

Abstract
The massive amount of information produced by ChIP-Seq, RNA-Seq, and other next-generation sequencing-based methods requires computational data analysis. However, biologists performing these experiments often lack training in bioinformatics. BioWardrobe aims to bridge this gap by providing a convenient user interface and by automating routine data-processing steps. This protocol details the use of BioWardrobe for identifying and visualizing ChIP-Seq peaks, calculating RPKMs, performing differential binding or gene expression analysis, and creating plots and heat maps. We specifically describe how to use BioWardrobe’s quality control measures for troubleshooting NGS-based experiments.
Keywords: Next-generation sequencing, ChIP-Seq, RNA-Seq, ATAC-Seq, DNase-Seq, RPKM, Peak calling, Heatmaps, Epigenomics, Transcriptomics
1. Introduction
The introduction of next-generation sequencing (NGS) led to the development of ChIP-Seq [1–3] (chromatin immunoprecipitation sequencing), RNA-Seq [4] (RNA sequencing), DNase-Seq [5] (DNase 1 sequencing), ATAC-Seq [6] (assay for transposase-accessible chromatin sequencing) and other “acronym”-Seqs. Due to the power of NGS, many methods previously used for a study of a single gene or locus can now provide similar (and often better controlled) data for the whole genome. However, with this remarkable power comes an obstacle that is forcing many scientists to avoid the modern methods: the need for computational data analysis. Indeed, the majority of these experiments can be reliably performed by experienced molecular biologists, but the data analysis requires computational knowledge that many biologists do not have. BioWardrobe aims to overcome this obstacle by providing a convenient browser-based graphical user interface and automated data analysis pipelines.
A majority of existing bioinformatics software require at least some programming expertise. Popular open source packages that are used for data analysis, such as HOMER [7] and Tuxedo Suite [8–11], have a command line interface. The user has to be technically adept and familiar with a Unix-like interface, and the packages have limited visualization options and no interactivity. The commercial software Strand NGS [12] and Partek [13] use a graphical interface and can be run on desktop computers; however, the sheer size of the files requires computational power and storage that is not available on a typical desktop computer. Web-based software, such as Illumina Basespace [14] and Galaxy [15] are more user friendly and allow for analysis and storage of data. However, even though the command line is not used, the user has to determine parameters for each tool, manage file format conversions and assemble pipelines, which requires almost as much knowledge as performing analysis in a command line interface. This can become cumbersome for users without a computational background.
BioWardrobe [16] overcomes these problems by allowing the users to download, visualize, and store data along with the analysis. The pipeline parameters are automatically chosen on the basis of the biological description of the experiment. The user-friendly web interface is primarily aimed at NGS analysis for biologists working in the epigenomics and transcriptional regulation field. The analysis routine is separated into two parts: basic and advanced analysis. For the semiautomated basic analysis, users need to provide the description of each experiment and the source of the data. BioWardrobe will download the data, map the reads, perform quality controls and provide initial output (e.g., peaks of enrichment). Advanced analysis allows for integration of multiple experiments and is guided by the user input. Overall, BioWardrobe improves accessibility of the NGS methodology by providing a convenient, browser-based graphical user interface and automated data analysis pipelines without requiring computational or programming expertise.
2. Materials
Hardware requirements. BioWardrobe can be installed on a Linux- or Mac-based server. For the human genome, we recommend at least an E5-based Mac Pro (or a Linux server) computer with at least 32 Gb of RAM and external (e.g., Thunderbolt or USB 3.0) storage array. We recommend at least 16 Tb of storage capacity for an average laboratory.
BioWardrobe installation. Though the user does not need to be a programming expert to perform data analysis, the native installation of BioWardrobe requires IT expertise and is not covered in this chapter. We recommend obtaining support from the institutional IT department or from a commercial service provider. BioWardrobe installation is described at https://biowardrobe.com and at https://github.com/Barski-lab/biowardrobe/wiki/Installing-BioWardrobe
BioWardrobe virtual machine. For evaluation purposes, users can quickly install a simplified version of BioWardrobe as a virtual machine. This image contains only the drosophila genome and can be run on desktop-class hardware (8 Gb RAM minimum). Virtual machine instructions are available here: https://biowardrobe.com/projects/wardrobe/wiki/Install_VirtualBox_image Please replace the link with https://github.com/Barski-lab/biowardrobe/wiki/VirtualBox-Instructions. For the production environment, we recommend using a native installation.
3. Methods
3.1. Basic Analysis
In basic analysis, the analytical pipeline is selected automatically on the basis of the experimental variables that are familiar to biologists: experiment type, library construction procedure, antibody, etc.
3.1.1. Adding Data
For loading the data, go to the “Data and Analysis” tab in BioWardrobe and click on “Experiments.” The experiments page shows all the existing data uploaded by the users. For performing a new analysis, click on the “New” button in the “Laboratory data” window.
- The Experimental Form: The “Experimental form” is where the information about the library is entered. The “General Info” tab has the following entries:
-
(a)Record basic information about the library in the “Experiment Description” section: cell type, conditions, the genome, etc. For ChIP/ATAC/DNase-Seq choose DNA-Seq as experiment type. Indicate if the experiment is paired or single read. For stranded RNA-Seq libraries choose RNA-Seq dUTP.
