

Abstract
Supramolecular assembly of peptide-based monomers into nanostructures offers many promising applications in advanced therapies. In this Tutorial Review, we introduce molecular designs to control the structure and potential biological function of supramolecular assemblies. An emphasis is placed on peptide-based supramolecular nanostructures that are intentionally designed to signal cells, either directly through the incorporation of amino acid sequences that activate receptors or indirectly by recruiting native signals such as growth factors. Additionally, we describe the use and future potential of hierarchical structures, such as single molecules that assemble into nanoscale fibers which then align to form macroscopic strings; the strings can then serve as scaffolds for cell growth, proliferation, and differentiation.
Introduction
Supramolecular polymerizations, in contrast to covalent polymerizations, use monomers that are connected through non-covalent interactions such as hydrogen bonds, π–π overlap, electrostatic interactions, and van der Waals interactions. These attractive forces are typically much weaker than covalent bonds, however non-covalent interactions are dynamic and monomer-to-supramolecular polymer transitions can be reversible. Supramolecular materials thus offer a number of appealing properties including the potential for self-healing, recyclability, and stimuli responsiveness. Additionally, directional interactions between monomers enable the formation of discrete nanostructures with a high degree of internal molecular order.1 Given this reversibility, it is not surprising that supramolecular polymerizations are integral to many cellular functions. For example, microtubules are filamentous structures formed by the supramolecular assembly of tubulin proteins located inside cells. The assembly-to-disassembly process is dynamic and reversible, which is critical for the microtubules’ role in organizing the cell interior and controlling the directional movement of intracellular particles and organelles.2 Inspired by the important role of supramolecular materials in biological systems, researchers have made extensive efforts to develop artificial analogs for therapeutic applications. Supramolecular biomaterials are unique because their dynamic nature potentially allows the constituent monomers to be rearranged and adapt themselves to physiological conditions, maximizing their biological function. In particular, peptide-based materials offer immense structural diversity and can be synthesized using well established, efficient methods, and are the focus of this Tutorial Review. Perhaps most important to the use of peptide-based supramolecular structures as biomaterials is the ability to easily incorporate amino acid sequences, or other biomolecular structures such as nucleic acids, fatty acids, and glycans, in order to trigger biochemical signaling in vivo. Supramolecular structures composed of peptides can also be highly biocompatible given their biomimetic chemical and mechanical properties and the non-toxic nature of byproducts when they biodegrade. Careful monomer design could even enable tuning of the rate of degradation. This Tutorial Review will highlight some of the recent developments in peptide-based supramolecular materials for therapeutic applications, particularly focusing on peptide structures that are designed to provide cell signals in addition to other functions.
While nearly all of the possible non-covalent interactions that can be used to induce supramolecular assembly have been applied to peptide-based systems, taking advantage of the natural propensity for hydrogen bonding within the secondary structure of peptides has been the natural choice for these materials. The use of peptide secondary structure motifs allows the rational design of low molecular weight peptides that assemble into nanostructures of varying morphologies, such as spheres, cylinders, sheets, and tubes.3 In many cases, however, hydrogen bonding is supplemented with an additional attractive interaction to induce assembly; arguably the most important of which is hydrophobic collapse by incorporating hydrophobic structural motifs onto the peptide. Among the earliest discovered4 and best studied systems in this class are peptide amphiphiles (PAs), which consist of a short peptide sequence linked to a hydrophobic alkyl tail (Fig 1a, example molecule 1). In 2001, the Stupp laboratory reported the first example of a PA molecule capable of forming high aspect ratio cylindrical nanofibers and hydrogels at pH values where charge density on the supramolecular assemblies was low.5 The filamentous assemblies form as a result of hydrophobic collapse of the tails and the formation of directional hydrogen bonds along the long axis of the nanofibers. A key component of the PA is a β-sheet forming amino acid sequence adjacent to the hydrophobic tail.6 Due to the significant interest in PAs for therapeutic applications as detailed in the following sections of this review, the structure and properties of PAs have been extensively studied, including many investigations from the Stupp laboratory. The internal dynamics of PA nanofibers were studied by means of electron paramagnetic resonance spectroscopy, revealing that the dynamic behavior changes from liquid to solid-like across their 10 nanometer cross-section.7 Similar techniques were also used to show that significant differences exist in water dynamics between fast-moving water in the hydrophobic interior of the PA nanofibers and slow-moving water on the surface, within a distance of 1.5 nanometers.8 These results indicate that water is not simply a solvent but rather an integral part of these peptide-based supramolecular nanostructures.
Fig. 1.
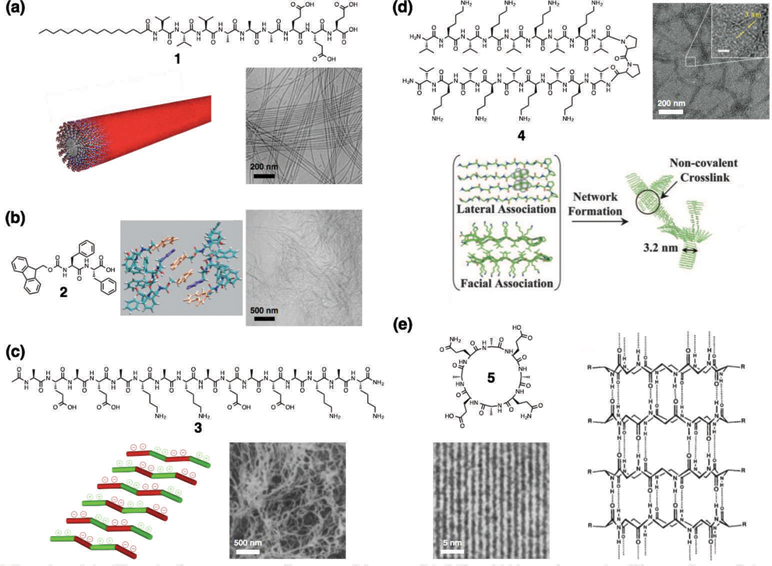
(a) Chemical structure of a peptide amphiphile (PA), schematic illustration of its supramolecular assembly into a cylindrical nanofiber, and cryogenic transmission electron microscopy (cryo-TEM) of nanofibers. Reprinted with permission from ref. 32 (Copyright 2015 Wiley-VCH). (b) Chemical structure of an Fmoc-dipeptide, schematic illustration of molecular packing (Fmoc groups: orange, phenyl groups: purple), and cryogenic scanning electron microscopy (cryo-SEM) of nanofibers. Reprinted with permission from ref. 15 (Copyright 2008 Wiley-VCH). (c) Chemical structure of the Z-DNA-binding mimetic amphipathic peptide, schematic illustration of its supramolecular assembly, and SEM of nanofibers. Reprinted with permission from ref. 16 (Copyright 1993 National Academy of Sciences). (d) Chemical structure of a β-hairpin peptide, schematic illustration of its folding and supramolecular assembly, and TEM of nanofibers. Reprinted with permission from ref. 19 (Copyright 2007 National Academy of Sciences). (e) Chemical structure of a cyclic peptide with alternating d- and l-amino acids and its tubular assembly, and TEM of closely packed nanotubes. Reprinted with permission from ref. 21 (Copyright 1993 Nature Publishing Group).
