial matrix stiffening result from impaired retinal endothelial NO and the associated increase in NF-κB activation. The underlying mechanotransduction pathway by which matrix stiffening regulates inflammatory processes, however, remains to be examined.
Current anti-inflammatory treatments for DR are limited to the use of steroids (52, 53). Although they work well in patients with DR who have late-stage macular edema, steroids exert side effects such as increased intraocular pressure and cataract formation (52, 54). Thus, there is an unmet need to identify new anti-inflammatory targets and therapies for more effective DR management. By highlighting the crucial role of subendothelial matrix stiffening in retinal EC activation, this study provides new mechanistic insights into the pathophysiology of inflammation associated with DR. These findings also form the basis for detailed in vivo studies that examine BM stiffness as a potential anti-inflammatory target for DR treatment. These animal studies will help determine the extent to which inhibition of BM stiffening suppresses diabetic retinal inflammation, both alone and in conjunction with existing DR therapies. Furthermore, future studies that examine the mechanotransduction pathways underlying the mechanical control of diabetic retinal inflammation may lead to the identification of entirely new class of molecular targets for more effective anti-inflammatory DR therapies.
Supplementary Material
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
Acknowledgments
This work was supported by Initial Complement Funds provided by the University of California, Riverside (UCR) Bourns College of Engineering (to K.G.) and U. S. National Institutes of Health, National Eye Institute Grant R01EY022327 (to A.D.). X.Y., H.A.S., and K.G. disclose a UCR invention. The remaining authors report no conflicts of interest.
Glossary
- AFM
atomic force microscope
- BAPN
β-aminopropionitrile
- BM
basement membrane
- BSA
bovine serum albumin
- DR
diabetic retinopathy
- EC
endothelial cell
- FBS
fetal bovine serum
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- HG
high glucose
- ICAM
intercellular adhesion molecule
- LOX
lysyl oxidase
- NG
normal glucose
- PAI
plasminogen activator inhibitor
- PBM
peripheral blood monocyte
- PFA
paraformaldehyde
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Antonetti D. A., Klein R., Gardner T. W. (2012) Diabetic retinopathy. N. Engl. J. Med. , 1227–1239 [DOI] [PubMed] [Google Scholar]
- 2.Rangasamy S., McGuire P. G., Das A. (2012) Diabetic retinopathy and inflammation: novel therapeutic targets. Middle East Afr. J Ophthalmol. , 52–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tang J., Kern T. S. (2011) Inflammation in diabetic retinopathy. Prog. Retin. Eye Res. , 343–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miyamoto K., Khosrof S., Bursell S. E., Rohan R., Murata T., Clermont A. C., Aiello L. P., Ogura Y., Adamis A. P. (1999) Prevention of leukostasis and vascular leakage in streptozotocin-induced diabetic retinopathy via intercellular adhesion molecule-1 inhibition. Proc. Natl. Acad. Sci. USA , 10836–10841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lutty G. A. (2013) Effects of diabetes on the eye. Invest. Ophthalmol. Vis. Sci. , ORSF81-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kowluru R. A., Kowluru A., Mishra M., Kumar B. (2015) Oxidative stress and epigenetic modifications in the pathogenesis of diabetic retinopathy. Prog. Retin. Eye Res. , 40–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chronopoulos A., Tang A., Beglova E., Trackman P. C., Roy S. (2010) High glucose increases lysyl oxidase expression and activity in retinal endothelial cells: mechanism for compromised extracellular matrix barrier function. Diabetes , 3159–3166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kagan H. M., Li W. (2003) Lysyl oxidase: properties, specificity, and biological roles inside and outside of the cell. J. Cell. Biochem. , 660–672 [DOI] [PubMed] [Google Scholar]
