

Abstract
MK2 and MK3 are downstream targets of p38 and ERK1/2. They control the mRNA stability of several inflammatory cytokines, including TNF-α and IL-10. Whereas MK2 is expressed ubiquitously, the expression of MK3 is restricted to muscle, liver, and heart tissues and T and NK cells. Using Mk-deficient and wild-type (WT) mice, we demonstrated an inhibitory effect of MK3, but not of MK2, on interferon (IFN)-γ expression in T and NK lymphocytes. The results provided evidence that the inhibitory effect of MK3 is based on negative feedback phosphorylation of p38 and ERK1/2, which causes decreased binding of Stat4 to the IFN-γ promoter and reduced expression of IFN-γ mRNA and protein. Consequently, all Mk3−/− mice challenged with the Th1-inducing influenza A virus (IAV) survived the WT LD50 virus dose. The reduced disease severity in the Mk3−/− mice was accompanied by a >10-fold reduction in viral lung titer and an increase in the number of activated NK cells and enhanced Th1 activation of CD4 T cells. Thus, our data describe the protein kinase MK3 as a novel regulator of the innate and adaptive immune responses.—Köther, K., Nordhoff, C., Masemann, D., Varga, G., Bream, J. H., Gaestel, M., Wixler, V., Ludwig, S. MAPKAP kinase 3 suppresses Ifng gene expression and attenuates NK cell cytotoxicity and Th1 CD4 T-cell development upon influenza A virus infection.
Keywords: interferon-γ, signal transduction, innate and adaptive immune response
Mitogen-activated protein kinase–activated protein kinases (MAPKAPKs) are downstream targets of the mitogen-activated protein kinases (MAPKs). They can be divided into 4 groups: the ribosomal S6 kinases, the mitogen- and stress-activated kinases, the MAPK-interacting kinases, and the MAPKAPKs (MKs). Members of the MK group include MK5, which is predominantly activated by the atypical MAPKs ERK3/4 (1, 2), and MK2 and MK3, which are primarily activated by the stress MAPKs p38 α/β (1, 3, 4) and also by ERK1/2 and JNK1/2 (3, 5). MK2 and MK3 are often referred to as isoenzymes, as they target similar downstream effectors and share 75% homology in their structure (1, 3). Both kinases are activated via phosphorylation of serine/threonine residues at the activation loop and in the C-terminal region (3, 6, 7). After phosphorylation, the activated kinases translocate from the nucleus to the cytoplasm (8, 9). MK3 shows a delayed cytosolic translocation (9), compared to MK2 (8), suggesting that its export is rather independent of the activation state and that activated MK3 may have some additional functions in the nucleus (9).
Several downstream effectors are known for MK2 and MK3. The most studied downstream targets are the heat-shock proteins 25 and 27 (Hsp25 and Hsp27), which, in their phosphorylated form, play an important role in the structural organization of the cytoskeleton (10). The mRNA-destabilizing protein tristetraprolin (TTP) is a further prominent substrate of both MK2 and MK3. Phosphorylation of TTP by the kinases results in enhanced mRNA stability of several pro- and anti-inflammatory cytokines, such as TNF-α, IL-10, and IL-6 (11–14). Another function common to MK2 and MK3 is their ability to regulate the chromatin association of proteins of the polycomb group (PcG; ref. 15).
MK3 displays a rather restricted pattern of expression in different tissues. Muscle, liver, and heart tissues and T and NK cells are the major sources of MK3 (5), whereas other tissues exhibit low MK3 expression levels (12). Despite its low expression level, MK3 can fulfill most of the MK2 functions, as evidenced by the aggravated phenotype of the Mk2-knockout mouse and the finding that overexpression of MK3 in Mk2−/− cells is associated with a regain of function (12). The presence of both kinases has also been reported to be essential for the stabilization of p38, as in the absence of either MK2 or MK3, the amount of p38 kinase is reduced (16).
MK2 and MK3 have also been implicated in several diseases. Indeed, crucial roles of the kinases in the development of collagen-induced arthritis (17), pulmonary fibrosis (18), and glomerulonephritis (19) have been described. These changes in disease outcome have been related to the TNF-α regulatory property of the kinases. Interestingly, in vitro experiments showed that MK2 and MK3 support influenza A virus (IAV) replication, but in a TNF-α-independent manner. IAV infection induces the activation of both MK2 and MK3, resulting in the formation of a tetrameric protein complex of MK2 or MK3 with p88rIPK, p58IPK, and the dsRNA-inducible protein kinase R (PKR), leading to an inhibition of PKR. Inactivation of PKR, in turn, leads to enhanced protein translation and the augmented synthesis of viral proteins (20).
Infection with IAV is a strong inducer of proinflammatory cytokine secretion and a Th1 T-cell response (21). Viral clearance is achieved by the interplay between innate and adaptive immune responses. The expression of cytokines, including interferon-γ (IFN-γ), has been shown to be highly upregulated on IAV infection and is beneficial for the induction of both innate and adaptive immune responses (22–24). However, the induction of IFN-γ in T and NK cells is counteracted by IAV, as infected NK cells produce less IFN-γ than noninfected NK cells (25). It is not yet clear how IAV decreases IFN-γ expression in NK cells, but a likely mechanism is via targeting the cellular signaling pathways involved in IFN-γ synthesis, such as the MAPK p38 or ERK1/2 pathway (26, 27).
We describe a novel function for MK3 as a virally induced inhibitor of IFN-γ expression that is mediated by the transcription factor Stat4. As a result, Mk3−/− mice, challenged with IAV infection, show a strong shift toward an innate NK cell and adaptive Th1 immune response and a resistance to IAV infection.
MATERIALS AND METHODS
Mice
All animal studies were performed in accordance with the German regulations of the Society for Laboratory Animal Science (GVSOLAS) and the European Health Law of the Federation of Laboratory Animal Science Associations (FELASA). The protocol was approved by the Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen (LANUV-NRW; Essen, Germany). Eight- to 10-wk-old pathogen-free, C57Bl/6 wild-type (WT), Mk2-deficient, (Mk2−/−), and Mk3-deficient (Mk3−/−) mice were used.
Isolation of NK and CD4+ T cells
NK and CD4+ T cells were isolated from the spleens of WT, Mk2−/−, and Mk3−/− mice (28). Briefly, the spleens from 3 mice were combined and minced through a 70 μm cell strainer, and, after lysis of the erythrocytes, the cells were passed through a sterile, prewetted nylon wool column and incubated for 50 min at 37°C in 5% CO2. Nonadherent cells were harvested, washed, and counted. The isolated NK cells were cultured in RPMI with 2 μM l-glutamine, 10% fetal bovine serum (FBS), 100 U/ml penicillin, 100 μg/ml streptomycin, 1× nonessential amino acids, 1 mM sodium pyruvate, and 10 mM β-mercaptoethanol at 37°C in 5% CO2, at a cell density of 1 × 106 cells/ml. For efficient growth, 2000 U/ml IL-2 [National Cancer Institute Biological Resources Branch (NCI-BRB) Preclinical Repository, Frederick, MD, USA] was added to the medium. Cultivation with high amounts of IL-2 generates lymphokine-activated killer (LAK) cells; FACS analysis revealed an 85–95% pure population of NK cells (data not shown). After cultivation for 6 d, the cells were harvested and kept for 3 h without IL-2. Quantitative reverse transcription polymerase chain reaction (qRT-PCR) analysis of IFN-γ mRNA revealed that the cells were no longer producing IFN-γ mRNA (data not shown). Next, the cells were seeded into 6-well plates at a density of 5 × 106 cells/well and stimulated with IL-2 (100 U/ml) + IL-12 (10 ng/ml) (Peprotech, Hamburg, Germany), left unstimulated, or preincubated for 30 min with different inhibitors (U0126, 2.5 μM; SB202190, 2.5 μM; and SP600125, 5 μM) and stimulated afterward with IL-2 + IL-12. U0126 represents an MEK1/2 inhibitor, whereas SB202190 and SP600125 are inhibitors of p38 and JNK1/2 MAP kinases, respectively. For mRNA stability experiments, cells were stimulated with IL-2 + IL-12 for 2 h and treated afterward with 2 mg/ml of actinomycin D for an additional 0, 15, 30, 45, or 60 min. Transfection of NK cells was performed with the AMAXA human T-Cell Nucleofector Kit (Lonza, Basel, Switzerland).
