

Abstract
Lamisil (terbinafine) may cause idiosyncratic liver toxicity through a proposed toxicological mechanism involving the reactive metabolite 6,6-dimethyl-2-hepten-4-ynal (TBF-A). TBF-A toxicological relevance remains unclear due to a lack of identification of pathways leading to and competing with TBF-A formation. We resolved this knowledge gap by combining computational modeling and experimental kinetics of in vitro hepatic N-dealkylation of terbinafine. A deep learning model of N-dealkylation predicted a high probability for N-demethylation to yield desmethyl-terbinafine followed by N-dealkylation to TBF-A and marginal contributions from other possible pathways. We carried out steady-state kinetic experiments with pooled human liver microsomes that relied on development of labeling methods to expand metabolite characterization. Those efforts revealed high levels of TBF-A formation and first order decay during metabolic reactions; actual TBF-A levels would then reflect the balance between those processes as well as reflect the impact of stabilizing adduction with glutathione and other biological molecules. Modeling predictions and experimental studies agreed on the significance of N-demethylation and insignificance of N-denaphthylation in terbinafine metabolism, yet differed on importance of direct TBF-A formation. Under steady-state conditions, the direct pathway was the most important source of the reactive metabolite with a Vmax/Km of 4.0 pmol/min/mg protein/μΜ in contrast to model predictions. Nevertheless, previous studies show that therapeutic dosing leads to accumulation of desmethyl-terbinafine in plasma, which means that likely sources for TBF-A would draw from metabolism of both the major metabolite and parent drug based on our modeling and experimental studies. Through this combination of novel modeling and experimental approaches, we are the first to identify pathways leading to generation of TBF-A for assessing its role in idiosyncratic adverse drug interactions.
Keywords: terbinafine, reactivity, liver toxicity, bioactivation, TBF-A
Graphic abstract
1.). INTRODUCTION
Lamisil (terbinafine) is an effective antifungal drug widely used for toe and fingernail infections (Abdel-Rahman & Nahata, 1997). The drug selectively accumulates in keratin-rich tissues to block fungal squalene monooxygenase activity and in doing so, creates a toxic level of squalene that kills the fungus (Ryder, 1989). When administered orally, terbinafine has relatively low rate of drug-drug interactions (Trepanier, Nafziger, Kearns, Kashuba, & Amsden, 1998), which is likely due to involvement of multiple cytochromes P450 in the drug metabolism (Schuster, 1987) (Vickers, et al., 1999) (Leyden, 1998). Nevertheless, terbinafine has recently been linked to CYP2D6 inhibition (Abdel-Rahman et al., 1999) (Madani, Barilla, Cramer, Wang, & Paul, 2002) prompting the U. S. Food and Drug Administration to recommend careful monitoring when co-administered with other CYP2D6 substrates (“Annotation of FDA Label for terbinafine and CYP2D6,” 2013) (Medication Guide: Lamisil (terbinafine hydrochloride) Tablets, 2016). Overall, the attractive properties of terbinafine have made it a popular treatment for onychomycosis, with 1.5 million prescriptions in the U.S. for 2010 alone according to IMS Health (Napodano, 2012). Despite success in the clinic, terbinafine use can induce adverse drug events. Symptoms include mild to severe gastrointestinal, skin, and taste disturbances (Hall, Monka, Krupp, & O’Sullivan, 1997) (Gupta, Sibbald, Knowles, Lynde, & Shear, 1997). In rare cases, compromised hepatobiliary function is possible but typically asymptomatic and reversible. For 1 of 45,000–54,000 patients, terbinafine induces symptomatic cholestatic injury that may require liver transplantation or cause death (Terbinafine product monograph: Canada, 1995). While rare, these serious adverse drug effects become significant considering the widespread use of oral terbinafine. Currently, precautionary liver function tests are part of the standard of care against terbinafine-induced liver toxicity. These precautionary tests may reduce the number of severe events, as therapy commonly increases markers of liver injury and terbinafine must be withdrawn (Ajit, Suvannasankha, Zaeri, & Munoz, 2003). Unfortunately, this type of monitoring reveals toxicity only after it occurs and thus is not predictive. The identification of the underlying mechanism for these adverse drug events associated with terbinafine might provide opportunities to identify patients at risk and thus better personalize treatment for improving health outcomes.
Drug-induced adverse events commonly involve an initiating event derived from metabolic activation of the drug into a reactive electrophilic metabolite (Attia, 2010). The metabolism of terbinafine includes extensive N-dealkylation, aliphatic hydroxylation and arene oxidation (Jensen, 1989) likely carried out by cytochromes P450 (Vickers, et al., 1999), yet early studies failed to detect 6,6-dimethylhept-2-ene-4-ynal (TBF-A), a potentially reactive allylic, propargylic aldehyde (Fig. 1). In 2001, Iverson and Uetrecht (Iverson & Uetrecht, 2001) trapped TBF-A with glutathione during microsomal reactions of terbinafine and validated the structure of conjugates using synthesized standards. The conjugative reaction occurred preferentially through a 1,6-Michael addition that then left a second electrophilic site available for 1,4-Michael addition with glutathione or cellular proteins. Importantly, glutathione conjugation of TBF-A was reversible so that the transport of the conjugates could lead to off-target effects. Based on these properties, the authors offered a provocative mechanism for terbinafine hepatotoxicity associated with adverse drug events for patients; in hepatocytes, terbinafine metabolism yields the TBF-A glutathione conjugate, which undergoes transport to the bile ducts where the reactive conjugate modifies hepatobiliary proteins such as bile acid transporters to induce liver dysfunction. Ultimately, this toxicological mechanism for terbinafine exposure depends on the presence and capacity of pathways to metabolically liberate the reactive TBF-A molecule from the drug. Thus, an understanding of the mechanism by which TBF-A forms from terbinafine is essential for being able to assess potential for patient variability in TBF-A accumulation and stratify risk for downstream liver toxicity.
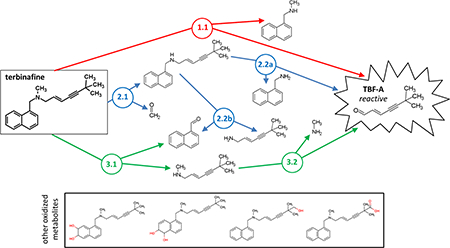
Fig. 1. N-Dealkylation of terbinafine pathways leading to formation of reactive TBF-A.

Three N-dealkylation pathways for terbinafine yield TBF-A (m/z 384, dansyl labeled). Pathway 1 (red) is a single step pathway yielding directly TBF-A and N-methyl-1-naphthyl methylamine (m/z 405, dansyl labeled) as a co-metabolite (step 1.1). Pathway 2 (blue) is a two-step pathway yielding first desmethyl terbinafine (m/z 511, dansyl labeled) and formaldehyde via N-demethylation (step 2.1), followed by generation of 1-naphthyl methylamine (m/z 391, dansyl labeled) and TBF-A from desmethyl terbinafine (step 2.2A). Pathway 3 (green) is a two-step pathway yielding first naphthaldehyde (m/z 404, dansyl labeled) and N-methyl-6,6-dimethyl-2-hepten-4- yn-1-amine (step 3.1), which undergoes N-dealkylation to yield TBF-A (step 3.2). There are three non-N-dealkylation primary metabolites of terbinafine from alternate pathways, i.e. hydroxyterbinafine (m/z 308) and two isomers of terbinafine dihydrodiol (m/z 326) that were also observed in this study (shown in grey box). Carboxyterbinafine has been reported by others (Vickers, et al., 1999) (Humbert, Cabiac, Denouel, & Kirkesseli, 1995) but was not observed under our steady state conditions.
