

Summary
Little is known about the in vivo impacts of targeted therapy on melanoma cell abundance and protein expression. Here, 21 antibodies were added to an established melanoma mass cytometry panel to measure 32 cellular features, distinguish malignant cells, and characterize dabrafenib and trametinib responses in BRAFV600mut melanoma. Tumor cells were biopsied before neoadjuvant therapy and compared to cells surgically resected from the same site after 4 weeks of therapy. Approximately 50,000 cells per tumor were characterized by mass cytometry and computational tools t-SNE/viSNE, FlowSOM, and MEM. The resulting single cell view of melanoma treatment response revealed initially heterogeneous melanoma tumors were consistently cleared of Nestin expressing melanoma cells. Melanoma cells subsets that persisted to week 4 were heterogeneous but expressed SOX2 or SOX10 proteins and specifically lacked surface expression of MHC I proteins by MEM analysis. Traditional histology imaging of tissue microarrays from the same tumors confirmed mass cytometry results, including persistence of NES-SOX10+ S100β+ melanoma cells. This quantitative single cell view of melanoma treatment response revealed protein features of malignant cells that are not eliminated by targeted therapy.
Keywords: Melanoma, mass cytometry, targeted therapy, kinase inhibitors, single cell
Introduction
The incidence of melanoma continues to rise at a faster rate than all other solid tumors (Erdmann et al., 2013). Among metastatic melanoma patients, the majority have a BRAFV600E mutation that induces oncogenic BRAF kinase activity and leads to enhanced survival and proliferation of melanoma cells (Hodis et al., 2012; Holmes, 2014). Despite the promising initial response of patients treated with inhibitors that target the oncogenic BRAFV600E mutation, most patients' tumors eventually develop resistance to these treatments (Van Allen et al., 2014; Wagle et al., 2011). Furthermore, a combined immunotherapy that targets PD-1 and CTLA-4 improves outcome, but only a fraction of patients obtain clinical benefit and this treatment has important side effects (Hamid et al., 2013; Hodi et al., 2010; Johnson et al., 2016a; Phan et al., 2003). Given the high rate of mutation and a broad range of genomic alterations observed after therapy in melanoma, cell diversity or intra-tumor cellular heterogeneity has also been implicated as a cellular mechanism of therapy escape (Johnson et al., 2017; Van Allen et al., 2014).
Study of melanoma and other solid tumors has increasingly moved towards approaches that monitor the collection of cell types within tumors including cancer cells, immune cells, fibroblasts, and other stromal cells (Irish, 2014). Studying the heterogeneous cells within patients' tumors could identify malignant or immunologic cell types that may predict treatment response or resistance (Johnson et al., 2016b). Only recently have studies begun to perform single cell analysis on matched tumors from patients before and after therapy (Hugo et al., 2015; Tirosh et al., 2016). These single cell studies have focused on RNA expression and use measurements of selected proteins as a confirmation tool. Pairing longitudinal studies with single cell analysis of proteins involved in melanoma cell identity and function could lead to a better understanding of the evolution of resistance and therapy evasion (Irish, 2014; Meacham and Morrison, 2013).
Technological advances have led to the development of several platforms made to dissect cell diversity. Single-cell genomic approaches enable a detailed evaluation of genomic and transcriptional features of cancer cells (Patel et al., 2014; Tirosh et al., 2016). While studies that utilize transcriptional profiling have identified resistant cells in relapsed tumors, these results don't align to identify a common resistant cell phenotype (Hugo et al., 2015; Tirosh et al., 2016). Furthermore, discrepancies between mRNA and protein expression indicate not all transcripts are regulated in a way that leads to detectable levels of protein (Koussounadis et al., 2015; Zhang et al., 2014). Mass cytometry uses metal labeled antibodies detected using time-of-flight mass spectrometry, permitting detection of more than 30 proteins per cell (Bendall et al., 2011; Bjornson et al., 2013). The use of metal labeled, rather than fluorophore-labeled, antibody tags significantly reduces issues with spectral overlap and cell autofluorescence (Leelatian et al., 2017a; Nicholas et al., 2016). Due to these advantages, mass cytometry has gained acceptance for the study of solid tissues (Wang et al., 2016; Wogsland et al., 2017). More recently, standardized methods have been developed to create viable single cell suspensions from solid tumors and tissues (Leelatian et al., 2017b).
This study introduces the application of mass cytometry to study the cell diversity of human melanoma tumors by measuring 32 proteins simultaneously before and during ongoing targeted therapy from the same tumor sites. This work aims to characterize and track changes to cancer cell phenotype that appear during combination BRAFV600E and MEK inhibition. By simultaneously measuring several proteins in tens of thousands of cells, mass cytometry could reveal novel features defining subsets that may be used for future therapeutic development (Irish, 2014; Spitzer and Nolan). Ideally, by revealing new cell types and their signature features, a single cell systems biology approach might both provide ways to track heterogeneous melanoma cell subsets and discover new hypotheses for cellular mechanisms of resistance.
Materials & Methods
Cell Culture and Cell Lines
MeWo, A2058, WM115, and SKMEL28 cells were grown in Minimum Essential Medium (Mediatech, Inc., Manassas, VA), supplemented with 10% fetal bovine serum (Gibco standard FBS, Life Technologies, Grand Island, NY), 1% penicillin (Gibco), and 1% streptomycin (Gibco). Jurkat T cells were grown in RPMI (Mediatech, Inc., Manassas, VA) supplemented with 10% fetal bovine serum (Gibco standard FBS, Life Technologies, Grand Island, NY), 1% penicillin (Gibco), and 1% streptomycin (Gibco). All cell lines were acquired as gifts from the laboratory of Dr. Vito Quaranta.
Tumor Collection and Dissociation
Lymph nodes and subcutaneous tumors from adults with metastatic melanoma were biopsied or surgically resected from 15 individual patients accordance with the Declaration of Helsinki. Institutional ethics approval was obtained from the Vanderbilt Institutional Review Board (project numbers 121165 and 030220). All patients had provided written informed consent prior to inclusion in the study. All patients in this study presented with either several subcutaneous legions or distal metastasis. All tumors within the study were thought to arise as cutaneous legions because no evidence of disease was present in mucosal epithelium or retinas. 11 patients received two weeks of BRAFV600E inhibitor dabrafenib followed by two weeks of dabrafenib and MEK inhibitor trametinib (Supplementary Table S1 and (Johnson et al., 2015)). Pretreatment (Pre-Tx) tumors were naïve to BRAFV600E inhibitor dabrafenib MEK inhibitor trametinib. More details of patients from this clinical study can be found with clinical trial code NCT01701037. Core biopsies were obtained before therapy and remaining tumors were surgically resected after four weeks of therapy. Tumor samples were enzymatically digested into a single-cell suspension and cryopreserved with techniques developed specifically to isolate viable melanoma tumor cells (Leelatian et al., 2017a; Leelatian et al., 2017b). Finally, before and after cryopreservation all samples were inspected with a hemocytometer and trypan blue staining (Thermo Fisher Scientific, Waltham, MA).
Tissue Microarrays and Immunohistochemistry
Melanoma tumors from the study were prepared and processed into TMAs by the VUMC Translational Pathology Shared Resource (TPSR) (Supplementary Table S1). Immunohistochemistry of serial sections (<10 μm) from TMAs was performed by the VUMC TPSR. Digital images were obtained with an Ariol SL-50 automated scanning microscope and the Leica SCN400 Slide Scanner from VUMC Digital Histology Shared Resource.
Fluorescence Flow Cytometry
Lives cells from patient 001 (MP-001) and Jurkat T cells were stained with fluorescent antibodies to analyze signaling status. Before stimulating cells for signaling, Alexa fluorophore 700 (Thermo Fisher Scientific, Waltham, MA) was added, as previously described (Irish and Doxie, 2014; Leelatian et al., 2017a). Alexa fluorophores can be used to test membrane permeability as a way to exclude dead and dying cells (Irish and Doxie, 2014; Krutzik and Nolan, 2006). Unstimulated cells and stimulated cells were allowed to rest in an incubator for 30 minutes in RPMI (Mediatech, Inc., Manassas, VA), supplemented with 10% fetal bovine serum (Gibco standard FBS, Life Technologies, Grand Island, NY). Stimulated cells were treated with 3.3 mM hydrogen peroxide for 4 minutes. After 34 minutes cells were fixed with 1.6% paraformaldehyde (Electron Microscopy Services, Fort Washington, PA) for 10 min at room temperature, washed with PBS (HyClone Laboratories, Logan, UT), pelleted at 800 × g, and permeabilized with 100% ice-cold methanol (Fisher Scientific, Waltham, MA) at −20°C overnight following established protocols (Irish et al., 2010; Krutzik and Nolan, 2003; Leelatian et al., 2015). Cells were washed twice with cell staining media composed of PBS and 1% BSA (Fisher Scientific, Waltham, MA) and pelleted at 800 × g. Each sample was stained with 100 μl staining media for 15 minutes at room temperature. MP-001 was stained with phospho-specific antibodies p-SRC, p-ERK, and p-AKT for 15 minutes (BD Biosciences, San Jose, CA and Cell Signaling Technology, Danvers, MA). Jurkat T cells were stained with p-SRC and p-ERK for 15 minutes (BD Biosciences, San Jose, CA). After staining, cells were washed twice with PBS, pelleted at 800 × g, and resuspended in PBS for analysis on a 5-laser LSRII (BD Biosciences, San Jose, CA) at the Vanderbilt Flow Cytometry Shared Resource.
