

Abstract
Acute myocardial infarction (AMI) is the leading cause of death worldwide. Its associated mortality, morbidity and complications have significantly decreased with the development of interventional cardiology and percutaneous coronary angioplasty (PCA) treatment, which quickly and effectively restore the blood flow to the area previously subjected to ischemia. Paradoxically, the restoration of blood flow to the ischemic zone leads to a massive production of reactive oxygen species (ROS) which generate rapid and severe damage to biomolecules, generating a phenomenon called myocardial reperfusion injury (MRI). In the clinical setting, MRI is associated with multiple complications such as lethal reperfusion, no-reflow, myocardial stunning, and reperfusion arrhythmias. Despite significant advances in the understanding of the mechanisms accounting for the myocardial ischemia reperfusion injury, it remains an unsolved problem. Although promising results have been obtained in experimental studies (mainly in animal models), these benefits have not been translated into clinical settings. Thus, clinical trials have failed to find benefits from any therapy to prevent MRI. There is major evidence with respect to the contribution of oxidative stress to MRI in cardiovascular diseases. The lack of consistency between basic studies and clinical trials is not solely based on the diversity inherent in epidemiology but is also a result of the methodological weaknesses of some studies. It is quite possible that pharmacological issues, such as doses, active ingredients, bioavailability, routes of administration, co-therapies, startup time of the drug intervention, and its continuity may also have some responsibility for the lack of consistency between different studies. Furthermore, the administration of high ascorbate doses prior to reperfusion appears to be a safe and rational therapy against the development of oxidative damage associated with myocardial reperfusion. In addition, the association with N-acetylcysteine (a glutathione donor) and deferoxamine (an iron chelator) could improve the antioxidant cardioprotection by ascorbate, making it even more effective in preventing myocardial reperfusion damage associated with PCA following AMI.
Keywords: Acute myocardial infarction, Repefusion injury, Oxidative stress, Ascorbate, N-acetylcysteine, Deferoxamine
Core tip: Acute myocardial infarction is the leading cause of death in the world. At least half of the resulting myocardial damage is associated with myocardial reperfusion. Myocardial reperfusion injury is associated with reactive oxygen species production and iron mobilization. Treatment with antioxidants such as ascorbate, N-acetylcysteine, and an iron chelator such as deferoxamine, could prevent the development of this damage.
INTRODUCTION
Acute myocardial infarction (AMI) is the leading cause of death worldwide, and it is associated with high morbidity and mortality. The AMI complications have significantly decreased with the development of interventional cardiology and percutaneous coronary angioplasty (PCA) treatment, which quickly and effectively restore the blood flow to the area previously subjected to ischemia[1]. Paradoxically, the restoration of blood flow to the ischemic zone leads to a massive production of reactive oxygen species (ROS), which generate rapid and severe damage to biomolecules, in a phenomenon called myocardial reperfusion injury (MRI)[2,3]. Sources of ROS in reperfusion include the predominant contribution of NADPH oxidases, which are present in many cell types in myocardial tissue. Other sources are xanthine oxidase, uncoupled eNOS and the mitochondrion[4]. In the clinical setting, MRI is associated with multiple complications such as lethal reperfusion, no-reflow phenomenon, myocardial stunning, and reperfusion arrhythmias (Figure 1).
Figure 1.
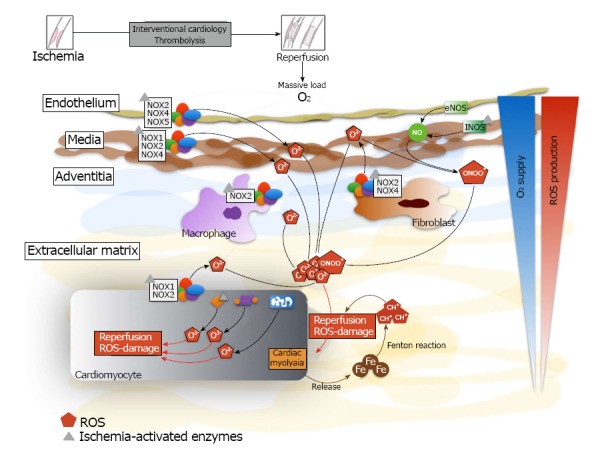
Generation of reactive oxygen species and mobilization of iron after myocardial reperfusion. There is a massive production of reactive oxygen species and iron mobilization by the different cellular types of the myocardial tissue. The iron reacts with superoxide anion to produce hydroxyl radical by the Fenton reaction. Inside cardiomyocyte, there is intracellular production of reactive oxygen species through NADPH oxidase, eNOS uncoupled, xanthine oxidase and mitochondrion. NOX: NADPH oxidase; ROS: Reactive oxygen species; Fe: Iron; eNOS: Endothelial nieric oxide synthases.
Despite significant advances in the understanding of the mechanisms accounting for MRI, it remains an unsolved problem. Although promising results have been obtained in experimental studies (mainly in animal models) these benefits have not been translated into clinical settings. Clinical trials have failed to find benefits from any therapy to prevent MRI, demonstrating a clear dissociation between the bench and the bedside[5].
Prevention of MRI in the clinical setting has intrinsic difficulties in its approach. First, any therapy oriented to MRI prevention must be administered prior to myocardial reperfusion (in other words, prior to PCA). In addition, it should be applied in doses high enough to counterbalance the rapid and massive ROS production following reperfusion. Moreover, there are many different visions regarding the best biomarker to define MRI in patients, and so clinical trials express their results with different outcomes (such as clinical outcomes, serum cardiac biomarkers, echocardiographic parameters, cardiac magnetic resonance, among many others) which makes the analyses even more difficult. All these elements have made it difficult to develop an effective therapy to prevent MRI in AMI patients. The present review focuses on the cellular and molecular mechanisms of oxidative-stress induced MRI during AMI, and the key points to develop an appropriate strategy to reduce oxidative damage derived from myocardial reperfusion.
PATHOPHYSIOLOGY
MRI is a clinical problem associated with procedures such as thrombolysis, angioplasty, and coronary bypass surgery, which are commonly used to re-establish the blood flow and minimize the damage to the heart due to severe myocardial ischemia[3]. There are three main hypotheses which have been proposed to explain the pathogenesis of ischemia reperfusion (IR) injury: oxidative stress, iron mobilization, and Ca2+-overload[6,7]. All of these mechanisms are most likely related, but it is not known whether they operate simultaneously or one precedes the other (Figure 2).
Figure 2.
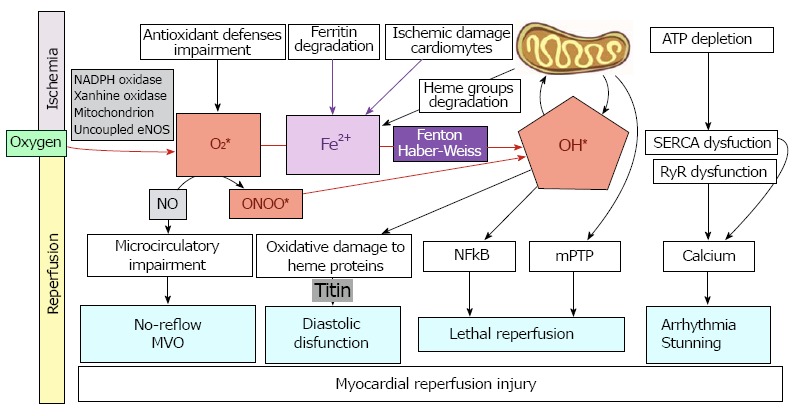
Role of reactive oxygen species and iron mobilization in myocardial reperfusion injury and its clinical implications. MVO: Microvascular obstruction; ONOO*: Peroxynitrite; NO: Nitric oxide; OH *: Radical hydroxyl; Fe: Iron; RyR: Ryanodine receptor channel; SERCA: Sarco/endoplasmic reticulum Ca2+-ATPase.
Oxidative stress
The level of myocardial tissue oxygenation increases following restoration of blood flow, which is initiated with a burst of ROS generation[8]; these ROS are the major initiators of myocardial damage in MRI[3]. Increased ROS production is mainly due to the activation of xanthine oxidase in endothelial cells, mitochondrial electron transport chain reactions in cardiomyocytes, and NADPH oxidase in inflammatory cells[9] (Figure 1).
Oxidative stress occurs when there is an imbalance between the generation of ROS and the antioxidant defense systems in the body so that the latter becomes overwhelmed[10]. ROS include hydrogen peroxide (H2O2), the superoxide radical anion, the hydroxyl radical (OH•), and peroxynitrite anion (ONOO-), and they have all been shown to increase with reperfusion[11] (Figure 2). As a result of lipid peroxidation, oxidation of DNA and proteins and membrane damage may take place. This leads to alterations in membrane permeability and to modifications of protein structure and functional changes[12].