As a tertiary amine, terbinafine undergoes metabolism to yield TBF-A through three main N-dealkylation pathways (Fig. 1). The direct formation of TBF-A is shown in Pathway 1 (colored red), while the other pathways involve two steps and branch off into competing pathways. In Pathway 2 (colored blue), the first step is N-demethylation to yield desmethyl-terbinafine, and subsequent metabolism yields TBF-A down one pathway. For Pathway 3 (colored green), terbinafine metabolism liberates N-methyl-6,6-dimethyl-2-hepten-4-yn-1-amine on the way to generating TBF-A in a second step. A previous in vitro kinetic study using human liver microsomes and recombinant cytochromes P450 reported formation of 11 terbinafine metabolites including three from the N-dealkylation pathways with the important exception of TBF-A, which escaped detection (Vickers, et al., 1999). In that study, the observed rates were combined and analyzed according to common reaction types (side chain oxidation, N-demethylation, deamination, dihydrodiol formation). Consequently, the efficiency of individual pathways, and their relative contributions to terbinafine metabolic activation to TBF-A and overall clearance remain unknown.
We resolved this knowledge gap by combining experimental and computational modeling of in vitro hepatic N-dealkylation of terbinafine. In previously published work, we modeled HLM N-dealkylations that revealed both amine and aldehyde metabolites and then predicted the subsequent reactivity of the aldehyde as a potential precursor for toxicity (Dang, Hughes, Miller, & Swamidass, 2018). We then modeled terbinafine metabolism to determine preferred N-dealkylation steps leading to TBF-A (Fig. 1). Of the three possible initial reactions, our model predicted the highest probability for N-demethylation of terbinafine to desmethyl-terbinafine (probability (P) of 0.79) (Reaction 2.1, blue, Fig. 1) and very low probabilities for the other reactions (P < 0.10) (Dang, Hughes, Miller, & Swamidass, 2018). Given those results, we modeled subsequent metabolism of desmethyl-terbinafine to yield TBF-A in Path 2.2A and the opposing Path 2.2B, whose probabilities were 0.35 and 0.21, respectively. The model findings suggest that Pathway 2 may be an effective route to generating TBF-A. Nevertheless, our modeling does not incorporate the impact of binding affinities and concentration as the driving force for these enzymatic processes.
In the present study, we complemented the computational modeling by carrying out steady-state kinetic studies with pooled human liver microsomes (HLM150) to determine the mechanisms and corresponding kinetic constants for terbinafine metabolism, and thus the concentration-dependent metabolic efficiency of N-dealkylation pathways. Steady-state conditions minimize secondary processes to determine mechanism and efficiency of single steps in metabolic pathways, which can then be put together to study the complete pathway. Initial metabolite profiling of terbinafine metabolism revealed low sensitivity and hence high variability with many metabolites while others were undetectable at all, such as TBF-A. As a solution, we developed in-house labeling methods for aldehydes with dansyl hydrazine and for amines with dansyl chloride, respectively. Through this effort, we improved sensitivity and quantitation of multiple metabolites including TBF-A. We measured steady-state metabolism of terbinafine and the major primary metabolite desmethyl-terbinafine as relevant N-dealkylation pathways leading to TBF-A. The resulting data were fit to the most statistically preferred kinetic mechanism and corresponding constants used to reveal the metabolic efficiency of the N-dealkylations leading to formation of TBF-A. Computational analyses were further used to model N-dealkylation pathways leading from terbinafine primary metabolites. Lastly, we compared and discussed the findings from our computational and experimental approaches for elucidating the potentially clinically relevant pathways leading to TBF-A formation.
2.). MATERIALS AND METHODS
2.1). Materials
All chemical solvents were purchased from Thermo-Fisher (Waltham, MA, USA); however, special chemicals were obtained from various commercial sources. Substrate terbinafine hydrochloride and its metabolites, i.e. N-desmethyl-terbinafine hydrochloride, N-methyl-1-naphthyl methylamine hydrochloride, 1-naphthyl methylamine, naphthoic acid, and 1-napthaldehyde were purchased from Millipore-Sigma-Aldrich (St. Louis, MO, USA), while E-6,6-dimethylhept-2-ene-4-ynal (TBF-A) was obtained from Toronto Research Chemicals (North York, ON, Canada). Millipore-Sigma-Aldrich was also the source of labeling agents and internals standards including dansyl chloride, dansyl hydrazine, fluoxetine hydrochloride, and dimethyl benzaldehyde. Lastly, human liver microsomes pooled from 150 donors (HLM150) were purchased from Corning (Corning, NY, USA) for metabolism studies. Corning states that their tissues are sourced from US-based Organ Procurement Organizations, which meet high ethical standards and strict traceability.
2.2). Steady-state metabolism of terbinafine and secondary metabolites
As a model for the average adult liver, we assessed terbinafine metabolism using HLM150. We initially carried out time course reactions to establish steady state conditions for metabolism of terbinafine and identify all measurable metabolites. These reactions contained 0.2 mg/mL protein for HLM150 and 500 μΜ substrate in 50 mM potassium phosphate buffer pH 7.4. Reactions were initiated upon addition of a NADPH regenerating system (2 μU μl-1 glucose-6-phosphate dehydrogenase, 10 mM glucose 6-phosphate, 2 mM MgCl2, 500 μM NADP+) and incubated at 37°C with shaking at 350 rpm. Identical mixtures without addition of NADPH regenerating system were also incubated as negative controls. At 0, 5, 10, 20, and 30 min, reaction aliquots were quenched by adding an 8-fold volume of ice cold acetonitrile containing two internal standards (200 μM fluoxetine and 0.5 μM dimethyl benzaldehyde) and incubated on ice for 5 min to optimize precipitation of proteins and phosphate buffer (Schellinger & Carr, 2004). After 2500 rpm centrifugation at 4°C for 15 min using a Beckman GPR Centrifuge, the supernatant was transferred to a 96 well half-volume microplate and evaporated to dryness using an Organomation Microvap Nitrogen Evaporator System (Organomation Associates, Inc, Berlin MA). Dried wells were then resuspended in mobile phase (20:80 water:acetonitrile + 0.01% formic acid) for LC-MS analysis.
The initial studies led to changes in the strategy for analyzing steady-state metabolism of terbinafine. First, the optimal conditions for steady-state studies were determined to be 0.1 mg/mL protein for HLM150, varying substrate, and NADPH regenerating system in 50 mM potassium phosphate buffer pH 7.4 at 37°C with shaking until quenching of the reaction at 30 min as described. Each set of steady-state reactions was performed in triplicate and replicated three to five times. Second, several important metabolites including TBF-A were undetectable or at the limit of detection. Consequently, we developed in-house methods using labeling compounds reported by others (Tomono, Miyoshi, & Ohshima, 2015) (Goehl, Sundaresan, & Prasad, 1979) (Gros & Labouesse, 1969) to improve and expand the analysis of amines and aldehydes (vide infra). Finally, the initial rates from these steady-state reactions were plotted against substrate concentration and then fit to the Michaelis-Menten equation (hyperbolic curve) and Hill equation (non-hyperbolic curve) using in GraphPad Prism 6.0 (GraphPad Software Inc., San Diego, CA). The best-fit and corresponding kinetic constants were determined using extra sum-of-squares F test.