Mass Cytometry
Live cells from tumors obtained from the same dissociation conditions as fluorescence flow cytometry analysis were stained for cell surface markers, fixed, permeabilized, and washed in concordance with established dissociation and mass cytometry protocols (Leelatian et al., 2015; Leelatian et al., 2017b). Rhodium intercalator was utilized as a membrane permeability reagent to computationally remove dead cells from the data (Ornatsky et al., 2008) (Fluidigm). Permeabilization with 0.02% Saponin (Millipore, Darmstadt, Germany) in PBS was also included before methanol permeabilization as part of an optimized multi-step protocol to detect SOX2 and MITF. Saponin staining was performed for 30 minutes with anti-SOX2 and MITF antibodies. Cell staining after methanol permeabilization was performed for 15 minutes at room temperature with iridium intercalator and metal-tagged antibodies (Supplemental Table S2). After staining, cells were washed once with PBS, once with deionized water, pelleted at 800 × g, resuspended in deionized water, and collected using a CyTOF 1 (Fluidigm) mass cytometer.
Data Analysis
Cytobank was used to store.fcs files and perform data analysis including viSNE gating of major tumor cell populations (Amir el et al., 2013; Kotecha et al., 2010). Statistical analysis of cells gated from samples before and after therapy was performed using commercial 2D graphing and statistics software GraphPad Prism. Tracking the percent abundance of cells from FlowSOM analysis was performed in STATA 14.2. MEM and FlowSOM analysis were performed in program language R. (Diggins et al., 2018; Diggins et al., 2017; Van Gassen et al., 2015b) (https://mem.vueinnovations.com).
Single Cell Analysis of Human Melanoma Tumors
Viable nucleated cells were gated with total histone H3 and rhodium intercalator for 7 sets of matched tumors (MP-029, MP-031, MP-032, MP-034, MP-055, MP-059, and MP-062) using established methods (Leelatian et al., 2017a; Leelatian et al., 2017b). The patient-specific viSNE analyses were performed with 28 of 32 markers to avoid channels potentially affected by gadolinium MRI contrast agent that the patients received before surgery (Supplementary Table S2). Major populations of cells were identified and quantified using expert gating performed on patient-specific viSNE maps of the pre- post-therapy tumors. (Supplementary Table S3).
To study how cellular heterogeneity was affected by therapy, cells were placed into a workflow that emphasized unsupervised subset identification, characterization, and data visualization with FlowSOM, MEM and viSNE analysis (Diggins et al., 2015; Diggins et al., 2018; Diggins et al., 2017; Van Gassen et al., 2015a). To identify subsets enriched before and after therapy, the viSNE analysis was performed using 17 markers with a variance greater than 0.2 (Supplementary Table S2). FlowSOM cluster analysis was conducted using t-SNE axes as inputs for clustering.
Additional patient's melanoma cells included within Figure 4 were gated as CD45 negative. Cells from these patients were analyzed using shared markers from the optimized melanoma mass cytometry panel (MP-004, MP-012, MP-019, MP-022, MP-023, MP-040, and MP-054) (Supplementary Table S2).
Fig. 4. Significant loss of Nestin protein expression was observed in melanoma cells at Week 4 following dabrafenib and trametinib.

(A) Box and whisker plots of mass cytometry data show the median intensity of 8 proteins for melanoma cell lines (A2058, MeWo, WM155, and SKMEL28), Pre-Tx therapy naïve melanoma tumors, and post-therapy Week 4 melanoma tumors. Melanoma tumor cells were gated as CD45lo/-.
Statistics
Non-parametric Kolmogorov-Smirnov tests with a significance threshold of p=0.05 were used to compare differences in median mass intensity in Figure 4. Wilcoxon signed rank tests with a significance threshold of p=0.05 were used to compare differences in cellular abundance of cell subsets from matched patient samples in Supplementary Fig. S2 and S3. Statistical tests were performed using commercial 2D graphing and statistics software GraphPad Prism.
Data Availability
Mass cytometry data for this manuscript can accessed via FlowRepository (https://flowrepository.org/).
Results
32-antibody mass cytometry panel and unsupervised computational analysis characterize protein expression in single cells from human melanoma tumors
Human melanoma tumor core biopsies and surgically resected human tumors were dissociated into single cell suspensions (Supplementary Table S1) (Leelatian et al., 2017a). Melanoma tissue samples comprising 14 sample pairs from 7 patients were placed into an analysis workflow to stain cell suspensions with an updated panel that contained 21 additional markers (Supplementary Table S2) (Leelatian et al., 2017a). Mass cytometry data generated from a pre-therapy (Pre-Tx) biopsy of tumor cells from melanoma patient (MP) number 59 (MP-059) and the general data analysis workflow are shown in Figure 1. Pre-Tx samples were from tumors naïve to BRAFV600E inhibitor dabrafenib and MEK inhibitor trametinib and were obtained just prior to the initiation of therapy, which culminated in surgical resection after four weeks (see Methods). Tumor heterogeneity was visualized using the Cytobank implementation of viSNE/t-SNE (Amir el et al., 2013), which organized cells in a two dimensional graph so that cells with similar protein expression patterns were placed near each other to form islands of phenotypically similar cells (Fig. 1A). Next, marker enrichment modeling (MEM, (Diggins et al., 2018; Diggins et al., 2017) objectively described the enriched protein features of the observed tumor cell types (Fig. 1B). The scale for MEM starts at +10 (most enriched), continues through 0 (no enrichment), and ends at -10 (most excluded). For example, one MEM label was ▲ CD45+3 CD49F+3 CD19+2 MHCI+1 ▼ SOX10-4 S100β-4 MCAM-2 CD44-2 BCAT-1 α-SMA-1 (Fig. 1B, MP-059 Pre-Tx tumor cell population #7), which indicted these cells were relatively enriched for B lineage CD19 and leukocyte CD45 proteins and specifically lacking melanoma cell proteins, including S100β, SOX10, and MCAM (Fig. 1B). This computational workflow using viSNE and MEM reduces bias that can arise in manual gating and characterizes phenotypically unusual cells that may be overlooked or hidden in traditional analyses (Diggins et al., 2015; Diggins et al., 2017; Irish, 2014).
Figure 1. A 32-antibody mass cytometry panel and unsupervised computational analysis characterize protein expression in single cells from human melanoma tumors.