For the isolation of CD4+ T cells, the nylon wool column–purified cells were separated by magnetic cell-sorting technology, with the Pan T Cell Isolation Kit II, with a CD8-rat antibody and rat-specific beads (Miltenyi Biotech, Bergisch-Galdbach, Germany), according to the manufacturer's instructions. After isolation, the CD4+ T cells were differentiated into Th1 or Th2 cell types under conditions described by Messi et al. (29). CD4+ T cells (1×106) were cultured for 7 d in 96-well plates in the presence of mouse T-activator CD3/CD28 Dynabeads (Life Technologies, GmbH, Darmstadt, Germany) in a 1:1 ratio. IL-2 (50 U/ml; NCI-BRB Preclinical Repository) was added for the last 2 d (neutral conditions). To induce a Th1 state, the cells were additionally cultured with recombinant IL-12 (5 ng/ml; PeproTech, Hamburg Germany) and anti-IL-4 antibody (10 μg/ml; eBioscience, Frankfurt, Germany); for Th2 conditions, IL-4 (20 ng/ml; eBioscience) and anti IFN-γ (10 μg/ml; eBioscience) antibodies were added.
Adherent cells
Cells of the Madin-Darby canine kidney II (MDCK II) line were grown in Dulbecco's modified Eagle's medium (PAA Laboratories, Pasching, Austria) supplemented with 10% FBS at 37°C in 5% CO2.
Virus and viral infections of mice
The recombinant influenza virus strain A/Puerto Rico/8/34 (PR8) was used for all experiments. It was generated as described elsewhere (30) and propagated on MDCK II cells. For mouse infections, the respective number of particles per mouse was resolved in 50 μl PBS. Before infection, the mice were anesthetized by the intraperitoneal application of a body weight-dependent mixture of 0.5% ketamine and 0.1% xylazine (Ceva Tiergesundheit GmbH, Düsseldorf, Germany). Afterward, 50 μl of virus suspension was applied intranasally. Body weight and survival were monitored daily. To determine the viral titer in the lungs, the number of infectious particles in 10% of lung homogenates was determined with a standard plaque assay.
RNA isolation, reverse transcription, and qRT-PCR
RNA was isolated with the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions, including on-column DNase digestion. Total RNA (1.5 μg) was reverse transcribed into cDNA by using 0.5 μg of oligo-dT primer and 200 U Revert Aid H-M-MuLV reverse transcriptase (Fermentas, St. Leon-Rot, Germany). The quantification of gene expression was achieved by real-time PCR with the Light Cycler 480 III (Roche, Basel, Switzerland). Changes in gene transcription were calculated as a difference between the transcription of the housekeeping gene GAPDH and the gene of interest using the 2−ΔΔCT method (31). Primers were the following: for mIFN-γ, 5′-TTGCCAAGTTTGAGGTCAACAA-3′ and 5′-TGGTGGACCACTCGGATGA-3′; for mTNFα, 5′-TCTTCTCATTCCTGCTTGTGG-3′ and 5′-GGTCTGGGCCATAGAACTGA-3′; for hMK3, 5′-ATGAGAACATGCACCATGGCAAGC-3′ and 5′-GGGCAATGTTATGGCTGTGCAGAA-3′; for mGM-CSF, 5′-GCATGTAGAGGCCATCAAAGA-3′ and 5′-CGGGTCTGCACACATGTTA-3′; for mGAPDH, 5′-ACAGCCGCATCTTCTTGTGCAGTG-3′ and 5′-GGCCTTGACTGTGCCGTTGAATTT-3′; for mCytochrome1, 5′-GCTACCCATGGTCTCATCGT-3′ and 5′-GAAACCCCTCCGAATGCT-3′. mGAPDH and mCytochrome1 served as the controls.
Analysis of BALF
Mice were euthanized, the trachea was exposed, a needle was inserted, and the lungs were washed 5 times with 500 μl PBS supplemented with 2 mM EDTA for each wash. The first lavage was always kept separate, to analyze soluble cytokines with the mouse Th1/Th2 10plex FlowCytomix Multiplex kit (eBioscience). The cells of the first lavage were resuspended in PBS, supplemented with 5% FBS, combined with the resuspended cells of the other 4 lavages/lung, and centrifuged at 360 g for 10 min at 4°C. After lysis of erythrocytes, the cells were counted with a Neubauer chamber and subsequently stained for analysis by flow cytometry (BD Biosciences, Heidelberg, Germany). For the detection of intracellular proteins, the cells were fixed with 2% PFA after the staining of the surface marker was performed, permeabilized using 0.5% saponin in PBS supplemented with 5% FBS, and stained with the corresponding antibodies. The number of immune cells was then assessed by gating on viable cells for the appropriate immune cell type and normalizing this number to the total cell count per sample. Analysis of the immune status of IAV-infected mice was determined with the following antibodies: F4/80-FITC, F4/80-PE, CD3ε-FITC, CD3ε-PE-Cy7, Dx5α-FITC, CD8-APC, IFN-γ-FITC, CD4-APC, and CD107a-PE (all from eBioscience); and CD45.2-FITC, CD8-PE, CD4-PE, IFN-γ-APC, and IL-4-PE (all from BD Pharmingen, Heidelberg, Germany).
Chromatin immunoprecipitation (ChIP) analysis
Isolated and cultured NK cells were stimulated with IL-2 + IL-12 for 2 h, fixed with 1% formaldehyde for 10 min, washed 2 times with ice-cold PBS, and lysed with RIPA buffer supplemented with a protease inhibitor cocktail and 1 mM sodium orthovanadate. DNA was sheared by sonication, and Stat4 was immunoprecipitated for 4 h with a specific monoclonal antibody (Santa Cruz Biotechnology, Heidelberg, Germany) or a control rabbit-IgG antibody coupled to protein A agarose (Roche, Mannheim, Germany). Aliquots of 10 μl of sheared DNA were put aside as input DNA and kept at 4°C. Afterward, nonbound proteins were washed away by centrifugation for 5 min using the following buffers: low-salt washing buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 150 mM NaCl, and 20 mM Tris-HCl, pH 8), high-salt washing buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 500 mM NaCl, and 20 mM Tris-HCl, pH 8), LiCl (0.25 M), and TE (pH 8). The precipitates were resuspended in elution buffer (1% SDS, 100 mM NaHCO3). DNA protein cross-links were reversed by incubating the precipitates with proteinase K for 2 h at 62°C. DNA was purified with the MolBio PCR Purification Kit (Dianova, Hamburg, Germany). The coimmunoprecipitated DNA was then analyzed by qRT-PCR with primers specific for the Stat4-binding site in the IFN-γ promoter region and normalized to the input controls. The amount of protein-bound DNA in comparison to total DNA per sample was calculated. Primers used for amplification of the IFN-γ promoter region were 5′-GTCGAAAGGAAACTCTAACATGCC-3′ and 5′-ATCAGCTGATGTGTCTTCTCTAGG-3′.