-
(b)Next, enter a short name for the experiment. This needs to be unique and descriptive because it will be used to identify experiment on the browser and in the advanced analysis. Along with the name, choose the folder where the data will be saved. Folders are managed by the administrators of each laboratory and can be shared with other laboratories.
-
(c)ChIP-Seq-specific parameters:
- Antibody: specifying antibody will instruct MACS2 [17] to identify either narrow (e.g., for TFs) or broad (e.g., for H3K27me3) peaks. The list of antibodies and what kind of peaks they produce can be edited in the Catalogs menu.
- Control for ChIP-Seq: Indicate that the library is an Input or IgG control by marking the checkbox. This will add the sample to the list of control experiments. Selecting a control experiment will instruct MACS2 to use it for peak calling.
-
(d)In the next section, provide BioWardrobe with the source of data. For “Download Type,” most users will select “Direct Link” to enter a URL for the file. Other options include “Upload directory” for local files or “Core facility” if it has been set up. BioWardrobe can process both compressed (gz, bz2) and uncompressed .fastq and .sra files. The URL should be a direct link to the file (i.e., entering this link in the browser should start the download). For paired-end experiments or single read experiments containing multiple fastq files separate the links with a semicolon (;). See the instructions for finding the link to sra files here: https://github.com/Barski-lab/biowardrobe/wiki/How-to-find-a-link-to-.sra-file-in-GEO
-
(e)The “Protocol” and “Notes” tabs are designed to enter the protocol and any additional information about the experiment, respectively.
-
(f)The “Advanced” tab contains additional parameters for ChIP-Seq experiments.
- The size of the fragment can be set in the “Expected Fragment Size” section. Normally the fragment size is determined automatically by MACS2, but the user can force BioWardrobe to use the fragment size specified here for both coverage and peak detection.
- This section also has the option of removing duplicates before analysis. Selecting this option will leave only one read aligned at the same position and strand (see Note 1).
- If poor quality bases are observed near the ends of the reads, they can be removed using the “Trim from the left” or “Trim from the right” options.
- The “Force to repeat experimental analysis” box needs to be checked if any of the parameters in the “Advanced” tab are changed after the initial analysis and the analysis has to be repeated.
-
(g)Click on “Save.” Typically, BioWardrobe takes ~2 h to perform the analysis.
-
(a)
3.1.2. Results of Basic Analysis
- After the analysis is complete, click on the experiment. The second tab is “Quality Control.” This tab shows various statistics and quality control measurements for the experiment (DNA-Seq/RNA-Seq). This tab also has the “Base Frequency plot” and the “QC” (quality control) sections.
-
(a)For DNA-Seq experiments (see Fig. 1a, b, Note 2), the statistics shown are:
- The number of reads—“Tags total.”
- The reads mapped to the genome—“Tags mapped,” shown in green in the pie chart.
- The reads that are mapped to multiple locations in the genome are discarded—“Multi-mapped reads,” shown in blue in the pie chart.
- Duplicates that are mapped, but removed if that option is selected in the “Advanced” tab—“Duplicates” (see Note 1).
- Reads that are not mapped—“Unmapped reads,” shown in red in the pie chart.
-
(b)For RNA-Seq experiments (see Fig. 1c, d, Note 3), the statistics shown are:
- The number of reads—“Tags total.”
- The reads mapped to the transcriptome in a unique way—“Tags mapped,” shown in green in the pie chart.
- The reads mapped to the genome outside the transcriptome, often because of DNA contamination as a result of insufficient digestion by DNAse—“Outside annotation,” shown in orange in the pie chart.
- The reads that can be mapped to multiple locations are discarded—“Multi-mapped reads,” shown in blue in the pie chart.
- Reads that are not mapped—“Unmapped reads,” shown in red in the pie chart.
- (c)
- (d)
-
(e)The “QC” section has a box plot showing the Phred quality scores for each base (see Note 6).
-
(f)These images can be downloaded by clicking on “Save chart.”
-
(a)
The “Genome browser” tab (see Fig. 3a–c): It displays read coverage and MACS2 peaks (for ChIP-Seq and similar libraries) on the local mirror University of California, Santa Cruz (UCSC) genome browser (see Note 7). All the data are uploaded to the browser and it can be used for performing “bioinformatics by eye”—comparing peaks between the experiments around the genome.
The “Run R” tab: The section “Default Result(s)” shows the various plots created by MACS2 for DNA-Seq experiments and shows the RKPM distribution and gene body density for RNA-Seq experiments. The results can be customized in the “Custom Result(s)” section, and the administrators also can edit the R code in the “Source” section (see Subheading 3.3).
The “RKPM list” tab (see Fig. 4): This window shows the RKPM values for genes for RNA-Seq data. These data can be saved as a .csv file by clicking on the “Save” option. The genome browser tab of the selected gene opens when the “Jump” button is clicked. The data can be presented for individual isoforms or summed up for genes or for common TSSs (see Note 8).