ROS sources: In pathophysiological conditions, there are many sources of ROS in myocardial tissue. The most important sources are NADPH oxidases (NOX), uncoupled eNOS, xanthine oxidases and the mitochondrion. NOX catalyzes the one electron reduction of O2 to generate super-oxide radical anion (O2•−), using NADPH as the source of electrons. This enzyme is largely present in the activated neutrophil, wherein it generates large amounts of toxic O2•− and other ROS important in bactericidal function[13]. Pathogenic roles of NOX-derived ROS are also verified in human IR injury in vivo[14]. It was recently reported that in isolated perfused murine hearts that NOX1 and/or NOX2 gene knock-out significantly attenuated MRI (by up to 50% of the final infarct size)[15], thus demonstrating the crucial importance of this enzyme in MRI.
The NO synthases (NOS) are a family of enzymes that convert the amino acid L-arginine to L-citrulline and NO. Endothelial NOS (eNOS) plays a major role in the regulation of vascular function. The eNOS may become uncoupled in the absence of the NOS substrate L-arginine or the cofactor BH4. Uncoupled eNOS results in the production of O2•− instead of NO[16-18]. This perpetuates a vicious cycle because peroxynitrite, the reaction product of superoxide and NO, leads to further eNOS uncoupling[19]. Furthermore, eNOS uncoupling may play a major role in MRI by increasing ROS production and limiting NO availability[20].
Xanthine oxidase is predominantly present in the vascular endothelium in the normal heart and generates O2•−, H2O2, and OH• as byproducts of its normal metabolic action[21]. Under pathological conditions, such as tissue ischemia, xanthine dehydrogenase can be converted to Xanthine oxidase. In IR this enzyme catalyzes the formation of uric acid with the coproduction of O2•−[22]. Superoxide release results in the recruitment and activation of neutrophils and their adherence to endothelial cells, which stimulates the formation of xanthine oxidase in the endothelium, with further O2•− production[23]
Mitochondria are cellular organelles involved in energy production, so any injury that they may suffer could cause impairment of cellular energy that could lead, depending on the intensity of the injury, to apoptosis or different levels of cellular damage. During ischemia, due to the lack of oxygen, the electron transport chain cannot function correctly and therefore ROS are produced at high levels. Additionally, ROS may cause oxidative damage of mitochondrial DNA, impairing mitochondrial function. This damage performs a positive feedback on ROS production that, at the same time, perpetuates mitochondrial damage and ROS synthesis. Oxidative injury to the mitochondrial membrane can also occur, resulting in membrane depolarization and the uncoupling of oxidative phosphorylation, with altered cellular respiration[24]. This can ultimately lead to mitochondrial damage, with release of cytochrome c, activation of caspases, and apoptosis[25].
RNS sources: The ROS are not solely responsible for free radical damage. Reactive nitrogen species (RNS), mainly peroxynitrite anions (ONOO-), also generate RNS-damage, thus producing nitrosative stress. Peroxynitrite results from the interaction between NO and the superoxide anion[4], and NO is synthesized mainly by nitric oxide synthases which have two isoforms in the cardiomyocyte: endothelial (eNOS) and inducible (iNOS). Oxidative and nitrosative damage causes the uncoupling of both NOS isoforms, resulting in the enhanced synthesis of O2•−[4].
Evidence supports the view that nitrosative stress plays an important role in the pathogenesis of MRI. While NO itself is not harmful, some of the reaction products (mainly OH•) resulting from high ONOO- formation in the cell are highly cytotoxic substances[26]. The production of O2•− is increased during reperfusion, which interacts with NO and leads to the formation of ONOO-, thus triggering the previously described phenomenon[27]. Peroxynitrite not only causes structural damage by attacking macromolecules, but it also leads to myocardial functional impairment[28]. The general view about the mechanisms that lead to nitrosative stress is that IR can induce iNOS expression and that the resulting high concentrations of NO can lead to cardiac injury[26]. The drop in NO concentration occurring during cardiac IR plays an important role in triggering the transcription nuclear factor kappaB (NF-κB) leading to activation and successive induction of iNOS expression during the reperfusion phase[29-31]. Figure 1 shows a diagram of ROS and RNS sources in myocardial tissue.
Iron mobilization
It has been postulated that iron homeostasis could play an important role in the development of MRI in the cardiomyocytes[32,33]. Free iron is deleterious for cells; thus generally it is bound to proteins forming complexes[34]. During ischemia, iron metabolism is impaired, and it is released as free iron. This catalytic free iron can generate ROS through the Fenton reaction, catalyzing the production of ·OH from H2O2 and O2•−[35]. It has been reported that susceptibility to injury from H2O2 in rat hearts is associated with the magnitude of the intracellular low molecular weight iron pool[36]. Some metals with redox properties have a well-documented role in the development of MRI[37,38]. Following reperfusion, both iron and copper are released to the coronary circulation[32] which can contribute to ROS generation (Figure 2). In patients with thalassemia the iron overload is related to arrhythmias and congestive heart failure, which is the main cause of death among these patients[39]. Iron chelation therapy has significantly improved the survival of patients with thalassemia[40], because iron chelators are effective and safe drugs to treat the iron poisoning[41].
Calcium homeostasis
Oxidative stress modifies phospholipids and proteins leading to lipid peroxidation and thiol-group oxidation; these changes are considered to alter membrane permeability and configuration in addition to producing functional modifications of various cellular proteins[42]. Oxidative stress may result in cellular defects including a depression in the sarcolemma Ca2+-pump ATPase that leads to a decreased Ca2+-efflux, and a depression in (Na + K)-ATPase activity that, in turn, leads to an increased Ca2+-influx[43]. Oxidative stress has also been reported to depress the sarcoplasmic reticulum Ca2+-pump ATPase (SERCA) and thus inhibit Ca2+ sequestration from the cytoplasm in cardiomyocytes[44]. The depression in Ca2+-regulatory mechanism by ROS ultimately results in intracellular Ca2+ ([Ca2+]i) overload and cell death. In addition, an increase in [Ca2+]i during ischemia induces the conversion of xanthine dehydrogenase to xanthine oxidase and subsequently results in increased production of O2•−[44].
Recently it has been shown that the function of the channel ryanodine receptor (RyR) is controlled by ROS[45]. It has been demonstrated that NADPH oxidase and the RyR channel could be located adjacent to each other in the T-tubules of cardiomyocytes[46]. Thus, the increase in ROS production after myocardial reperfusion could lead to an increase in RyR channel function, resulting in an intracellular calcium overload, thereby causing activation of pro-apoptotic intracellular pathways, necrosis, and electromechanical alteration. All these mechanisms are summarized in Figure 3.
Figure 3.
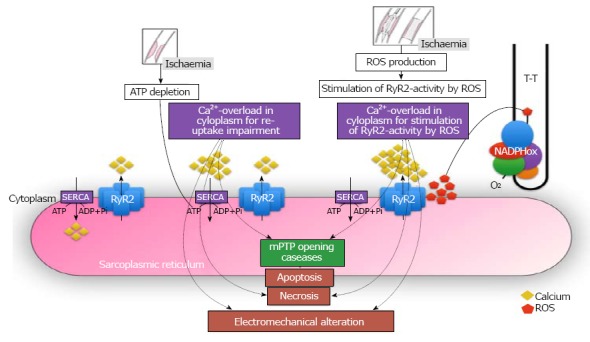
Central role of calcium in the electro-mechanical dissociation of cardiomyocyte after myocardial reperfusion. RyR: Ryanodine receptor channel; SERCA: Sarco / endoplasmic reticulum Ca2+-ATPase; mPTP: Mitochondrial permeability transition pore; Ca: Calcium; ROS: Reactive oxygen species.
Redox-sensitive signaling pathways: Not only do ROS exert their actions by directly modifying organic molecules, but ROS are also involved in the regulation of the expression of several genes[47]. NF-ĸB and AP-1, both of which can experience ROS-mediated activation, stimulate the transcription of several protein mediators, for example, proinflammatory cytokines that activate several cell death pathways[48]. The role of cytokines, chemokines, leukocytes, and acute-phase proteins such as high-sensitivity C-reactive protein in the pathogenesis of MRI has been reported in several studies[49,50]. Oxidative stress, ROS and inflammation are linked in a way that is very difficult to dissect. These phenomena have important molecular bridges that are activated in the presence of ROS[51], leading to the activation of multiple mechanisms that cause heart tissue remodeling and therefore enhance the susceptibility to rhythm disorders. Among those molecules, the most studied has been the transcriptional factor NF-ĸB, a factor that responds to changes of the cellular oxidative state, ischemia-reperfusion, and inflammatory molecules[52]. When NF-ĸB is activated, for example in the presence of ROS by phosphorylation of its inhibitory cofactor (Iĸ-B), it bonds to a DNA response element and promotes the transcription of genes involved in inflammatory and pro-fibrotic response, for example IL-6, which transforms growth factors TGF-β and TNF-α[53]. Those molecules act in various tissues, but particularly in the heart, producing extracellular matrix remodeling and fibrosis (structural remodeling), which changes the electrophysiological properties of the heart. Several studies have associated NF-ĸB activation with cardiac dysfunction, ventricular hypertrophy, and maladaptive cardiac growth[54] (Figure 2).