The assembly pathways of PA supramolecular structures have also been studied in two ways, the first comparing the competing solubilizing ability of the solvent mixture.9 The second used the ionic strength of the solution to tune the charge density at the PA surface, and thus control the balance between the attractive forces resulting from hydrogen bonding and hydrophobic collapse with charge repulsion due to the surface charges.10 The results of the second study imply that pH could be used to influence PA assembly, and indeed pH is one of the external stimuli that PAs have been designed to respond to (often reversibly). Dynamic structural changes to PAs in response to changes in pH have been used for targeted cancer therapy with PA 6 (Fig. 2a),11 as well as a pH-dependent contrast agent for bioimaging using noninvasive magnetic resonance imaging (MRI) with PA 7 (Fig. 2b).12 Inspired by cellular control of protein function, PA structure has also been reversibly controlled using enzymes that can recognize a peptide sequence on PA 8 and either phosphorylate or cleave a phosphate group, thereby changing the monomer structure and thus the nature of the supramolecular assembly (Fig. 2c).13
Fig. 2.
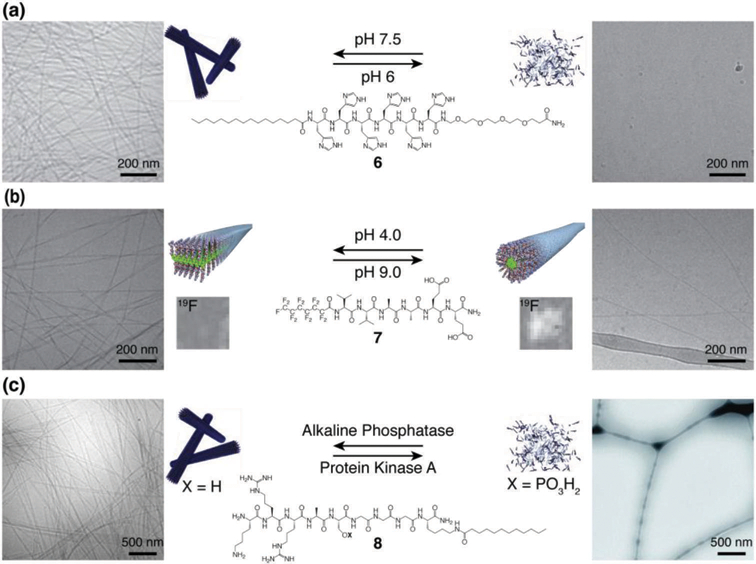
Structures of reversibly responsive PAs with schematic illustrations and cryogenic transmission electronic micrographs of their supramolecular assemblies. (a) pH-responsive histidine-based PA 6 at pH 7.5 (left) and pH 6.0 (right), showing the assembled fibers and dissembled states, respectively. (b) pH-responsive PA 7 with a perfluorinated tail at pH 4.0 (left) and pH 9.0 (right), showing the assembled ribbons and fibers, respectively. 19F MRI solution images of the assemblies are shown above the schematic illustrations. (c) Enzyme-responsive PA 8 in the de-phosphorylated form after reaction with alkaline phosphatase (left) and phosphorylated form (right) after conversion with protein kinase A, showing the assembled fibers and dissembled states, respectively. Adapted with permission from ref. 11, ref. 12, and ref. 13 (Copyright (a, b) 2014 American Chemical Society, (c) 2011 Royal Society of Chemistry).
Another series of well-studied peptide-based supramolecular structures in the class that utilize hydrophobic collapse along with hydrogen bonding includes short peptides in which polar fragments of the amino acid backbone are linked to hydrophobic groups such as N-(fluorenyl-9-methoxycarbonyl) (Fmoc) either as side groups or as N-capping aromatic groups, (Fig. 1b, example molecule 2). In 2003, Xu and coworkers reported Fmoc-dipeptides that assemble into nanofibers and form hydrogels.14 Ulijn’s group has extensively studied these structures and their mechanism of assembly.15 Using Fourier transform infrared spectroscopy (FT-IR) and wide-angle X-ray scattering (WAXS) they discovered that in addition to the anti-parallel β-sheet interactions between peptides, the π–π interactions between Fmoc groups are also crucial for the formation of nanofibers and represent another attractive force beyond the hydrophobic collapse.
Amphipathic peptides are an additional example of peptide-based supramolecular architectures in the class that utilize hydrophobic collapse. These peptides are composed of alternating hydrophobic and hydrophilic amino acid residues that possess strong propensities to form β-sheets, displaying hydrophobic and hydrophilic faces that can further assemble into fibrous architectures. Inspired by the Z-DNA-binding peptide sequence found in a yeast protein, Zhang et al. reported the first example of an amphipathic peptide which is composed of a self-complementary ionic peptide with alternating alanine-glutamic acid (AG) and alanine-lysine (AK) sequences that stack to form nanofibers (Fig. 1c, example molecule 3).16 They also discovered that the nanostructures can be stabilized by addition of salts to form a macroscopic membrane. Hamley and coworkers synthesized hybrid copolymers containing blocks of self-complementary amphipathic peptide and poly(ethylene glycol), and investigated their supramolecular assembly in response to pH and temperature changes.17 Similarly, Saiani and coworkers reported amphipathic peptides functionalized with thermoresponsive poly(N-isopropylacrylamide) and demonstrated the controllable thermal and mechanical properties of the resulting hydrogels.18 Schneider and Pochan developed a β-hairpin peptide that consists of an alternating valine-lysine (VK) sequence connected through a diproline linker (Fig. 1d, example molecule 4).19 Using circular dichroism (CD) spectroscopy and FT-IR, they found that at low pH individual peptides are unstructured, affording a low-viscosity solution. In contrast, under basic conditions intramolecular folding takes place causing formation of an amphiphilic β-hairpin that is prone to assemble into supramolecular nanofibers. Hartgerink and coworkers reported multidomain-type peptides with an ABA block motif, where the A blocks consist of oligolysines (K) while the central B block is composed of glutamine-leucine (QL) repeats.20 Using CD spectroscopy, FT-IR, and cryogenic transmission electron microscopy (cryo-TEM), they discovered that the relative sizes of the A and B blocks determines the secondary structure of the peptides, which then dictates the resulting supramolecular nanostructure. In this system, block A consists of positively charged lysine residues, which can frustrate self-assembly of the B block as a result of electrostatic repulsion. Ghadiri and coworkers reported an eight-residue cyclic peptide with alternating d- and l-amino acids that forms supramolecular nanotubes (Fig. 1e, example molecule 5).21 The nanotubes exhibit a strong propensity to form crystalline bundles. Using FT-IR, TEM, and electron diffraction, they found that the cyclic peptide forms lateral hydrogen bonds with a periodicity of 4.73 Å, corresponding to an antiparallel β-sheet between cyclic peptides in which neighboring macrocycles are rotated 180° with respect to each other.
The Ulijn group has recently developed methods to search for peptide sequences that are likely to assemble using high-throughput, dynamic peptide libraries (Fig. 3).22 The process starts with mixtures of dipeptides as the input (Fig. 3a, input dyads). Upon addition of thermolysin, a relatively nonspecific protease, a dynamic exchange of peptide sequences occurs (Fig. 3a, “exchange and selection”). Over time, the peptides that give rise to the formation of assembling structures are amplified because of the thermodynamic stabilization that results from the supramolecular assembly (Fig. 3b). When the system reaches a steady state and no further change in composition is observed, peptides with the greatest propensity to assemble are characterized using spectroscopy and microscopy (Fig. 3a, “evaluate”). Furthermore, modifications of the reaction conditions can promote or reduce certain supramolecular interactions to induce the formation of specific nanostructures (Fig. 3a, “modify conditions”).
Fig. 3.