- Islands list: DNA-seq data have the “Islands list” tab (see Note 9) with the list of islands detected by MACS2, their position, and other statistics. The table also contains the information regarding nearby genes.
-
(a)If an island has more than one summit, each entry is shown separately with the summit coordinates as “start” and “end” along with the position of the island.
-
(b)If the box “Show unique islands” is selected, each island is shown only once and the summits are not shown.
-
(c)The “region” section has information about the islands, if they are in the promoter region, upstream of the promoter, or in an exon, intron, or intergenic region.
-
(d)The definition of promoter (radius around the TSS) can be changed in the heading area of the tab. Any changes made will also be reflected in the “Islands Distribution Plot.”
-
(e)Peaks can be filtered by specifying a minimum p/q/fold enrichment value.
-
(f)The “get fasta” button is used to obtain the sequence under the peak in fasta format. The “Fasta region” specifies the radius around the summit to report. Selecting 0 will produce the sequence under the whole peak.
-
(g)The table can be saved in the .csv format by clicking the “Save” button.
-
(a)
Average Tag Density (see Fig. 5a, b): The “Average Tag Density” tab shows the density profiles for a DNA-seq experiment around all the annotated transcription start sites (TSS) (see Note 10).
The “Islands Distribution” (see Fig. 6): The “Islands Distribution” window shows the distribution of the island over the promoter, upstream, exonic, intronic, and intergenic regions (assigned in this order). It also displays the percentage of distribution in each region on the diagram (see Note 11).
Fig. 1.
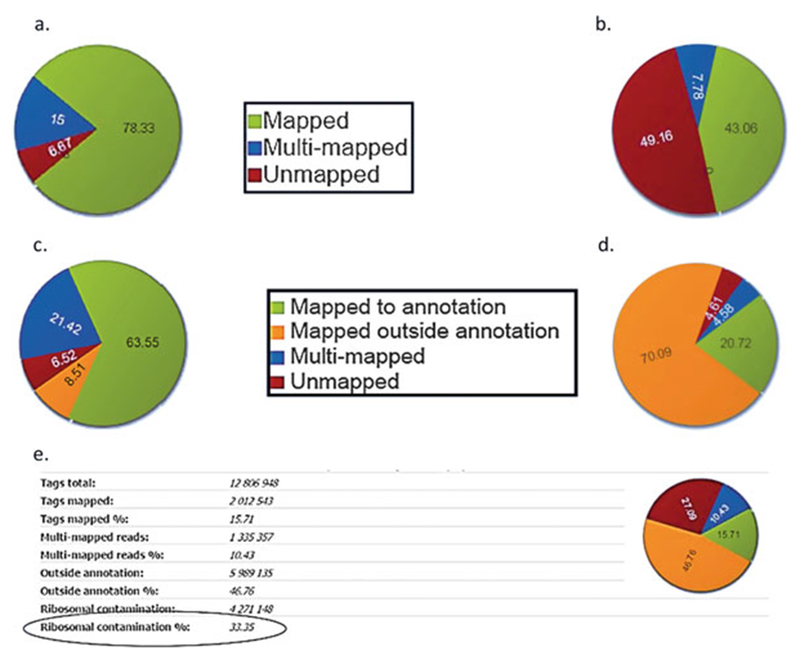
Mapping statistics (a) and (b) ChIP-Seq mapping statistics show mapped reads in green, multi-mapped reads in blue and unmapped reads in red. A high percentage of unmapped reads may indicate contamination with adapter dimers—see Fig. 2c. (c) and (d) RNA-Seq mapping statistics shows reads mapped to annotation in green, multi-mapped reads in blue, reads mapped outside annotation in orange and unmapped reads in red. High percentage of reads mapped outside annotation may indicate DNA contamination. (e) Ribosomal RNA contamination: High percentage of reads mapping to rDNA repeat indicate problems with mRNA isolation
Fig. 2.