Western blot
Isolated and cultured NK cells were stimulated with IL-2 + IL-12 for up to 2 h. At the indicated time points, the cells were washed twice with PBS and subsequently lysed with RIPA buffer. Supernatants were cleared by centrifugation at 20,000 g, 4°C for 10 min. Total protein lysate (20 μg) was separated by 10% SDS-PAGE. Western blot images were taken with LumiImager Stella 3200 (Raytest GmbH, Schraubenhardt, Germany). Band intensities were quantified by using AIDA image-analysis software (Raytest). The following antibodies were used to detect phosphorylated and nonphosphorylated forms of proteins in Western blot analysis: anti-phospho-p38 (Thr180/Tyr181) and anti-phospho-p42/p44 (Thr202/Tyr204) (New England Biolabs, Frankfurt-am-Main, Germany); anti-phospho-Stat1 (Tyr701), anti-phospho-Stat3 (Tyr705), anti-ERK2, and anti-p38 (Santa Cruz Biotechnology); anti-phospho-JNK1/2 (Thr183/Tyr185) (BD Pharmingen); and anti-β-actin (Sigma-Aldrich, Taufkirchen Germany).
Statistical analysis
Data are shown as means ± sem, unless specified otherwise. Statistical analysis was performed with the Kruskal-Wallis test if >2 groups were compared, the Mann-Whitney test if 2 groups were compared, or 2-way ANOVA if >1 parameter changed in the compared groups. GraphPad Prism software (GraphPad, San Diego, CA, USA) was used for statistical calculations, with P < 0.05 regarded as statistically significant. N indicates the number of independent biological experiments; n indicates the number of animals analyzed per group.
RESULTS
MK3 negatively regulates IFN-γ mRNA expression
The regulation of mRNA stability of proinflammatory cytokines is a general function of both MK2 and MK3. However, the expression pattern of MK3 is very restricted compared with the ubiquitous expression of MK2. T and NK lymphocytes exhibit high levels of MK3 (data not shown), suggesting a specific role for MK3 in these cells. To explore whether these kinases play a role in the stabilization of the type II IFN-γ, a key effector cytokine of cell-mediated immunity, we isolated NK cells from C57Bl/6 WT, Mk2−/−, and Mk3−/− mice and stimulated these cells with a mixture of IL-2 and IL-12 to induce IFN-γ expression. Surprisingly, the Mk3−/− NK cells showed a significantly higher amount of synthesized IFN-γ mRNA than did the WT or Mk2−/− NK lymphocytes (Fig. 1A). This effect of MK3 was specific for IFN-γ, as deletion of Mk3 had no influence on the amount of TNF-α or GM-CSF mRNA, and Mk2 deficiency resulted, as expected (12), in the reduction of the levels of both TNF-α and GM-CSF in NK cells (Fig. 1A). These results demonstrate an inhibitory rather than a stabilizing effect of MK3 on IFN-γ transcripts and suggest a novel function for MK3 in lymphoid cells. Of note, this effect does not appear to be influenced by MK2. To confirm the functional significance of MK3 in the regulation of IFN-γ mRNA stability or synthesis, a functionally active (MK3) or kinase-dead (K>M) form of human MK3 (3) was transiently overexpressed in Mk3−/− mouse NK lymphocytes, and the amount of synthesized IFN-γ mRNA in these cells was compared with empty-vector-transfected NK cells (Fig. 1B). Whereas reconstitution of mouse Mk3−/− cells with human MK3 resulted in down-regulation of IFN-γ mRNA, expression of the kinase-inactive MK3 enzyme hardly influenced it. The increased synthesis of IFN-γ mRNA in Mk3−/− NK cells compared to WT cells also led to increased production of IFN-γ protein, as flow cytometry analysis of intracellular IFN-γ protein showed. Early on, nonstimulated Mk3−/− NK lymphocytes included a higher number of IFN-γ+ cells, as well as more intracellular IFN-γ protein compared with WT cells, and the difference increased further during stimulation of NK cells with the IL-2 and IL-12 cytokines (Fig. 1C).
Figure 1.
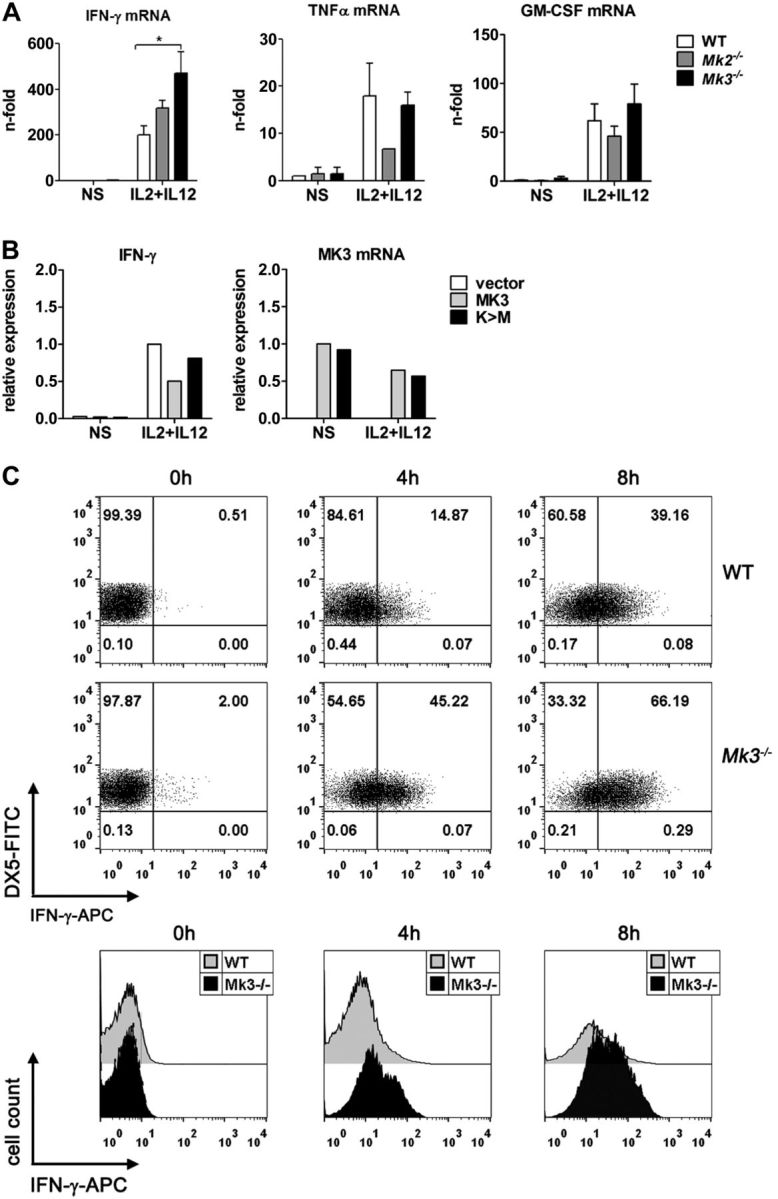
MK3 inhibits IFN-γ mRNA production in NK cells. A) NK cells from C57Bl/6 WT, Mk2−/−, and Mk3−/− mice were isolated, cultured, and stimulated with IL-2 + IL-12 for 3 h, and the total mRNA was isolated and analyzed for the expression of IFN-γ, TNF-α, and GM-CSF mRNA by qRT-PCR. Results are shown as means ± sem (N=3–4). Cells from 3 animals were pooled in each experiment. *P < 0.05, Kruskal-Wallis test. B) NK cells from Mk3−/− mice were isolated, cultured, and transfected for 24 h with an empty control vector or plasmids encoding for WT (MK3) or kinase-dead (K>M) recombinant human MK3. The cells were then stimulated with IL-2 + IL-12 for 3 h, and the total mRNA was analyzed for expression of mouse IFN-γ and human MK3 mRNA by qRT-PCR. The experiment was performed twice, with similar results. Because of the transfection particularity of primary cells, a variation in induction of IFN-γ mRNA was observed between experiments; thus, representative data of only 1 experiment are shown. NS, nonstimulated. C) NK cells from WT and Mk3−/− mice were isolated and stimulated with IL-2 + IL-12 for the indicated times, after which the cells were analyzed by flow cytometry for expression of NK-specific Dx5 and intracellular IFN-γ proteins. Numbers in the quadrants of the dot-plot graphs represent the percentage of cells in each. Representative images of 3 repeated experiments are shown.