Exposure to low-to-moderate ROS levels should trigger a survival response and reinforce ROS scavengers of the antioxidant defense system to elicit a cardioprotective effect for myocardial reperfusion. The molecular mechanism responsible for this adaptive change involves enhanced antioxidant activity achieved by up-regulating several housekeeping genes partly under the control of Nrf2 (nuclear factor-erythroid 2-related factor 2); Nrf2 is normally sequestered in the cytosol by Keap[55]. Upon oxidative stimulation, Nrf2 oxidizes or covalently modifies Keap1 thiol groups, which dissociate from Keap1 and undergo nuclear translocation. In the nucleus, Nrf2 binds to antioxidant response elements in target gene promoters[56], which increase the expression of antioxidant enzymes. It has been demonstrated that the constitutive levels/activities of a number of important antioxidants and phase 2 enzymes, such as CAT, GSH-Px, glutathione reductase, glutathione transferase, NADPH-quinone oxidoreductase 1, and heme oxygenase-1 in primary cardiomyocytes are dependent on Nrf2 status. In addition, Nrf2 diminishes the susceptibility of cardiomyocytes to injury elicited by oxidants and electrophilic species[57], making the Nrf2 signaling pathway an important mechanism for myocardial cytoprotection. It is of interest to note that ROS levels could be responsible for the activation of NF-κB and/or Nrf2 pathways.
Clinical implications: Myocardial damage caused by ischemia-reperfusion events are mainly associated with four clinical conditions: lethal reperfusion, myocardial stunning, no-reflow phenomenon, and reperfusion arrhythmias (Figure 2).
Lethal reperfusion is a paradoxical type of MRI caused by the restoration of coronary blood flow after an ischemic episode. It is defined as the death of cardiomyocytes that were viable immediately before myocardial reperfusion. Its main manifestation is as an increased infarct size due to reperfusion, a condition mainly associated with AMI[3]. In the late fifties, it was suggested that myocardial reperfusion contributes part of the histological damage associated with ischemia-reperfusion models. However, for decades it was very complex to determine the precise evolution of necrosis along the transition from ischemia to reperfusion in myocardial tissue[58]. Nowadays, the harmful effects of myocardial reperfusion damage, also known as lethal reperfusion injury, are considered to involve myocardial cell death derived from the restoration of blood flow subsequent to an ischemic process, and to act through mechanisms strongly associated with oxidative stress[3].
Reperfusion arrhythmias clinically represent a major comorbidity of AMI with an 88.7% occurrence rate in certain small clinical trials with continuous monitoring[59]. In addition, postoperative atrial fibrillation (POAF), the most common reperfusion arrhythmia associated with cardiac surgeries, has an incidence ranging between 20%-40%[60]. Myocardial stunning, despite being a reversible damage, is the cause of an impaired ventricular function that leads to increased morbidity. It is derived from a short-term ischemia-reperfusion process that was first reported in the early 1930s[61]. Myocardial stunning is present to a greater or lesser extent in all survivors of AMI. In the late 1980s evidence began to appear suggesting an important role of oxidative stress in the development of myocardial stunning, proposing that the main injury pathway could be an altered calcium homeostasis associated with sarcoplasmic reticulum damage[62]. More recently, clinical studies have strengthened this hypothesis[63], and it has been reported in animal models that interventions aimed to improve antioxidant defenses attenuate myocardial stunning[64,65].
The no-reflow phenomenon is an impaired myocardial perfusion of a specific segment of the coronary system that is not associated with an angiographic occlusion of the respective vessel[66]. Vascular and endothelial damage can occur after the reperfusion of previously blocked coronary circulation. It can be exhibited as a microvascular dysfunction after restoring the flow during either angioplasty or thrombolysis, thus leading to the development of the no-reflow phenomenon[67]. The presence of coronary microvascular dysfunction and this phenomenon are associated with larger infarct size, lower left ventricular ejection fraction, adverse left ventricular remodeling in the remote stage of myocardial infarction, and increased incidences of heart failure and death, compared with patients without no-reflow phenomenon[68]. Some studies using animal models showed that antioxidant strategies are able to reduce this phenomenon[69-71], and this data is consistent with a small clinical trial finding that antioxidant depletion is associated with no-reflow phenomenon in AMI[72]. In addition, recent research in rabbits shows that the suppression of the oxidative stress-sensitive transcription factor NF-ĸB, a key mediator of inflammation in cardiovascular systems, reduces myocardial no-reflow phenomenon[73].
Recently, our group has reported major clinical benefits with the use of antioxidants in pathologies associated with myocardial reperfusion, such as POAF and AMI. With regard to POAF, we documented a significant decrease in the incidence of this arrhythmia in patients undergoing cardiac surgery with extracorporeal circulation after administration of ascorbate, alpha-tocopherol, and omega-3 polyunsaturated fatty acids, which was accompanied by a significant decrease in oxidative stress biomarkers in auricular tissue and peripheral blood[60].
ROLE OF ANTIOXIDANTS
Despite a molecular basis and in vitro evidence supporting the use of antioxidants to prevent MRI, clinical evidence continues to be controversial. In the clinical setting, impaired micro-circulatory reperfusion was improved by ascorbate infusion in patients undergoing elective PCA[74]. Similar results were recently reported by our group[75]. These results suggest a positive role of antioxidants in counteracting the deleterious effects of oxidative stress on microvascular function. On the other hand, the ROS scavenger edaravone when administered to patients with AMI immediately prior to reperfusion, significantly reduced infarct size and reperfusion arrhythmias[76,77]. Also some experimental studies reported that the use of deferoxamine (DFO) and N-acetylcysteine (NAC) could improve microvascular dysfunction[78,79].
Carotenoids represent another potential pharmacological alternative in the management of MRI[80]. Carotenoids are a widely distributed group of fat-soluble pigments which exert antioxidant, anti-inflammatory, and antiproliferative properties[81]. Several experimental data support potential role of carotenoids in this pathological condition: Tong et al[81] demonstrated that pretreatment with lycopene reduced cardiomyocyte death induced by ischemia/reoxygenation in vitro, and also reduced myocardial infarct size in an in vivo model of AMI[82]. Another carotenoid, crocetin, protected against myocardial reperfusion injury in vivo by inhibiting ROS production, reducing eNOS expression and myocardium apoptosis[82]. All-trans retinoic acid presented also protective activity against reperfusion injury both in vitro and in vivo, probably by down-regulating MAPK signaling[84]. Despite the fact that carotenoids have been useful in preventing MRI in experimental studies and have arisen as a promising pharmacological alternative, further clinical studies and randomized clinical trials are required.
In the following paragraphs we will discuss a new hypothesis for the prevention of MRI through the combined use of ascorbate, NAC, and DFO prior to reperfusion in order to strengthen antioxidant defense systems and so prevent oxidative damage (Figure 4).
Figure 4.
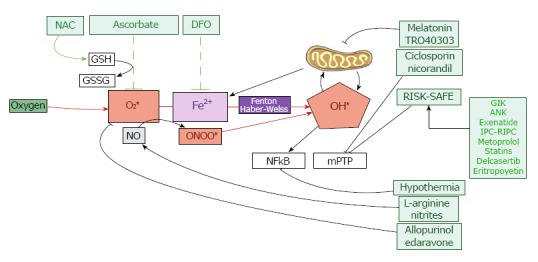
Experimental, pharmacological and clinical approaches to prevent myocardial reperfusion injury at cellular level. RISK: Reperfusion injury salvage kinase pathway; SAFE: Survivor activating factor enhancement pathway; GSH: Reduced glutathione; GSSG: Oxidized glutathione; NAC: N-acetylcysteine; DFO: deferoxamine; ONOO*: Peroxynitrite; NO: Nitric oxide; OH *: Radical hydroxyl; mPTP: Mitochondrial permeability transition pore.
Ascorbate
The basis of this hypothesis is to achieve high plasma levels of ascorbate prior to reperfusion in order to strengthen the antioxidant defense system of myocardial tissue. Thus, when oxygen suddenly arrives to the previously ischemia-damaged myocardial tissue-which is the primary substrate for the production of the highly reactive superoxide anion radical-ascorbate may efficiently reduce ROS and prevent oxidative damage[5,8]. To support this hypothesis, we will discuss the main actions of this antioxidant and its pharmacokinetic properties.
Ascorbate is an essential antioxidant that performs its roles in different cell locations by acting in water-soluble components[85,86]. The most studied mechanism in which ascorbate acts is partly based on its ability to directly reduce ROS[87-89]. Besides its ROS scavenger actions, ascorbate exerts a complex modulation of numerous enzymes involved in ROS production, endothelial dysfunction, platelet aggregation, and smooth muscle cell tone[90-92]. The four most important mechanisms in which ascorbate modulates the endothelial function are NADPH down-regulation, and the up-regulation of eNOS, phospholipase A2, and antioxidant enzymes. NADPH oxidase, the most important superoxide source in the cardiovascular system, can be directly down-regulated by ascorbate[91,92]. The mechanism behind this effect has not been completely elucidated. It has been reported that ascorbate could be involved in the transcriptional and post-transcriptional modulation of NADPH oxidase[89,93] as well as in its synthesis[94]. In the presence of oxidative stress, eNOS is mostly in its uncoupled form which leads to endothelial dysfunction. In this context, ascorbate has been shown to increase eNOS activity, by preventing the oxidation of tetrahydrobiopterin and by inhibiting the p47phox subunit expression[95]. Therefore, ascorbate increases NO synthesis, reduces ROS formation and contributes to vascular tone regulation[96-98]. In relation to the up-regulation of antioxidant enzymes, some studies have demonstrated a positive correlation between antioxidant vitamin and antioxidant enzyme activity, particularly SOD. The mechanisms underlying these findings are not well explained, but it is plausible to hypothesize the existence of transcriptional and post-transcriptional events involved in the up-regulation of those antioxidant enzymes[92].