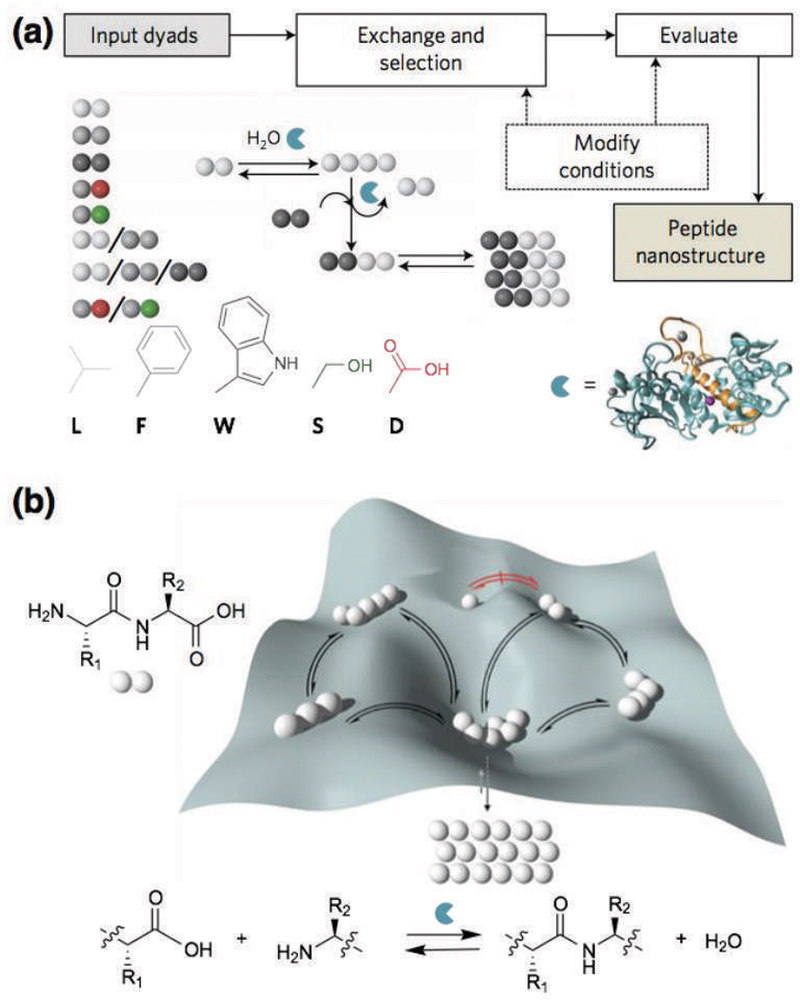
(a) Schematic illustration of the dynamic peptide library system to search for supramolecular materials, which involves mixtures of dipeptides (input dyads), dynamic exchange of peptide sequences catalyzed by enzymatic condensation, hydrolysis, and transacylation, with the most-stable assembling structure eventually emerging (peptide nanostructure). The reaction conditions may be modified to promote or reduce certain supramolecular interactions and thermodynamic selection. (b) Potential energy surface that shows the formation of peptide oligomers. The depth of the wells represents the relative stability of the assembling peptides formed. Reprinted with permission from ref. 22 (Copyright 2016 Nature Publishing Group).
Growth factor mimetic and targeted peptide supramolecular materials
Growth factors are proteins that are essential for regulating important biological events such as cell proliferation, migration, and differentiation.23 For instance, vascular endothelial growth factor (VEGF) and fibroblast growth factor (FGF) are important in the formation of blood vessels (angiogenesis), and bone morphogenetic protein (BMP) is known to mediate bone formation (osteogenesis). Peptide supramolecular materials that present growth factor-mimetic structures or materials that are capable of binding to growth factors on their surface can potentially improve cell signaling and therefore enhance the replacement and the repair of damaged tissues. Moreover, development of such materials could allow minimal use of exogenous growth factors and thereby reduce treatment cost, batch-to-batch variation, potential biological contamination, and, most importantly, dangerous side effects.
Motivated by the great demand for the development of novel therapies for cardiovascular disease, a worldwide leading cause of morbidity and mortality, Hartgerink and coworkers recently reported an amphipathic multidomain peptide 9 for angiogenesis.24 The peptide consists of a lysine and three serine-leucine (SL) repeats flanking both sides of the protease cleavage sequence leucine-arginine-glycine (LRG), which is conjugated to a VEGF-mimetic sequence at one of the far lysines (Fig. 4a). The alternating hydrophobic and hydrophilic sequence drives the multidomain peptides to associate into bilayers of antiparallel β-sheets that form a nanofiber hydrogel in the presence of PO43– anions (Fig. 4b–4d). In vitro experiments using human umbilical vein endothelial cells showed that the VEGF-mimetic multidomain peptide activates VEGFR1, VEGFR2, and NP-1 receptors, which is indicative of vasculogenic receptor activation. In vivo angiogenesis experiments demonstrated that the hydrogel is rapidly infiltrated with host cells and that by day 7, numerous large microvessels were observed. Furthermore, their follow-up work using a peripheral artery disease mouse model demonstrated the successful preservation of tissue structure and function (Fig. 4e).25
Fig. 4.
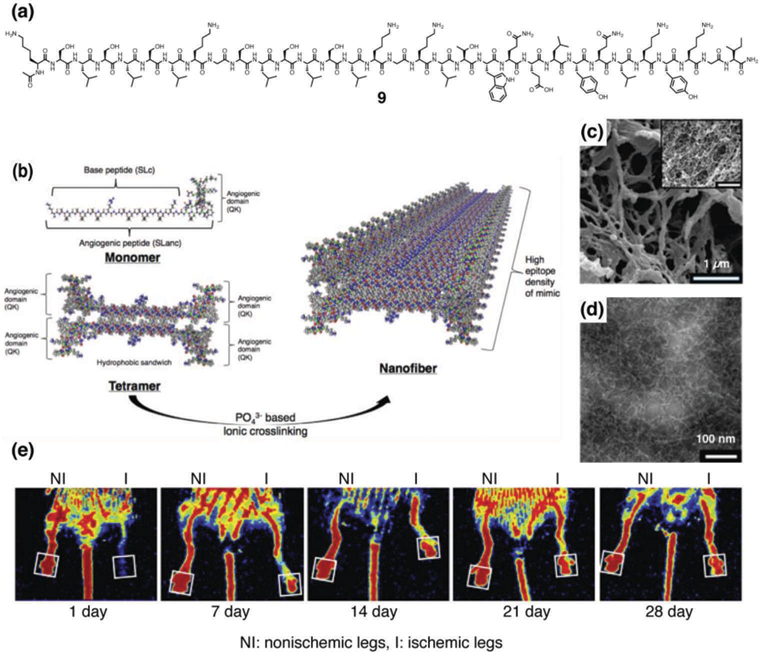
(a) Chemical structure of a vascular endothelial growth factor (VEGF)-mimetic multi-domain peptide 9. (b) Schematic illustrations of supramolecular assembly of 9 to form nanofibers. (c) SEM image of nanofibers formed by 9. (d) TEM of nanofibers formed by 9. (e) Recovery from hind limb ischemia after treatment with VEGF-mimetic multidomain peptide 9. Laser Doppler perfusion imaging showed rapid restoration of blood flow to the foot pad (boxed region) in VEGF-mimetic multidomain peptide-treated 13-month old mice. Reprinted with permission from ref. 24 and ref. 25 (Copyright 2015 American Chemical Society, 2016 Elsevier).