2.3). Labeling aldehydes with dansyl hydrazine
Given the initial absence of observable TBF-A in microsomal reactions, we developed a labeling method based on reports by others (Tomono, Miyoshi, & Ohshima, 2015) (Goehl, Sundaresan, & Prasad, 1979) using dansyl hydrazine to trap carbonyls like 1-naphthaldehyde and TBF-A into relatively stable hydrazones to improve their detection. The bulky group increased the mass of the analyte for more effective detection by MS. Quenched microsomal reaction supernatants were mixed with 1/10 volume of 55 mM dansyl hydrazine in acetonitrile and 1% acetic acid and then stored in the dark. After one hr, mixtures were evaporated to dryness using an Organomation Microvap Nitrogen Evaporator System (Organomation Associates, Inc, Berlin MA) and then resuspended in mobile phase (20:80 water:acetonitrile + 0.01% formic acid) for LC-MS analysis. Samples were stored in mobile phase at 4° C for at least two hr prior to injection to reach equilibrium. Dimethyl benzaldehyde present in the microsomal reaction quench served as an internal standard and positive control for the dansyl hydrazine labeling reaction. Samples were analyzed by our LC-MS approach by targeting masses for dansyl hydrazone derivatives of expected aldehydes (see Results, subsection 3.1).
2.4). Labeling amines with dansyl chloride
The analysis of N-methyl-1-naphthyl methylamine and 1-naphthyl methylamine was limited by low sensitivity, and thus, we adapted a method described by others (Gros & Labouesse, 1969) to label primary and secondary amines using dansyl chloride and improve detection as described for the dansyl hydrazones. Quenched microsomal reaction supernatants were dried down using Organomation Microvap Nitrogen Evaporator System (Organomation Associates, Inc, Berlin MA), and a dansyl chloride solution was prepared by combining 20 mM dansyl chloride in acetone, with 25 mM bicarbonate pH 8.3, and water at a 3:2:1 ratio. Dried wells were resuspended using 60μL of the dansyl chloride solution for labeling. After 15 min, labeling reactions were quenched by adding 10 μL 0.1 M sodium hydroxide. Samples were dried and resuspended in mobile phase (20:80 water:acetonitrile + 0.01% formic acid) for LC-MS analysis. Fluoxetine present in the microsomal reaction quench served as an internal standard and positive control for the dansyl chloride labeling reaction. Samples were analyzed by our LC-MS approach by targeting masses for dansylated derivatives of expected amines (see Results, subsection 3.1).
2.5). UHPLC-MS analysis of metabolic reactions
We employed an LC-MS method to resolve and quantitate analytes from the reactions based on m/z and co-elution with authentic standards. Reaction metabolites were separated by a Cortecs C-18 2.7 μm column (4.6 × 50 mm) using a Waters Acquity Arc UHPLC system and detected by a Waters Acquity QDa single quadrupole MS system (Waters, Milford, MA). The mobile phase consisted of Solvents A (0.01% formic acid/water) and B (0.01% formic acid/acetonitrile). A gradient method started with 65% Solvent A for 1 min, decreased to 20% over 5 min and held for 2 min. Solvent A was then increased back to 65% over 1 min and held for the remainder of the run. The total flow rate was 0.5 mL/min, and the total run time per sample was 15 min. The QDa was set with a cone voltage of 20V to detect masses from 150 to 650 in positive ion mode as reported by others (Vickers, et al., 1999). Analyte responses were normalized to internal standards and quantitated relative to authentic metabolite standards. Standard curves were used for quantitation of analytes.
As a complement to those efforts, we analyzed labeled TBF-A by mass spectrometry to determine the parent masses and fragmentation patterns for TBF-A adducts with dansyl hydrazine to validate purported structures. Samples were injected onto an Agilent Technologic 1290 Infinity HPLC using a gradient flow of 0.35 mL/min Solvent A (0.01% formic acid/water) and B (acetonitrile) through an InfinityLab Poroshell 120 EC-C-18 column (2.1 × 150 mm, 2.7 Micron). The method started at 40% Solvent B for 1 min, increased to 100% B over a 9 min period and was held for 1 min before being returned to 40% B over a 1 min period. Analytes were scanned with Agilent Technologic 6490 Triple Quad LC/MS. ESI source was operated in positive ion mode, and ion spectra were acquired in full scan mode monitoring the m/z range of 50–1000 amu. Subsequently, product ion spectra were generated from the theoretical precursor ion m/z 384 monitoring for fragmentation by collision-induced dissociation (20 eV).
2.6). Assessment of competing reactions during terbinafine metabolism
Modeling and experimental studies described and scaled reaction progression differently, which impacted the interpretation of their findings alone and in combination. For modeling efforts, the likelihood for N-dealkylations was dependent on the diversity and number of reactions represented in the training set. By contrast, reaction progression in experimental studies was determined by the rate of the reaction (Vmax) and binding interactions (Km) dependent on substrate concentration. Those kinetic values provided critical mechanistic insights on the type of forces driving reaction progression and the influence of substrate structure on them. Substrate binding to the enzyme is essential for making the chemical step possible and thus concentration should frame the analysis of reaction progression. As a strategy to assess and compare individual reactions, we limited conditions to a constant and sub-saturating substrate concentration, so that [S] << Km. The rate of the reaction defined by the Michaelis-Menten equation then was reduced to Vmax/Km, i.e. the catalytic efficiency. The use of this kinetic parameter is conditional; substrate concentrations cannot be the same at all reaction steps of a metabolic pathway and thus this approach could only be applied to assess competing reactions at an individual pathway step. This limitation equally applied to model predictions that lacked the dependence of the reaction rate on substrate concentration.
Model predictions and experimental values also differed in scaling data for individual reactions that would confound a direct comparison of the respective findings. Model probabilities ranged from 0 to 1.0, while kinetic constants were continuous variables. These qualities would scale differently the magnitude of substrate structure effects on reaction progress making direct comparisons of findings from computational and experimental studies not possible. As a solution, we calculated fractional contributions of competing reactions at each reaction step to assess the relative dominance of pathway branches during metabolism. The modeled probability for an individual reaction was divided by the sum of probabilities for all competing pathways to yield its fractional contribution to overall substrate metabolism. Similarly, the Vmax/Km for an individual reaction at a step in the pathway was divided by the sum of all values of competing reactions to determine its fractional contribution to substrate metabolism. Despite these efforts, neither experimental nor modeling approaches lent themselves to assessing the metabolic flux of terbinafine through Pathways 1, 2 and 3 to yield TBF-A; the modeling lacked a way to account for the dependence of reactions on concentration and experimental studies were carried out under steady-state conditions at equilibrium. Nevertheless, we developed robust strategies for studying partitioning at individual steps of metabolism and comparing insights gained from modeling predictions and experimental observations.