Ascorbate counteracts and prevents the oxidation of lipids, proteins, and DNA, subsequently protecting their structure and biological function. Together with glutathione, ascorbate constitutes a primary line of defense against ROS[99]. Ascorbate, in aqueous compartments, can recycle α-tocopherol in membranes by reducing the α-tocopheroxyl radical back to α-tocopherol[100]. Accordingly, ascorbate has been shown to recycle α-tocopherol in lipid bilayers[101] and erythrocytes[95].
Ascorbate scavenging is concentration-dependent and requires intravenous administration. This is necessary because ascorbate concentration in plasma is tightly controlled and an excess of ascorbate is excreted as a function of dosage. In fact, even with supplementation approaching maximally tolerated doses, ascorbate plasma concentrations are always < 250 µmol/L. By contrast, intravenously injected ascorbate can safely lead to concentrations of 25-30 mmol/L[102]. It is of interest to mention that intra-arterial administration of high doses of ascorbate has been demonstrated to abolish both in vivo and in vitro effects of the superoxide anion with respect to the impairment of vascular endothelial function in patients with essential hypertension[103]. Unfortunately, oral doses are not enough to scavenge superoxide anions, thus a beneficial effect should not be expected.
Our group recently developed a randomized clinical trial in patients with AMI undergoing PCA, where massive doses of ascorbate (or placebo) were administered prior to PCA. Patients treated with ascorbate prior to myocardial reperfusion showed a better recovery of ejection fraction at 2-3 mo (measured by cardiac magnetic resonance) and significantly higher myocardial perfusion after PCA (TIMI-myocardial perfusion grade) than placebo patients, with no differences in infarct size[75] (Table 1).
Table 1.
Clinical trials
| Study details | Country | n Intervention | Main findings | Ref. | ||
| AA | Ascorbate previous to elective coronary angioplasty | Italy | 28 | 28 | Decrease in oxidative stress and improves reperfusion parameters | [74] |
| Ascorbate previous to primary coronary angioplasty in patients with AMI | Chile | 53 | 46 | Improve ventricular function and reperfusion No differences in infarct size | [75] | |
| NAC | N-acetylcysteine previous and after primary coronary angioplasty in patients with AMI | Germany | 126 | 126 | Decrease in oxidative stress No differences in infarct size | [105] |
| N-acetylcysteine and nitroglycerine previous to primary coronary angioplasty in patients with AMI | Australia | 67 | 65 | Decrease in infarct size and cardiac damage biomarkers | [116] | |
| DFO | Deferoxamine previous and after coronary angioplasty in patients with AMI | Australia | 28 | 32 | Decrease in oxidative stress No differences in infarct size | [114] |
Main clinical studies that have used ascorbate, N-acetylcysteine or deferoxamine to prevent reperfusion injury in patients affected by acute myocardial infarction and treated with coronary angioplasty. AA: Ascorbate; NAC: N-acetylcysteine; DFO: Deferoxamine; IR: Ischemia reperfusion; AMI: Acute myocardial infarction.
N-acetyl-L-cysteine
Ascorbate consumes glutathione (GSH) to exert its antioxidant activity. High doses of ascorbate might be associated with a decrease in cellular GSH reserves[5]. For this reason, N-acetyl-L-cysteine (NAC) - a known GSH-donor-may also have synergistic effects with high doses of ascorbate. In the following paragraphs, we will discuss the potential role of NAC in preventing MRI.
Despite numerous studies and a prolonged track record of clinical trials, the effects of NAC are clouded in controversy and its pharmacological mechanism has not yet been fully clarified. However, there is plenty of evidence regarding its mechanism of action. First of all, NAC’s main feature, and also the most studied one, is its capacity to act as a precursor for synthesis of GSH, thus replenishing GSH that has become depleted through the use of this peptide in detoxification routes[104]. However, it is vital to think of NAC as a pro-drug, because actions that are driven by this drug are dependent on its successful conversion to the antioxidant and detoxifying agent, GSH. Another frequently mentioned property of NAC is its intrinsic antioxidant activity. Nevertheless, the evidence regarding the antioxidant potential of NAC suggests that it does not have a noteworthy direct antioxidant activity[105].
NAC acts indirectly through chelation of metal ions such as catalytic iron[106,107] giving it the capability of mediating Fenton’s reaction, thus ameliorating the possibility of the formation of hydroxyl radicals. This property is due to the fact that NAC forms conjugates with some metals. However, the importance of this mechanism in driving any protective effects compared to intracellular GSH replenishing is still unclear. Current evidence agrees on the capability of NAC to act as an inhibitor of NF-κB[108], a transcription factor that plays a critical role in inflammation, immunity, cell proliferation, differentiation, and survival. In conclusion, molecular mechanisms by which NAC exerts its diverse effects are complex and still unclear. Although it has been shown that NAC interacts with numerous biochemical pathways, its main mechanism involves serving as a precursor of cysteine and replenishing cellular GSH levels[104].
NAC has been widely used in different experimental and clinical settings to counteract oxidative stress. It has been demonstrated that NAC in combination with nitroglycerin and streptokinase is associated with significantly less oxidative stress and improved preservation of left ventricular function[109]. However, it has also been reported that a high-dose of NAC prior to PCA, although it reduces oxidative stress, does not provide an additional advantage in the prevention of MRI[110]. Additionally, an interesting study published in 2006 shows that administration of NAC in combination with streptokinase significantly diminishes oxidative stress and improves left ventricular function in patients with AMI[111]. A recent study using a rat model of myocardial ischemia-reperfusion injury demonstrates that treatment with continuous infusion of NAC (150 mg/kg per hour) starting 30 min before occlusion and lasting for 2 h (or until 1 h after the start of reperfusion) produces a significant limitation of the infarct and allows the recovery of the decreased total glutathione when compared to control[112]. Recently has been published the NACIAM trial by Pasupathy et al[113], that demonstrated a protective effect with the use of high doses of NAC in combination with a nitric oxide donor in patients with AMI (Table 1). This important study shows that NAC has a powerful protective effect when used in combination and previous to myocardial reperfusion. In summary, due to the known antioxidant and cardioprotective effect and its role as GSH-donor, it is plausible to suggest that NAC might have a synergistic effect with high doses of ascorbate and deferoxamine to prevent MRI.
Deferoxamine
Given the known role of iron in the lethal reperfusion, iron chelators have been tested to ameliorate this injury. One of the most frequently used drugs for this purpose is DFO. The first reports of its use to improve cardiac function in myocardium iron overload by directly removing iron from the myocardium[114] date from 1980s[115]. In animal models of AMI, the use of DFO has exhibited positive results. Some studies performed in dogs reported a decrease in the infarct size when they used DFO during the reperfusion, suggesting that iron-catalyzed production of ROS contributes to cardiomyocyte necrosis in the setting of MRI[116,117]. Studies have described improved recovery of myocardial function after ischemia, by using iron chelation[36,118]. The results obtained from animal models of MRI have suggested the use of iron chelators in the human model with partial results to date. Paraskevaidis et al[119] suggested DFO infusion was able to reduce myocardial stunning after elective coronary artery bypass grafting and to improve long-term ejection fraction. In a recent clinical study, Chan et al[120] randomized patients with STEMI to intravenous deferoxamine before coronary angioplasty and then for 12 h vs placebo (Table 1). The serum iron levels and lipid peroxidation biomarkers were reduced in the DFO-group without differences in the infarct size. The role of iron and ascorbate in the MRI has become of increasing interest in the last few years. It has been demonstrated that the combined use of DFO and ascorbate prevent reperfusion arrhythmias[121].
As has been previously discussed, cumulated evidence from both experimental and clinical studies leads us to support the view that a novel combined antioxidant strategy could limit MRI and its consequences. This novel hypothesis is based on the combined use of antioxidants prior to the reperfusion therapy in order to limit the oxidative challenge during reperfusion. The key points of this novel intervention are: (1) To achieve high plasma concentrations of ascorbate through massive intravenous doses to counteract the ROS and RNS production; (2) the use of NAC to prevent GSH depletion; and (3) the use of DFO to diminish the catalytic free iron levels in order to prevent the ROS production by the Fenton reaction.
Accordingly, in our laboratory recent studies of the murine Langendorff model have been conducted to determine the effect of antioxidants in MRI. We are now studying the effect of ascorbate, NAC, and DFO used alone and in association. Under these conditions, we expect a lower vulnerability of the myocardial tissue to the reperfusion injury associated with oxidative stress. This protective effect could be expressed by a lower infarct size, reduced post-reperfusion arrhythmias and myocardial stunning occurrence, and improved microvascular function. Finally, at present, there is no evidence available from any trial that has applied this antioxidant protocol to diminish MRI. Table 1 shows a summary of the main clinical studies that have used antioxidants to prevent MRI in patients with AMI.