Heparan-sulfate proteoglycans are known to bind to angiogenesis promoting growth factors such as VEGF and FGF, to protect the growth factors from degradation, increase local concentrations of growth factors, and enhance growth factor-receptor interactions to stimulate signaling for angiogenesis.26 To mimic the structure and function of heparin, Guler and coworkers developed PA 10 which is decorated with carboxylic acid, hydroxyl, and sulfonate groups (Fig. 5a).27 Using CD, SEM, and TEM, they confirmed that PA 10 forms supramolecular nanofibers with β-sheet peptide secondary structures (Fig. 5b and 5c). The in vitro angiogenesis assay was performed using human umbilical vein endothelial cells, and capillary-like structures were observed when the cells were cultured on a hydrogel formed by PA 10, even without addition of any exogenous growth factors (Fig. 5d). In vivo bioactivity was then evaluated using a rat corneal micropocket neovascularization assay, and a combination of PA 10 nanofiber gel with growth factors (VEGF/FGF-2) showed significantly larger vascularized area compared to the sole growth factor solution (Fig. 5e–5g). To study the interaction between growth factors and the PA nanofibers more in detail, they carried out ELISA-based binding assays for a variety of growth factors including VEGF, FGF-2, BMP-2, hepatocyte growth factor, and nerve growth factor.28 Compared to the control PA which lacks the carboxylic acid, hydroxyl, and sulfonate groups, PA 10 showed an enhanced binding capability to VEGF, FGF-2, and hepatocyte growth factor. Quite strikingly, VEGF121, which lacks the heparin-binding domain, did not show any binding to the nanofibers formed by PA 10, indicating that the sulfonate groups on the PA nanofiber are important for the binding of VEGF. Furthermore, they demonstrated that a hydrogel formed by nanofibers of PA 10 containing side groups present in heparin such as sulfonate moieties can also induce osteogenesis.29 An ELISA assay showed that PA 10 binds to BMP-2 much stronger than the control PA without the heparin-mimetic functional groups. When osteoblast-like Saos-2 cells were cultured on the nanofibers of PA 10, a significant enhancement in alkaline phosphatase expression was observed compared to the cells cultured on a tissue culture plate control or PAs lacking sulfonate groups at day seven, indicating that PA 10 may enhance osteogenic maturation. The mineral deposition ability of osteoblast-like cells was then evaluated by SEM, Alizarin red-S staining, and energy disperse X-ray analysis spectroscopy, and the presence of calcium and phosphate was predominantly observed. More recently, their follow-up work using a rabbit tibial bone model demonstrated that a hydrogel formed by PA 10 nanofibers enhances bone regeneration and biomineralization in vivo (Fig. 5h–5l).30 The treatment of bone tissue defects is a major challenge in orthopedics, and peptide materials that enhance osteogenesis can potentially replace the use of autograft and solve the risks of infections, donor site morbidity, and transplant rejections.31
Fig. 5.
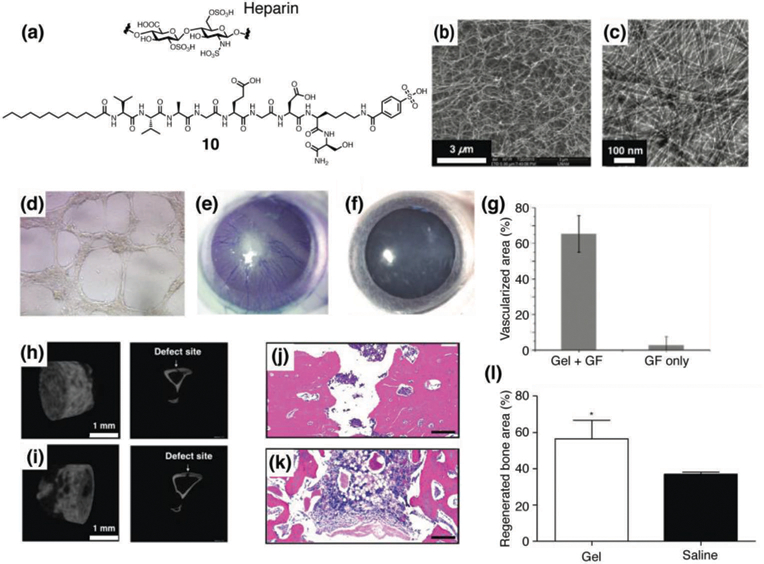
(a) Chemical structures of heparin and a heparin-mimetic PA 10. (b) SEM image of nanofibers formed by 10. (c) TEM of nanofibers formed by 10. (d) In vitro angiogenesis assay. Bright field image of HUVECs cultured on nanofiber matrix formed by PA 10 (100x magnification). (e–g) Evaluation of in vivo bioactivity by corneal angiogenesis assay. (e) Injection of hydrogel formed by 1wt% of PA 2 with 10 ng of VEGF and bFGF-induced vascularization in cornea. (f) Application of growth factor solution (10 ng of VEGF and bFGF) in physiological saline (without PA gel) did not induce vascularization. (g) Ratio of vascularized area to total area was calculated for both groups. (h–i) Representative µCT images of bones treated with (h) PA 10 hydrogel and (i) saline solution after 4 weeks. (j–l) Histological evaluation of a tibial defect model treated with (j) PA 10 hydrogel and (k) saline solution after 4 weeks stained with H&E. Scale bars are 200 µm. (l) Regenerated bone areas were quantified through the histological evaluation of H&E results. Reprinted with permission from ref. 27 and ref. 30 (Copyright 2011 American Chemical Society, 2016 Royal Society of Chemistry).
Pursuing another strategy to develop peptide materials that bind to BMP-2, the Stupp laboratory used phage display to identify a BMP-2 binding peptide sequence, and the binding sequence was incorporated into PA supramolecular nanostructures through co-assembly with a second PA lacking the binding sequence.32 Surface plasmon resonance was used to determine the binding constant of the PA to BMP-2, and the PA displayed two orders of magnitude higher value than that of a diluent PA lacking the BMP-2 binding sequence. Using a rat posterolateral lumbar intertransverse spinal fusion model, the potential of the PA to promote BMP-2-induced spinal fusion was investigated in vivo. PA nanofiber gels displaying BMP-2-binding sequences exhibited enhanced spinal fusion rates compared to controls, which effectively decreased the required dose of BMP-2 by 10-fold. Remarkably, a 42% fusion rate was observed for gels containing the bioactive PA nanofibers even without the addition of any exogenous BMP-2, suggesting that the nanofibers may potentially recruit endogenous growth factors.
In yet another approach to develop peptide supramolecular materials binding to BMP-2, Stupp and coworkers designed a series of glycopeptide amphiphiles 11–14 that form supramolecular nanofibers (Fig. 6a and 6b).33 The objective of this work was to use the glycopeptide nanofibers as mimics of the highly complex sulfated polysaccharides known to bind hundreds of proteins in biology26 by using an actual sulfated glucosamine monosaccharide rather than just sulfonate groups. In particular, PA 11 displays a trisulfated monosaccharide on the surface of nanofibers to mimic the highly sulfated sugars of heparin. Surface plasmon resonance was used to determine that PA 11 can bind to five critical growth factor proteins with different polysaccharide binding domains: BMP-2, BMP-4, FGF-1, FGF-2 and VEGF. SAXS and CD spectroscopy confirmed that growth factor binding does not disrupt the filamentous shape of the nanostructures or their internal β-sheet secondary structure. Given the known molecular exchange dynamics in PA nanostructures,34 and the presence of water within the fibers,8 one can expect adaptive binding of the growth factors involving spatial reconfiguration of PA molecules and water in contact with the growth factor. An important achievement in this work was the demonstration that BMP-2 binds to the PA nanofibers in the way it occurs in biological systems, namely through its heparin binding domain, a fact that could be critical for the nanofibers to amplify signaling in vivo. When C2C12 mouse myoblast progenitor cells were cultured with BMP-2 in the presence of PA 11 nanofibers, a significant enhancement in alkaline phosphatase expression was observed compared to PAs 12–14, indicating osteogenic differentiation (Fig. 6c). Western blot was used to confirm the osteogenic differentiation occurred specifically through the BMP signal transduction (Fig. 6d). Beyond amplifying signaling of BMP-2 significantly in vitro, the sulfated glycopeptide nanostructures of PA 11 also promoted bone regeneration in vivo using a model for the clinically-important objective of spinal fusion with a dose of BMP-2 100-fold lower than the value required for the control animals (Fig. 6e–6g). PA nanofibers with only negatively charged groups but lacking the sulfated monosaccharide yielded significantly lower rates of spinal fusion. These highly bioactive sulfated glycopeptide nanostructures may enable a variety of additional therapies in the future involving other proteins.