3.). RESULTS
3.1). Labeling significantly expanded observable terbinafine metabolites
Terbinafine underwent extensive metabolism by HLM150 leading to ten individual metabolites detected by MS (Fig. 1). In the positive ion mode, direct analysis of the initial time course data revealed the presence of terbinafine (m/z 292), desmethyl-terbinafine (m/z 278, unlabeled; m/z 511, labeled), N-methyl-1-naphthyl methylamine (m/z 172, unlabeled; m/z 405, labeled), 1-naphthyl methylamine (m/z 158, unlabeled; m/z 391, labeled), hydroxyterbinafine (m/z 308), and terbinafine dihydrodiols (two isomers m/z 326). Due to a lack of commercially available standards, hydroxyterbinafine and dihydrodiol identification relied on m/z responses and resolution of isomers as described previously by others (Vickers, et al., 1999). Among the metabolites, N-methyl-1-naphthyl methylamine and 1-napthyl methylamine were detectable in reactions only at high terbinafine levels (>100 μΜ). Importantly, TBF-A was not observed at all. In negative ion mode, we observed only trace amounts of naphthoic acid formed during the reaction. We improved significantly sensitivity for metabolites through labeling approaches that enabled the first stable trapping of TBF-A. Through dansyl hydrazine labeling, we observed all three aldehydes from the reaction including 1-naphthaldehyde (m/z 404, labeled), TBF-A (m/z 384, labeled) and formaldehyde (m/z 278, labeled), which were not detected in the unlabeled reaction analysis. Given the novelty and significance of TBF-A, we confirmed the structure of the purported hydrazone by LC-MS/MS to observe fragmentation of the theoretical precursor ion of m/z 384 (data not shown). Dansylation of amines using dansyl chloride improved sensitivity for N-methyl-1-naphthyl methylamine and 1-naphthyl methylamine by at least 1000-fold based on the standard curve slopes. This outcome made it possible to carry out kinetics of the former while only trace amounts of the latter were detected. Of the N-dealkylation metabolites (Fig. 1), N-methyl-6,6-dimethyl-2-hepten-4- yn-1-amine and 6,6-dimethyl-2-hepten-4-yn-1-amine were not detected in reactions despite the labeling techniques.
3.2). Establishing steady-state conditions revealed TBF-A decay
We determined steady-state conditions in a two-step process. We initially assessed the linearity of metabolite formation during the reaction as a function of time and then chose a single time point within that range to validate linearity with response to protein concentration. The time courses for metabolites except TBF-A were linear up to 40 min (Fig. 2.A). During the reaction, TBF-A levels initially rose, gradually crested, and then began to decrease, suggesting a significant process of elimination. Given its reactivity, we investigated whether TBF-A levels decreased in the presence of active or heat-inactivated HLMs as well as the dependency of decay on NADPH. TBF-A decayed rapidly in buffer solution and accelerated in the presence of HLM. There were no differences among reactions with HLMs indicating a lack of significance contributions from enzymes on decay (Fig. 2.B). Subsequent studies revealed the exponential rate of decay depended on TBF-A concentration consistent with a first order process (Fig. 2.C). This observation explains the observed increase in loss of TBF-A during the time course, because higher concentration would increase the rate of decay. Efforts to identify possible products from the decay even in the absence of HLM were unsuccessful. For kinetic studies, we carried out reactions at a 30 min reaction time to maximize yields, and hence sensitivity, while under steady-state conditions. Under those conditions, observed rates for desmethyl-terbinafine were linear as a function of protein concentration from 0.05 to 0.3 mg/mL (Fig. 2.D), so that 0.1 mg/mL was chosen for steady-state studies. Lastly, steady-state reactions under these conditions yielded only primary metabolites, indicating no significant secondary metabolism.
Fig. 2. Reaction conditions for formation of TBF-A and co-metabolites.

While establishing steady-state conditions, TBF-A decay was observed under reaction conditions. Unless otherwise noted, conditions were 500 μΜ substrate in 0.5 mg/mL HLM initiated with NADPH and incubated at 37°C for 30 min. (A) Linear formation of N-methyl-1-naphthyl methylamine (red circle) from terbinafine observed for 40 min. TBF-A (orange triangle) levels increased, plateaued and then decreased during that time. (B) TBF-A depletion observed in potassium phosphate buffer only (green circle), HLM plus NADPH (red square), HLM without NADPH (blue triangle), and heat inactivated HLM (purple diamond). (C) Decay rates for TBF-A over 30 min in buffer increased linearly as a function of starting TBF-A concentration. (D) Linear formation of desmethyl-terbinafine (N-dealkylation metabolite) observed from terbinafine as a function of HLM concentration from 0.025 to 0.2 mg/mL.
3.3). Terbinafine N-dealkylation through Pathway 1 efficiently yielded TBF-A
For Pathway 1, N-dealkylation of terbinafine yields TBF-A and N-methyl-1-naphthyl methylamine. Kinetic profiles for the co-metabolites are shown in Fig. 3.A and 3.B and were fit best to the Michaelis-Menten mechanism yielding kinetic constants summarized in Table 1. Due to decay, TBF-A kinetics would inherently underestimate the true kinetics for the reaction. By contrast, the rates for N-methyl-1-naphthyl methylamine directly reflect the efficiency of this pathway to TBF-A formation. Based on the fits of the data, the apparent Vmax for TBF-A was six-fold lower and the Km two-fold lower than that observed for N-methyl-1-naphthyl methylamine. Nevertheless, the combination of these differences resulted in a more than two-fold underestimation of efficiency of the path when measuring TBF-A directly.
Fig. 3. Steady-state kinetic profiles for Pathway 1 of terbinafine N-dealkylation.

N-Dealkylation of terbinafine yielded metabolic kinetics for two metabolites derived from Pathway 1 as illustrated in Fig. 1. The kinetic profiles include those for (A) TBF-A (dansyl hydrazine labeled) and (B) N-methyl-1-naphthyl methylamine (dansyl chloride labeled). Both sets of data were fit best to the Michaelis-Menten equation (p < 0.05), and the corresponding constants reported in Table 1. Twelve experimental reactions were carried out with terbinafine as substrate. Reaction conditions and data analysis were carried out as described in Materials and Methods.
Table 1.
Michaelis-Menten Kinetic Constants for Terbinafine N-Dealkylation Pathways.a
| Steady-state kinetic constants for individual metabolites | ||||||||
|---|---|---|---|---|---|---|---|---|
| Pathway | Step | Substrate | Vmaxb | Km (μΜ) | Vmax/Km | Vmaxb | Km (μΜ) | Vmax/Km |
| Pathway 1 | 1 | terbinafine | TBF-Ac | N-methyl-1 -naphthyl-methylamine | ||||
| 33.7 ± 1.4 | 20.7 ± 4.2 | 1.63 | 218 ± 24 | 54.6 ± 17 | 3.99 | |||
| Pathway 2 | 2 | terbinafine | desmethyl-terbinafine | Formaldehyde | ||||
| 344 ± 16 | 30.1 ± 6.2 | 11.4 | high background masking rates | |||||
| 2A | desmethyl-terbinafine | TBF-Ac | 1-naphthyl-methylamine | |||||
| 132 ± 15 | 133 ± 32 | 1.05 | below limit of quantitatione | |||||
| 2B | desmethyl-terbinafine | 1-naphthaldehyde | 6,6-dimethyl-2-hepten-4-yn-1-amine | |||||
| 19.1 ± 1.4 | 68.4 ± 15 | 0.28 | undetected (unlabeled or labeled) | |||||
| Pathway 3d | 3 | terbinafine | 1-naphthaldehyde | N-methyl-6,6-dimethyl-2-hepten-4-yn-1-amine | ||||
| 6.38 ± 0.33 | 45.9 ± 9.7 | 0.14 | undetected (unlabeled or labeled) | |||||
Data fit best to the Michaelis-Menten equation over the Hill equation (P < 0.05). Values shown with standard deviation from mean.