CONCLUSION
There is major evidence with respect to the contribution of oxidative stress to MRI in cardiovascular diseases. Despite the many significant advances in the understanding of the mechanisms of MRI, it remains an unsolved problem. There is a lack of consistency between basic studies and clinical trials aimed to reduce MRI through antioxidant therapies. Although promising results have been obtained in experimental studies (mainly in animal models), these benefits have not been translated into clinical settings. It is noteworthy that the administration of high ascorbate doses prior to reperfusion and also NAC administration appear to be safe and rational therapies against the development of oxidative damage associated with myocardial reperfusion. Furthermore, ascorbate association with NAC and DFO could improve the beneficial effect of ascorbate, making it even more effective in preventing myocardial reperfusion damage associated with PCA following AMI.
Footnotes
Conflict-of-interest statement: No potential conflicts of interest.
Manuscript source: Invited manuscript
Peer-review started: March 28, 2018
First decision: April 11, 2018
Article in press: May 10, 2018
Specialty type: Cardiac and cardiovascular systems
Country of origin: Chile
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B
Grade C (Good): C, C
Grade D (Fair): 0
Grade E (Poor): 0
P- Reviewer: Ciccon MM, Sun CK, Schoenhagen P S- Editor: Ji FF L- Editor: A E- Editor: Wu YXJ
Contributor Information
Jaime González-Montero, Molecular and Clinical Pharmacology Program, Institute of Biomedical Sciences, Faculty of Medicine, University of Chile, Santiago 70058, Chile.
Roberto Brito, Molecular and Clinical Pharmacology Program, Institute of Biomedical Sciences, Faculty of Medicine, University of Chile, Santiago 70058, Chile; Internal Medicine Department, University of Chile, Clinical Hospital, Santiago 70058, Chile.
Abraham IJ Gajardo, Molecular and Clinical Pharmacology Program, Institute of Biomedical Sciences, Faculty of Medicine, University of Chile, Santiago 70058, Chile; Internal Medicine Department, University of Chile, Clinical Hospital, Santiago 70058, Chile.
Ramón Rodrigo, Molecular and Clinical Pharmacology Program, Institute of Biomedical Sciences, Faculty of Medicine, University of Chile, Santiago 70058, Chile. rrodrigo@med.uchile.cl.
References
- 1.Roe MT, Halabi AR, Mehta RH, Chen AY, Newby LK, Harrington RA, Smith SC Jr, Ohman EM, Gibler WB, Peterson ED. Documented traditional cardiovascular risk factors and mortality in non-ST-segment elevation myocardial infarction. Am Heart J. 2007;153:507–514. doi: 10.1016/j.ahj.2006.12.018. [DOI] [PubMed] [Google Scholar]
- 2.Vanden Hoek TL, Li C, Shao Z, Schumacker PT, Becker LB. Significant levels of oxidants are generated by isolated cardiomyocytes during ischemia prior to reperfusion. J Mol Cell Cardiol. 1997;29:2571–2583. doi: 10.1006/jmcc.1997.0497. [DOI] [PubMed] [Google Scholar]
- 3.Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–1135. doi: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]
- 4.Rodrigo R. Oxidative stress and antioxidants: their role in human disease. Nova Biomedical Books, 2009 [cited 2018 Mar 26]: 358 [Google Scholar]
- 5.Rodrigo R, Libuy M, Feliú F, Hasson D. Molecular basis of cardioprotective effect of antioxidant vitamins in myocardial infarction. Biomed Res Int. 2013;2013:437613. doi: 10.1155/2013/437613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferrari R. The role of mitochondria in ischemic heart disease. J Cardiovasc Pharmacol. 1996;28 Suppl 1:S1–10. doi: 10.1097/00005344-199600003-00002. [DOI] [PubMed] [Google Scholar]
- 7.Jahangiri A, Leifert WR, Kind KL, McMurchie EJ. Dietary fish oil alters cardiomyocyte Ca2+ dynamics and antioxidant status. Free Radic Biol Med. 2006;40:1592–1602. doi: 10.1016/j.freeradbiomed.2005.12.026. [DOI] [PubMed] [Google Scholar]
- 8.Rodrigo R, Prieto JC, Castillo R. Cardioprotection against ischaemia/reperfusion by vitamins C and E plus n-3 fatty acids: molecular mechanisms and potential clinical applications. Clin Sci (Lond) 2013;124:1–15. doi: 10.1042/CS20110663. [DOI] [PubMed] [Google Scholar]
- 9.Chamiec T, Herbaczyńska-Cedro K, Ceremuzyński L. Effects of antioxidant vitamins C and E on signal-averaged electrocardiogram in acute myocardial infarction. Am J Cardiol. 1996;77:237–241. doi: 10.1016/s0002-9149(97)89385-x. [DOI] [PubMed] [Google Scholar]
- 10.Juránek I, Bezek S. Controversy of free radical hypothesis: reactive oxygen species--cause or consequence of tissue injury? Gen Physiol Biophys. 2005;24:263–278. [PubMed] [Google Scholar]
- 11.Eaton P, Clements-Jewery H. Peroxynitrite: in vivo cardioprotectant or arrhythmogen? Br J Pharmacol. 2008;155:972–973. doi: 10.1038/bjp.2008.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zimmerman JJ. Defining the role of oxyradicals in the pathogenesis of sepsis. Crit Care Med. 1995;23:616–617. doi: 10.1097/00003246-199504000-00003. [DOI] [PubMed] [Google Scholar]
- 13.Brandes RP, Kreuzer J. Vascular NADPH oxidases: molecular mechanisms of activation. Cardiovasc Res. 2005;65:16–27. doi: 10.1016/j.cardiores.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 14.Loukogeorgakis SP, van den Berg MJ, Sofat R, Nitsch D, Charakida M, Haiyee B, de Groot E, MacAllister RJ, Kuijpers TW, Deanfield JE. Role of NADPH oxidase in endothelial ischemia/reperfusion injury in humans. Circulation. 2010;121:2310–2316. doi: 10.1161/CIRCULATIONAHA.108.814731. [DOI] [PubMed] [Google Scholar]
- 15.Braunersreuther V, Montecucco F, Asrih M, Pelli G, Galan K, Frias M, Burger F, Quinderé AL, Montessuit C, Krause KH, et al. Role of NADPH oxidase isoforms NOX1, NOX2 and NOX4 in myocardial ischemia/reperfusion injury. J Mol Cell Cardiol. 2013;64:99–107. doi: 10.1016/j.yjmcc.2013.09.007. [DOI] [PubMed] [Google Scholar]
- 16.Vásquez-Vivar J, Kalyanaraman B, Martásek P, Hogg N, Masters BS, Karoui H, Tordo P, Pritchard KA Jr. Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc Natl Acad Sci USA. 1998;95:9220–9225. doi: 10.1073/pnas.95.16.9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xia Y, Zweier JL. Direct measurement of nitric oxide generation from nitric oxide synthase. Proc Natl Acad Sci USA. 1997;94:12705–12710. doi: 10.1073/pnas.94.23.12705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chalupsky K, Cai H. Endothelial dihydrofolate reductase: critical for nitric oxide bioavailability and role in angiotensin II uncoupling of endothelial nitric oxide synthase. Proc Natl Acad Sci USA. 2005;102:9056–9061. doi: 10.1073/pnas.0409594102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schulz E, Jansen T, Wenzel P, Daiber A, Münzel T. Nitric oxide, tetrahydrobiopterin, oxidative stress, and endothelial dysfunction in hypertension. Antioxid Redox Signal. 2008;10:1115–1126. doi: 10.1089/ars.2007.1989. [DOI] [PubMed] [Google Scholar]
- 20.Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, Mitch WE, Harrison DG. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest. 2003;111:1201–1209. doi: 10.1172/JCI14172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Friedl HP, Smith DJ, Till GO, Thomson PD, Louis DS, Ward PA. Ischemia-reperfusion in humans. Appearance of xanthine oxidase activity. Am J Pathol. 1990;136:491–495. [PMC free article] [PubMed] [Google Scholar]
- 22.Granger DN. Role of xanthine oxidase and granulocytes in ischemia-reperfusion injury. Am J Physiol. 1988;255:H1269–H1275. doi: 10.1152/ajpheart.1988.255.6.H1269. [DOI] [PubMed] [Google Scholar]