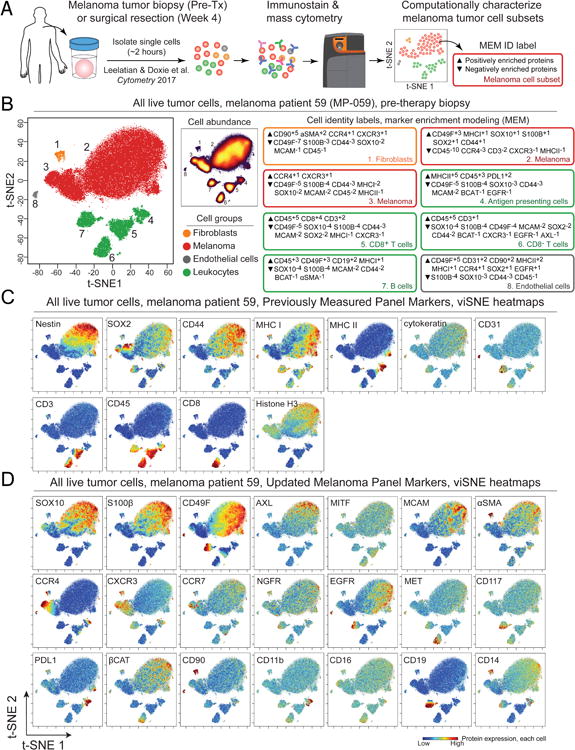
(A) An overview of the analysis approach is shown. Melanoma tumor samples from patients enrolled in Phase II interventional study NCT01701037 were obtained as a biopsy prior to neoadjuvant therapy (Pre-Tx) using dabrafenib and trametinib therapy or from surgical resection after 4 weeks of therapy (Week 4). Viable single cells were isolated following established protocols (Leelatian et al., 2016; Leelatian et al., 2017b), analyzed by mass cytometry, and characterized using a modular, unsupervised computational workflow created for melanoma from dimensionality reduction tool t-SNE/viSNE (Amir el et al., 2013), clustering tool FlowSOM (Van Gassen et al., 2015a), and cell subset identification and protein enrichment characterization with MEM (Diggins et al., 2017). In (B), viSNE plots and MEM labels used to identify and characterize cells from melanoma tumors are shown. viSNE plots display all live cells from one Pre-Tx melanoma tumor (MP-059) arranged according to protein expression and shaded based on cell type (B, left) or abundance (B, middle, density plot). MEM labels quantified protein enrichment (▲ up to +10) or specific absence (▼ down to -10) in the indicated tumor cell subset. For example, melanoma cells in subset 2 had the label ▲CD49F+3 MHCI+1 SOX10+1 S100β+1 SOX2+1 CD44+1 ▼CD45-10 CCR4-3 CD3-2 CXCR3-1 MHC II-1, which identified this population as melanoma cells based on enrichment of SOX2, S100β, and SOX10 proteins and a lack of MHC II protein expression. In (C) and (D), viSNE plots show the same cells from (B) but display per-cell expression of the indicated protein on a rainbow scale where red indicates high expression and blue indicates low expression. Antibodies for protein targets listed in (C) comprised a validated backbone melanoma cell identity panel (Leelatian et al., 2016). (D) An additional 21 proteins were added to the melanoma mass cytometry panel here to characterize features of melanoma cell subsets.
From the protein expression and cellular abundance data in the viSNE and MEM analysis, cells were classified into one of the five major groups (Supplementary Table S3). The most abundant stromal populations were defined as follows: 1) leukocytes (CD45hi and MHC class I), 2) fibroblasts (Thy-1/CD90, α-SMA), 3) endothelial cells (PECAM/CD31 and MHC class I) and 4) epithelial cells (cytokeratin and MHC class I) (Fig. 1B, 1C, and 1D). The panel was also developed to detect epithelial cells, however, epithelial cells were not detected within the tumors of this study (Supplementary Table S3). Cells negative for markers of stromal cell populations were identified as melanoma cells. Melanoma cells varied in phenotype, however, these cells reliably displayed features consistent with melanoma cell identity including loss of MHC class I or expression of Nestin, SOX10, SOX2, MCAM, or S100β (Fig. 1B, 1C, and 1D).
In addition to the melanoma and tumor cell identity markers in the mass cytometry panel used here (Figure 1), panel design experiments also included analysis of phospho-proteins that had been previously validated for fluorescence and mass cytometry (Irish et al., 2004; Irish et al., 2010; Krutzik and Nolan, 2003). For example, phospho-flow analysis of viable post-treatment melanoma tumor cells obtained at following dabrafenib and trametinib revealed essentially no remaining phosphorylation of ERK1/2 or other kinases (Supplementary Fig. S1). Furthermore, stimulation with 3.3 mM peroxide, a positive control for phosphorylation that inhibits protein tyrosine phosphatase activity in leukocytes and melanoma cells (Irish et al., 2010; Polikowsky et al., 2015), revealed melanoma cell signaling potential was suppressed (Supplementary Fig. S1). Melanoma cells obtained after BRAF and MEK inhibitor treatment were viable according to two standard tests, hemocytometer analysis of trypan blue exclusion and flow cytometer analysis of Alexa fluorophore exclusion (see Methods). These results demonstrated a profound lack of signaling capacity in melanoma cells following BRAF and MEK inhibitor treatment. Because observed signaling responses were effectively suppressed after kinase inhibitor therapy, antibodies detecting phospho-proteins were not included in the optimized melanoma mass cytometry antibody panel (Supplementary Table S2), which was tailored to focus on features of melanoma cell identity, neural stem and progenitor cells, trafficking, and immune interaction.
BRAF and MEK inhibitor treatment alters tumor phenotype through elimination or expansion of distinct melanoma cell subsets
Analysis of changes in the major stromal cell types from melanoma tumors was compared before and after therapy. The stromal population was quantified from the expert drawn gates of 14 individual viSNE analyses of cells from 7 matched tumor samples obtained Pre-Tx or following surgical resection after BRAF and MEK targeted therapy (Week 4). No significant difference was found between bulk populations of Pre-Tx and Week 4 fibroblasts, endothelial cells, or leukocytes (Supplementary Fig. S2). Previous reports have shown changes in subsets of infiltrating T cells (Frederick et al., 2013). A relative increase in CD3 T cells and cytotoxic CD8+ T cells was observed in tumors following therapy (p=0.02 for both, Supplementary Table S3).
To establish a methodology to identify features of cell populations that escaped targeted therapy, Pre-Tx and Week 4 tumor cells from MP-059 were analyzed simultaneously in a new viSNE map (Fig. 2A). Features for viSNE analysis were selected based on a variance of 0.2 or greater across 14 melanoma tissue samples (Supplementary Table S2) following established methods (Irish et al., 2004; Irish et al., 2010). Cells from this viSNE analysis were next characterized by MEM to quantify feature enrichment and phenotypic stability over time during treatment (Fig. 2B).
Figure 2. BRAF and MEK inhibitor treatment alters tumor phenotype through elimination or expansion of distinct melanoma cell subsets in MP-059.
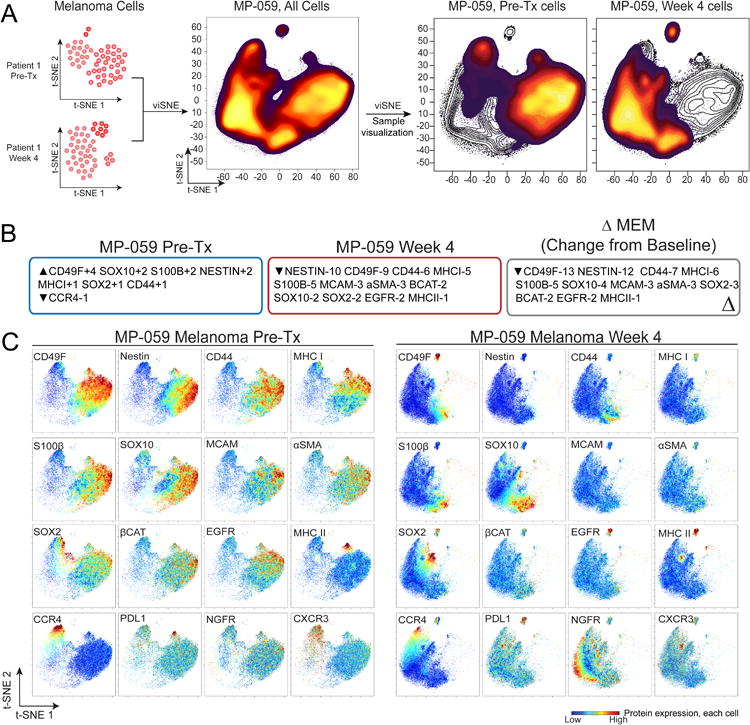
(A) viSNE density plots show phenotypic change over time for a single melanoma tumor (MP-059). Pre-Tx and Week 4 melanoma cells were computationally separated from other stromal cell types as in Figure 1 and combined for a new viSNE analysis. (B) MEM labels quantify enrichment of 17 of 32 measured proteins in Pre-Tx and Week 4 melanoma cells from one tumor (MP-059). Features for MEM analysis were selected with a variance > 0.2 from 7 pairs of Pre-Tx and Week 4 tumors. Only significantly enriched proteins are shown. The overall change in tumor phenotype (ΔMEM) was calculated by subtracting Week 4 MEM enrichment scores from those measured in the Pre-Tx sample. (C) viSNE plots show protein expression in cells (heat) from the combined viSNE analysis of MP-059 melanoma cells from Pre-Tx and Week 4. The 16 proteins with the greatest difference in expression across melanoma cells are shown.