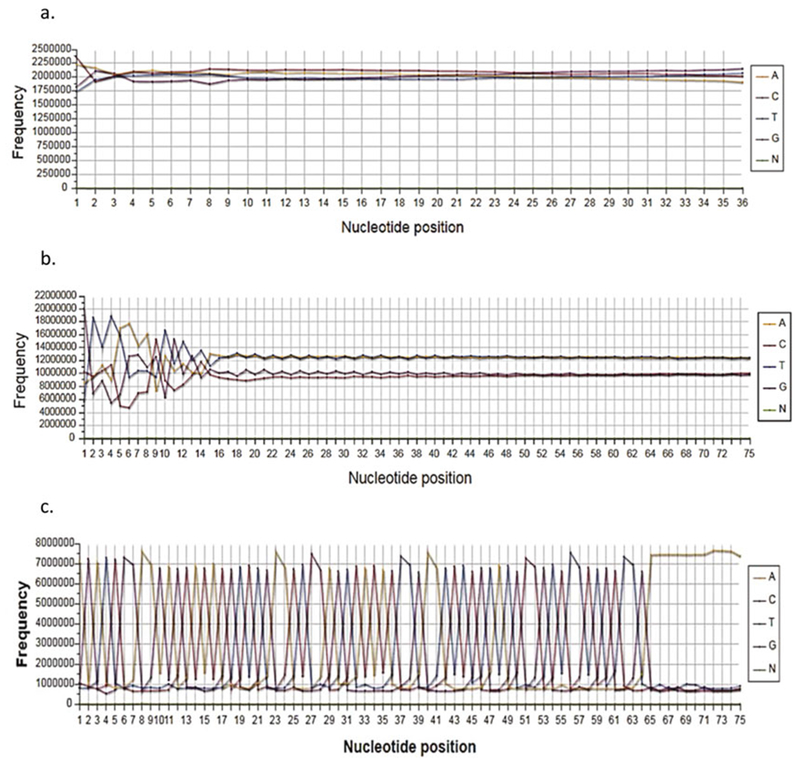
Base frequency plot (a) Absence of AT bias in H3K4me3 ChIP-Seq reads suggests enrichment of H3K4me3 in genic areas. For RNA-Seq such plot would indicate a good library without gDNA contamination. (b) For RNA-Seq AT bias indicates DNA contamination. (c) The spiky plots indicate adapter contamination during library construction
Fig. 3.
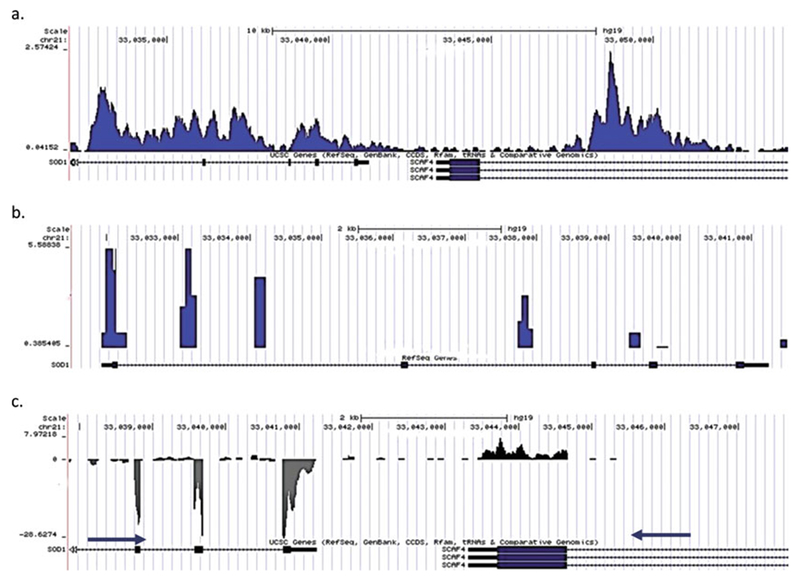
(a) Genome browser tab showing H3K4me3 peaks at the ACTB promoter. (b) The presence of tall square peaks in the genome browser before removing duplicates indicates over amplification. (c) Customized display for showing strand-specific RNA-Seq tag density
Fig. 4.
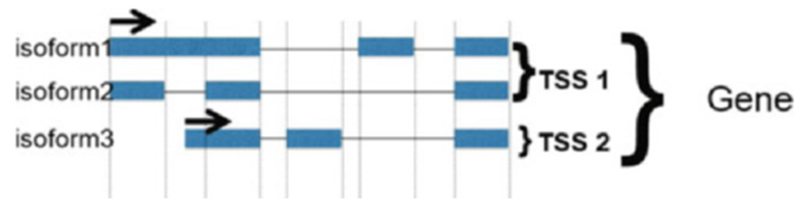
RPKMS can be grouped by isoforms, a common Transcription Start Site (TSS) or genes
Fig. 5.