MK3 regulates the transcription of IFN-γ
The high level of IFN-γ mRNA in Mk3−/−-stimulated cells suggests that MK3 either inhibits the transcription of the Ifng gene or destabilizes its mRNA post-transcriptionally. To distinguish between the two possibilities, we first compared the stability of IFN-γ mRNA in WT and Mk3−/− NK cells. Therefore, NK cells were stimulated with the IFN-γ-inducing cytokines IL-2 and IL-12 for 2 h, then treated with the transcriptional inhibitor actinomycin D for 15, 30, 45, and 60 min. IFN-γ mRNA decay in the Mk3−/− cells was not slower than in the WT cells but was even faster, indicating that MK3 protects rather than destabilizes the IFN-γ mRNA (Fig. 2A). The mRNA of the housekeeping gene GAPDH, which was analyzed as a control in the same experiment, showed no reduction in its amount throughout the whole experiment and no difference between the WT and Mk3−/− cells (Fig. 2B). Altogether, our results suggest an important role for MK3 in the regulation of IFN-γ expression and further indicate that it occurs at the level of transcription rather than post-transcriptionally. To further demonstrate this finding, we studied the dependency of IFN-γ promoter activity on the expression of MK3. Jurkat T cells, stably transduced with either MK3-specific shRNA or scrambled control RNA (scRNA), were further transfected with a reporter gene construct harboring a 751-bp IFN-γ promoter sequence in front of the luciferase gene. The cells were then stimulated with PMA + ionomycin, to mimic T-cell activation by TCR/CD28 costimulation. Luciferase activity was measured 16 h later. The results showed that the IFN-γ promoter was significantly more active in cells with a reduced amount of MK3, thus demonstrating the inhibitory effect of MK3 on the IFN-γ promoter (Supplemental Fig. S1).
Figure 2.

Mk3 deficiency leads to enhanced activation of ERK1/2 and p38 and induces a stronger binding of Stat4 to the IFN-γ promoter, but does not alter IFN-γ mRNA stability. A, B) NK cells from WT and Mk3−/− mice were stimulated with IL-2 + IL-12 for 3 h and treated with actinomycin D at the indicated time points. Amounts of IFN-γ (A) and GAPDH (B) mRNA were determined by qRT-PCR (N=3, with NK cells pooled from 3 animals in each experiment). Statistical analysis, 2-way ANOVA. C) NK cells isolated from C57Bl/6 WT and Mk3−/− mice were preincubated for 30 min with either DMSO or the indicated inhibitor and stimulated for 3 h with IL-2 + IL-12. IFN-γ mRNA was analyzed by qRT-PCR (N=3–4, with NK cells pooled from 3 animals in each experiment). *P < 0.05, Kruskal-Wallis test. D) NK cells were isolated from C57Bl/6 WT and Mk3−/− mice, cultured, and stimulated for the indicated times with IL-2 + IL-12 and subsequently analyzed for the activation of ERK1/2 and p38 by Western blot (N=3). E) p38 data from D were quantified with AIDA software (Raytest) **P < 0.01; 2-way ANOVA (N=3). F) Isolated NK cells from C57Bl/6 WT or Mk3−/− mice were stimulated with IL-2 + IL-12 for the indicated times, and the cell lysates were subsequently analyzed by Western blot for activation of JNK, Stat1, and Stat3. G) ChIP assays were performed on isolated NK cells of C57Bl/6 WT and Mk3−/− mice using Stat4 antibody or an IgG control antibody. Coprecipitated IFN-γ promoter DNA was analyzed with qRT-PCR and compared to the whole amount of the IFN-γ promoter DNA in the samples. DNA amount bound to Stat4 in unstimulated WT cells was arbitrarily set at 1 (N=4–5, with cells pooled from 3 animals in each experiment). *P < 0.05; Mann-Whitney test.
MK3 is a downstream target of 3 MAPK family members: p38, ERK1/2, and JNK1/2 (3). An involvement of these kinases, especially of p38 and ERK1/2, in the regulation of the IFN-γ promoter has been reported (26, 27). To study which of the 3 MAPK pathways is responsible for the MK3-mediated regulation of the IFN-γ promoter, WT or Mk3−/− NK cells were preincubated for 30 min with pathway-specific inhibitors to block the activation of p38, JNK1/2, or ERK1/2. The cells were subsequently stimulated for 3 h with IL-2 + IL-12, and mRNA levels of IFN-γ were analyzed (Fig. 2C). Whereas in the WT cells, only the inhibition of p38 kinase led to a strong reduction of IFN-γ mRNA synthesis, in the Mk3−/− cells, inhibition of each of the 3 MAPKs reduced the transcription of IFN-γ, essentially abolishing the enhancing effect of the Mk3 deficiency. The p38 inhibitor showed the strongest effect on IFN-γ induction, but inhibition of ERK1/2 and JNK1/2 in Mk3−/− cells also had a clear effect. The strong inhibition of IFN-γ synthesis by U0126 in the absence of MK3 was surprising, as MK3 is thought to be a downstream target of ERK1/2, but U0126 is an MEK1/2 inhibitor; this assumes a feedback link of MK3 to ERK1/2 kinase activity.
The activation of the IFN-γ promoter by p38 and ERK1/2 kinases is well described (26, 27). Whereas p38 acts mainly via phosphorylation of the Stat4 transcription factor at S721 (32), the effect of ERK has been shown to be Stat4 independent (33). Our inhibitor studies showed that, in WT cells, the production of IFN-γ mRNA is predominantly dependent on p38 activity, but it is also controlled by the activity of other MAPKs in Mk3−/− cells. We therefore studied whether MAP kinases were differentially activated in WT and Mk3−/− NK cells after stimulation with IL-2 + IL-12. Indeed, phosphorylation of ERK1/2 was significantly stronger in the Mk3−/− cells than in the WT cells, although the maximum was reached in both cell types at the same time, very rapidly after stimulation (Fig. 2D; top panel). The phosphorylation/activation kinetic of p38 kinase was distinct from that of ERK1/2. The phosphorylation of p38 rose slowly, reaching the highest values only in the later stages of cytokine stimulation (Fig. 2D; bottom panel), but the phosphorylation level in the MK3-deficient cells was significantly elevated compared to that in the WT cells when normalized to expressed p38. It is of note that the expression of p38 is impaired in the absence of MK2 or MK3 (12); therefore, we normalized the p38 phosphorylation/activity to the available amount of the p38 protein (Fig. 2E). Surprisingly, JNK showed no or only very weak changes in phosphorylation status during IL-2 + IL-12 stimulation, but was functional, as it could be easily phosphorylated in these cells after anisomycin treatment for 30 min (Fig. 2F). Finally, to exclude the possibility that the observed difference in the IFN-γ induction was due to different stimulation of JAK/Stat pathways, we compared the phosphorylation of Stat1 and Stat3 in WT and Mk3−/− NK cells during their stimulation with the IL-2 and IL-12 cytokines. Results presented in Fig. 2F show that phosphorylation of the Stat transcription factors in WT and Mk3−/− NK cells was similar.
To further explore whether the enhanced relative activity of p38 in Mk3−/− cells is also reflected in IFN-γ promoter activity, we analyzed the binding of Stat4, a major transcription factor of the IFN-γ promoter and a target of p38 kinase, to the IFN-γ promoter by ChIP assay. NK cells from WT and Mk3−/− mice were stimulated for 2 h with IL-2 + IL-12, after which the DNA-protein interactions were cross-linked, and the Stat4 protein was immunoprecipitated. Subsequently, the amount of IFN-γ promoter DNA bound to precipitated Stat4 transcription factor was analyzed by qRT-PCR in comparison to the total amount of IFN-γ promoter DNA in the samples. We observed an increment in binding of Stat4 to the IFN-γ promoter, after stimulation of WT NK cells with the cytokines, but the Stat4 binding in the Mk3-deficient cells was further augmented compared to that in the WT cells (Fig. 2G), suggesting again that MK3 regulates the IFN-γ expression at the transcriptional level and that it exerts an inhibitory effect.