Analysis with viSNE and MEM indicated Pre-Tx cells were enriched for neural crest stem cell markers Nestin, SOX10, SOX2, CD49F, and melanoma diagnostic biomarker S100β (Ordonez, 2014). In contrast, Week 4 cells lost Nestin and CD49F expression and expressed other markers of neural crest identity including NGFR, SOX10, and SOX2. Furthermore, Week 4 cells specifically lacked immune interaction protein MHC I (Fig. 2C) (Garcia-Lora et al., 2003). The overall change in tumor phenotype was calculated as change (Δ) in MEM (ΔMEM) by subtracting Week 4 MEM enrichment scores from those measure in the Pre-Tx sample (Diggins et al., 2018). ΔMEM analysis revealed Nestin, a neural cell filament (Park et al., 2010), and CD49F, a stem-cell niche associated integrin (Krebsbach and Villa-Diaz, 2017), were the melanoma cell features whose enrichment changed the most from Pre-Tx to Week (Fig. 3). These results provided a quantitative workflow to characterize and track changes in tumor heterogeneity.
Fig. 3. Combined viSNE analysis of melanoma tumors identifies patient specific changes in tumor phenotype following BRAF and MEK inhibitor treatment.
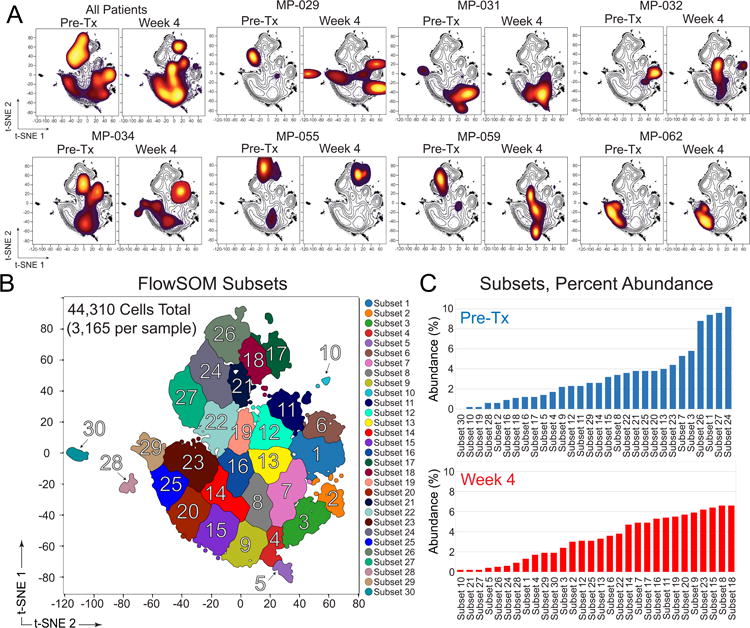
(A) viSNE density plots show phenotype and abundance of melanoma cells from paired samples of Pre-Tx and Week 4 tumors (N = 7 tumors). A combination viSNE density plot (upper left) shows all patients combined in two plots at Pre-Tx and Week 4. (B) FlowSOM cluster analysis of all patients (Pre-tx and Week 4 combined, as in A) automatically identified phenotypically distinct subsets. (C) Abundance (% of Pre-Tx or % of Week 4 cells) for all subsets identified by FlowSOM is shown, organized from least to most common in Pre-Tx or Week 4 melanoma tumors. Subset labels from (B) and (C) are the same and additional information on patients and subset phenotypes is shown in Supplementary Table S4 and Supplementary Figure S5.
Combined viSNE analysis of melanoma tumors identifies patient-specific changes in tumor phenotype following BRAF and MEK inhibitor treatment
To systematically identify subset phenotypes that escape therapy, matched tumors were placed into an analysis workflow that identifies and characterizes subset phenotypes from a sequential viSNE, FlowSOM, and MEM analysis (Supplementary Fig. S4A). Using this unsupervised approach minimizes the influence of human bias between users and could reveal information about populations that may be overlooked with biaxial gating alone. Equal numbers of melanoma cells from 7 pairs of samples Pre-Tx and Week 4 melanoma tumors were next analyzed in one new viSNE map (Fig. 3). Analysis of the pre-therapy samples within one viSNE map made it apparent that melanoma cell phenotypes were largely distinct for each patient Pre-Tx (Fig. 3A). Some tumors were relatively homogenous in phenotype (Fig. 3A, MP-029, MP-032, and MP-062), whereas other tumors contained multiple phenotypically distinct cell types that formed well-separated ‘islands’ in the common viSNE map (Fig. 3A, MP-031, MP-034, MP-055, and MP-059). These results were observed when heat plots of cell density for each sample were overlaid on a black and white contour plot showing the density of all cells. Analysis of Week 4 cells for each patient revealed the emergence of subset phenotypes that did not exist before therapy (Fig. 3A, MP-029, MP-032, MP-034, and MP-059), or little change from the pre-existing phenotype (Fig. 3A, MP-031, MP-055, and MP-062).
An unsupervised analysis approach was next used to identify melanoma cell subsets that evaded therapy. FlowSOM, a machine learning tool based on self-organizing maps (Van Gassen et al., 2015b), was used to automatically identify cell subsets within the common viSNE map of Pre-Tx and Week 4 (Fig. 3B). Manual analysis by experts was conducted prior to FlowSOM analysis and revealed approximate 30 phenotypically distinct cell types within the combined set of Pre-Tx and Week 4 melanoma tumors. FlowSOM analysis was used to automatically identify 30 phenotypically distinct cell subsets in Pre-Tx and Week 4 tumor samples (Fig. 3B) and then percent abundance for each cell subset was quantified (Fig. 3C, Supplementary Table S4).
Cell subsets were categorized into three groups by comparing Pre-Tx and Week 4 subset abundance (Supplementary Figure S4). An intra-tumoral cell subset was considered to have regressed when >75% of its cells were observed Pre-Tx, meaning that it decreased in abundance by at least 50% on treatment. In contrast, a subset was considered to have emerged when >75% of its cells were seen at Week 4. All other subsets demonstrated less than 50% change from Pre-Tx to Week 4 and were considered to have persisted (Supplementary Figure S4). Thus, the terms regressing, persisting, and emerging used here refer to changes in the abundance of cells within a single tumor and are not comparable to overall clinical assessments of patient responses to treatment. Once subsets were categorized, MEM was used to identify common phenotypic signatures of each melanoma cell group. Compared to the other subsets, regressing melanoma cell subsets (Fig 3C, e.g. subsets 24, 27, 1, and 26) were enriched for neural crest stem cell proteins Nestin, CD49F, SOX2, and SOX10, melanocyte biomarker S100β, and immune interaction protein MHC I (Supplementary Table S4 and Supplementary Fig. S4). In contrast, the emerging melanoma cell subsets especially lacked Nestin and CD49F (Supplementary Table S4 and Supplementary Fig. S4). The phenotypes of the persisting cells were more heterogeneous, and no proteins were especially enriched in these cells (e.g., Supplementary Figure S4, SOX2, SOX10, and CD49F). However, persisting cells contrasted with both regressing and emerging cell subsets, in that they most commonly lacked MHC class I protein expression (Supplementary Figure S4 and Supplementary Table S4).
Significant loss of Nestin protein expression was observed in melanoma cells at Week 4 following dabrafenib and trametinib
Since the melanoma cell subsets with the greatest changes in abundance on treatment also shared similar phenotypes, median protein expression was next analyzed in aggregate comparisons of all Pre-Tx cells to all Week 4 cells. Unpaired comparisons of 4 melanoma cell lines, 14 Pre-Tx samples, and 11 Week 4 samples showed that median Nestin protein expression was significantly lower in Week 4 samples than in Pre-Tx samples (p=0.04, Fig. 4A). Melanoma cell lines generally expressed high levels of Nestin protein (MeWo, A2058, and WM115), although SKMEL28 cells were an exception and only moderately expressed Nestin. CD49F, MCAM, MHC II, and CCR4 were also expressed in Pre-Tx melanoma cells, but no statistically significant difference in median protein expression was observed between Pre-Tx and Week 4 (Fig. 4). Paired comparison of the 21 most highly expressed melanoma cell proteins in Pre-Tx and Week 4 samples also showed significant decreases in median expression of Nestin, MHC class I, CD49F, and EGFR protein (Supplementary Figure S6). Features previously associated with response (MITF) or resistance (AXL, EGFR) were expressed at low levels in tumor samples and did not change significantly over time (Supplementary Fig S6).