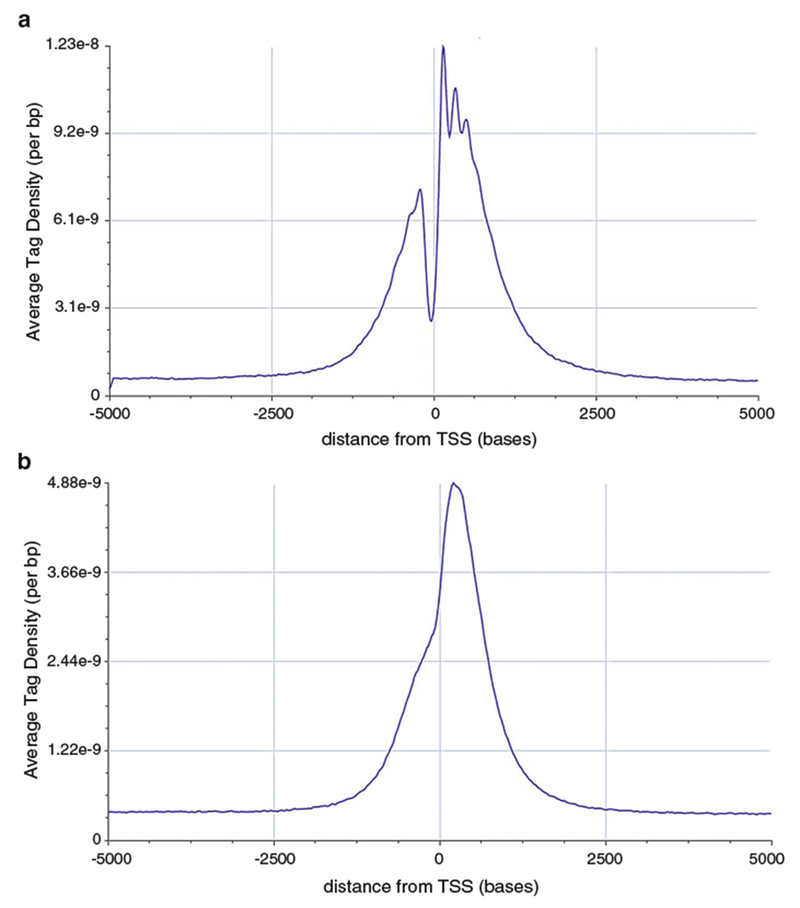
Average tag density profiles (a) and (b) Show the enrichment of H3K4me3 around the Transcription Start Site. Note that the signal-to-noise ratio is different in (a) and (b), suggesting that experiment (a) worked much better. Direct peak height comparison between the peaks on the browser may be ill-advised. Also note that the resolution in (a) is better due to better fragmentation
Fig. 6.
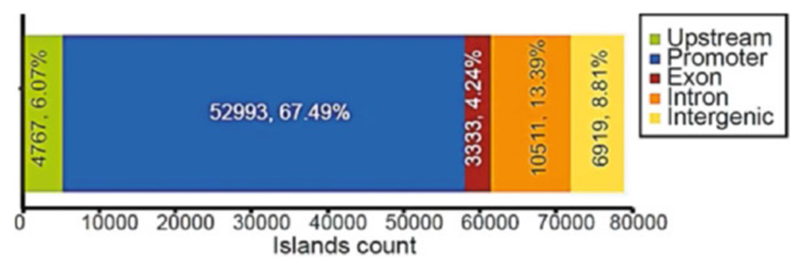
Islands distribution shows the distribution of H3K4me3 around different genomic regions. From this figure, we can conclude that H3K4me3 is enriched around the promoter regions
3.2. Advanced Analysis
If the quality of experimental data is satisfactory, the users can proceed to further analysis. BioWardrobe’s advanced analysis options allow the user to integrate data from several experiments to perform differential expression or binding analysis and create average tag density profiles and heatmaps.
To start the analysis, go to the “Data & Analysis” menu and click on “Analyze” → “Project designer”.
A new project can be created by typing the project name in the field on the top left.
After entering the name of the new project and hitting enter, the project file will be created. Upon clicking it, the various analysis options will become available. This includes “Genes Lists,” “R language processing,” “ATDP & Heatmaps,” “DESeq” [18, 19], and “MAnorm” [20] (see below).
3.2.1. Gene Lists
The “Gene Lists” function allows the user to create and manage lists of genes for future analysis. Experimental raw data can be added to the project by dragging the library from the middle pane. Here gene lists can be created by filtering RNA-Seq data on the basis of gene expression level in one or several experiments.
3.2.2. DESeq
The “DESeq” function (see Note 12) can be used to perform the differential gene expression analysis. In order to define replicates, a user can create experiment groups and drag the replicate experiments into these groups. After the conditions are defined, DESeq analysis can be performed.
Click on “DESeq.”
Select the conditions (groups of replicates) to use and populate them by dragging libraries from the RNA-Seq data tab.
To initiate the analysis click the “+” icon next to one of the groups. Enter a name for the analysis and specify the conditions to be compared in the “DESeq input” field. Series type is used when more than two conditions are compared. Assuming that there are three conditions (1, 2, and 3), selecting “Pairwise series” will perform all pairwise comparisons (1–2, 1–3, 2–3), whereas “Time series” will perform 1–2 and 2–3 and “Kinetics series” will perform 1–2 and 1–3 comparisons. “Annotation grouping” specifies the annotation to be used for comparison—(isoforms/genes/common TSS—see Note 8).
After all the conditions have been set up, click “Run.”
Once analysis is complete the results of DESeq will appear and can be saved as an excel file by clicking on “Save.”
The results can be filtered and gene sets can be created by choosing the options in the “Filter” section. Filtering can be done on the basis of raw/adjusted p-value, RPKMs or chromosome using logical operators (AND/OR).
3.2.3. ATDP and Heatmaps
The “ATDP & Heatmaps” function serves to produce the average tag density profiles and heat maps to compare the chromatin environment at different gene sets. The gene sets created while doing the DESeq analysis can be viewed in this window. ChIP-Seq data can be added here by dragging and dropping the data.
Average tag density profiles are used to analyze the differential enrichment of certain modifications around TSS or gene bodies. To create one, click the graphs icon next to one of libraries.
In the “ATDP input,” the plots can be added along with the names that will be displayed in the plot legend.