- 23.Wang X, Han M, Bao J, Tu W, Dai Z. A superoxide anion biosensor based on direct electron transfer of superoxide dismutase on sodium alginate sol-gel film and its application to monitoring of living cells. Anal Chim Acta. 2012;717:61–66. doi: 10.1016/j.aca.2011.12.045. [DOI] [PubMed] [Google Scholar]
- 24.Nathan AT, Singer M. The oxygen trail: tissue oxygenation. Br Med Bull. 1999;55:96–108. doi: 10.1258/0007142991902312. [DOI] [PubMed] [Google Scholar]
- 25.Macdonald J, Galley HF, Webster NR. Oxidative stress and gene expression in sepsis. Br J Anaesth. 2003;90:221–232. doi: 10.1093/bja/aeg034. [DOI] [PubMed] [Google Scholar]
- 26.Darra E, Rungatscher A, Carcereri de Prati A, Podesser BK, Faggian G, Scarabelli T, Mazzucco A, Hallström S, Suzuki H. Dual modulation of nitric oxide production in the heart during ischaemia/reperfusion injury and inflammation. Thromb Haemost. 2010;104:200–206. doi: 10.1160/TH09-08-0554. [DOI] [PubMed] [Google Scholar]
- 27.Rodrigo R, Vinay J, Castillo R, Cereceda M, Asenjo R, Zamorano J, Araya J, Castillo-Koch R, Espinoza J, Larraín E. Use of vitamins C and E as a prophylactic therapy to prevent postoperative atrial fibrillation. Int J Cardiol. 2010;138:221–228. doi: 10.1016/j.ijcard.2009.04.043. [DOI] [PubMed] [Google Scholar]
- 28.Ferdinandy P, Danial H, Ambrus I, Rothery RA, Schulz R. Peroxynitrite is a major contributor to cytokine-induced myocardial contractile failure. Circ Res. 2000;87:241–247. doi: 10.1161/01.res.87.3.241. [DOI] [PubMed] [Google Scholar]
- 29.Kanno S, Lee PC, Zhang Y, Ho C, Griffith BP, Shears LL 2nd, Billiar TR. Attenuation of myocardial ischemia/reperfusion injury by superinduction of inducible nitric oxide synthase. Circulation. 2000;101:2742–2748. doi: 10.1161/01.cir.101.23.2742. [DOI] [PubMed] [Google Scholar]
- 30.Suzuki H, Colasanti M. Cross-talk between constitutive and inducible nitric oxide synthases. Circulation. 2001;103:E81–E81. doi: 10.1161/01.cir.103.14.e81. [DOI] [PubMed] [Google Scholar]
- 31.Kitamoto S, Egashira K, Kataoka C, Koyanagi M, Katoh M, Shimokawa H, Morishita R, Kaneda Y, Sueishi K, Takeshita A. Increased activity of nuclear factor-kappaB participates in cardiovascular remodeling induced by chronic inhibition of nitric oxide synthesis in rats. Circulation. 2000;102:806–812. doi: 10.1161/01.cir.102.7.806. [DOI] [PubMed] [Google Scholar]
- 32.Chevion M, Jiang Y, Har-El R, Berenshtein E, Uretzky G, Kitrossky N. Copper and iron are mobilized following myocardial ischemia: possible predictive criteria for tissue injury. Proc Natl Acad Sci USA. 1993;90:1102–1106. doi: 10.1073/pnas.90.3.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Korkmaz S, Barnucz E, Loganathan S, Li S, Radovits T, Hegedus P, Zubarevich A, Hirschberg K, Weymann A, Puskás LG, et al. Q50, an iron-chelating and zinc-complexing agent, improves cardiac function in rat models of ischemia/reperfusion-induced myocardial injury. Circ J. 2013;77:1817–1826. doi: 10.1253/circj.cj-12-1162. [DOI] [PubMed] [Google Scholar]
- 34.Esposito BP, Breuer W, Sirankapracha P, Pootrakul P, Hershko C, Cabantchik ZI. Labile plasma iron in iron overload: redox activity and susceptibility to chelation. Blood. 2003;102:2670–2677. doi: 10.1182/blood-2003-03-0807. [DOI] [PubMed] [Google Scholar]
- 35.Merkofer M, Kissner R, Hider RC, Brunk UT, Koppenol WH. Fenton chemistry and iron chelation under physiologically relevant conditions: Electrochemistry and kinetics. Chem Res Toxicol. 2006;19:1263–1269. doi: 10.1021/tx060101w. [DOI] [PubMed] [Google Scholar]
- 36.Voogd A, Sluiter W, Koster JF. The increased susceptibility to hydrogen peroxide of the (post-)ischemic rat heart is associated with the magnitude of the low molecular weight iron pool. Free Radic Biol Med. 1994;16:453–458. doi: 10.1016/0891-5849(94)90122-8. [DOI] [PubMed] [Google Scholar]
- 37.Pucheu S, Coudray C, Tresallet N, Favier A, de Leiris J. Effect of iron overload in the isolated ischemic and reperfused rat heart. Cardiovasc Drugs Ther. 1993;7:701–711. doi: 10.1007/BF00877824. [DOI] [PubMed] [Google Scholar]
- 38.Tang WH, Wu S, Wong TM, Chung SK, Chung SS. Polyol pathway mediates iron-induced oxidative injury in ischemic-reperfused rat heart. Free Radic Biol Med. 2008;45:602–610. doi: 10.1016/j.freeradbiomed.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 39.Zurlo MG, De Stefano P, Borgna-Pignatti C, Di Palma A, Piga A, Melevendi C, Di Gregorio F, Burattini MG, Terzoli S. Survival and causes of death in thalassaemia major. Lancet. 1989;2:27–30. doi: 10.1016/s0140-6736(89)90264-x. [DOI] [PubMed] [Google Scholar]
- 40.Xia S, Zhang W, Huang L, Jiang H. Comparative efficacy and safety of deferoxamine, deferiprone and deferasirox on severe thalassemia: a meta-analysis of 16 randomized controlled trials. PLoS One. 2013;8:e82662. doi: 10.1371/journal.pone.0082662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoffbrand AV, Taher A, Cappellini MD. How I treat transfusional iron overload. Blood. 2012;120:3657–3669. doi: 10.1182/blood-2012-05-370098. [DOI] [PubMed] [Google Scholar]
- 42.Hool LC. The L-type Ca(2+) channel as a potential mediator of pathology during alterations in cellular redox state. Heart Lung Circ. 2009;18:3–10. doi: 10.1016/j.hlc.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 43.Dixon IM, Hata T, Dhalla NS. Sarcolemmal Na(+)-K(+)-ATPase activity in congestive heart failure due to myocardial infarction. Am J Physiol. 1992;262:C664–C671. doi: 10.1152/ajpcell.1992.262.3.C664. [DOI] [PubMed] [Google Scholar]
- 44.Sasaki M, Joh T. Oxidative stress and ischemia-reperfusion injury in gastrointestinal tract and antioxidant, protective agents. J Clin Biochem Nutr. 2007;40:1–12. doi: 10.3164/jcbn.40.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Donoso P, Sanchez G, Bull R, Hidalgo C. Modulation of cardiac ryanodine receptor activity by ROS and RNS. Front Biosci (Landmark Ed) 2011;16:553–567. doi: 10.2741/3705. [DOI] [PubMed] [Google Scholar]
- 46.Prosser BL, Ward CW, Lederer WJ. X-ROS signaling: rapid mechano-chemo transduction in heart. Science. 2011;333:1440–1445. doi: 10.1126/science.1202768. [DOI] [PubMed] [Google Scholar]
- 47.Kim YH, Lim DS, Lee JH, Shim WJ, Ro YM, Park GH, Becker KG, Cho-Chung YS, Kim MK. Gene expression profiling of oxidative stress on atrial fibrillation in humans. Exp Mol Med. 2003;35:336–349. doi: 10.1038/emm.2003.45. [DOI] [PubMed] [Google Scholar]
- 48.Bowie A, O’Neill LA. Oxidative stress and nuclear factor-kappaB activation: a reassessment of the evidence in the light of recent discoveries. Biochem Pharmacol. 2000;59:13–23. doi: 10.1016/s0006-2952(99)00296-8. [DOI] [PubMed] [Google Scholar]
- 49.Chung MK, Martin DO, Sprecher D, Wazni O, Kanderian A, Carnes CA, Bauer JA, Tchou PJ, Niebauer MJ, Natale A, et al. C-reactive protein elevation in patients with atrial arrhythmias: inflammatory mechanisms and persistence of atrial fibrillation. Circulation. 2001;104:2886–2891. doi: 10.1161/hc4901.101760. [DOI] [PubMed] [Google Scholar]
- 50.Lamm G, Auer J, Weber T, Berent R, Ng C, Eber B. Postoperative white blood cell count predicts atrial fibrillation after cardiac surgery. J Cardiothorac Vasc Anesth. 2006;20:51–56. doi: 10.1053/j.jvca.2005.03.026. [DOI] [PubMed] [Google Scholar]
- 51.Pavlović D, Đorđević V, Kocić G. A “cross-talk” between oxidative stress and REDOX cell signaling. Med Biol. 2002;9:131–137. [Google Scholar]
- 52.Chandra J, Samali A, Orrenius S. Triggering and modulation of apoptosis by oxidative stress. Free Radic Biol Med. 2000;29:323–333. doi: 10.1016/s0891-5849(00)00302-6. [DOI] [PubMed] [Google Scholar]