Units are pmol/min/mg protein.
Kinetic results reflect underestimation of rates due to competing decay process as shown in Fig 2.
Pathway steps 3A and 3B not studied due to absence of authentic standards and low efficiency of previous step to obviate the significance of this pathway for TBF-A.
Limit of quantitation calculated as standard deviation of response divided by slope of standard curve.
3.4). First step of terbinafine N-dealkylation through Pathway 2 was most efficient
For Pathway 2, TBF-A formation requires a two-step process. Initial N-dealkylation of terbinafine produces formaldehyde and desmethyl-terbinafine. Despite our best efforts, background formaldehyde levels remained too high to accurately measure formation rates during metabolism. Desmethyl-terbinafine rates could be determined from unlabeled and labeled reactions; however, the dansyl hydrazine labeling reaction interfered with quantitation of the metabolite for reactions at higher terbinafine concentrations through unknown processes (data not shown). Fig. 4.A displays the resulting kinetic profile and Table 1 lists the corresponding constants for what was the most efficient N-dealkylation reaction for terbinafine. Subsequent metabolism of desmethyl-terbinafine branches off into two directions. One path leads to TBF-A and 1- naphthyl methylamine (Pathway 2.2A, Fig. 1); however, only TBF-A levels were consistently measurable, resulting in the kinetic profile in Fig. 4.B. Based on the kinetic constants (Table 1), this step is much less efficient than the initial N-dealkylation and the analogous step in Pathway 1 leading to TBF-A. The competing path (Pathway 2.2B, Fig. 1) leads to 1-naphthaldehyde and 6,6-dimethyl-2-hepten-4-yn-1-amine, although only kinetics for 1-naphthaldehyde were measurable (Fig. 4.C; Table 1). Despite a similar Km to other reactions, the Vmax was one of the lowest, indicating poor metabolic efficiency. By comparison, Pathway 2.2A is more efficient that Pathway 2.2B; however, the actual difference in efficiency is much higher due to the underestimation of TBF-A yields.
Fig. 4. Steady-state kinetic profiles for Pathway 2 of terbinafine N-dealkylation.

N-Dealkylation of terbinafine and desmethyl-terbinafine yielded metabolic kinetics for three metabolites derived from Pathway 2 as illustrated in Fig. 1. The kinetic profiles include those for (A) desmethyl-terbinafine (dansyl chloride labeled) from terbinafine, (B) TBF-A (dansyl hydrazine labeled) from desmethyl-terbinafine (Path 2.2A), and (C) naphthaldehyde (dansyl hydrazine labeled) from desmethyl-terbinafine (Path 2.2B). All sets of data were fit best to the Michaelis-Menten equation (p < 0.05), and the corresponding constants reported in Table 1. Nine experimental reactions were carried out with terbinafine and desmethyl-terbinafine as substrate. Reaction conditions and data analysis were carried out as described in Materials and Methods.
3.5). Terbinafine N-dealkylation through Pathway 3 was the least efficient
Like Pathway 2, this pathway requires two steps to generate TBF-A. The N-dealkylation step generates naphthaldehyde and N-methyl-6,6-dimethyl-2-hepten-4-yn-1-amine, although only the aldehyde was detectable at levels necessary for determining reaction rates. The kinetic profile for the reaction is shown in Fig. 5 and the corresponding constants in Table 1. Despite similarities to Path 2.2B, N-denaphthylation was about two-fold less efficient due mostly to a lower Vmax. The overall kinetics of this reaction indicated it to be the most inefficient metabolic step among all analyzed in this study. Subsequent reactions would then be irrelevant to terbinafine metabolism yielding TBF-A; thus, further studies were not carried out.
Fig. 5. Steady-state kinetic profiles for Pathway 3 of terbinafine N-dealkylation.

N-Dealkylation of terbinafine yielded metabolic kinetics for one metabolite derived from Pathway 3 as illustrated in Fig. 1. The kinetic profile for naphthaldehyde (dansyl hydrazine labeled) is shown. Data were fit best to the Michaelis-Menten equation (p < 0.05), and the corresponding constants reported in Table 1. Nine experimental reactions were carried out with terbinafine as substrate. Reaction conditions and data analysis were carried out as described in Materials and Methods.
3.6). Structure-function relationships identified for terbinafine N-dealkylations
Information gained from computational modeling and experimental studies provided an opportunity to assess the impact of structure on the metabolism of terbinafine derivatives. Among all three pathways (Fig. 6), modeled N-dealkylation to yield TBF-A increased in probability with decreasing terbinafine substituents. Direct formation of TBF-A was the least probable (Pathway 1.1, P 0.06), yet the probability increased significantly after N-demethylation (Pathway 2.2a, P 0.35) and much more in the absence of the bulky naphthyl moiety (Pathway 3.2, P 0.74). N-Demethylation reactions for Pathways 2.1 and 3.2 were consistently highly probable (P > 0.74). Like reactions yielding TBF-A, N-denapthylation probability was low (Pathway 3.1, P 0.09), but increased when the methyl group was absent (Pathway 2.2b, P 0.21). Compared to the parent drug, N-dealkylation of the terbinafine diol metabolites slightly decreased in probability for all steps. By contrast, pathways for terminal hydroxyl-or carboxylterbinafine significantly decreased in probability especially with respect to N- demethylation and N-dealkylation leading to TBF-A. Overall, probabilities for reactions generally increased with fewer and less bulky N-alkyl groups.
Fig. 6. An overview of the kinetic efficiencies, model predictions and fractional contributions for terbinafine N-dealkylation pathways.

Reactions are labeled by pathway number, reaction number, and branch designation if applicable (e.g. 2.2a). The table lists information gained from experimental and computational modeling studies as well as their limitations (see Results for details). These data include the experimentally measured Vmax/Km values, modeled reaction probabilities, and fractional contributions for each reaction at the pathway branching point based on the respective data sets. The model was also used to predict probabilities for N-dealkylations of known primary metabolites of terbinafine, for which we have no experimental kinetics. *TBF-A efficiencies are underestimated due to decay kinetics competing with those for formation of TBF-A.
Due to technical challenges, an analysis of structure-function relationships with experimental data was limited to only two of the N-dealkylation reaction types for terbinafine (Fig. 6). While semi-quantitative, we compared kinetics for TBF-A formation in Pathways 1.1 and 2.2a based on the use of a similar methodology for analysis of the reaction kinetics. Given that condition, the rate of TBF-A formation increased after removal of the terbinafine methyl group, but substrate binding (Km) increased more comparatively, leading to a decreased overall catalytic efficiency. By contrast, the loss of the methyl group doubled the efficiency of N-denaphthylation from 0.14 to 0.28 mainly due to a higher rate of turnover. No experiments were carried out on the oxidized primary metabolites of terbinafine due to their lack of commercial availability.