Click “Run.” The plots will be generated within a few minutes. The three tabs will show “Average Tag Density” around TSS, “Gene Body Average Tag Density” and “Tag Density Heatmaps.” Plots can be saved in .svg format using the save button.
In the “Average Tag Density” tab the difference in the modification level between the gene sets can be further analyzed using the Mann–Whitney–Wilcoxon (MWW) test. To perform the statistical analysis, highlight the area around TSS (or within gene body) that will be compared. After confirming the coordinates, BioWardrobe will produce the box-plot and the matrix of MWW p-values indicating whether the tag densities are significantly different between the gene sets.
The “Tag Density Heatmaps” tab will show tag density for individual genes within the same groups. These can be used to correlate gene expression data with data for several modifications in several conditions.
In the “Tag Density Heatmaps” section, the color scale and order of the genes can be changed by using the buttons above the graphs.
3.2.4. MAnorm
“MAnorm” (see Note 13) can be used to obtain differential ChIP-Seq enrichment (different levels of binding between the data sets compared). Data can be added by dragging and dropping.
To perform analysis by MAnorm, enter the name that will be assigned to the result of the analysis.
In the “MAnorm Input” section, add the data sets that are going to be compared.
Click on “Run.” The analysis can take up to an hour.
The results can be saved using the “Save” option. They can be filtered using the p values and rescaled M (log fold change) value.
3.3. Customizable Analysis with R
BioWardrobe allows advanced users to add customizable analysis using precalculated values from the BioWardrobe database. R scripts can be used in both basic and advanced analysis. Additionally, an R library that allows users to access the data stored in BioWardrobe from the R environment is provided.
3.3.1. Basic Analysis with R
- After the basic analysis of an experiment is completed, the R analysis tab becomes visible. Switching to the R tab triggers the scripts to run. BioWardrobe checks the time stamps of the last script edit and last run and reinitiates a run if the script was edited after the completion of the last run. There are two scripts available, default and custom. Default scripts will be run on every experiment in the database.
-
(a)Open an experiment after the analysis is finished.
-
(b)Switch to the R tab. If it is the first time that the R tab is accessed after analysis, the system will run the scripts. If not, it will show the results.
-
(a)
- To edit default scripts, there is a “source” subtab on the R tab. Select a script to edit, “Default” or “Custom.”
-
(a)When script editing is finished, press the “Apply” button at the top left. It saves the script and updates the time stamp.
-
(b)Close the experiment window and open it again.
-
(c)Switch to the R tab and BioWardrobe will run the new script.
-
(a)
3.3.2. Advanced Analysis with R
- Select the “R language processing” in “Advanced Analysis.” This option can be used to run preconfigured R scripts that use data from more than a single experiment in BioWardrobe. “The R language processing” panel has a tree view with two main folders, “Raw Data” and “R Results.” The “Raw Data” folder is the list of experiments used in the current project.
-
(a)To perform analysis, click the “R” icon and in the window that appears, type the name that will be assigned to the result.
-
(b)The “Predefined R script” then has to be selected (the current version of BioWardrobe has “IDR” and “PCA” analysis).
-
(c)In the “R arguments” section, experiments to be analyzed can be selected.
-
(d)Run the experiment by selecting the “Run” button.
-
(a)
3.3.3. BioWardrobe R Library
Advanced users can access BioWardrobe’s precalculated data in R by installing and using a BioWardrobe R library on the same system that has BioWardrobe installed.
- To install BioWardrobe R library:
-
(a)Clone BioWardrobe repository from GitHub by “git clone https://github.com/CCH-MC/biowardrobe”.
-
(b)Go into scripts/R folder by “cd scripts/R”.
-
(C)Run “R CMD INSTALL wardrobe.”
-
(a)
- To use BioWardrobe R library:
-
(a)Run R.
-
(b)To load the library in R, type “library(wardrobe)”.
-
(c)To access the data set from an experiment with an ID (1000 for instance), type “experiment<-wardrobe (id=1000)”.
-
(d)Now the experiment variable contains all the available information:
- experiment$uid: experiment internal UID—string.
- experiment$isRNA: numeric 1/0, if 1 it is an RNA-Seq experiment.
- RNA-Seq specific
- experiment$dataseti: ’data.frame’—table with RPKM values counted against isoforms.
- experiment$datasetg: ’data.frame’—table with RPKM values counted against genes.
- experiment$datasetc: ’data.frame’—table with RPKM values counted against common TSS.
- DNA-Seq specific
- experiment$dataset: ’data.frame’—MACS output table.
- experiment$fragmentsize: numeric—ChIP-seq library fragment size that was used in the calculations.
- experiment$isPair: 1/0—1 if it is a pair-end experiment.
- experiment$isDUTP: 1/0—1 if it is a dUTP experiment.
- experiment$tagsmapped: integer—the number of tags mapped to the reference genome.
- experiment$db: string—UCSC database name: e.g., mm10/hg19/rn5/xenTro3.
- experiment$annotation: string—UCSC annotation table; for instance, using “refGene,” one can select data from experiment$db.experiment$annotation to view an annotation for the reference genome.