- 53.Liakopoulos OJ, Schmitto JD, Kazmaier S, Bräuer A, Quintel M, Schoendube FA, Dörge H. Cardiopulmonary and systemic effects of methylprednisolone in patients undergoing cardiac surgery. Ann Thorac Surg. 2007;84:110–8; discussion 118-9. doi: 10.1016/j.athoracsur.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 54.Opie LH, Commerford PJ, Gersh BJ, Pfeffer MA. Controversies in ventricular remodelling. Lancet. 2006;367:356–367. doi: 10.1016/S0140-6736(06)68074-4. [DOI] [PubMed] [Google Scholar]
- 55.Rodrigo R, Libuy M, Feliú F, Hasson D. Oxidative stress-related biomarkers in essential hypertension and ischemia-reperfusion myocardial damage. Dis Markers. 2013;35:773–790. doi: 10.1155/2013/974358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kobayashi M, Yamamoto M. Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation. Antioxid Redox Signal. 2005;7:385–394. doi: 10.1089/ars.2005.7.385. [DOI] [PubMed] [Google Scholar]
- 57.Zhu H, Jia Z, Misra BR, Zhang L, Cao Z, Yamamoto M, Trush MA, Misra HP, Li Y. Nuclear factor E2-related factor 2-dependent myocardiac cytoprotection against oxidative and electrophilic stress. Cardiovasc Toxicol. 2008;8:71–85. doi: 10.1007/s12012-008-9016-0. [DOI] [PubMed] [Google Scholar]
- 58.Piper HM, García-Dorado D, Ovize M. A fresh look at reperfusion injury. Cardiovasc Res. 1998;38:291–300. doi: 10.1016/s0008-6363(98)00033-9. [DOI] [PubMed] [Google Scholar]
- 59.Tatli E, Alicik G, Buturak A, Yilmaztepe M, Aktoz M. Arrhythmias following revascularization procedures in the course of acute myocardial infarction: are they indicators of reperfusion or ongoing ischemia? ScientificWorldJournal. 2013;2013:160380. doi: 10.1155/2013/160380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rodrigo R, Korantzopoulos P, Cereceda M, Asenjo R, Zamorano J, Villalabeitia E, Baeza C, Aguayo R, Castillo R, Carrasco R, et al. A randomized controlled trial to prevent post-operative atrial fibrillation by antioxidant reinforcement. J Am Coll Cardiol. 2013;62:1457–1465. doi: 10.1016/j.jacc.2013.07.014. [DOI] [PubMed] [Google Scholar]
- 61.Tennant R, Wiggers CJ. The effects of coronary occlusion on myocardial contraction. Am J Physiol Content. American Physiological Society. 1935;112:351–361. [Google Scholar]
- 62.Bolli R. Mechanism of myocardial “stunning”. Circulation. 1990;82:723–738. doi: 10.1161/01.cir.82.3.723. [DOI] [PubMed] [Google Scholar]
- 63.Laky D, Parascan L, Cândea V. Myocardial stunning. Morphological studies in acute experimental ischemia and intraoperatory myocardial biopsies. Rom J Morphol Embryol. 2008;49:153–158. [PubMed] [Google Scholar]
- 64.Kals J, Starkopf J, Zilmer M, Pruler T, Pulges K, Hallaste M, Kals M, Pulges A, Soomets U. Antioxidant UPF1 attenuates myocardial stunning in isolated rat hearts. Int J Cardiol. 2008;125:133–135. doi: 10.1016/j.ijcard.2007.01.032. [DOI] [PubMed] [Google Scholar]
- 65.Crystal GJ, Malik G, Yoon SH, Kim SJ. Isoflurane late preconditioning against myocardial stunning is associated with enhanced antioxidant defenses. Acta Anaesthesiol Scand. 2012;56:39–47. doi: 10.1111/j.1399-6576.2011.02583.x. [DOI] [PubMed] [Google Scholar]
- 66.Kloner RA, Ganote CE, Jennings RB. The “no-reflow” phenomenon after temporary coronary occlusion in the dog. J Clin Invest. 1974;54:1496–1508. doi: 10.1172/JCI107898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Movahed MR, Butman SM. The pathogenesis and treatment of no-reflow occurring during percutaneous coronary intervention. Cardiovasc Revasc Med. 2008;9:56–61. doi: 10.1016/j.carrev.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 68.Bouleti C, Mewton N, Germain S. The no-reflow phenomenon: State of the art. Arch Cardiovasc Dis. 2015;108:661–674. doi: 10.1016/j.acvd.2015.09.006. [DOI] [PubMed] [Google Scholar]
- 69.Oyanagui Y, Sato S. Superoxide dismutases and anti-oxidants protected mice from no-reflow and necrotic damage induced by ischemia. Free Radic Res Commun. 1993;18:147–157. doi: 10.3109/10715769309147488. [DOI] [PubMed] [Google Scholar]
- 70.Shimizu M, Sjöquist PO, Wang QD, Rydén L. Effects of the angiotensin AT1 receptor blocker candesartan on myocardial ischemic/reperfusion injury. J Am Soc Nephrol. 1999;10 Suppl 11:S137–S142. [PubMed] [Google Scholar]
- 71.Molyneux CA, Glyn MC, Ward BJ. Oxidative stress and cardiac microvascular structure in ischemia and reperfusion: the protective effect of antioxidant vitamins. Microvasc Res. 2002;64:265–277. doi: 10.1006/mvre.2002.2419. [DOI] [PubMed] [Google Scholar]
- 72.Matsumoto H, Inoue N, Takaoka H, Hata K, Shinke T, Yoshikawa R, Masai H, Watanabe S, Ozawa T, Yokoyama M. Depletion of antioxidants is associated with no-reflow phenomenon in acute myocardial infarction. Clin Cardiol. 2004;27:466–470. doi: 10.1002/clc.4960270809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zeng M, Yan H, Chen Y, Zhao HJ, Lv Y, Liu C, Zhou P, Zhao B. Suppression of NF-κB reduces myocardial no-reflow. PLoS One. 2012;7:e47306. doi: 10.1371/journal.pone.0047306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Basili S, Tanzilli G, Mangieri E, Raparelli V, Di Santo S, Pignatelli P, Violi F. Intravenous ascorbic acid infusion improves myocardial perfusion grade during elective percutaneous coronary intervention: relationship with oxidative stress markers. JACC Cardiovasc Interv. 2010;3:221–229. doi: 10.1016/j.jcin.2009.10.025. [DOI] [PubMed] [Google Scholar]
- 75.Ramos C, Brito R, González-Montero J, Valls N, Gormaz JG, Prieto JC, Aguayo R, Puentes Á, Noriega V, Pereira G, Palavecino T, Rodrigo R. Effects of a novel ascorbate-based protocol on infarct size and ventricle function in acute myocardial infarction patients undergoing percutaneous coronary angioplasty. Arch Med Sci. 2017;13:558–567. doi: 10.5114/aoms.2016.59713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Valls N, Gormaz JG, Aguayo R, González J, Brito R, Hasson D, Libuy M, Ramos C, Carrasco R, Prieto JC, et al. Amelioration of persistent left ventricular function impairment through increased plasma ascorbate levels following myocardial infarction. Redox Rep. 2016;21:75–83. doi: 10.1179/1351000215Y.0000000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tsujita K, Shimomura H, Kaikita K, Kawano H, Hokamaki J, Nagayoshi Y, Yamashita T, Fukuda M, Nakamura Y, Sakamoto T, et al. Long-term efficacy of edaravone in patients with acute myocardial infarction. Circ J. 2006;70:832–837. doi: 10.1253/circj.70.832. [DOI] [PubMed] [Google Scholar]
- 78.Defraigne JO, Pincemail J, Detry O, Franssen C, Meurisse M, Limet R. Preservation of cortical microcirculation after kidney ischemia-reperfusion: value of an iron chelator. Ann Vasc Surg. 1994;8:457–467. doi: 10.1007/BF02133066. [DOI] [PubMed] [Google Scholar]
- 79.Brunet J, Boily MJ, Cordeau S, Des Rosiers C. Effects of N-acetylcysteine in the rat heart reperfused after low-flow ischemia: evidence for a direct scavenging of hydroxyl radicals and a nitric oxide-dependent increase in coronary flow. Free Radic Biol Med. 1995;19:627–638. doi: 10.1016/0891-5849(95)00077-b. [DOI] [PubMed] [Google Scholar]
- 80.Ciccone MM, Cortese F, Gesualdo M, Carbonara S, Zito A, Ricci G, De Pascalis F, Scicchitano P, Riccioni G. Dietary intake of carotenoids and their antioxidant and anti-inflammatory effects in cardiovascular care. Mediators Inflamm. 2013;2013:782137. doi: 10.1155/2013/782137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tong C, Peng C, Wang L, Zhang L, Yang X, Xu P, Li J, Delplancke T, Zhang H, Qi H. Intravenous Administration of Lycopene, a Tomato Extract, Protects against Myocardial Ischemia-Reperfusion Injury. Nutrients. 2016;8:138. doi: 10.3390/nu8030138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang Y, Chung SJ, McCullough ML, Song WO, Fernandez ML, Koo SI, Chun OK. Dietary carotenoids are associated with cardiovascular disease risk biomarkers mediated by serum carotenoid concentrations. J Nutr. 2014;144:1067–1074. doi: 10.3945/jn.113.184317. [DOI] [PubMed] [Google Scholar]