Fig. 6.
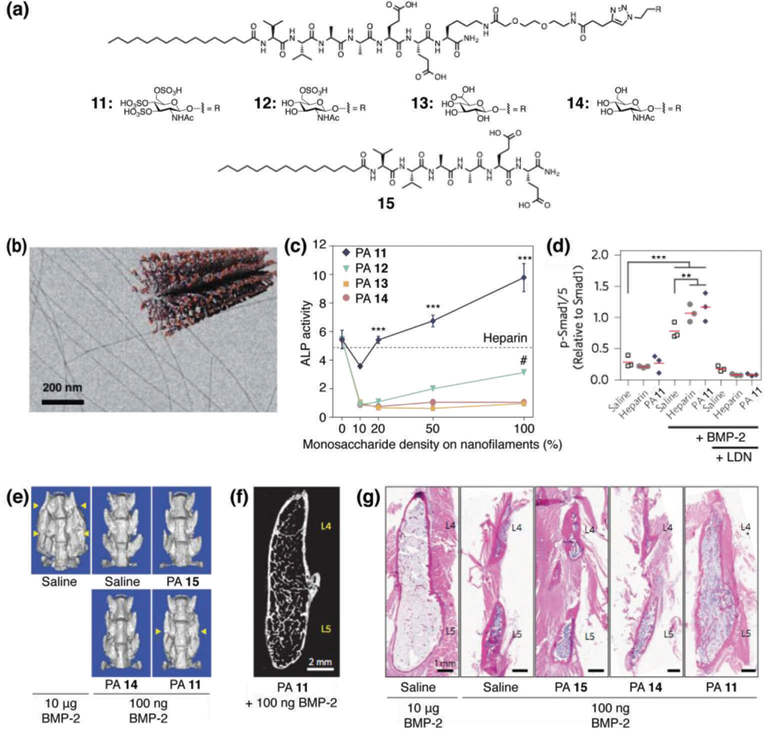
(a) Chemical structures of glycol-PAs 11–14 and backbone PA 15 lacking the glycol unit. (b) Cryogenic TEM image and a schematic illustration of PA 13 to form nanofibers. (c) Plot of alkaline phosphatase (ALP) activity in C2C12 cells treated with BMP-2 and glycol-PAs 11–14 as a function of increasing monosaccharide density on the nanostructures (treatment with heparin is indicated by the dashed line). (d) Quantification of Western blots of C2C12 cells stimulated for 3 h with BMP-2 revealing the effect of heparin or PA 11 nanofibers on Smad phosphorylation. Cells were also treated with LDN to inhibit BMP-2 signaling. (e) Representative volume renderings from µCT (yellow arrows indicate fusion). (f) Digital sagittal section through the fusion mass from an animal treated with 100 ng BMP-2 and PA 11 nanostructures. (g) Representative sagittal cross-sectional images of L4–L5 posterolateral spine specimens with H&E staining. Reprinted with permission from ref. 33 (Copyright 2017 Nature Publishing Group).
Peptide supramolecular materials inspired by cell adhesion molecules
Cell adhesion molecules play critical roles in cell attachment and maturation through interaction with integrin receptors on the cell membrane.35 Incorporation of bioactive peptide sequences found in cells adhesion molecules, such as arginine-glycine-aspartic acid-serine (RGDS) found in fibronectin, and isoleucine-lysine-valine-alanine-valine (IKVAV) found in laminin, into peptide supramolecular scaffolds has been a well-established approach to develop artificial extracellular matrices that can signal cells. For example, Hartgerink and coworkers developed an amphipathic multidomain peptide functionalized with the RGDS epitope that forms supramolecular nanofibers, and demonstrated the potential of its hydrogel as a delivery agent for stem cell secretome.36 In another example, Gouveia et al. developed a PA that incorporated an MMP1 cleavage site adjacent to the RGDS sequence and showed that coatings of this PA could support human corneal stromal fibroblasts growth.37 Wang and coworkers synthesized an amphipathic arginine-alanine-aspartic acid-alanine (RADA) repeating peptide that incorporated an IKVAV sequence, and characterized its supramolecular nanofibers using AFM and CD. The hydrogel formed by the IKVAV-peptide nanofiber was used to encapsulate neural stem cells and injected into the brain using a rat model. The authors reported that the nanofibers improved survival of the cells and reduced formation of glial astrocytes.38
More recently, researchers explored peptide supramolecular materials bearing peptides present in tenascin. The tenascin family is a series of extracellular matrix glycoproteins that show high expression rates during the development or regenerations of neurons, bones, and blood vessels.39 Among them, tenascin-C is a common protein in both neural and bone tissues, interacting with a variety of extracellular matrix molecules and cell surface molecules including integrin and regulating cell adhesion, spreading, and proliferation. Inspired by the biological functions of tenascin-C in nature, Sever et al. developed a PA molecule that incorporates a tenascin-C derived functional sequence VFDNFVLKK.40 Using SEM and CD spectroscopy, the tenascin-C mimetic PA was confirmed to form nanofibers with a predominantly β-sheet secondary structure when co-assembled with its non-bioactive diluent PA. When rat mesenchymal stem cells were cultured with the co-assembly of tenascin-C mimetic PA and diluent PA, they observed cell adhesion and spreading, as well as an enhanced expression of a variety of osteogenic markers at the mRNA level, indicating osteogenic differentiation of the stem cells. The osteogenic differentiation was further supported by an alkaline phosphatase assay and Alizarin red-S staining. These results demonstrated that incorporation of tenascin-C into peptide materials is a promising strategy for regenerative medicine.
As mentioned above, tenascin-C is also known to be an important molecule in the extracellular matrix of the central nervous system, which plays a role in spinal cord regeneration after injury and guides neural progenitor cells during the development of brain. To explore the potential of PAs for the treatment of spinal cord injury, Berns et al. designed a tenascin-C mimetic PA that forms supramolecular nanofibers.41 Neuronal cells were encapsulated in hydrogels of aligned nanofibers formed by the tenascin-C-mimetic PA, and enhanced neurite outgrowth was observed. Furthermore, these PAs promoted migration of neural stem cells when cultured on nanofiber coatings. This work was recently followed with related in vivo studies by Kuhn and coworkers that showed a 24-fold increase in redirected neuroblasts in rats injected with the tenascin-C mimetic PA compared with controls, with no significant increase in neuroinflammatory response.42 The ability of aligned PA nanofibers to promote migration opens the possibility to guide endogenous or transplanted neural stem cells to regenerate dysfunctional areas of the central nervous systems.