Parallel mass cytometry and immunohistochemical analysis show loss of Nestin protein expression in melanoma cells at Week 4 following dabrafenib and trametinib
Standard formalin fixed, paraffin-embedded archival tissue blocks of the same tumor specimens studied by mass cytometry were available for four tumors. However, images from immunohistochemistry (IHC) staining of tissue will include all cells, whereas the mass cytometry analyses here used gating to analyze proteins specifically within melanoma cells. To compare mass cytometry findings to traditional IHC, mass cytometry data were re-analyzed to calculate the percentage of Nestin expressing cells within all live tumor cells (Fig. 5). Next, hematoxylin counterstain and expression of Nestin, SOX10, and S100β were evaluated by IHC in subcellular sections (<10 μm) of tissue microarrays (TMAs) from four tumors also analyzed by mass cytometry. A comparable loss of Nestin expression between Pre-Tx and Week 4 was apparent in both mass cytometry and IHC (Fig. 5C). Furthermore, Week 4 melanoma cell abundance and Nestin heterogeneity were also observed to be comparable in both mass cytometry and IHC in all TMAs (Fig. 5C, SOX10 and S100β expression in Nestin negative cells). While Nestin was most abundant Pre-Tx, different intensities in Nestin expression among neighboring melanoma cells also provided another indicator of intra-tumor cellular heterogeneity (e.g. Patient 22, Fig. 5C and Supplementary Fig. S7).
Fig. 5. Parallel mass cytometry and immunohistochemical analysis show loss of Nestin protein expression in melanoma cells at Week 4 following dabrafenib and trametinib.
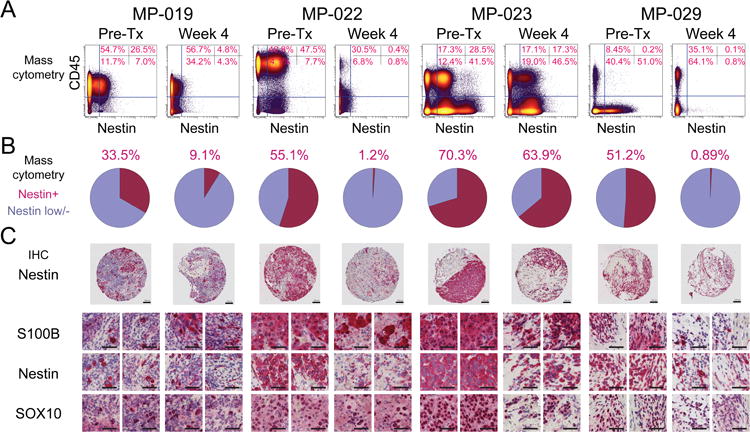
(A) To compare mass cytometry findings to traditional IHC, which includes all tumor cells in an image, mass cytometry data were re-gated to include all live cells. Traditional biaxial plots of mass cytometry data shown CD45 protein expression (leukocytes) versus Nestin protein expression Pre-Tx and at Week 4 following treatment for four tumors. (B) Pie graphs show the percentage of cells expressing Nestin observed by mass cytometry in each tumor Pre-Tx and at Week 4. (C) Paraffin-embedded core biopsies from tissue microarrays of the same tumors as in (A) were analyzed by immunohistochemistry for Nestin protein and, in in higher magnification views, for Nestin and traditional histology markers of melanoma, S100β and SOX10. Serial slices are shown for two fields of view per TMA per time point. Scale bars indicate 100 μm in all images.
To investigate these results further, Kaplan-Meier statistical analysis was performed on 11 patient sets, which revealed no significant association between Nestin or CD49F expression and outcome (Supplementary Fig. S8). Analysis of tumor size suggested that low expression of Nestin and CD49F might be more common in larger tumors (Supplementary Fig. S9). Additional analysis of publicly available single-cell RNA-Seq data from therapy naïve and relapse samples revealed increased Nestin transcript expression at the time of relapse in some samples (Supplementary Fig. S10) (Tirosh et al., 2016). Taken together with the protein measurements made at Week 4 here, these findings suggest that melanoma cells lacking Nestin protein after treatment may be capable of repopulating the Nestin-expressing melanoma cell population by the time of relapse.
Discussion
These results reveal the loss of Nestin-expressing tumor cells, a hallmark of aggressive, metastatic disease, immediately after targeted BRAFV600E and MEK inhibitor therapy. Nestin expression has previously been associated with advanced stage melanoma, stem-ness, and dissemination of circulating melanoma cells (Akiyama et al., 2013; Fusi et al., 2010; Ladstein et al., 2014). However, Nestin negative cells persisted and emerged after therapy, and these cells maintained other features of neural crest identity, including SOX2, CD49F, and SOX10 expression (Simoes-Costa and Bronner, 2015). To date, the depletion of Nestin has been shown to produce invasive phenotypes in melanoma, however, a direct link between MAPK inhibition and melanoma phenotype has not been previously established (Lee et al., 2014). Three central elements of the approach here were the focus on single cell measurements of core cell identity proteins, comparison of cells from one tumor over time, and unsupervised computational analysis. This approach was especially useful in revealing phenotypically distinct populations of melanoma cells present in resected Week 4 tumor samples. The melanoma cells observed at Week 4 contrasted significantly with the cells observed in the same tumors prior to the start of BRAF and MEK inhibitor treatment (Fig. 3, Fig. 4, and Supplementary Fig. 4) and with cells characterized at the time of relapse in other studies (Hugo et al., 2015; Tirosh et al., 2016). Thus, while relapse is likely driven by diverse mechanisms among patients, the populations of cells initially involved in evading a single treatment may have common, targetable features.
While the selection of stem-like subsets has been implicated as the primary means of relapse in other cancers, cellular plasticity and therapy-induced reprogramming could be responsible for the shift in phenotype in melanoma (Holzel et al., 2013). Neural cell plasticity and therapy-induced reprogramming have previously been observed in models of melanoma, including the emergence of melanoma tumors that display a dependence on MAPK inhibition to support tumor growth (Das Thakur et al., 2013; Handoko et al., 2013; Sun et al., 2014). IHC data published previously for tumors from patients on this trial showed a decrease in Ki67 positive cells to an average of 2.9% at Week 4 (Johnson et al., 2015). This result indicates that proliferation is largely absent by Week 4 and suggests it is unlikely that the changes in tumor composition observed here can be explained solely by the growth of an intrinsically-resistant subset. Furthermore, analysis of therapy naïve and relapse samples suggests that the Pre-Tx cellular composition of the tumor recovers after the end of targeted therapy and prior to the time of relapse (Tirosh et al., 2016).
While the increase in T cell infiltration suggested a robust response to therapy, melanoma cells were not completely eradicated (Frederick et al., 2013). Melanoma subsets observed after therapy were either emerging novel subsets not present before therapy or persisting MHC class I negative cells. Lack of MHC class I expression has been implicated in impaired immune surveillance (Garcia-Lora et al., 2003). It was also apparent that individual melanoma cells almost never co-expressed Nestin and PD-L1 (N = 7 pairs, Supplementary Figure 5). At the sample level, low median PD-L1 expression was seen on some melanoma cell subsets (e.g. Subsets 1, 6, 11, Supplementary Figure S4). In these subsets of PD-L1 expressing melanoma cells, a deficit in Nestin protein expression was observed. Furthermore, in subsets where median Nestin expression was higher (e.g. subsets 27, 26, and 24), median PD-L1 expression was low to zero. Taken together, loss of Nestin might represent a transient identity influenced by therapy, and loss or a sustained lack of MHC proteins coupled with PD-L1 expression could facilitate evasion from the immune system to enable a broader range of genomic and non-genomic alterations to arise at relapse (Hugo et al., 2015; Johnson et al., 2014). Targeting antitumor immunity after this therapeutic window could be a favorable period to kill residual cells before tolerance develops into clinical resistance.
The results presented here introduce a longitudinal mass cytometry study of human melanoma tumors and establish a reference for future longitudinal solid tumor studies. The measurement of 32 markers of cell identity, trafficking, and immune interaction was especially valuable in clarifying the identity of melanoma cells which displayed unexpected phenotypes, such as the abnormal neural crest phenotype that persisted following treatment. The observed loss of Nestin and persistence of MHC class I negative subsets in patients could have negative consequences for other targeted therapies and immunotherapy. With the range of therapeutic options for melanoma rapidly growing, combining clinical studies with single cell analysis could identify novel features to track clinical responses and reveal unexpected consequences of treatment.
Supplementary Material
Supplementary Figure S1. Little or no phosphorylation of ERK, SRC, or AKT was observed in melanoma tumor cells after BRAK and MEK inhibitor therapy. Post-therapy BRAF and MEK inhibitor-treated melanoma tumor cells and signaling positive control Jurkat T cells were stained with phospho-specific antibodies to quantify signaling potential. In (A) Jurkat T cells show high basal phosphorylation and induction of ERK and SRC phosphorylation following stimulation by phosphatase inhibitor peroxide (3.3 mM for 4 minutes). In (B) melanoma tumor cells from patient MP-001 showed low basal and potentiated signaling activity in unstimulated cells (top row) or following peroxide stimulation (bottom row).