- 9.Mammoto A., Mammoto T., Kanapathipillai M., Wing Yung C., Jiang E., Jiang A., Lofgren K., Gee E. P., Ingber D. E. (2013) Control of lung vascular permeability and endotoxin-induced pulmonary oedema by changes in extracellular matrix mechanics. Nat. Commun. , 1759 [DOI] [PubMed] [Google Scholar]
- 10.Ghosh K., Ingber D. E. (2007) Micromechanical control of cell and tissue development: implications for tissue engineering. Adv. Drug Deliv. Rev. , 1306–1318 [DOI] [PubMed] [Google Scholar]
- 11.Ghosh K., Thodeti C. K., Dudley A. C., Mammoto A., Klagsbrun M., Ingber D. E. (2008) Tumor-derived endothelial cells exhibit aberrant Rho-mediated mechanosensing and abnormal angiogenesis in vitro. Proc. Natl. Acad. Sci. USA , 11305–11310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scholl Z. N., Yang W., Marszalek P. E. (2014) Chaperones rescue luciferase folding by separating its domains. J. Biol. Chem. , 28607–28618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kothapalli D., Liu S. L., Bae Y. H., Monslow J., Xu T., Hawthorne E. A., Byfield F. J., Castagnino P., Rao S., Rader D. J., Puré E., Phillips M. C., Lund-Katz S., Janmey P. A., Assoian R. K. (2012) Cardiovascular protection by ApoE and ApoE-HDL linked to suppression of ECM gene expression and arterial stiffening. Cell Reports , 1259–1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ito H., Akiyama H., Iguchi H., Iyama K., Miyamoto M., Ohsawa K., Nakamura T. (2001) Molecular cloning and biological activity of a novel lysyl oxidase-related gene expressed in cartilage. J. Biol. Chem. , 24023–24029 [DOI] [PubMed] [Google Scholar]
- 15.Bondareva A., Downey C. M., Ayres F., Liu W., Boyd S. K., Hallgrimsson B., Jirik F. R. (2009) The lysyl oxidase inhibitor, beta-aminopropionitrile, diminishes the metastatic colonization potential of circulating breast cancer cells. PLoS One , e5620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beacham, D. A., Amatangelo, M. D., and Cukierman, E. (2007) Preparation of extracellular matrices produced by cultured and primary fibroblasts. Curr. Protoc. Cell Biol. Chapter 10, Unit 10.9 doi:10.1002/0471143030.cb1009s33 [DOI] [PubMed] [Google Scholar]
- 17.Wang Y. L., Pelham R. J. Jr (1998) Preparation of a flexible, porous polyacrylamide substrate for mechanical studies of cultured cells. Methods Enzymol. , 489–496 [DOI] [PubMed] [Google Scholar]
- 18.Yeung T., Georges P. C., Flanagan L. A., Marg B., Ortiz M., Funaki M., Zahir N., Ming W., Weaver V., Janmey P. A. (2005) Effects of substrate stiffness on cell morphology, cytoskeletal structure, and adhesion. Cell Motil. Cytoskeleton , 24–34 [DOI] [PubMed] [Google Scholar]
- 19.Orr A. W., Sanders J. M., Bevard M., Coleman E., Sarembock I. J., Schwartz M. A. (2005) The subendothelial extracellular matrix modulates NF-kappaB activation by flow: a potential role in atherosclerosis. J. Cell Biol. , 191–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pober J. S., Sessa W. C. (2007) Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. , 803–815 [DOI] [PubMed] [Google Scholar]
- 21.Palamakumbura A. H., Trackman P. C. (2002) A fluorometric assay for detection of lysyl oxidase enzyme activity in biological samples. Anal. Biochem. , 245–251 [DOI] [PubMed] [Google Scholar]
- 22.Fogelgren B., Polgár N., Szauter K. M., Ujfaludi Z., Laczkó R., Fong K. S., Csiszar K. (2005) Cellular fibronectin binds to lysyl oxidase with high affinity and is critical for its proteolytic activation. J. Biol. Chem. , 24690–24697 [DOI] [PubMed] [Google Scholar]