CD4+ T cells deficient in MK3 show a stronger Th1 response than do WT cells
Apart from NK cells, T lymphocytes express high amounts of IFN-γ when activated by antigen exposure. The amount of secreted IFN-γ defines whether T lymphocytes will develop into a Th1 or Th-2 type and is therefore also decisive for the development of a pro- or anti-inflammatory immune response. A proinflammatory Th1 immune response resulted in increased amounts of secreted IFN-γ, whereas an anti-inflammatory Th2 response is marked by the secretion of IL-4 and IL-10 (34). As MK3 is highly expressed in T lymphocytes, it is conceivable that the knockout of Mk3 leads not only to an enhanced production of IFN-γ but also to a stronger Th1 response. To test this hypothesis, we isolated CD4 T cells from WT and Mk3−/− mice and cultured them under neutral or Th1- or Th-2-inducing conditions. After 2 d of stimulation, the supernatants were analyzed for the amount of secreted IFN-γ, and the cells were additionally stained for intracellular IFN-γ. In agreement with our assumption, the Mk3−/− CD4 T cells secreted more soluble IFN-γ than did the WT CD4 T lymphocytes (Fig. 3A), and, in addition, more CD3+CD4+ Mk3−/− cells were found to be positive for intracellular IFN-γ (Fig. 3B). Interestingly, stimulated Mk3−/− T lymphocytes showed a significantly higher number of IFN-γ-positive cells, even when they were kept under neutral conditions.
Figure 3.

CD4+ T cells from Mk3−/− mice respond with a stronger induction of IFN-γ in comparison to WT mice. A, B) Splenic CD4+ T cells from C57Bl/6 WT and Mk3−/− mice were isolated and activated with anti-CD3+anti-CD28 Dynabeads (Life Technologies) for 5 d, after which they were stimulated with IL-2 (neutral), IL-2 + IL-12 (Th1), or IL-2 + IL-4 (Th2) for a further 2 d. Supernatants were then analyzed for secreted IFN-γ (A), and the cells for intracellular expression of synthesized IFN-γ (B). Mean intracellular IFN-γ level of WT cells cultured under neutral conditions was assigned a value of 1 (B). *P < 0.05. C) After activation with anti-CD3+anti-CD28 Dynabeads, CD4+ T cells were incubated with DMSO or the U0126 inhibitor for 30 min before stimulation with IL-2 (neutral), IL-2 + IL-12 (Th1), or IL-2 + IL-4 (Th2) for 48 h, and the supernatants were analyzed for secreted amounts of IFN-γ (N=3, with cells pooled from 3 animals in each experiment). Statistical analysis, Mann-Whitney test.
To delineate the importance of ERK1/2 in MK3-mediated regulation of Th1/Th2 development, CD4+ cells were skewed to neutral, Th1, or Th2 differentiation in the presence of the MEK-specific inhibitor U0126. Analysis of the T-cell supernatants 2 d later showed a significant reduction in the amount of secreted IFN-γ in WT and Mk3−/− cells (Fig. 3C), underscoring the importance of ERK1/2 kinases in the induction of IFN-γ per se, as well as its cooperative action with MK3 in this process. However, IFN-γ secretion was not completely inhibited by U0126, indicating that other signaling pathways are also involved.
Deficiency of Mk3 is protective against lethal IAV infection
Our in vitro results showed that in the absence of MK3, NK and T cells produce more IFN-γ, and T lymphocytes display stronger Th1 development. This finding in turn suggests that Mk3-knockout mice should be more resistant than WT mice to certain pathogens that induce a strong Th1 immune response. To test this possibility experimentally, we infected C57Bl/6 WT and Mk3−/− mice with IAV. This disease is well studied, and the recovery from IAV infection is believed to require the development of a strong Th1 immune response (21). First, we infected WT and Mk3−/− mice with different doses of the H1N1 virus PR8, to determine whether they differ in their sensitivity to the virus (Fig. 4A, B). We observed that the LD50 for the Mk3−/− mice [2500 plaque-forming units (pfu)] was >8-fold higher than for the WT mice (300 pfu). A more severe development of the disease in the WT mice, when compared to that in the Mk3−/− animals, was also clearly seen when monitoring their body weight changes and survival (Fig. 4C, D) or the virus titers in the lung (Fig. 5A, B) of infected mice after infection with 1 LD50 for the WT mice. Although all the Mk3−/− mice survived the infection and lost only ∼8% of their body weight, the surviving WT mice showed an up to 25% body weight loss. In accordance with these data, influenza viruses propagated much more efficiently in the lungs of the WT mice than in the Mk3−/− mice (Fig. 5A, B). In WT mice, viral replication was faster, and higher titers were reached. Furthermore, viral clearance was delayed in the WT compared to the Mk3−/− mice; we were unable to detect any viral particles in the Mk3-deficient mice at 9 d postinfection (p.i.) but still could detect some viral particles in the WT mice (Fig. 5A, B).
Figure 4.

Loss of Mk3 enhances survival of IAV-infected mice. A, B) C57Bl/6 WT (A) and Mk3−/− (B) mice were infected with different doses of IAV to determine the LD50. For the WT animals, doses between 1 × 102 and 1 × 103 particles/mouse were used; for the Mk3−/− mice, doses between 1 × 102 and 1 × 104 particles/mouse. Average survival per group is shown. C, D) C57Bl/6 WT and Mk3−/− mice were infected with 1 LD50 of WT mice (300 particles/mouse) and the survival rate (C) and body weight (D) were monitored for up to 18 d (N=2; n=4–6 animals/group). ***P < 0.001; 2-way ANOVA.
Figure 5.

IAV infection results in reduced viral titer and an increase in secreted IFN-γ in the lungs of Mk3−/− mice. C57Bl/6 WT and Mk3−/− mice were infected with IAV PR8 (1 LD50 of WT mice; 300 particles/mouse). A, B) Viral lung titers were determined at d 1 p.i. (A) or 6 and 9 d p.i. (B). Viral titers of individual mice are shown. **P < 0.01, ***P < 0.001; Mann-Whitney test. C–E) WT and Mk3−/− mice were infected with 300 particles of IAV PR8, and BAL fluids of the infected mice were analyzed at 6 d p.i. ror secreted IFN-γ (C), IL-6 (D), and MCP-1 (E). Mean values of WT mice were arbitrarily taken as unity (N=3, with ≥3 animals/group in each experiment). *P < 0.05, Mann-Whitney test. F) C57Bl/6 WT and Mk3−/− mice were infected with 300 particles of IAV PR8 per mouse. At 1 and 2 d p.i., the animals received either control IgG or IFN-γ specific antibodies (1 mg/kg, intraperitoneally) and at 3 d p.i., the lung viral titers were determined. Viral titers of individual animals, as well as means ± sem, are shown. *P < 0.05; Kruskal-Wallis test.
As our in vitro data showed that MK3 deficiency results in enhanced expression of IFN-γ in stimulated lymphoid cells, it was intriguing to see whether this also happened in vivo after infection of mice. Therefore, the amount of secreted IFN-γ was measured in the bronchoalveolar fluid on different days after IAV infection. The concentration of IFN-γ in BALF reached the maximum at 6 d p.i. in both mouse types (data not shown), but the amount of secreted IFN-γ in the lungs of the infected Mk3−/− mice was almost twice as high as that detected in the WT mice (Fig. 5C). It is fascinating that the elevated level of cytokine production was restricted to IFN-γ, as other cytokines important for the disease outcome, such as IL-6 or MCP-1 (Fig. 5D, E) and TNF-α or IL-17 (data not shown), showed no difference between the infected WT and Mk3−/− mice.