Supplementary Figure S2. Targeting BRAFV600E and MEK in melanoma did not significantly change the abundance of stromal leukocytes, fibroblasts, or endothelial cells. Sample level statistical analysis of 14 tumors viSNE gated cell populations (7 Pre-Tx tumors and 7 matched Week 4 post-therapy tumors). Wilcoxon signed rank tests were performed with a threshold of p=0.05.
Supplementary Figure S3. Targeting BRAFV600E and MEK in melanoma tumors increased CD3+ T cell and CD8+ effector T cell infiltration. Sample level statistical analysis of 14 tumors % CD3 expressing T cells and % CD8 T cells (7 Pre-Tx tumors and 7 matched Week 4 post-therapy tumors). Wilcoxon signed rank tests were performed with a significance threshold of p=0.05 (p=0.02, and p=0.02).
Supplementary Figure S4. FlowSOM and MEM analysis quantitatively characterized features of melanoma subsets before and after therapy. (A) Subsets identified from a common viSNE map of all patients were identified with FlowSOM. (B) Marker enrichment modeling (MEM) analysis quantitatively labeled 30 cell subsets with 17 markers with the highest variance for melanoma cells across patients. Represented alongside MEM analysis are two additional heat maps of the percent abundance and median intensity for the same subsets.
Supplementary Figure S5. Visualization of cell phenotypes before and after therapy in patients with viSNE analysis. A viSNE analysis of all Pre-Tx and Week 4 melanoma cells from 7 matched samples. The viSNE plots display protein expression as heat for proteins with the greatest variance across patient samples.
Supplementary Figure S6. Median intensity for all features in Pre-Tx and Week 4 melanoma cells from all tumors studied with the optimized mass cytometry panel (Supplementary Table S2). Aggregate analysis of median intensity (arcsinh scale) for 20 measured proteins in melanoma cells gated as in Figure 1 from 14 tumor samples representing matched pairs of Pre-Tx and Week 4 from 7 individual patients. These graphs display additional data for samples shown in Figure 3 and Supplementary Figure S4 (e.g. AXL, MITF, and EGFR displayed here). Wilcoxon signed rank tests were performed and p-values less than 0.05 are shown.
Supplementary Figure 7. IHC of Nestin expression showed intra-tumor cellular diversity that was comparable to mass cytometry. Frozen, fixed, and paraffin embedded core biopsies at three points of treatment were used to acquire TMA's (tissue microarrays). Subcellular sections from the TMA <10 μm were used for immunohistochemistry of Nestin. Nestin expression was found to be high, medium or negative for tumor cells within several regions (blue=high, green=mid, yellow=negative).
Supplementary Figure S8. Kaplan-Meier curves for survival and progression in melanoma patients. Kaplan-Meier statistical analysis of 11 Pre-Tx tumors CD45 low/negative cells divided into two groups by median Nestin or CD49F expression. Patients with high expression of Nestin and CD49F did not have better overall survival and time to progression.
Supplementary Figure S9. Tumor volume plotted against median Nestin or median CD49F protein expression in melanoma cells. Dot plots show eleven patients' Pre-Tx tumor volume compared to the median level Nestin protein expression or the median level of CD49F protein expression.
Supplementary Figure S10. mRNA expression for Nestin, CD49F, SOX10, SOX2, MHC I (HLA-A), and MHC I (HLA-B) was not significantly decreased at the time of relapse in data from Tirosh et al. Box and whisker plots are pooled mRNA expression from 12 tumors and 6 patients' melanoma cells published by Tirosh et al., Science 2016. Tumors were therapy naïve or at the time of relapse following MAPK inhibitor treatment, in contrast with the time of surgical resection following 4 weeks of treatment, as here. The expression level of proteins that changed significantly here was quantified as Ep(I)=log2(TPM(I)+1) where (I) is a set of cells and TPM is transcript per million. Wilcoxon signed rank tests were performed with a threshold of p=0.05.
Significance.
This study creates a 32-marker mass cytometry panel for melanoma and applies it to deeply characterize melanoma cell protein expression signatures before and after neoadjuvant therapy targeting BRAF and MEK. Targeted therapy is revealed to clear Nestin-expressing melanoma cells, which are considered a hallmark of aggressive, metastatic melanoma. Persisting subsets after targeted therapy also lacked surface expression of MHC I, which may inform strategies for combining targeted therapy and immunotherapy. This study reveals new in vivo biology of melanoma cells that can be used as a reference point for patient-derived xenograft and cell line research models.
Acknowledgments
This study was supported by NIH/NCI T32 CA009592 (D.B.D), R25 GM062459 (D.B.D), R25 CA136440 (K.E.D.), F31 CA199993 (A.R.G.), R00 CA143231 (J.M.I.), the Vanderbilt-Ingram Cancer Center (VICC, P30 CA68485), Vanderbilt Medical Scholars (J.S.G) and VICC Ambassadors.
Footnotes
Disclosure Of Potential Conflicts Of Interest: Competing Financial Interests Statement: J.M.I. is co-founder and board member and Cytobank Inc. and received research support from Incyte Corp and Janssen.
Author contributions: All authors discussed data visualization, contributed intellectually to the manuscript, and approved the final manuscript. M.C.K., J.M.I., D.B.D., J.A.S., and K.B.D. initially designed clinical research. M.C.K. and J.A.S. designed the clinical trial, consented and enrolled patients, and M.C.K. collected tissue for research. D.B.D., J.M.I., A.R.G., and M.C.K. designed mass cytometry panels. D.B.D. created and characterized custom conjugated mass cytometry antibodies. D.B.D., A.R.G., C.E.R. collected, normalized, and annotated mass cytometry data. D.B.D., K.E.D., A.R.G., J.S.G., and J.M.I. contributed statistical and computational analyses of mass cytometry. D.B.D., K.B.D., M.C.K., and J.M.I. contributed to histology experiments with tissue microarrays. D.B.D., A.R.G., J.S.G, and J.M.I. prepared figures. D.B.D., A.R.G., and J.M.I. drafted the manuscript.
References
- Akiyama M, Matsuda Y, Ishiwata T, Naito Z, Kawana S. Nestin is highly expressed in advanced-stage melanomas and neurotized nevi. Oncology reports. 2013;29:1595–9. doi: 10.3892/or.2013.2287. [DOI] [PubMed] [Google Scholar]
- Amir El AD, Davis KL, Tadmor MD, Simonds EF, Levine JH, Bendall SC, Shenfeld DK, Krishnaswamy S, Nolan GP, Pe'er D. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nature biotechnology. 2013;31:545–52. doi: 10.1038/nbt.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendall SC, Simonds EF, Qiu P, Amir El AD, Krutzik PO, Finck R, Bruggner RV, Melamed R, Trejo A, Ornatsky OI, et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science (New York, NY) 2011;332:687–96. doi: 10.1126/science.1198704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjornson ZB, Nolan GP, Fantl WJ. Single-cell mass cytometry for analysis of immune system functional states. Current opinion in immunology. 2013;25:484–94. doi: 10.1016/j.coi.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das Thakur M, Salangsang F, Landman AS, Sellers WR, Pryer NK, Levesque MP, Dummer R, Mcmahon M, Stuart DD. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature. 2013;494:251–5. doi: 10.1038/nature11814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diggins KE, Ferrell PB, Jr, Irish JM. Methods for discovery and characterization of cell subsets in high dimensional mass cytometry data. Methods (San Diego, Calif) 2015;82:55–63. doi: 10.1016/j.ymeth.2015.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diggins KE, Gandelman JS, Roe CE, Irish JM. Generating Quantitative Cell Identity Labels with Marker Enrichment Modeling (MEM) In: J. Paul Robinson, managing editor … [et al], editor. Current protocols in cytometry / editorial board. Vol. 83. 2018. pp. 10.21.1–10.21.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diggins KE, Greenplate AR, Leelatian N, Wogsland CE, Irish JM. Characterizing cell subsets using marker enrichment modeling. Nature methods. 2017;14:275–278. doi: 10.1038/nmeth.4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdmann F, Lortet-Tieulent J, Schuz J, Zeeb H, Greinert R, Breitbart EW, Bray F. International trends in the incidence of malignant melanoma 1953-2008--are recent generations at higher or lower risk? International journal of cancer. Journal international du cancer. 2013;132:385–400. doi: 10.1002/ijc.27616. [DOI] [PubMed] [Google Scholar]
- Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, Mitra D, Boni A, Newton LP, Liu C, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:1225–31. doi: 10.1158/1078-0432.CCR-12-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusi A, Ochsenreither S, Busse A, Rietz A, Keilholz U. Expression of the stem cell marker nestin in peripheral blood of patients with melanoma. The British journal of dermatology. 2010;163:107–14. doi: 10.1111/j.1365-2133.2010.09779.x. [DOI] [PubMed] [Google Scholar]
- Garcia-Lora A, Algarra I, Garrido F. MHC class I antigens, immune surveillance, and tumor immune escape. Journal of cellular physiology. 2003;195:346–55. doi: 10.1002/jcp.10290. [DOI] [PubMed] [Google Scholar]
- Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. The New England journal of medicine. 2013;369:134–44. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handoko HY, Boyle GM, Ferguson B, Muller HK, Soyer HP, Walker GJ. Plasticity of melanoma in vivo: murine lesions resulting from Trp53, but not Cdk4 or Arf deregulation, display neural transdifferentiation. Pigment cell & melanoma research. 2013;26:731–4. doi: 10.1111/pcmr.12124. [DOI] [PubMed] [Google Scholar]
- Hodi FS, O'day SJ, Mcdermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. Improved survival with ipilimumab in patients with metastatic melanoma. The New England journal of medicine. 2010;363:711–23. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251–63. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes D. The cancer that rises with the Sun. Nature. 2014;515:S110–S111. doi: 10.1038/515S110a. [DOI] [PubMed] [Google Scholar]
- Holzel M, Bovier A, Tuting T. Plasticity of tumour and immune cells: a source of heterogeneity and a cause for therapy resistance? Nature reviews. Cancer. 2013;13:365–76. doi: 10.1038/nrc3498. [DOI] [PubMed] [Google Scholar]
- Hugo W, Shi H, Sun L, Piva M, Song C, Kong X, Moriceau G, Hong A, Dahlman KB, Johnson DB, et al. Non-genomic and Immune Evolution of Melanoma Acquiring MAPKi Resistance. Cell. 2015;162:1271–85. doi: 10.1016/j.cell.2015.07.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irish JM. Beyond the age of cellular discovery. Nature immunology. 2014;15:1095–7. doi: 10.1038/ni.3034. [DOI] [PubMed] [Google Scholar]
- Irish JM, Doxie DB. High-dimensional single-cell cancer biology. Current topics in microbiology and immunology. 2014;377:1–21. doi: 10.1007/82_2014_367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irish JM, Hovland R, Krutzik PO, Perez OD, Bruserud O, Gjertsen BT, Nolan GP. Single cell profiling of potentiated phospho-protein networks in cancer cells. Cell. 2004;118:217–28. doi: 10.1016/j.cell.2004.06.028. [DOI] [PubMed] [Google Scholar]
- Irish JM, Myklebust JH, Alizadeh AA, Houot R, Sharman JP, Czerwinski DK, Nolan GP, Levy R. B-cell signaling networks reveal a negative prognostic human lymphoma cell subset that emerges during tumor progression. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:12747–54. doi: 10.1073/pnas.1002057107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AS, Crandall H, Dahlman K, Kelley MC. Preliminary results from a prospective trial of preoperative combined BRAF and MEK-targeted therapy in advanced BRAF mutation-positive melanoma. Journal of the American College of Surgeons. 2015;220:581–93.e1. doi: 10.1016/j.jamcollsurg.2014.12.057. [DOI] [PubMed] [Google Scholar]
- Johnson DB, Balko JM, Compton ML, Chalkias S, Gorham J, Xu Y, Hicks M, Puzanov I, Alexander MR, Bloomer TL, et al. Fulminant Myocarditis with Combination Immune Checkpoint Blockade. N Engl J Med. 2016a;375:1749–1755. doi: 10.1056/NEJMoa1609214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DB, Estrada MV, Salgado R, Sanchez V, Doxie DB, Opalenik SR, Vilgelm AE, Feld E, Johnson AS, Greenplate AR, et al. Melanoma-specific MHC-II expression represents a tumour-autonomous phenotype and predicts response to anti-PD-1/PD-L1 therapy. Nature communications. 2016b;7:10582. doi: 10.1038/ncomms10582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DB, Pectasides E, Feld E, Ye F, Zhao S, Johnpulle R, Merritt R, Mcdermott DF, Puzanov I, Lawrence D, et al. Sequencing Treatment in BRAFV600 Mutant Melanoma: Anti-PD-1 Before and After BRAF Inhibition. Journal of immunotherapy (Hagerstown, Md: 1997) 2017;40:31–35. doi: 10.1097/CJI.0000000000000148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson GL, Stuhlmiller TJ, Angus SP, Zawistowski JS, Graves LM. Molecular Pathways: Adaptive Kinome Reprogramming in Response to Targeted Inhibition of the BRAF-MEK-ERK Pathway in Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014 doi: 10.1158/1078-0432.CCR-13-1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotecha N, Krutzik PO, Irish JM. Web-based analysis and publication of flow cytometry experiments. In: J. Paul Robinson, managing editor … [et al.], editor. Current protocols in cytometry / editorial board. Chapter 10. 2010. p. Unit10.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koussounadis A, Langdon SP, Um IH, Harrison DJ, Smith VA. Relationship between differentially expressed mRNA and mRNA-protein correlations in a xenograft model system. Scientific reports. 2015;5:10775. doi: 10.1038/srep10775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebsbach PH, Villa-Diaz LG. The Role of Integrin alpha6 (CD49f) in Stem Cells: More than a Conserved Biomarker. Stem cells and development. 2017;26:1090–1099. doi: 10.1089/scd.2016.0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krutzik PO, Nolan GP. Intracellular phospho-protein staining techniques for flow cytometry: monitoring single cell signaling events. Cytometry. Part A : the journal of the International Society for Analytical Cytology. 2003;55:61–70. doi: 10.1002/cyto.a.10072. [DOI] [PubMed] [Google Scholar]
- Krutzik PO, Nolan GP. Fluorescent cell barcoding in flow cytometry allows high-throughput drug screening and signaling profiling. Nature methods. 2006;3:361–8. doi: 10.1038/nmeth872. [DOI] [PubMed] [Google Scholar]
- Ladstein RG, Bachmann IM, Straume O, Akslen LA. Nestin expression is associated with aggressive cutaneous melanoma of the nodular type. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2014;27:396–401. doi: 10.1038/modpathol.2013.151. [DOI] [PubMed] [Google Scholar]
- Lee CW, Zhan Q, Lezcano C, Frank MH, Huang J, Larson AR, Lin JY, Wan MT, Lin PI, Ma J, et al. Nestin depletion induces melanoma matrix metalloproteinases and invasion. Laboratory investigation; a journal of technical methods and pathology. 2014;94:1382–95. doi: 10.1038/labinvest.2014.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leelatian N, Diggins KE, Irish JM. Characterizing Phenotypes and Signaling Networks of Single Human Cells by Mass Cytometry. Methods in molecular biology. 2015;1346:99–113. doi: 10.1007/978-1-4939-2987-0_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leelatian N, Doxie DB, Greenplate AR, Mobley BC, Lehman JM, Sinnaeve J, Kauffmann RM, Werkhaven JA, Mistry AM, Weaver KD, et al. Single cell analysis of human tissues and solid tumors with mass cytometry. Cytometry B Clin Cytom. 2016 doi: 10.1002/cyto.b.21481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leelatian N, Doxie DB, Greenplate AR, Mobley BC, Lehman JM, Sinnaeve J, Kauffmann RM, Werkhaven JA, Mistry AM, Weaver KD, et al. Single cell analysis of human tissues and solid tumors with mass cytometry. Cytometry Part B, Clinical cytometry. 2017a;92:68–78. doi: 10.1002/cyto.b.21481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leelatian N, Doxie DB, Greenplate AR, Sinnaeve J, Ihrie RA, Irish JM. Preparing Viable Single Cells from Human Tissue and Tumors for Cytomic Analysis. Current protocols in molecular biology. 2017b;118:25C 1 1–25C 1 23. doi: 10.1002/cpmb.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501:328–337. doi: 10.1038/nature12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas KJ, Greenplate AR, Flaherty DK, Matlock BK, Juan JS, Smith RM, Irish JM, Kalams SA. Multiparameter analysis of stimulated human peripheral blood mononuclear cells: A comparison of mass and fluorescence cytometry. Cytometry. Part A : the journal of the International Society for Analytical Cytology. 2016;89:271–80. doi: 10.1002/cyto.a.22799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordonez NG. Value of melanocytic-associated immunohistochemical markers in the diagnosis of malignant melanoma: a review and update. Human pathology. 2014;45:191–205. doi: 10.1016/j.humpath.2013.02.007. [DOI] [PubMed] [Google Scholar]
- Ornatsky OI, Lou X, Nitz M, Schafer S, Sheldrick WS, Baranov VI, Bandura DR, Tanner SD. Study of cell antigens and intracellular DNA by identification of element-containing labels and metallointercalators using inductively coupled plasma mass spectrometry. Analytical chemistry. 2008;80:2539–47. doi: 10.1021/ac702128m. [DOI] [PubMed] [Google Scholar]