The Stupp laboratory has also studied how cellular response is impacted by the spatial presentation of an epitope group on the surface of a supramolecular assembly. The RGDS epitope was systematically separated from the surface of the PA nanofibers by a linker made of one, three, or five glycine residues (Fig. 7a–7d), and it was found that the PA with the longest linker length displayed the strongest effect on cell morphology with a strong adhesion between cells and substrate (Fig. 7e and 7f).43 PAs with a rigid linker were also synthesized using three aryl amides, and it was found that unlike linker length, changes in the molecular flexibility had minimal influence on cell behavior. The high persistence length nanofibers used in this study, as opposed to common flexible polymers, allowed them to conclude that epitope topography at the nanoscale has a strong influence on its bioactive properties. Another related factor in the interaction of PA nanofibers with proteins is the architecture of the PA backbone, for example linear vs. branched. This effect was previously studied by Guler et al. in the context of biotin interaction with the avidin receptor, and branched biotinylated PAs presenting two ligands per molecule were found to exhibit enhanced receptor recognition relative to linear ones.44
Fig. 7.
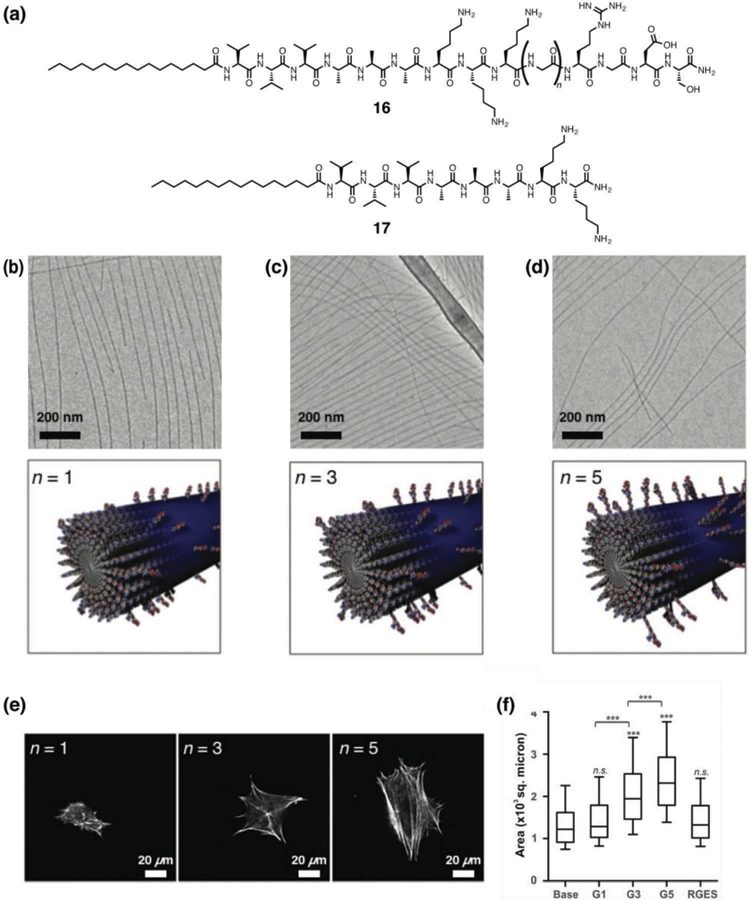
(a) Chemical structures of RGDS epitope-presenting PA 16 in which a glycine linker of variable length (n = 1, 3, and 5) presents the epitope, and base PA 17 lacking the RGDS sequence. (b–d) Cryogenic TEM images and schematic illustrations of RGDS epitope-presenting PAs co-assembled with base PA 17 at a 1:9 weight ratio. (b) PA 16 (n = 1), (c) PA 16 (n = 3) and (d) PA 16 (n = 5). (e) Representative images of 3T3 fibroblasts cultured on PA-coated substrates for 5 h and stained with phalloidin for actin filaments. (f) Cell morphology on PA substrates, compared by measuring the projected cell area. Reprinted with permission from ref. 43 (Copyright 2015 Royal Society of Chemistry).
Hierarchical ordering of supramolecular nanostructures
The majority of peptide-based structures, including many of those described above, assemble into nanomaterials with little order beyond the nanoscale. In comparison, biological systems have evolved to precisely control structure from the atomic level to the nanoscale and up to macroscopic assemblies of cells and tissues. Reproducing the hierarchical ordering that is so evident in biology within supramolecular nanostructures remains a longstanding goal for the field and is critical for materials that are intended to interface with living systems for medical applications. Methods to design, control, and study higher levels of assembly within supramolecular nanostructures are becoming more common and are a critical area of research for these materials to reach their potential in therapeutics. In this section, we highlight only a few of these methods to control the hierarchical assembly of peptide–based supramolecular assemblies, and the corresponding effect it has on regenerative medicine.
As described above, peptide amphiphiles form long, one-dimensional nanofibers upon annealing. However, under the right processing conditions some PA sequences, such as C16V3A3E3, form a strongly birefringent liquid.45 Importantly, this liquid crystal can be macroscopically aligned as shown in Fig. 8a–8b by extruding it into a salt solution if provided enough shear and extensional force, for example by manually dragging the gel from a pipette or extruding it from a print nozzle. This results in noodle-like viscoelastic strings that consist of PA fibers that are hierarchically ordered from the assembled fibers to the aligned strings that can extend to centimeters (Fig. 8c). These aligned noodle-like strings are obvious targets for interfacing cells or organisms with hierarchical macroscopic PA structures. By combining human mesenchymal stem cells with the PA solution prior to extrusion from a pipette into a salt solution, the cells were encapsulated within the gelled strings. The cells began to elongate along the long axis of the string within 12 hours, potentially due to contact guidance along the aligned noodle. The string was also formed in the presence of HL-1 cardiomyocytes, cells that require extensive cell-cell contacts to propagate their spontaneous electrical activity, which were found to proliferate and to fill the structure. Measuring intracellular calcium concentration using fluorescence showed spontaneous electrical contact after six days, implying pockets of significant cell-cell interaction, and after ten days the action potentials were able to pass along the entire macroscopic string. These results have inspired further research in using these noodle-like strings to template neuron growth,46 and for the delivery of signals to promote regeneration, such as sonic hedgehog to encourage regrowth of the cavernous nerve.47 Recently, strings of varying stiffness were used to encapsulate muscle stem cells, with stiffness corresponding to the degree of cell alignment.48 These PA string scaffolds enhanced myogenic progenitor cell survival and growth and were able to induce differentiation and development. Of particular interest is that this system showed promise in vivo with muscle stem cells engraftment in mice. Groups have taken inspiration from other hierarchical materials, such as collagen, to create ordered supramolecular peptide systems across scales. An example is work by Chung and coworkers, who developed a collagen-like PA through the addition of a twelve amino acid hydroxyapatite-binding sequence to a PA nanofiber.49 They observed hierarchical ordering on the microscale, which also impacted the macroscopic properties of the materials. Importantly, these materials showed promise for directed growth of bone cells and hydroxyapatite mineral.
Fig. 8.