Previously, we showed that IAV infection induces the activation of the p38-MK3 signaling axis, resulting in inhibition of PKR and, consequently, enhanced viral replication. Accordingly, knockout of MK3 leads to reduced viral replication in vitro (20). Thus, we wished to explore whether the decreased viral replication in the lungs of Mk3−/− mice is driven by changed PKR activity or increased IFN-γ expression. Therefore, we analyzed the IAV replication in the lungs of WT and Mk3−/− mice in the presence of IFN-γ-neutralizing antibodies. As the data in Fig. 5F show, intraperitoneal application of anti- IFN-γ but not of control IgG antibodies increased viral titers in the lungs of the Mk3−/− mice, underscoring the high importance of IFN-γ for the attenuated viral replication in Mk3−/− mice. Finally, we “reconstituted” the Mk3−/− phenotype in WT animals by applying soluble IFN-γ to IAV-infected WT mice. Comparing viral titer in the lungs of these mice with those in the Mk3−/− mouse lungs that were treated with a solvent control showed that increasing the amount of IFN-γ in infected WT mice indeed decreased viral replication, albeit the reduction of viral titers in lungs of the IFN-γ-treated WT mice was not as pronounced as that in the Mk3-deficient mice (Supplemental Fig. S2).
Altogether, the in vivo obtained data are congruent with our in vitro results and strongly suggest that the decreased viral propagation in the Mk3−/− mice is mainly due to the increased IFN-γ synthesis and a stronger proinflammatory immune response against the replicating virus in these mice.
Mk3 deficiency promotes activation of NK and CD8+ lymphocytes
To analyze the immune status of WT and Mk3−/− mice during IAV infection, flow cytometry was used to assess which immune cells are present in the bronchoalveolar space. The number of cells that infiltrated the bronchoalveolar space steadily increased after IAV infection, but no significant difference between the WT and Mk3−/− mice was observed (Fig. 6A). In addition, we did not observe significant differences for all other cell types, including CD45.2+ leukocytes (Fig. 6B), F4/80+ macrophages (Fig. 6C), CD3−Dx5+ NK cells (Fig. 6D), CD4+ T cells (Fig. 6E), or CD8+ T cells (Fig. 6F), between 0 and 9 d p.i. These results imply that the observed elevated level of IFN-γ in the BALF of IAV-infected Mk3−/− mice is due to elevated production of the cytokine rather than to the elevated number of immigrated immune cells. Flow cytometry of NK cells as well as CD4+ and CD8+ lymphocytes for intracellular IFN-γ confirmed this suggestion, revealing an enhanced number of IFN-γ+ Th cells, cytotoxic T cells, and NK cells in infected lungs of Mk3−/− mice (Fig. 6G–I).
Figure 6.

Mk3 deficiency leads to an increased expression of IFN-γ by activated NK and T cells after IAV infection, but does not affect the total number of lung-infiltrating cells. C57Bl/6 WT and Mk3−/− mice were infected with 300 particles of IAV PR8. At 1, 3, 6, and 9 d p.i., the BAL fluid was analyzed for the content of the indicated cells by flow cytometry: total cell count (A); CD45.2+ (B); F4/80+ (C); CD3−/Dx5+ (D); CD3+/CD4+ (E); CD3+CD8+ (F); (G–I) intracellular IFN-γ+ NK-cell (G) and CD4+ (H) and CD8+ (I) T-cell subpopulations; and cytotoxic active NK cells (J). N = 3, with BAL cells pooled from 3 animals in each experiment. Statistical analysis, 2-way ANOVA.
IFN-γ provokes the maturation of native NK lymphocytes to cytotoxic NK cells. To explore whether their amount is enhanced in Mk3−/− compared to infected WT lungs, NK lymphocytes from both types of mice were compared for expression of the CD107a marker, the surface exposure of which is characteristic of active cytotoxic NK cells. The number of NK cells expressing the CD107a receptor on their surface was larger in the Mk3−/− than in the WT lungs, especially at later stages of IAV infection (Fig. 6J), showing that more cytotoxic active NK cells were detectable in Mk3-deficient mice.
DISCUSSION
In this report, we have shown that the protein kinase MK3 is an important regulator of IFN-γ expression in NK and T lymphocytes. MK3 inhibits the Stat4-mediated transcription of the Ifng gene, but not the stability of its mRNA. We further demonstrated that the absence of MK3 enhances polarization of CD4+ T cells to a Th1 type. Finally, we illustrated by the infection of Mk3−/− and WT mice with IAV that Mk3 deficiency is beneficial for anti-influenza defense, as it facilitated the maturation of cytotoxic NK and CD8+ lymphocytes due to enhanced IFN-γ expression.
MK3 is highly expressed in T and NK lymphocytes, suggesting that it plays an essential role in these cells. The common cytokine synthesized by both cell types is IFN-γ, which is important for their development into cytotoxic cells. The mRNA stability of IFN-γ (35) is regulated similarly to that of TNF-α (11, 12), in which both MK2 and MK3 are involved. However, comparison of the IFN-γ mRNA half-life in WT and Mk3-deficient NK cells revealed only a weak mRNA-stabilizing effect of MK3. The major effect of MK3 on IFN-γ expression was observed at the transcriptional level. Because the main inducer of the IFN-γ promoter is the transcription factor Stat4, the functional activity of which is also essential for the development of Th1 responses in vivo (36, 37), we analyzed whether MK3 influences the Stat4-mediated activity of the IFN-γ promoter. Our experiments showed that MK3 reduced the binding of the activated Stat4 to the IFN-γ promoter, as absence of MK3 significantly enhanced the promoter activity (Supplemental Fig. S1). Furthermore, reporter gene assays revealed that the inhibitory effect of MK3 was linked to a 751-bp IFN-γ promoter sequence proximal to the gene coding region, which was described by Soutto et al. (38) as a region containing the Stat4-binding site. Whether MK3 inhibits the binding of Stat4 to the IFN-γ promoter directly, or via its phosphorylation, has still to be determined.
IFN-γ expression by Th1 effector T cells as well as activation of Stat4 is mainly induced via phosphorylation and activation of p38 MAPK (27), but the involvement of ERK1/2 in promoting IFN-γ transcription has been described, as well (33). Consistent with these data, we observed here that the enhanced production of IFN-γ in Mk3−/− NK cells was abrogated when either the p38 or ERK1/2 signaling cascades were inhibited. This indicates that both cascades are involved in the IFN-γ promoter suppression by MK3. Interestingly, the phosphorylation of both p38 and ERK1/2, after the stimulation of the NK cells with IL-2 + IL-12, was much stronger in the Mk3-deficient cells. This result demonstrated that MK3 has an inhibitory effect on p38 and ERK1/2 and further suggests that MK3 exerts its inhibitory effect on the IFN-γ promoter indirectly, just by inhibiting the activity of p38 and ERK1/2 kinases, thereby diminishing the phosphorylation/activation of the Stat4 transcription factor. Notably, a similar observation was recently published by Prickaerts et al. (39), demonstrating that MK3 controls the expression of the ATF3 transcription factor via a negative regulatory feedback on MAPKs.