3.7). Metabolic reactions strongly favored over others in terbinafine N-dealkylation
We leveraged findings from computational and experimental studies to determine relative significance of competing reactions at each metabolic step by calculating their fractional contributions. At the onset of metabolism, computational modeling strongly favored partitioning to Pathway 2 with minor competition from alternate pathways (Fig.6). The resulting major metabolite desmethyl-terbinafine then favored N-dealkylation to TBF-A over N-denaphthylation. The minimal partitioning through the first step of Pathway 3 led to a similarly low preference for generation of TBF-A. Based on experimental data, nearly three-fourths of terbinafine underwent N-demethylation in Pathway 2 and one-fourth N-dealkylation to yield TBF-A directly in Pathway 1 (Fig. 6). Terbinafine N-denaphthylation in Pathway 3 was negligible. We applied a similar strategy to assess the contributions of competing pathways to desmethyl-terbinafine metabolism in Pathway 2. The reaction leading to TBF-A was preferred most (80%) of the time compared to N-denaphthylation. Taken together, these findings indicate preferential steps leading to TBF-A formation but importantly cannot predict the concentration-dependent flux to TBF-A through these multi-step pathways.
3.8). Non-dealkylation pathways for terbinafine involved high affinity pathways
Despite the focus on N-dealkylation pathways, observation of hydroxyterbinafine and terbinafine dihydrodiol metabolites (Fig. 1) during steady-state reactions provided the first insights on their metabolic pathways. In the absence of standards, it was only possible to fit the observed response rates as a function of terbinafine concentration. In following, we determined the metabolic mechanism and kinetic constant reflecting enzyme-substrate interactions but not the maximal rate. The kinetic profiles were measured from at least three experimental replicates and fit best to the Michaelis- Menten mechanism (p < 0.05) (Fig. 7.A and 7.B). The Km for hydroxyterbinafine was 10.9 + 6.1 μΜ, while that for the dihydrodiols were 15.3 + 5.1 and 18.6 + 5.5 μM, respectively. Similarly, we determined the Km for dihydriodiols to be 22.1 + 4.2 μM for the metabolism of desmethyl-terbinafine (Fig. 7.C). Desmethyl hydroxyterbinafine was not detected as a metabolite from the reaction though.
Fig. 7. Steady-state kinetic profiles for metabolites of other oxidation pathways of terbinafine metabolism.

Steady-state reactions yielded several metabolites from oxidative pathways not involving N-dealkylation. No quantitative standards for the products were available, so data was reported based on MS peak area. No labeling was used for detection of these metabolites. Terbinafine reactions yielded profiles for (A) hydroxyterbinafine and (B) two isomers of terbinafine dihydrodiol (m/z). (C) Desmethyl-terbinafine reactions yielded profiles for two isomers of desmethyl-terbinafine dihydrodiol, (m/z 312) not shown in Fig. 1. Three experimental reactions were carried out for each substrate. Reaction conditions were carried out as described in Materials and Methods.
4.). DISCUSSION
4.1). Development of novel methods was necessary to study TBF-A
Knowledge of drug metabolism led to speculation over thirty years ago on the possibility of terbinafine metabolism yielding reactive, toxic metabolites (Jensen, 1989) (Battig, Nefzger, & Schulz, 1987), yet the likelihood for bioactivation pathways remained elusive until now. In this study, we combined computational modeling and experimental approaches to assess the significance of three N-dealkylation pathways for terbinafine to generate the reactive alpha, beta unsaturated aldehyde TBF-A (Fig. 1). Rather than speculation, our modeling of metabolic pathways yielded probabilities for each reaction step that shed light on competition for alternate branches within the pathways leading to TBF-A. Despite high probabilities predicted for TBF-A formation, multiple in vivo (Jensen, 1989) (Humbert, Cabiac, Denouel, & Kirkesseli, 1995) (Kovarik, Mueller, Zehender, Denouel, Caplain, & Millerioux, 1995) and in vitro (Vickers, et al., 1999) studies, including the current one, failed to directly observe TBF-A or any possible downstream metabolites. The first evidence for TBF-A required trapping with glutathione (Iverson & Uetrecht, 2001). The reversibility of the resulting adduct formation indicated only a fraction of the metabolite was observable at any given point. A more quantitative approach would be necessary to more accurately measure TBF-A levels formed during metabolism. We addressed this problem by developing a novel trapping method to create a stable dansylated hydrazone of TBF-A. Dansyl hydrazine trapping revealed unexpectedly high levels of TBF-A formation and importantly, its subsequent decay during metabolic reactions.
4.2). TBF-A spontaneously decayed in solution
TBF-A decay likely explains why the reactive metabolite has escaped detection for so long; knowledge of the mechanism of decay is then necessary for understanding how the process impacts TBF-A levels and its role in toxicity. TBF-A spontaneously decayed in buffered solution and slightly accelerated in the presence of HLM150 regardless of whether or not the enzymes were heat inactivated, indicating non-enzymatic processes at work. The first order process could be explained through two mechanisms. First, TBF-A may react with itself, perhaps by forming a dimer. Alternatively, TBF-A may react with water. This bimolecular process would appear as a pseudo-first order process due to high bulk water concentration (55 M). A plausible reaction between water and TBF-A is hydration of the double bond to yield an aldol as reported for acrolein (Aizenbud, Aizenbud, Reznick, & Avezov, 2016) - another alpha, beta unsaturated aldehyde. Unfortunately, we were not able to detect a TBF-A aldol or any of its possible degradative products. Nevertheless, the formation of transient, stable TBF-A species would decrease the rate of decay and hence increase its half-life. The spontaneous decay of TBF-A did not occur in methanol used to prepare our stock solutions. This observation may reflect the formation of a stabilizing hemiacetal as observed for formaldehyde. Similarly, the formation of a reversible glutathione adduct with TBF-A reported by Iverson and Uetrecht (Iverson & Uetrecht, 2001) may stabilize the reactive metabolite against decay and thus create an accumulation of reactive metabolite that could be transferred or released by glutathione to cause damage to proteins. Such a mechanism would have potential toxicological relevance given the high levels of glutathione (~7 mM) in the liver (Wahllander, Soboll, & Sies, 1979) capable of driving equilibrium to adduct formation.
4.3). TBF-A formation was significant during terbinafine metabolism
Our trapping of TBF-A as a stable hydrazone led to its first reported kinetics. The initial time course studies for terbinafine metabolism revealed the impact of first order decay on TBF-A levels as they rose and then fell while other metabolites increased linearly over the same time frame. Consequently, studies using late time points would significantly underestimate TBF-A levels, which may explain very low levels of the glutathione adduct of TBF-A reported previously from a 1 hr reaction and high microsomal protein levels (Iverson & Uetrecht, 2001). The decay process induced an underestimation of TBF-A kinetics and thus, we sought an alternate, possibly more accurate way to assess TBF-A formation relying on kinetics for the amine co-metabolites in Pathway 1.1 and 2.2a (Fig. 1). Unfortunately, those amines demonstrated low sensitivity and high variability and in fact, neither of them has been reported in other studies (Vickers, et al., 1999) (Jensen, 1989) (Kovarik, Mueller, Zehender, Denouel, Caplain, & Millerioux, 1995). As an analytical solution, we developed a novel dansyl chloride approach that increased sensitivity to the metabolites 1000-fold, making quantitative kinetic studies possible. Kinetics for N-methyl-1-naphthyl methylamine during terbinafine metabolism confirmed measurable and relatively high rates of formation for these pathways missed in previous studies. While the strategy worked in that case, we had less success with reactions using desmethyl-terbinafine as a substrate. The low efficiency of this reaction led only to measurable kinetics by TBF-A formation. These relatively efficient metabolic processes yielding TBF-A confirmed the high probabilities for those reaction steps predicted by our modeling efforts. During the reaction, the measured levels of TBF-A would reflect the balance between formation of the reactive metabolite and its decay as well as the mitigating effects of processes stabilizing TBF-A in solution.