- experiment$alias: string—a short experiment name.
- experiment$bamfile: string—the full path to the bam file.
- experiment$fastgz: string the full path to the fastq bzipped file.
-
(a)
4 Notes
Removal of duplicates: Many bioinformaticians prefer to remove duplicate reads under the assumption that those are PCR duplicates. BioWardrobe provides the ability to remove duplicates by checking “Remove duplicates” in the “Advanced” tab. We recommend to first perform the analysis without removing duplicates and to check the results on the browser. The presence of tall, square peaks will indicate over-amplification of the PCR library (Fig. 3b). Please note that MACS2, which is used to call peaks, removes excess duplicates automatically and thus peak calling will be affected only slightly.
Mapping statistics (ChIP-seq): For ChIP-Seq, mapping is performed with BowTie and allows up to one mismatch. BioWardrobe shows the percentage of reads that are mapped to a unique location, mapped to multiple locations or are unmapped (Fig. 1a, b). Only uniquely mapped reads are used for downstream analysis. Results of mapping depend on several biological and technical variables including quality of sequencing, read length, contamination and biological features of the target protein. For a typical ChIP-Seq experiment, 70–80% of reads are uniquely mapped. An increased fraction of unmapped reads may be caused by a bad sequencing run (also see base quality plot—Note 5), contamination with adapter dimers (base frequency plot will be spiky—Fig. 2c), very short fragments such that the second adapter is included in the read (trimming on 3′ end before alignment may help) or contamination with the DNA/library from another organism. An increased fraction of multi-mapped reads may indicate that the protein of interest (e.g., H3K9me3) is recruited to repetitive areas of the genome.
Mapping statistics (RNA-seq): For RNA-Seq, mapping is performed with RNA-STAR, which is provided with transcriptome annotation. BioWardrobe shows the percentage of reads that are mapped to a unique location within annotation or outside of annotation, mapped to multiple locations or are unmapped. For stranded (dUTP) RNA-Seq, only reads mapped to the correct strand are considered to be within annotation. Only reads that are mapped to annotations are used in RPKM calculations, but all uniquely mapped reads are displayed on the browser. Typically for unstranded polyA RNA-Seq, ~15% of reads are uniquely mapped outside of annotation (Fig. 1c, d). A higher percentage of reads mapped outside of the transcriptome may indicate contamination with genomic DNA (Fig. 1d). In this case, DNase treatment needs to be added to the RNA extraction protocol. For total transcriptome/RiboZero experiments, the percentage of the reads mapped outside of annotation is much higher due to the presence of unspliced transcripts.
rRNA contamination: Given that 85–95% of total RNA is rRNA, researchers use means such as oligodT hybridization or RiboZero/Rybominus hybridization to enrich for mRNAs. Ribosomal RNA contamination shows the percentage of the reads that can be mapped to rDNA repeats and shows whether an mRNA isolation step worked successfully. For human samples constructed using the oligo-dT approach, rRNA contamination is typically <2%; for a RiboZero-like approach, the rRNA contamination is ~4–10% (Fig. 1e).
Base frequency plots are very simple yet useful for troubleshooting an experiment. The human genome is normally AT-rich, but genes are not. Thus, for ChIP-Seq, the absence of an AT bias in these base frequency plots suggests enrichment in the vicinity of genic areas (Fig. 2a). Conversely, being derived from genes, RNA-Seq reads typically do not have an AT bias; thus, having an AT bias may indicate DNA contamination during library construction (Fig. 2b). DNaseI treatment may be used during RNA purification to get rid of genomic DNA. The spiky plot in Fig. 2c is characteristic of adapter contamination in the library and suggests that the adapter/insert ratio during ligation needs to be decreased. This problem will not affect results, but the experiment will require more sequencing since a large fraction of the reads will be unproductively used on adapter dimers.
Base quality box plots show the Phred scores at each base as reported by the sequencer in the fastq file. The sequencing quality typically decreases toward the end of the run and may result in reduced mapping. If needed, the reads can be trimmed on either end in the “Advanced” tab of the Experiment form. Typically, Illumina sequencers report scores in the 30–40 range. Phred quality scores represent the probability of error in matching the bases during sequencing. Higher scores represent a greater chance that the base call is correct.
Genome browser: For ChIP-Seq, we display results on the browser as coverage by fragments normalized to the number of millions of reads mapped (Fig. 3a). For single-read sequencing, the fragments are estimated by extending reads in the 3′ direction to average fragment length (determined by MACS2). For paired-end experiments, actual fragment lengths are used. For both single and paired read RNA-Seq, coverage by actual reads is shown. In the case of stranded RNA-Seq, we employ a custom modification of the UCSC browser that displays strand-specific coverage (Fig. 3c).
RPKMs are estimated for transcripts using a custom BioWardrobe algorithm. The results can be summed up for transcripts using a common TSS, which is useful for the study of transcriptional regulation, or for genes, which is useful for functional analysis, such as Gene Ontology (Fig. 4). Raw read numbers can also be viewed by adding the read number column to the RPKM table.