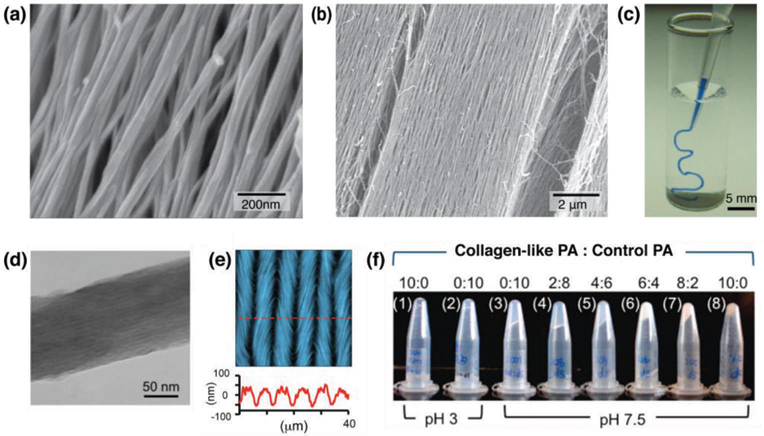
(a, b) SEM images of aligned PA fibers within macroscopic strings made by dragging annealed PA solutions into a calcium chloride solution. (c) A PA solution dyed with trypan blue extruded into phosphate-buffered saline solution after annealing. (d) Transmission electron micrograph of precipitated collagen-like PA. (e) AFM image (top) and linecut (bottom) of periodic microstructure of collagen-like PA, showing a periodic, undulating pattern. (f) Photos of mixtures of collagen-like PA and non-collagen-like control PA (1 wt% on DI water), ranging from pure collagen-like PA (1, 8) to pure control PA (2, 3). Reprinted with permission from ref. 45 (a-c) and ref. 49 (d-f) (Copyright 2010 Nature Publishing Group, 2015 American Chemical Society).
Another example of hierarchical ordering in aligned supramolecular assembly was recently reported by Li and Hartgerink using multidomain peptides with the structure K2(SLZL)3K2.50 In a similar fashion to that described above, the peptide was extruded through a pipette into a salt solution, resulting in highly aligned strings of gelled peptide. The macroscopic hydrogels could be centimeters in length and strong enough to be manipulated with tweezers. The presence of dihydroxyphenylalanine in the structure was critical to the formation of the hydrogels, which was attributed to both the increased solvation and unique bidentate hydrogen bonding possible with dihydroxyphenylalanine compared with phenylalanine or tyrosine. Dihydroxyphenylalanine also enabled the covalent capture of the gel through the addition of an oxidant such as ortho-quinone, forming mechanically strong strings that could be successfully transferred to media.
Conclusion
The work described here demonstrates the structural and functional diversity of peptide-based supramolecular assemblies. Nature uses a combination of supramolecular and covalent peptide polymers to achieve many of the functions critical to life, and we are just now beginning to understand how to leverage these materials for biomedical functions. Learning how to control these materials will be critical to their use in regenerative medicine and generally for the development of novel therapies.
Key learning points.
Balance of different intermolecular interactions between peptide-based monomers determine the supramolecular nanostructures.
Control over the dynamics of supramolecular materials offer novel structures and functions.
Supramolecular extracellular matrix mimics have great potential in tissue regeneration.
Peptide-based supramolecular materials can be designed to bind or mimic growth factor proteins for regenerative medicine.
Supramolecular nanomaterials can be organized into hierarchically ordered structures across length scales.
Acknowledgments
The authors are grateful for support of the work discussed herein by grants from Department of Energy (DE-FG02–00ER45810), the DOE-funded Center for Bio-Inspired Energy Science, an Energy Frontier Research Center (DE-SC0000989), the National Institutes of Health (National Institute of Dental and Craniofacial Research 5R01DE015920, Bioengineering Research Partnerships 5R01EB003806 and 5R01HL116577, Center of Cancer Nanotechnology Excellence F5U54CA151880, Project Parent Grant P01HL108795), and the National Science Foundation (DMR-1508731).
Author Biography
Kohei Sato earned a B.S. in chemistry in 2009 from Chiba University. He completed his Ph.D. on organic synthesis and supramolecular chemistry under the guidance of Prof. Takuzo Aida at the University of Tokyo in 2014. After carrying out postdoctoral research in the laboratory of Prof. Samuel Stupp at Northwestern University, he became an assistant professor at the Tokyo Institute of Technology in 2018.
Mark P. Hendricks received a B.S. in chemistry from Harvey Mudd College in 2010 and a Ph.D. from Columbia University in 2015. He worked under Prof. Jonathan Owen as a graduate student, studying the synthesis of semiconducting nanocrystals. Following postdoctoral research in the laboratory of Prof. Samuel Stupp at Northwestern University he started as an assistant professor of chemistry at Whitman College in 2018.
Liam C. Palmer earned a B.S. in Chemistry from the University of South Carolina in 1999. He completed his Ph.D. on molecular encapsulation under the supervision of Prof. Julius Rebek, Jr. at The Scripps Research Institute in 2005. He is now a research associate professor in the Department of Chemistry and the Director of Research for the Simpson Querrey Institute at Northwestern University.
Samuel I. Stupp received a B.S. in chemistry from the University of California at Los Angeles and a Ph.D. in materials science and engineering from Northwestern University in 1977. He was a member of the faculty at Northwestern until 1980 and then spent 18 years at the University of Illinois at Urbana−Champaign before returning to Northwestern in 1999. He is currently Board of Trustees Professor of Materials Science & Engineering, Chemistry, Medicine, and Biomedical Engineering and serves as the Director of the Simpson Querrey Institute at Northwestern University. His group’s research is focused on the design of new materials through supramolecular chemistry and self-assembly, with emphasis on functions of interest in biomaterials for advanced medicine and energy relevant materials.
References
- 1.Aida T, Meijer EW and Stupp SI, Science, 2012, 335, 813–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tabony J, Biol. Cell, 2006, 98, 603–617. [DOI] [PubMed] [Google Scholar]
- 3.Hamley IW, Angew. Chem. Int. Ed. Engl, 2014, 53, 6866–6881. [DOI] [PubMed] [Google Scholar]
- 4.Berndt P, Fields GB and Tirrell M, J Am Chem Soc, 1995, 117, 9515–9522. [Google Scholar]
- 5.Hartgerink JD, Beniash E and Stupp SI, Science, 2001, 294, 1684–1688. [DOI] [PubMed] [Google Scholar]
- 6.Paramonov SE, Jun H-W and Hartgerink JD, J Am Chem Soc, 2006, 128, 7291–7298. [DOI] [PubMed] [Google Scholar]
- 7.Ortony JH, Newcomb CJ, Matson JB, Palmer LC, Doan PE, Hoffman BM and Stupp SI, Nat. Mater, 2014, 13, 812–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ortony JH, Qiao B, Newcomb CJ, Keller TJ, Palmer LC, Deiss-Yehiely E, Olvera M Cruz de la, Han S and Stupp SI, .J Am. Chem. Soc, 2017, 139, 8915–8921. [DOI] [PubMed] [Google Scholar]
- 9.Korevaar PA, Newcomb CJ, Meijer EW and Stupp SI, J. Am. Chem. Soc, 2014, 136, 8540–8543. [DOI] [PubMed] [Google Scholar]