4.4). Modeling and experimental designs impacted the interpretation of results
The differences in how modeling and experimental studies described and scaled reaction progression impacted the interpretation of their findings alone and in combination. Modeling efforts generated a probability for N-dealkylation to occur based on patterns of known reactions in a training set. By contrast, experimental studies measured the significance of reaction progression through the rate of the reaction (Vmax) and binding interactions (Km) dependent on substrate concentration. Those metrics provided additional insights on how substrate structures impacted the types of forces contributing to the reaction. The observed rate of reaction depends on concentration to saturate the enzyme. Thus, we limited conditions to a constant and sub-saturating substrate concentration, so that the rate of the reaction was reduced to the catalytic efficiency (Vmax/Km) for assessing and comparing individual reactions. When considering metabolic pathways, substrate concentration is not the same, making our approach only suitable for assessing competing reactions at an individual pathway step. Similarly, such an analysis was necessary for modeling predictions given the lack of consideration of the concentration as a driving force in probabilities for reaction progression. Moreover, modeling and experimental approaches scaled values differently, impacting their interpretation and comparison as a function of substrate structure. As a solution, we calculated fractional contributions using modeling and experimental data for competing reactions at each reaction step to assess the relative dominance of pathway branches during metabolism. Despite these advantages, the avoidance of concentration in the analyses precluded an assessment of the metabolic flux of terbinafine through Pathways 1,2 and 3 to yield TBF-A. Nevertheless, our robust strategies provided ways to study partitioning at individual metabolic steps and compare insights gained from modeling predictions and experimental observations.
4.5). TBF-A derived mainly from Pathway 1 under steady-state conditions
Modeling predictions and experimental studies agreed on the significance of N-demethylation (Pathway 2) and insignificance of N-denaphthylation (Pathway 3) in terbinafine metabolism, yet differed on the importance of direct TBF-A formation through Pathway 1 (Fig. 6). According to terbinafine reaction kinetics, direct formation of TBF-A in Pathway 1 was the most important source of the reactive metabolite under steady-state conditions. The efficiency of the reaction and fractional contributions was only three-fold less than that for the first step of Pathway 2. Desmethyl-terbinafine may be the major metabolite from the terbinafine reaction, but its subsequent metabolism to yield TBF-A is less efficient than the parent drug, making contributions minimal. Moreover, steady-state conditions obviate significant product (desmethyl-terbinafine) accumulation, which would further decrease the catalytic potential to ultimately generate the reactive metabolite through Pathway 2. Consequently, the partitioning of steps in terbinafine metabolism suggests that TBF-A formation through Pathway 2 is not as significant as Pathway 1 under steady-state conditions. By comparison, the N-dealkylation model correctly predicted a high probability for terbinafine metabolism down Pathway 2 yet significantly underpredicted the likelihood for Pathway 1. In fact, the direct formation of TBF-A was the least probable N-dealkylation pathway. For Pathways 3 and 2.2b, both modeling and experimental studies reported poor N-denaphthylation of terbinafine and desmethyl-terbinafine. These outcomes may reflect sufficient representation of N-demethylations and N-denaphthylations in the training set, but a lack of N-dealkylations yielding a relatively long alkyl metabolite. Given this insight, we can expand the number and diversity of those types of reactions and retrain the model to improve accuracy. Taken together, these efforts revealed the importance of direct formation of TBF-A during terbinafine metabolism as a potential initiator of toxicity.
4.5). Two TBF-A pathways may be relevant under in vivo conditions
The emphasis on terbinafine metabolism describes only part of the story about its reactive metabolite; in practice, patients dose terbinafine daily leading to the accumulation of metabolites due to variations in their absorption, distribution, metabolism, excretion, and transport. In fact, plasma levels of terbinafine and desmethyl-terbinafine are approximately equivalent (Kovarik, Mueller, Zehender, Denouel, Caplain, & Millerioux, 1995); it is possible that metabolic Pathways 1.1 and 2.2a simultaneously contribute to TBF-A generation in patients (Fig. 6). The similarity in concentration would translate into an almost two-fold higher contribution from Pathway 1 due to its higher efficiency for that metabolic step. Moreover, clinical studies also reported carboxyterbinafine and desmethyl-carboxyterbinafine in plasma (Humbert, Cabiac, Denouel, & Kirkesseli, 1995) that may also undergo metabolism to yield TBF-A. These alternative oxidation pathways were not investigated experimentally in this study due to the emphasis on competing N-dealkylation pathways that lead to the reactive metabolite TBF-A. Modeling studies suggested the introduction of a negatively-charged group at the terminus of the molecule significantly decreased the probability of metabolism. This observation seems reasonable given that cytochromes P450 typically prefer hydrophobic substrates. Based on these collective studies, if the likelihood for TBF-A generation is so high, then why is toxicity from this reactive metabolite not more commonly observed? The answer may lie in our observation of a high decay rate for TBF-A. Toxicity in patients may then depend on the balance between TBF-A formation and decay for TBF-A; a lack of understanding the factors impacting that balance would make subsequent adverse drug events with terbinafine appear idiosyncratic.
4.6). General lessons about reactive metabolites
In the case of terbinafine, several factors confounded an accurate determination of its reactive metabolite. TBF-A eluded direct observation by LC coupled to MS and spectroscopic detection based on its physico-chemical properties such (Vickers, et al., 1999). A labeling method would be necessary to reveal its presence but glutathione was not an effective trap for it especially given its reversibility (Iverson & Uetrecht,2001). Despite the capture of the reactive metabolite, its decay further restricted the conditions for observing and studying TBF-A properties. Taken together, these limitations in experimental design and instrumentation highlight challenges in demonstrating how potentially important reactive metabolites in the clinic can be missed by experimental approaches.