IFN-γ and Stat4 are key players in the differentiation of naive T cells into the Th1 state (36). In agreement with the data showing that MK3 inhibits the Stat4-mediated expression of IFN-γ, we observed a clear shift toward the Th1 response of Mk3−/− T lymphocytes when stimulated with IL-2 + IL-12. Moreover, many Mk3-deficient T cells were transformed into a Th1 state, even when they were kept in neutral differentiation conditions or were stimulated with IL-4, the classic inducer of Th2 differentiation. This suggests that the Mk3-deficient mice should have an advantage being challenged with Th1 inducing pathogens, but at the same time implies that the absence of MK3 should aggravate chronic infections, as was indeed described for glomerulonephritis (19). In accordance with this suggestion, our experiments demonstrated a clear protection of Mk3−/− mice against acute IAV infection. The viral titers in the lungs of Mk3−/− animals were decreased, the survival rate was increased, and the body weight loss was minor when compared to the weight loss of the infected WT mice. The measured parameters went along well with the enhanced amounts of secreted IFN-γ in the BAL fluid of Mk3−/− lungs, as well as with significantly increased numbers of IFN-γ positive T and NK cells in these animals, which of course positively influences the antiviral immune response. In agreement with this, injection of anti-IFN-γ antibodies into Mk3−/− mice resulted in an increase in viral titer. To the contrary, application of soluble IFN-γ into WT mice decreased viral propagation. Thus, our data are consistent with the published data showing a protective role of IFN-γ in the early stages of IAV infection (24) and the promoting effect of IFN-γ in induction of the innate and adaptive immune responses (23). In addition to the observed enhanced immune cell response, the dsRNA inducible PKR also may contribute to the decrease in viral titers, especially at 1 d p.i., as it was reported that absence of MK3 favored a stronger activation of the PKR in cell culture, resulting in reduced viral titers (20).
Propagation of influenza viruses strongly depends on host cell signaling and being displayed in cells; influenza viruses actively induce several signaling pathways, including p38 and ERK1/2 MAPK cascades and the MK3 as substrate of them (40). It is intriguing to speculate that the IAV-mediated activation of MK3 is responsible for the still unresolved inhibition of IFN-γ expression in IAV-infected NK cells (25).
A further striking finding of this work was that MK3 differed significantly in the mode of action from its close homologue MK2, as only MK3, but not MK2, regulated IFN-γ expression (Fig. 1A). The data reported herein indicate that MK2 and MK3 may even have distinct functions in the regulation of the immune response. Although Mk2 but not Mk3 deficiency destabilized the TNF-α mRNA and inhibited its translation (11, 12), thus supporting the development of proinflammatory and chronic immune reactions, the Mk3-, but not the Mk2-deficiency, favored the development of effective innate NK and adaptive Th1 immune responses, due to decreased inhibition of the Ifng gene. Although the anti-inflammatory role of MK2 is described for several diseases, including LPS-induced septic shock, inflammation-mediated arthritis, and lung fibrosis, little is known so far about MK3 (17–19).
The data in this report demonstrate for the first time that MK3 has an important inhibitory role in the development of IFN-γ-mediated innate and adaptive immune responses and suggest a new role for MK3, which is distinct from MK2. Further, our data imply that MK3 inhibition is a promising approach to enhancing the Th1 immune response to disease-inducing pathogens, such as those in IAV infections. Establishment of MK3 as a therapeutic target would have the advantage of specifically upregulating IFN-γ, without disturbing the expression of other cytokines and would allow a prevention of a cytokine overflow, which is the major danger on infection with the highly pathogenic H5N1 IAV.
Supplementary Material
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
Acknowledgments
The authors acknowledge Ludmilla Wixler (University of Münster, Germany) for generation of the IFN-γ reporter gene construct.
This work was supported by the Interdisciplinary Center of Clinical Research (IZKF) at the University of Münster, (grants Lud2/010/11 and Lud2/017/13), as well as by the Graduate School GRK1409 and the Collaborative Research Center SFB1009, funded by the Deutsche Forschungsgemeinschaft (DFG).
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- ChIP
- chromatin immunoprecipitation
- FBS
- fetal bovine serum
- Hsp
- heat-shock protein
- IAV
- influenza A virus
- IFN-γ
- interferon-γ
- MAPK
- mitogen-activated protein kinase
- MK
- mitogen-activated protein kinase–activated protein kinase
- MDCK
- Madin-Darby canine kidney
- p.i.
- postinfection
- pfu
- plaque-forming unit
- PKR
- protein kinase R
- PR8
- Puerto Rico/8/34
- qRT-PCR
- quantitative reverse transcription polymerase chain reaction
- TTP
- tristetraprolin
- WT
- wild-type
REFERENCES
- 1.Gaestel M. (2006) MAPKAP kinases—MKs—two's company, three's a crowd. Nat. Rev. Mol. Cell Biol. , 120–130 [DOI] [PubMed] [Google Scholar]
- 2.Seternes O. M., Mikalsen T., Johansen B., Michaelsen E., Armstrong C. G., Morrice N. A., Turgeon B., Meloche S., Moens U., Keyse S. M. (2004) Activation of MK5/PRAK by the atypical MAP kinase ERK3 defines a novel signal transduction pathway. EMBO J. , 4780–4791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ludwig S., Engel K., Hoffmeyer A., Sithanandam G., Neufeld B., Palm D., Gaestel M., Rapp U. R. (1996) 3pK, a novel mitogen-activated protein (MAP) kinase-activated protein kinase, is targeted by three MAP kinase pathways. Mol. Cell. Biol. , 6687–6697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rouse J., Cohen P., Trigon S., Morange M., Alonso-Llamazares A., Zamanillo D., Hunt T., Nebreda A. R. (1994) A novel kinase cascade triggered by stress and heat shock that stimulates MAPKAP kinase-2 and phosphorylation of the small heat shock proteins. Cell , 1027–1037 [DOI] [PubMed] [Google Scholar]
- 5.Sithanandam G., Latif F., Duh F. M., Bernal R., Smola U., Li H., Kuzmin I., Wixler V., Geil L., Shrestha S. (1996) 3pK, a new mitogen-activated protein kinase-activated protein kinase located in the small cell lung cancer tumor suppressor gene region. Mol. Cell. Biol. , 868–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ben-Levy R., Leighton I. A., Doza Y. N., Attwood P., Morrice N., Marshall C. J., Cohen P. (1995) Identification of novel phosphorylation sites required for activation of MAPKAP kinase-2. EMBO J. , 5920–5930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Engel K., Schultz H., Martin F., Kotlyarov A., Plath K., Hahn M., Heinemann U., Gaestel M. (1995) Constitutive activation of mitogen-activated protein kinase-activated protein kinase 2 by mutation of phosphorylation sites and an A-helix motif. J. Biol. Chem. , 27213–27221 [DOI] [PubMed] [Google Scholar]
- 8.Engel K., Kotlyarov A., Gaestel M. (1998) Leptomycin B-sensitive nuclear export of MAPKAP kinase 2 is regulated by phosphorylation. EMBO J. , 3363–3371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zakowski V., Keramas G., Kilian K., Rapp U. R., Ludwig S. (2004) Mitogen-activated 3p kinase is active in the nucleus. Exp. Cell Res. , 101–109 [DOI] [PubMed] [Google Scholar]
- 10.Clifton A. D., Young P. R., Cohen P. (1996) A comparison of the substrate specificity of MAPKAP kinase-2 and MAPKAP kinase-3 and their activation by cytokines and cellular stress. FEBS Lett. , 209–214 [DOI] [PubMed] [Google Scholar]