- 10.Tantakitti F, Boekhoven J, Wang X, Kazantsev RV, Yu T, Li J, Zhuang E, Zandi R, Ortony JH, Newcomb CJ, Palmer LC, Shekhawat GS, Olvera M Cruz de la, Schatz GC and Stupp SI, Nat. Mater, 2016, 15, 469–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moyer TJ, Finbloom JA, Chen F, Toft DJ, Cryns VL and Stupp SI, J. Am. Chem. Soc, 2014, 136, 14746–14752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Preslar AT, Tantakitti F, Park K, Zhang S, Stupp SI and Meade TJ, ACS Nano, 2016, 10, 7376–7384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Webber MJ, Newcomb CJ, Bitton R and Stupp SI, Soft Matter, 2011, 7, 9665–9672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Y, Gu H, Yang Z and Xu B, J. Am. Chem. Soc, 2003, 125, 13680–13681. [DOI] [PubMed] [Google Scholar]
- 15.Smith AM, Williams RJ, Tang C, Coppo P, Collins RF, Turner ML, Saiani A and Ulijn RV, Adv. Mater, 2008, 20, 37–41. [Google Scholar]
- 16.Zhang S, Holmes T, Lockshin C and Rich A, Proc. Natl. Acad. Sci. U.S.A, 1993, 90, 3334–3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hamley IW, Ansari IA, Castelletto V, Nuhn H, Rösler A and Klok H-A, Biomacromolecules, 2005, 6, 1310–1315. [DOI] [PubMed] [Google Scholar]
- 18.Stoica F, Alexander C, Tirelli N, Miller AF and Saiani A, Chem. Commun, 2008, 0, 4433–4435. [DOI] [PubMed] [Google Scholar]
- 19.Haines-Butterick L, Rajagopal K, Branco M, Salick D, Rughani R, Pilarz M, Lamm MS, Pochan DJ and Schneider JP, Proc. Natl. Acad. Sci. U.S.A, 2007, 104, 7791–7796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dong H, Paramonov SE, Aulisa L, Bakota EL and Hartgerink JD, J. Am. Chem. Soc, 2007, 129, 12468–12472. [DOI] [PubMed] [Google Scholar]
- 21.Ghadiri MR, Granja JR, Milligan RA and McRee DE, Nature, 1993, 366, 324–327. [DOI] [PubMed] [Google Scholar]
- 22.Pappas CG, Shafi R, Sasselli IR, Siccardi H, Wang T, Narang V, Abzalimov R, Wijerathne N and Ulijn RV, Nat. Nanotechnol, 2016, 11, 960–967. [DOI] [PubMed] [Google Scholar]
- 23.Barrientos S, Stojadinovic O, Golinko MS, Brem H and Canic MT, Wound Repair and Regeneration, 2008, 16, 585–601. [DOI] [PubMed] [Google Scholar]
- 24.Kumar VA, Taylor NL, Shi S, Wang BK, Jalan AA, Kang MK, Wickremasinghe NC and Hartgerink JD, ACS Nano, 2015, 9, 860–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumar VA, Liu Q, Wickremasinghe NC, Shi S, Cornwright TT, Deng Y, Azares A, Moore AN, Acevedo-Jake AM, Agudo NR, Pan S, Woodside DG, Vanderslice P, Willerson JT, Dixon RA and Hartgerink JD, Biomaterials, 2016, 98, 113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu D and Esko JD, Annu. Rev. Biochem, 2014, 83, 129–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mammadov R, Mammadov B, Toksoz S, Aydin B, Yagci R, Tekinay AB and Guler MO, Biomacromolecules, 2011, 12, 3508–3519. [DOI] [PubMed] [Google Scholar]
- 28.Mammadov R, Mammadov B, Guler MO and Tekinay AB, Biomacromolecules, 2012, 13, 3311–3319. [DOI] [PubMed] [Google Scholar]
- 29.Kocabey S, Ceylan H, Tekinay AB and Guler MO, Acta Biomater, 2013, 9, 9075–9085. [DOI] [PubMed] [Google Scholar]
- 30.Tansik G, Kilic E, Beter M, Demiralp B, Sendur GK, Can N, Ozkan H, Ergul E, Guler MO and Tekinay AB, Biomater. Sci, 2016, 4, 1328–1339. [DOI] [PubMed] [Google Scholar]
- 31.Griffin KS, Davis KM, McKinley TO, Anglen JO, Chu T-MG, Boerckel JD and Kacena MA, Clinic Rev. Bone Miner. Metab, 2015, 13, 232–244. [Google Scholar]
- 32.Lee SS, Hsu EL, Mendoza M, Ghodasra J, Nickoli MS, Ashtekar A, Polavarapu M, Babu J, Riaz RM, Nicolas JD, Nelson D, Hashmi SZ, Kaltz SR, Earhart JS, Merk BR, McKee JS, Bairstow SF, Shah RN, Hsu WK and Stupp SI, Adv. Healthcare Mater, 2015, 4, 131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee SS, Fyrner T, Chen F, Álvarez Z, Sleep E, Chun DS, Weiner JA, Cook RW, Freshman RD, Schallmo MS, Katchko KM, Schneider AD, Smith JT, Yun C, Singh G, Hashmi SZ, McClendon MT, Yu Z, Stock SR, Hsu WK, Hsu EL and Stupp SI, Nat. Nanotechnol, 2017, 12, 821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.da Silva RMP, van der Zwaag D, Albertazzi L, Lee SS, Meijer EW and Stupp SI, Nat. Commun, 2016, 7, 11561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Albelda SM and Buck CA, FASEB J, 1990, 4, 2868–2880. [PubMed] [Google Scholar]
- 36.Bakota EL, Wang Y, Danesh FR and Hartgerink JD, Biomacromolecules, 2011, 12, 1651–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gouveia RM, Hamley IW and Connon CJ, J. Mater. Sci-Mater. M, 2015, 26, 242. [DOI] [PubMed] [Google Scholar]
- 38.Cheng T-Y, Chen M-H, Chang W-H, Huang M-Y and Wang T-W, Biomaterials, 2013, 34, 2005–2016. [DOI] [PubMed] [Google Scholar]
- 39.Jones FS and Jones PL, Developmental Dynamics, 2000, 218, 235–259. [DOI] [PubMed] [Google Scholar]
- 40.Sever M, Mammadov B, Guler MO and Tekinay AB, Biomacromolecules, 2014, 15, 4480–4487. [DOI] [PubMed] [Google Scholar]
- 41.Berns EJ, Álvarez Z, Goldberger JE, Boekhoven J, Kessler JA, Kuhn HG and Stupp SI, Acta Biomater, 2016, 37, 50–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Motalleb R, Berns EJ, Patel P, Gold J, Stupp SI and Kuhn HG, J. Tissue Eng. Regen. Med, 12, e2123–e2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sur S, Tantakitti F, Matson JB and Stupp SI, Biomater. Sci, 2015, 3, 520–532. [DOI] [PubMed] [Google Scholar]
- 44.Guler MO, Soukasene S, Hulvat JF and Stupp SI, Nano Lett, 2005, 5, 249–252. [DOI] [PubMed] [Google Scholar]
- 45.Zhang S, Greenfield MA, Mata A, Palmer LC, Bitton R, Mantei JR, Aparicio C, de la Cruz MO and Stupp SI, Nat. Mater, 2010, 9, 594–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Berns EJ, Sur S, Pan L, Goldberger JE, Suresh S, Zhang S, Kessler JA and Stupp SI, Biomaterials, 2014, 35, 185–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Choe S, Veliceasa D, Bond CW, Harrington DA, Stupp SI, McVary KT and Podlasek CA, Acta Biomater, 2016, 32, 89–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sleep E, Cosgrove BD, McClendon MT, Preslar AT, Chen CH, Sangji MH, Pérez CMR, Haynes RD, Meade TJ, Blau HM and Stupp SI, Proc. Natl. Acad. Sci. U.S.A, 2017, 114, E7919–E7928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jin H-E, Jang J, Chung J, Lee HJ, Wang E, Lee S-W and Chung W-J, Nano Lett, 2015, 15, 7138–7145. [DOI] [PubMed] [Google Scholar]
- 50.Li I-C and Hartgerink JD, J. Am. Chem. Soc, 2017, 139, 8044–8050. [DOI] [PMC free article] [PubMed] [Google Scholar]