4.7). Concluding remarks
We are the first to identify pathways leading to generation of the reactive metabolite TBF-A as a potential initiator of idiosyncratic adverse drug interactions. We achieved this goal through a combination of novel tools in modeling and experimental approaches. The N-dealkylation model predicted a high likelihood for TBF-A formation among three possible pathways, yet experimental evidence for reactive metabolites required a trapping method to stabilize it for quantitative studies as well as revealing a critical dynamic for steady-state TBF-A levels. A comparison of the information gained from modeling and experimental approaches posed challenges due to their differences in type and scaling of information; however, the calculation of fractional contributions provided a solution for comparing competing reactions at each reaction step. Based on experimental results, modeling the pathways was accurate for the major N- demethylation step but less so for N-dealkylations yielding TBF-A. Training with a larger more diverse set of N-dealkylations could resolve this shortcoming. In the end, the kinetics revealed that TBF-A formation is not a minor pathway but a relatively significant one when compared to competing reactions. On-going studies are focusing on identifying specific cytochrome P450 isozymes responsible for these reactions. These isozymes and their activity are integral to full understanding of TBF-A generation and vary in the population due to clinical factors such as genetic polymorphisms, sex, age, race, ethnicity, and pathophysiological conditions. Taken together, our novel design of modeling and experimental studies provided a more powerful approach to detecting and quantitating metabolites leading to a more accurate assessment of possible critical pathways and corresponding kinetics that would have otherwise been missed.
Acknowledgments:
Research reported in this publication was supported by the National Library of Medicine of the National Institutes of Health under award numbers R01LM012222 and R01LM012482. Computations were performed using the facilities of the Washington University Center for High Performance Computing that was partially funded by National Institutes of Health (NIH) grant numbers 1S10RR022984–01A1 and 1S10OD018091–01. Na Le Dang was partially supported by NIH Medical Scientist Training Program grant GM07200. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflicts of Interest Statement:
The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest: none
REFERENCES
- Abdel-Rahman S, & Nahata M (1997). Oral terbinafine: A new antifungal agent. Ann. Pharmacother, 31, 445–456. [DOI] [PubMed] [Google Scholar]
- Aizenbud D, Aizenbud I, Reznick A, & Avezov K (2016). Acrolein—an α,β-Unsaturated Aldehyde: A Review of Oral Cavity Exposure and Oral Pathology Effects. Rambam Maimonides Med J, 7, e0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajit C, Suvannasankha A, Zaeri N, & Munoz S (2003). Terbinafine-associated hepatotoxicity. Am. J. Med. Sci, 325, 292–295. [DOI] [PubMed] [Google Scholar]
- Annotation of FDA Label for terbinafine and CYP2D6. (2013). Retrieved from https://www.pharmgkb.org/label/PA166104822
- Attia S (2010). Deleterious effects of reactive metabolites. Oxid. Med. Cell Longev, 3, 238–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battig F, Nefzger M, & Schulz G (1987). Major biotransformation routes of some allylamine antimycotics. Barcelona: J. R. Prous Science Publishers, S. A. [Google Scholar]
- Dang N, Hughes T, Miller G, & Swamidass S (2018). Computationally assessing the bioactivation of drugs by N-dealkylation. Chem. Res. Toxicol, 31, 68–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goehl T, Sundaresan G, & Prasad V (1979). Fluorometric high-pressure liquid chromatographic determination of hydrocortisone in human plasma. J. Pharm. Sci, 68, 1374–1376. [DOI] [PubMed] [Google Scholar]
- Gros C, & Labouesse B (1969). Study of the Dansylation Reaction of Amino Acids, Peptides and Proteins. European J. Biochem, 7, 463–470. [DOI] [PubMed] [Google Scholar]
- Gupta A, Sibbald R, Knowles S, Lynde C, & Shear N (1997). Terbinafine therapy may be associated with the development of psoriasis de novo or its exacerbation Four case reports and a review of drug-induced psoriasis. J. Am. Acad. Dermatol, 36, S858–862. [DOI] [PubMed] [Google Scholar]
- Hall M, Monka C, Krupp P, & O’Sullivan D (1997). Safety of oral terbinafine: Results of a postmarketing surveillance study of 25884 patients. Arch. Dermatol, 133, 1213–1219. [DOI] [PubMed] [Google Scholar]
- Humbert H, Cabiac M, Denouel J, & Kirkesseli S (1995). Pharmacokinetics of terbinafine and of its five main metabolites in plasma and urine, following a single oral dose in healthy subjects. Biopharm Drug Dispos, 16, 685–694. [DOI] [PubMed] [Google Scholar]
- Iverson S, & Uetrecht J (2001). Identification of a reactive metabolite of terbinafine: Insights into terbinafine-induced hepatotoxicity. Chemical Research in Toxicology. [DOI] [PubMed] [Google Scholar]
- Jensen J (1989). Clinical pharmacokinetics of terbinafine (Lamisil). Clinical and Experimental Dermatology. [DOI] [PubMed] [Google Scholar]
- Kovarik J, Mueller E, Zehender H, Denouel J, Caplain H, & Millerioux L (1995). Multiple-dose pharmacokinetics and distribution in tissue of terbinafine and metabolites. Antimicrob. Agents Chemother, 39, 2739–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyden J (1998). Pharmacokinetics and pharmacology of terbinafine and itraconazole. Drug Metab. Dispos, 27, S42–47. [DOI] [PubMed] [Google Scholar]
- Madani S, Barilla D, Cramer J, Wang Y, & Paul C (2002). Effect of Terbinafine on the Pharmacokinetics and Pharmacodynamics of Desipramine in Healthy Volunteers Identified as Cytochrome, 1211–1218. https://doi.Org/10.1177/0091270002238763 [DOI] [PubMed]
- Medication Guide: Lamisil (terbinafine hydrochloride) Tablets. (2016). East Hanover, New Jersey: Retrieved from https://www.fda.gov/downloads/Drugs/DrugSafety/UCM524717.pdf [Google Scholar]
- Napodano J (2012). Anacor Looks Right For A Long-Term Play. Retrieved from Seeking Alpha. [Google Scholar]
- Ryder N (1989). The mechanism of action of terbinafine. Clin. Exp. Dermatol, 14, 110–113. [DOI] [PubMed] [Google Scholar]
- Schellinger A, & Carr P (2004). Solubility of buffers in aqueous-organic eluents for reversed-phase liquid chromatography. LCGC North America, 22, 544–548. [Google Scholar]
- Schuster I (1987). Metabolic degradation of terbinafine in liver microsomes from man, guinea pig and rat, in Recent Trends in the Discovery, Development and Evaluation of Antifungal Agents (Fromtling RA ed.). Barcelona: JR Prous Science Publishers. [Google Scholar]
- Terbinafine product monograph: Canada. (1995). 1–33. Dorval, Quebec.
- Tomono S, Miyoshi N, & Ohshima H (2015). Comprehensive analysis of the lipophilic reactive carbonyls present in biological specimens by LC/ESI-MS/MS. J. Chromatogr. B, 988, 149–156. [DOI] [PubMed] [Google Scholar]
- Trépanier E, Nafziger A, Kearns G, Kashuba A, & Amsden G (1998). Absence of effect of terbinafine on the activity of CYP1A2, NAT-2, and Xanthine oxidase. J. Clin. Pharmacol, 38, 424–428. [DOI] [PubMed] [Google Scholar]
- Vickers A, Sinclair J, Zollinger M, Heitz F, Gl??nzel U, Johanson L, et al. (1999). Multiple cytochrome P-450s involved in the metabolism of terbinafine suggest a limited potential for drug-drug interactions. Drug Metabolism and Disposition. [PubMed] [Google Scholar]
- Wahllander A, Soboll S, & Sies H (1979). Hepatic mitochondrial and cytosolic glutathione content and the subcellular distribution of GSH-S-transferases. FEBS Letters, 97, 138–140. [DOI] [PubMed] [Google Scholar]