- 11.Hitti E., Iakovleva T., Brook M., Deppenmeier S., Gruber A. D., Radzioch D., Clark A. R., Blackshear P. J., Kotlyarov A., Gaestel M. (2006) Mitogen-activated protein kinase-activated protein kinase 2 regulates tumor necrosis factor mRNA stability and translation mainly by altering tristetraprolin expression, stability, and binding to adenine/uridine-rich element. Mol. Cell. Biol. , 2399–2407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ronkina N., Kotlyarov A., Dittrich-Breiholz O., Kracht M., Hitti E., Milarski K., Askew R., Marusic S., Lin L. L., Gaestel M., Telliez J. B. (2007) The mitogen-activated protein kinase (MAPK)-activated protein kinases MK2 and MK3 cooperate in stimulation of tumor necrosis factor biosynthesis and stabilization of p38 MAPK. Mol. Cell. Biol. , 170–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tudor C., Marchese F. P., Hitti E., Aubareda A., Rawlinson L., Gaestel M., Blackshear P. J., Clark A. R., Saklatvala J., Dean J. L. (2009) The p38 MAPK pathway inhibits tristetraprolin-directed decay of interleukin-10 and pro-inflammatory mediator mRNAs in murine macrophages. FEBS Lett. , 1933–1938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao W., Liu M., D'Silva N. J., Kirkwood K. L. (2011) Tristetraprolin regulates interleukin-6 expression through p38 MAPK-dependent affinity changes with mRNA 3′ untranslated region. J. Interferon Cytokine Res. , 629–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Voncken J. W., Niessen H., Neufeld B., Rennefahrt U., Dahlmans V., Kubben N., Holzer B., Ludwig S., Rapp U. R. (2005) MAPKAP kinase 3pK phosphorylates and regulates chromatin association of the polycomb group protein Bmi1. J. Biol. Chem. , 5178–5187 [DOI] [PubMed] [Google Scholar]
- 16.Kotlyarov A., Yannoni Y., Fritz S., Laass K., Telliez J. B., Pitman D., Lin L. L., Gaestel M. (2002) Distinct cellular functions of MK2. Mol. Cell. Biol. , 4827–4835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hegen M., Gaestel M., Nickerson-Nutter C. L., Lin L. L., Telliez J. B. (2006) MAPKAP kinase 2-deficient mice are resistant to collagen-induced arthritis. J. Immunol. , 1913–1917 [DOI] [PubMed] [Google Scholar]
- 18.Liu T., Warburton R. R., Guevara O. E., Hill N. S., Fanburg B. L., Gaestel M., Kayyali U. S. (2007) Lack of MK2 inhibits myofibroblast formation and exacerbates pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. , 507–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guess A. J., Ayoob R., Chanley M., Manley J., Cajaiba M. M., Agrawal S., Pengal R., Pyle A. L., Becknell B., Kopp J. B., Ronkina N., Gaestel M., Benndorf R., Smoyer W. E. (2013) Crucial roles of the protein kinases MK2 and MK3 in a mouse model of glomerulonephritis. PloS One , e54239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luig C., Kother K., Dudek S. E., Gaestel M., Hiscott J., Wixler V., Ludwig S. (2010) MAP kinase-activated protein kinases 2 and 3 are required for influenza A virus propagation and act via inhibition of PKR. FASEB J. , 4068–4077 [DOI] [PubMed] [Google Scholar]
- 21.Johnson P. A., Conway M. A., Daly J., Nicolson C., Robertson J., Mills K. H. (2000) Plasmid DNA encoding influenza virus haemagglutinin induces Th1 cells and protection against respiratory infection despite its limited ability to generate antibody responses. J. Gen. Virol. , 1737–1745 [DOI] [PubMed] [Google Scholar]
- 22.Baumgarth N., Brown L., Jackson D., Kelso A. (1994) Novel features of the respiratory tract T-cell response to influenza virus infection: lung T cells increase expression of gamma interferon mRNA in vivo and maintain high levels of mRNA expression for interleukin-5 (IL-5) and IL-10. J. Virol. , 7575–7581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baumgarth N., Kelso A. (1996) In vivo blockade of gamma interferon affects the influenza virus-induced humoral and the local cellular immune response in lung tissue. J. Virol. , 4411–4418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weiss I. D., Wald O., Wald H., Beider K., Abraham M., Galun E., Nagler A., Peled A. (2010) IFN-gamma treatment at early stages of influenza virus infection protects mice from death in a NK cell-dependent manner. J. Interferon Cytokine Res. , 439–449 [DOI] [PubMed] [Google Scholar]
- 25.Guo H., Kumar P., Moran T. M., Garcia-Sastre A., Zhou Y., Malarkannan S. (2009) The functional impairment of natural killer cells during influenza virus infection. Immunol. Cell. Biol. , 579–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mainiero F., Gismondi A., Soriani A., Cippitelli M., Palmieri G., Jacobelli J., Piccoli M., Frati L., Santoni A. (1998) Integrin-mediated ras-extracellular regulated kinase (ERK) signaling regulates interferon gamma production in human natural killer cells. J. Exp. Med. , 1267–1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rincon M., Enslen H., Raingeaud J., Recht M., Zapton T., Su M. S., Penix L. A., Davis R. J., Flavell R. A. (1998) Interferon-gamma expression by Th1 effector T cells mediated by the p38 MAP kinase signaling pathway. EMBO J. , 2817–2829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bream J. H., Curiel R. E., Yu C. R., Egwuagu C. E., Grusby M. J., Aune T. M., Young H. A. (2003) IL4 synergistically enhances both IL2- and IL12-induced IFN-gamma expression in murine NK cells. Blood , 207–214 [DOI] [PubMed] [Google Scholar]
- 29.Messi M., Giacchetto I., Nagata K., Lanzavecchia A., Natoli G., Sallusto F. (2003) Memory and flexibility of cytokine gene expression as separable properties of human T(H)1 and T(H)2 lymphocytes. Nat. Immunol. , 78–86 [DOI] [PubMed] [Google Scholar]
- 30.Hoffmann E., Neumann G., Kawaoka Y., Hobom G., Webster R. G. (2000) A DNA transfection system for generation of influenza A virus from eight plasmids. Proc. Natl. Acad. Sci. U. S. A. , 6108–6113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods , 402–408 [DOI] [PubMed] [Google Scholar]
- 32.Visconti R., Gadina M., Chiariello M., Chen E. H., Stancato L. F., Gutkind J. S., O'Shea J. J. (2000) Importance of the MKK6/p38 pathway for interleukin-12-induced STAT4 serine phosphorylation and transcriptional activity. Blood , 1844–1852 [PubMed] [Google Scholar]
- 33.Kondadasula S. V., Roda J. M., Parihar R., Yu J., Lehman A., Caligiuri M. A., Tridandapani S., Burry R. W., Carson W. E. (2008) Colocalization of the IL12 receptor and FcgammaRIIIa to natural killer cell lipid rafts leads to activation of ERK and enhanced production of interferon-gamma. Blood , 4173–4183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paul W. E., Seder R. A. (1994) Lymphocyte responses and cytokines. Cell , 241–251 [DOI] [PubMed] [Google Scholar]
- 35.Ogilvie R. L., Sternjohn J. R., Rattenbacher B., Vlasova I. A., Williams D. A., Hau H. H., Blackshear P. J., Bohjanen P. R. (2009) Tristetraprolin mediates interferon-gamma mRNA decay. J. Biol. Chem. , 11216–11223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaplan M. H., Sun Y. L., Hoey T., Grusby M. J. (1996) Impaired IL12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature , 174–177 [DOI] [PubMed] [Google Scholar]
- 37.Young H. A., Bream J. H. (2007) IFN-gamma: recent advances in understanding regulation of expression, biological functions, and clinical applications. Curr. Top. Microbiol. Immunol. , 97–117 [DOI] [PubMed] [Google Scholar]
- 38.Soutto M., Zhang F., Enerson B., Tong Y., Boothby M., Aune T. M. (2002) A minimal IFN-gamma promoter confers Th1 selective expression. J. Immunol. , 4205–4212 [DOI] [PubMed] [Google Scholar]
- 39.Prickaerts P., Niessen H. E., Mouchel-Vielh E., Dahlmans V. E., van den Akker G. G., Geijselaers C., Adriaens M. E., Spaapen F., Takihara Y., Rapp U. R., Peronnet F., Voncken J. W. (2012) MK3 controls polycomb target gene expression via negative feedback on ERK. Epigenetics Chromatin , 12–8935-8935-8912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ludwig S., Pleschka S., Planz O., Wolff T. (2006) Ringing the alarm bells: signalling and apoptosis in influenza virus infected cells. Cell. Microbiol. , 375–386 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.