- Park D, Xiang AP, Mao FF, Zhang L, Di CG, Liu XM, Shao Y, Ma BF, Lee JH, Ha KS, et al. Nestin is required for the proper self-renewal of neural stem cells. Stem cells (Dayton, Ohio) 2010;28:2162–71. doi: 10.1002/stem.541. [DOI] [PubMed] [Google Scholar]
- Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan GQ, Yang JC, Sherry RM, Hwu P, Topalian SL, Schwartzentruber DJ, Restifo NP, Haworth LR, Seipp CA, Freezer LJ, et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:8372–7. doi: 10.1073/pnas.1533209100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polikowsky HG, Wogsland CE, Diggins KE, Huse K, Irish JM. Cutting Edge: Redox Signaling Hypersensitivity Distinguishes Human Germinal Center B Cells. Journal of immunology (Baltimore, Md: 1950) 2015;195:1364–1367. doi: 10.4049/jimmunol.1500904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoes-Costa M, Bronner ME. Establishing neural crest identity: a gene regulatory recipe. Development (Cambridge, England) 2015;142:242–57. doi: 10.1242/dev.105445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer Matthew h, Nolan Garry p. Mass Cytometry: Single Cells. Many Features Cell. 165:780–791. doi: 10.1016/j.cell.2016.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Wang L, Huang S, Heynen GJ, Prahallad A, Robert C, Haanen J, Blank C, Wesseling J, Willems SM, et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature. 2014;508:118–22. doi: 10.1038/nature13121. [DOI] [PubMed] [Google Scholar]
- Tirosh I, Izar B, Prakadan SM, Wadsworth MH, 2nd, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science (New York, NY) 2016;352:189–96. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Allen EM, Wagle N, Sucker A, Treacy DJ, Johannessen CM, Goetz EM, Place CS, Taylor-Weiner A, Whittaker S, Kryukov GV, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer discovery. 2014;4:94–109. doi: 10.1158/2159-8290.CD-13-0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gassen S, Callebaut B, Van Helden MJ, Lambrecht BN, Demeester P, Dhaene T, Saeys Y. FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytometry Part A : the journal of the International Society for Analytical Cytology. 2015a doi: 10.1002/cyto.a.22625. [DOI] [PubMed] [Google Scholar]
- Van Gassen S, Callebaut B, Van Helden MJ, Lambrecht BN, Demeester P, Dhaene T, Saeys Y. FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytometry. Part A : the journal of the International Society for Analytical Cytology. 2015b;87:636–45. doi: 10.1002/cyto.a.22625. [DOI] [PubMed] [Google Scholar]
- Wagle N, Emery C, Berger MF, Davis MJ, Sawyer A, Pochanard P, Kehoe SM, Johannessen CM, Macconaill LE, Hahn WC, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29:3085–96. doi: 10.1200/JCO.2010.33.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YJ, Golson ML, Schug J, Traum D, Liu C, Vivek K, Dorrell C, Naji A, Powers AC, Chang KM, et al. Single-Cell Mass Cytometry Analysis of the Human Endocrine Pancreas. Cell metabolism. 2016;24:616–626. doi: 10.1016/j.cmet.2016.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wogsland CE, Greenplate AR, Kolstad A, Myklebust JH, Irish JM, Huse K. Mass Cytometry of Follicular Lymphoma Tumors Reveals Intrinsic Heterogeneity in Proteins Including HLA-DR and a Deficit in Nonmalignant Plasmablast and Germinal Center B-Cell Populations. Cytometry Part B, Clinical cytometry. 2017;92:79–87. doi: 10.1002/cyto.b.21498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Wang J, Wang X, Zhu J, Liu Q, Shi Z, Chambers MC, Zimmerman LJ, Shaddox KF, Kim S, et al. Proteogenomic characterization of human colon and rectal cancer. Nature. 2014;513:382–7. doi: 10.1038/nature13438. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1. Little or no phosphorylation of ERK, SRC, or AKT was observed in melanoma tumor cells after BRAK and MEK inhibitor therapy. Post-therapy BRAF and MEK inhibitor-treated melanoma tumor cells and signaling positive control Jurkat T cells were stained with phospho-specific antibodies to quantify signaling potential. In (A) Jurkat T cells show high basal phosphorylation and induction of ERK and SRC phosphorylation following stimulation by phosphatase inhibitor peroxide (3.3 mM for 4 minutes). In (B) melanoma tumor cells from patient MP-001 showed low basal and potentiated signaling activity in unstimulated cells (top row) or following peroxide stimulation (bottom row).
Supplementary Figure S2. Targeting BRAFV600E and MEK in melanoma did not significantly change the abundance of stromal leukocytes, fibroblasts, or endothelial cells. Sample level statistical analysis of 14 tumors viSNE gated cell populations (7 Pre-Tx tumors and 7 matched Week 4 post-therapy tumors). Wilcoxon signed rank tests were performed with a threshold of p=0.05.
Supplementary Figure S3. Targeting BRAFV600E and MEK in melanoma tumors increased CD3+ T cell and CD8+ effector T cell infiltration. Sample level statistical analysis of 14 tumors % CD3 expressing T cells and % CD8 T cells (7 Pre-Tx tumors and 7 matched Week 4 post-therapy tumors). Wilcoxon signed rank tests were performed with a significance threshold of p=0.05 (p=0.02, and p=0.02).
Supplementary Figure S4. FlowSOM and MEM analysis quantitatively characterized features of melanoma subsets before and after therapy. (A) Subsets identified from a common viSNE map of all patients were identified with FlowSOM. (B) Marker enrichment modeling (MEM) analysis quantitatively labeled 30 cell subsets with 17 markers with the highest variance for melanoma cells across patients. Represented alongside MEM analysis are two additional heat maps of the percent abundance and median intensity for the same subsets.
Supplementary Figure S5. Visualization of cell phenotypes before and after therapy in patients with viSNE analysis. A viSNE analysis of all Pre-Tx and Week 4 melanoma cells from 7 matched samples. The viSNE plots display protein expression as heat for proteins with the greatest variance across patient samples.
Supplementary Figure S6. Median intensity for all features in Pre-Tx and Week 4 melanoma cells from all tumors studied with the optimized mass cytometry panel (Supplementary Table S2). Aggregate analysis of median intensity (arcsinh scale) for 20 measured proteins in melanoma cells gated as in Figure 1 from 14 tumor samples representing matched pairs of Pre-Tx and Week 4 from 7 individual patients. These graphs display additional data for samples shown in Figure 3 and Supplementary Figure S4 (e.g. AXL, MITF, and EGFR displayed here). Wilcoxon signed rank tests were performed and p-values less than 0.05 are shown.
Supplementary Figure 7. IHC of Nestin expression showed intra-tumor cellular diversity that was comparable to mass cytometry. Frozen, fixed, and paraffin embedded core biopsies at three points of treatment were used to acquire TMA's (tissue microarrays). Subcellular sections from the TMA <10 μm were used for immunohistochemistry of Nestin. Nestin expression was found to be high, medium or negative for tumor cells within several regions (blue=high, green=mid, yellow=negative).
Supplementary Figure S8. Kaplan-Meier curves for survival and progression in melanoma patients. Kaplan-Meier statistical analysis of 11 Pre-Tx tumors CD45 low/negative cells divided into two groups by median Nestin or CD49F expression. Patients with high expression of Nestin and CD49F did not have better overall survival and time to progression.
Supplementary Figure S9. Tumor volume plotted against median Nestin or median CD49F protein expression in melanoma cells. Dot plots show eleven patients' Pre-Tx tumor volume compared to the median level Nestin protein expression or the median level of CD49F protein expression.
Supplementary Figure S10. mRNA expression for Nestin, CD49F, SOX10, SOX2, MHC I (HLA-A), and MHC I (HLA-B) was not significantly decreased at the time of relapse in data from Tirosh et al. Box and whisker plots are pooled mRNA expression from 12 tumors and 6 patients' melanoma cells published by Tirosh et al., Science 2016. Tumors were therapy naïve or at the time of relapse following MAPK inhibitor treatment, in contrast with the time of surgical resection following 4 weeks of treatment, as here. The expression level of proteins that changed significantly here was quantified as Ep(I)=log2(TPM(I)+1) where (I) is a set of cells and TPM is transcript per million. Wilcoxon signed rank tests were performed with a threshold of p=0.05.
Data Availability Statement
Mass cytometry data for this manuscript can accessed via FlowRepository (https://flowrepository.org/).