- 23.Lefer D. J., Jones S. P., Girod W. G., Baines A., Grisham M. B., Cockrell A. S., Huang P. L., Scalia R. (1999) Leukocyte-endothelial cell interactions in nitric oxide synthase-deficient mice. Am. J. Physiol. , H1943–H1950 [DOI] [PubMed] [Google Scholar]
- 24.Peng H. B., Libby P., Liao J. K. (1995) Induction and stabilization of I kappa B alpha by nitric oxide mediates inhibition of NF-kappa B. J. Biol. Chem. , 14214–14219 [DOI] [PubMed] [Google Scholar]
- 25.De Caterina R., Libby P., Peng H. B., Thannickal V. J., Rajavashisth T. B., Gimbrone M. A. Jr., Shin W. S., Liao J. K. (1995) Nitric oxide decreases cytokine-induced endothelial activation; nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J. Clin. Invest. , 60–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hartge M. M., Unger T., Kintscher U. (2007) The endothelium and vascular inflammation in diabetes. Diab. Vasc. Dis. Res. , 84–88 [DOI] [PubMed] [Google Scholar]
- 27.Shirwany N. A., Zou M. H. (2012) Vascular inflammation is a missing link for diabetes-enhanced atherosclerotic cardiovascular diseases. Front. Biosci. (Landmark Ed.) , 1140–1164 [DOI] [PubMed] [Google Scholar]
- 28.Chang K. C., Tseng C. D., Chou T. F., Cho Y. L., Chi T. C., Su M. J., Tseng Y. Z. (2006) Arterial stiffening and cardiac hypertrophy in a new rat model of type 2 diabetes. Eur. J. Clin. Invest. , 1–7 [DOI] [PubMed] [Google Scholar]
- 29.Oxlund H., Rasmussen L. M., Andreassen T. T., Heickendorff L. (1989) Increased aortic stiffness in patients with type 1 (insulin-dependent) diabetes mellitus. Diabetologia , 748–752 [DOI] [PubMed] [Google Scholar]
- 30.Huynh J., Nishimura N., Rana K., Peloquin J. M., Califano J. P., Montague C. R., King M. R., Schaffer C. B., Reinhart-King C. A. (2011) Age-related intimal stiffening enhances endothelial permeability and leukocyte transmigration. Sci. Transl. Med. , 112ra122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levental K. R., Yu H., Kass L., Lakins J. N., Egeblad M., Erler J. T., Fong S. F., Csiszar K., Giaccia A., Weninger W., Yamauchi M., Gasser D. L., Weaver V. M. (2009) Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell , 891–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goldsbury, C. S., Scheuring, S., and Kreplak, L. (2009) Introduction to atomic force microscopy (AFM) in biology. Curr. Prot. Protein Sci. Chapter 17, Unit 17 17.11-19 [DOI] [PubMed] [Google Scholar]
- 33.Trache, A., and Meininger, G. A. (2008) Atomic force microscopy (AFM). Curr Prot. Microbiol. Chapter 2, Unit 2C.2 [DOI] [PubMed] [Google Scholar]
- 34.Lehenkari P. P., Charras G. T., Nesbitt S. A., Horton M. A. (2000) New technologies in scanning probe microscopy for studying molecular interactions in cells. Expert Rev. Mol. Med. , 1–19 [DOI] [PubMed] [Google Scholar]
- 35.Halfter W., Oertle P., Monnier C. A., Camenzind L., Reyes-Lua M., Hu H., Candiello J., Labilloy A., Balasubramani M., Henrich P. B., Plodinec M. (2015) New concepts in basement membrane biology. [E-pub ahead of print] FEBS J. 10.1111/febs.13495 [DOI] [PubMed] [Google Scholar]
- 36.Davis G. E., Senger D. R. (2005) Endothelial extracellular matrix: biosynthesis, remodeling, and functions during vascular morphogenesis and neovessel stabilization. Circ. Res. , 1093–1107 [DOI] [PubMed] [Google Scholar]
- 37.Roy S., Maiello M., Lorenzi M. (1994) Increased expression of basement membrane collagen in human diabetic retinopathy. J. Clin. Invest. , 438–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ljubimov A. V., Burgeson R. E., Butkowski R. J., Couchman J. R., Zardi L., Ninomiya Y., Sado Y., Huang Z. S., Nesburn A. B., Kenney M. C. (1996) Basement membrane abnormalities in human eyes with diabetic retinopathy. J. Histochem. Cytochem. , 1469–1479 [DOI] [PubMed] [Google Scholar]