Island list: Peak calling is performed by MACS2 software using either a narrow or broad peak function. The user can select whether narrow or broad peak calling is used for each antibody in the antibody catalog. MACS2 can also be instructed to use input control for peak calling by designating an input sample as a control and then selecting it in the “Experiment form” tab. The island table lists peak coordinates, nearest gene, peak location relative to it and p/q-values for each peak. Sequences under the peaks can be obtained by clicking the “fasta” button located above the table. These sequences can be used to identify overrepresented motifs using tools such as MEME-ChIP [21] or PScan [22].
Average tag density profiles can be used to assess the enrichment obtained in the experiment. This is particularly useful for TSS-proximal modifications such as H3K4me or ATAC-seq. The ratio of the signal at the TSS to the signal away from the TSS is a good indication of quality immunoprecipitation (Fig. 5).
Peak distribution plots show the distribution of peaks between genomic areas (i.e., promoter, upstream, etc.). The definition (radius around TSS) of the promoter can be adjusted in the island tab.
DEseq2: To identify differentially expressed genes, BioWardrobe uses DESeq2. The comparisons can be set up for genes, common TSS or isoforms. DESeq2 performs a pairwise comparison using the raw read numbers for each expression unit. For convenience, BioWardrobe provides average RPKM values for each condition. Since the normalization used in DESeq2 is different from RPKM, the fold change reported by DESeq2 will not necessarily match the fold change between average RPKMs. We recommend filtering analysis results on the basis of p-adj and log fold change (LOGR).
MAnorm is used to perform differential enrichment analysis. The analysis can take up to an hour. In addition to displaying the islands, the table shows the neighboring genes and where the island is located relative to these genes. We recommend filtering the list on the basis of both the p-value and rescaled M (log2 fold change). Unlike some other programs, MAnorm adjusts for differential enrichment between experiments by assuming that the true intensities of most common peaks are the same between two ChIP-Seq samples. We consider this to be a very important feature that overrides some of MAnorm’s deficiencies: replicates cannot be used for MAnorm, and MAnorm analysis is not commutative (see Wiki at biowardrobe.com for further discussion).
References
- 1.Barski A, Cuddapah S, Cui K et al. (2007) High-resolution profiling of histone methylations in the human genome. Cell 129:823–837. 10.1016/j.cell.2007.05.009 [DOI] [PubMed] [Google Scholar]
- 2.Mikkelsen TS, Ku M, Jaffe DB et al. (2007) Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448:553–560. 10.1038/nature06008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robertson G, Hirst M, Bainbridge M et al. (2007) Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Methods 4:651–657. 10.1038/nmeth1068 [DOI] [PubMed] [Google Scholar]
- 4.Mortazavi A, Williams BA, McCue K et al. (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5:621–628. 10.1038/nmeth.1226.nmeth.1226 [pii] [DOI] [PubMed] [Google Scholar]
- 5.Boyle AP, Davis S, Shulha HP et al. (2008) High-resolution mapping and characterization of open chromatin across the genome. Cell 132:311–322. 10.1016/j.cell.2007.12.014.S0092-8674(07)01613-3 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buenrostro JD, Giresi PG, Zaba LC et al. (2013) Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10:1213–1218. 10.1038/nmeth.2688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heinz S, Benner C, Spann N et al. (2010) Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38:576–589. 10.1016/j.molcel.2010.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10:R25 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Trapnell C, Roberts A, Goff L et al. (2012) Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7:562–578. 10.1038/nprot.2012.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trapnell C, Pachter L, Salzberg SL (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25:1105–1111. 10.1093/bioinformatics/btp120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trapnell C, Hendrickson DG, Sauvageau M et al. (2013) Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol 31:46–53. 10.1038/nbt.2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Strand NGS: http://www.strand-ngs.com
- 13.Partek: http://www.partek.com/
- 14.Illumina Basespace. https://basespace.illumina.com.
- 15.Goecks J, Nekrutenko A, Taylor J, Galaxy Team (2010) Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol 11:R86 10.1186/gb-2010-11-8-r86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kartashov AV, Barski A (2015) BioWardrobe: an integrated platform for analysis of epigenomics and transcriptomics data. Genome Biol 16:158 10.1186/s13059-015-0720-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y, Liu T, Meyer CA et al. (2008) Model-based analysis of ChIP-Seq (MACS). Genome Biol 9:R137 10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome Biol 11:R106 10.1186/gb-2010-11-10-r106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shao Z, Zhang Y, Yuan G-C et al. (2012) MAnorm: a robust model for quantitative comparison of ChIP-Seq data sets. Genome Biol 13: R16 10.1186/gb-2012-13-3-r16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Machanick P, Bailey TL (2011) MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics 27:1696–1697. 10.1093/bioinformatics/btr189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zambelli F, Pesole G, Pavesi G (2009) Pscan: finding over-represented transcription factor binding site motifs in sequences from co-regulated or co-expressed genes. Nucleic Acids Res 37:W247–W252. 10.1093/nar/gkp464 [DOI] [PMC free article] [PubMed] [Google Scholar]