- 39.Chronopoulos A., Trudeau K., Roy S., Huang H., Vinores S. A., Roy S. (2011) High glucose-induced altered basement membrane composition and structure increases trans-endothelial permeability: implications for diabetic retinopathy. Curr. Eye Res. , 747–753 [DOI] [PubMed] [Google Scholar]
- 40.To M., Goz A., Camenzind L., Oertle P., Candiello J., Sullivan M., Henrich P. B., Loparic M., Safi F., Eller A., Halfter W. (2013) Diabetes-induced morphological, biomechanical, and compositional changes in ocular basement membranes. Exp. Eye Res. , 298–307 [DOI] [PubMed] [Google Scholar]
- 41.Shimizu M., Minakuchi K., Moon M., Koga J. (1997) Difference in interaction of fibronectin with type I collagen and type IV collagen. Biochim. Biophys. Acta , 53–61 [DOI] [PubMed] [Google Scholar]
- 42.Grant M. B., Ellis E. A., Caballero S., Mames R. N. (1996) Plasminogen activator inhibitor-1 overexpression in nonproliferative diabetic retinopathy. Exp. Eye Res. , 233–244 [DOI] [PubMed] [Google Scholar]
- 43.Azad N., Agrawal L., Emanuele N. V., Klein R., Bahn G. D., McCarren M., Reaven P., Hayward R., Duckworth W., Group V. S.; VADT Study Group (2014) Association of PAI-1 and fibrinogen with diabetic retinopathy in the Veterans Affairs Diabetes Trial (VADT). Diabetes Care , 501–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marchina E., Barlati S. (1996) Degradation of human plasma and extracellular matrix fibronectin by tissue type plasminogen activator and urokinase. Int. J. Biochem. Cell Biol. , 1141–1150 [DOI] [PubMed] [Google Scholar]
- 45.Brownlee M. (2005) The pathobiology of diabetic complications: a unifying mechanism. Diabetes , 1615–1625 [DOI] [PubMed] [Google Scholar]
- 46.Mammoto A., Connor K. M., Mammoto T., Yung C. W., Huh D., Aderman C. M., Mostoslavsky G., Smith L. E., Ingber D. E. (2009) A mechanosensitive transcriptional mechanism that controls angiogenesis. Nature , 1103–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zheng L., Howell S. J., Hatala D. A., Huang K., Kern T. S. (2007) Salicylate-based anti-inflammatory drugs inhibit the early lesion of diabetic retinopathy. Diabetes , 337–345 [DOI] [PubMed] [Google Scholar]
- 48.Iliaki E., Poulaki V., Mitsiades N., Mitsiades C. S., Miller J. W., Gragoudas E. S. (2009) Role of alpha 4 integrin (CD49d) in the pathogenesis of diabetic retinopathy. Invest. Ophthalmol. Vis. Sci. , 4898–4904 [DOI] [PubMed] [Google Scholar]
- 49.Palenski T. L., Sorenson C. M., Sheibani N. (2013) Inflammatory cytokine-specific alterations in retinal endothelial cell function. Microvasc. Res. , 57–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kolluru G. K., Siamwala J. H., Chatterjee S. (2010) eNOS phosphorylation in health and disease. Biochimie , 1186–1198 [DOI] [PubMed] [Google Scholar]
- 51.Li Q., Verma A., Han P. Y., Nakagawa T., Johnson R. J., Grant M. B., Campbell-Thompson M., Jarajapu Y. P., Lei B., Hauswirth W. W. (2010) Diabetic eNOS-knockout mice develop accelerated retinopathy. Invest. Ophthalmol. Vis. Sci. , 5240–5246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Das A., McGuire P. G., Rangasamy S. (2015) Diabetic macular edema: pathophysiology and novel therapeutic targets. Ophthalmology , 1375–1394 [DOI] [PubMed] [Google Scholar]
- 53.Bandello F., Preziosa C., Querques G., Lattanzio R. (2014) Update of intravitreal steroids for the treatment of diabetic macular edema. Ophthalmic Res. , 89–96 [DOI] [PubMed] [Google Scholar]
- 54.O’Day R. F., Barthelmes D., Zhu M., Wong T. Y., McAllister I. L., Arnold J. J., Gillies M. C. (2014) Intraocular pressure rise is predictive of vision improvement after intravitreal triamcinolone acetonide for diabetic macular oedema: a retrospective analysis of data from a randomised controlled trial. BMC Ophthalmol. , 123 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.