- 83.Wang Y, Sun J, Liu C, Fang C. Protective effects of crocetin pretreatment on myocardial injury in an ischemia/reperfusion rat model. Eur J Pharmacol. 2014;741:290–296. doi: 10.1016/j.ejphar.2014.07.052. [DOI] [PubMed] [Google Scholar]
- 84.Zhu Z, Zhu J, Zhao X, Yang K, Lu L, Zhang F, Shen W, Zhang R. All-Trans Retinoic Acid Ameliorates Myocardial Ischemia/Reperfusion Injury by Reducing Cardiomyocyte Apoptosis. PLoS One. 2015;10:e0133414. doi: 10.1371/journal.pone.0133414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Levine M, Rumsey SC, Daruwala R, Park JB, Wang Y. Criteria and recommendations for vitamin C intake. JAMA. 1999;281:1415–1423. doi: 10.1001/jama.281.15.1415. [DOI] [PubMed] [Google Scholar]
- 86.Wang X, Quinn PJ. The location and function of vitamin E in membranes (review) Mol Membr Biol. 2000;17:143–156. doi: 10.1080/09687680010000311. [DOI] [PubMed] [Google Scholar]
- 87.Heller R, Werner-Felmayer G, Werner ER. Alpha-Tocopherol and endothelial nitric oxide synthesis. Ann N Y Acad Sci. 2004;1031:74–85. doi: 10.1196/annals.1331.007. [DOI] [PubMed] [Google Scholar]
- 88.Heller R, Werner-Felmayer G, Werner ER. Antioxidants and endothelial nitric oxide synthesis. Eur J Clin Pharmacol. Springer-Verlag. 2006;62(S1):21–28. [Google Scholar]
- 89.Gille L, Staniek K, Nohl H. Effects of tocopheryl quinone on the heart: model experiments with xanthine oxidase, heart mitochondria, and isolated perfused rat hearts. Free Radic Biol Med. 2001;30:865–876. doi: 10.1016/s0891-5849(01)00475-0. [DOI] [PubMed] [Google Scholar]
- 90.Ramlawi B, Otu H, Mieno S, Boodhwani M, Sodha NR, Clements RT, Bianchi C, Sellke FW. Oxidative stress and atrial fibrillation after cardiac surgery: a case-control study. Ann Thorac Surg. 2007;84:1166–1172; discussion 1172-1173. doi: 10.1016/j.athoracsur.2007.04.126. [DOI] [PubMed] [Google Scholar]
- 91.Newaz MA, Yousefipour Z, Nawal NN. Modulation of nitric oxide synthase activity in brain, liver, and blood vessels of spontaneously hypertensive rats by ascorbic acid: protection from free radical injury. Clin Exp Hypertens. 2005;27:497–508. doi: 10.1081/CEH-200067681. [DOI] [PubMed] [Google Scholar]
- 92.Guney M, Oral B, Demirin H, Karahan N, Mungan T, Delibas N. Protective effects of vitamins C and E against endometrial damage and oxidative stress in fluoride intoxication. Clin Exp Pharmacol Physiol. 2007;34:467–474. doi: 10.1111/j.1440-1681.2007.04596.x. [DOI] [PubMed] [Google Scholar]
- 93.Gille L, Gregor W, Staniek K, Nohl H. Redox-interaction of alpha-tocopheryl quinone with isolated mitochondrial cytochrome bc1 complex. Biochem Pharmacol. 2004;68:373–381. doi: 10.1016/j.bcp.2004.03.031. [DOI] [PubMed] [Google Scholar]
- 94.Ulker S, McKeown PP, Bayraktutan U. Vitamins reverse endothelial dysfunction through regulation of eNOS and NAD(P)H oxidase activities. Hypertension. 2003;41:534–539. doi: 10.1161/01.HYP.0000057421.28533.37. [DOI] [PubMed] [Google Scholar]
- 95.May JM, Qu ZC, Mendiratta S. Protection and recycling of alpha-tocopherol in human erythrocytes by intracellular ascorbic acid. Arch Biochem Biophys. 1998;349:281–289. doi: 10.1006/abbi.1997.0473. [DOI] [PubMed] [Google Scholar]
- 96.Taddei S, Virdis A, Ghiadoni L, Salvetti A. Endothelial dysfunction in hypertension: fact or fancy? J Cardiovasc Pharmacol. 1998;32 Suppl 3:S41–S47. [PubMed] [Google Scholar]
- 97.Newaz MA, Nawal NN, Rohaizan CH, Muslim N, Gapor A. alpha-Tocopherol increased nitric oxide synthase activity in blood vessels of spontaneously hypertensive rats. Am J Hypertens. 1999;12:839–844. doi: 10.1016/s0895-7061(99)00022-9. [DOI] [PubMed] [Google Scholar]
- 98.Wu F, Schuster DP, Tyml K, Wilson JX. Ascorbate inhibits NADPH oxidase subunit p47phox expression in microvascular endothelial cells. Free Radic Biol Med. 2007;42:124–131. doi: 10.1016/j.freeradbiomed.2006.10.033. [DOI] [PubMed] [Google Scholar]
- 99.Gao F, Yao CL, Gao E, Mo QZ, Yan WL, McLaughlin R, Lopez BL, Christopher TA, Ma XL. Enhancement of glutathione cardioprotection by ascorbic acid in myocardial reperfusion injury. J Pharmacol Exp Ther. 2002;301:543–550. doi: 10.1124/jpet.301.2.543. [DOI] [PubMed] [Google Scholar]
- 100.Packer JE, Slater TF, Willson RL. Direct observation of a free radical interaction between vitamin E and vitamin C. Nature. 1979;278:737–738. doi: 10.1038/278737a0. [DOI] [PubMed] [Google Scholar]
- 101.Niki E, Noguchi N, Tsuchihashi H, Gotoh N. Interaction among vitamin C, vitamin E, and beta-carotene. Am J Clin Nutr. 1995;62:1322S–1326S. doi: 10.1093/ajcn/62.6.1322S. [DOI] [PubMed] [Google Scholar]
- 102.Levine M, Padayatty SJ, Espey MG. Vitamin C: a concentration-function approach yields pharmacology and therapeutic discoveries. Adv Nutr. 2011;2:78–88. doi: 10.3945/an.110.000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Schneider MP, Delles C, Schmidt BM, Oehmer S, Schwarz TK, Schmieder RE, John S. Superoxide scavenging effects of N-acetylcysteine and vitamin C in subjects with essential hypertension. Am J Hypertens. 2005;18:1111–1117. doi: 10.1016/j.amjhyper.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 104.Rushworth GF, Megson IL. Existing and potential therapeutic uses for N-acetylcysteine: the need for conversion to intracellular glutathione for antioxidant benefits. Pharmacol Ther. 2014;141:150–159. doi: 10.1016/j.pharmthera.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 105.Winterbourn CC, Metodiewa D. Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic Biol Med. 1999;27:322–328. doi: 10.1016/s0891-5849(99)00051-9. [DOI] [PubMed] [Google Scholar]
- 106.Lodge JK, Traber MG, Packer L. Thiol chelation of Cu2+ by dihydrolipoic acid prevents human low density lipoprotein peroxidation. Free Radic Biol Med. 1998;25:287–297. doi: 10.1016/s0891-5849(98)00048-3. [DOI] [PubMed] [Google Scholar]
- 107.Joshi D, Mittal DK, Shrivastava S, Shukla S. Protective role of thiol chelators against dimethylmercury induced toxicity in male rats. Bull Environ Contam Toxicol. 2010;84:613–617. doi: 10.1007/s00128-010-9982-3. [DOI] [PubMed] [Google Scholar]
- 108.Lu Y, Qin W, Shen T, Dou L, Man Y, Wang S, Xiao C, Li J. The antioxidant N-acetylcysteine promotes atherosclerotic plaque stabilization through suppression of RAGE, MMPs and NF-κB in ApoE-deficient mice. J Atheroscler Thromb. 2011;18:998–1008. doi: 10.5551/jat.8870. [DOI] [PubMed] [Google Scholar]
- 109.Arstall MA, Yang J, Stafford I, Betts WH, Horowitz JD. N-acetylcysteine in combination with nitroglycerin and streptokinase for the treatment of evolving acute myocardial infarction. Safety and biochemical effects. Circulation. 1995;92:2855–2862. doi: 10.1161/01.cir.92.10.2855. [DOI] [PubMed] [Google Scholar]
- 110.Thiele H, Hildebrand L, Schirdewahn C, Eitel I, Adams V, Fuernau G, Erbs S, Linke A, Diederich KW, Nowak M, et al. Impact of high-dose N-acetylcysteine versus placebo on contrast-induced nephropathy and myocardial reperfusion injury in unselected patients with ST-segment elevation myocardial infarction undergoing primary percutaneous coronary intervention. The LIPSIA-N-ACC (Prospective, Single-Blind, Placebo-Controlled, Randomized Leipzig Immediate PercutaneouS Coronary Intervention Acute Myocardial Infarction N-ACC) Trial. J Am Coll Cardiol. 2010;55:2201–2209. doi: 10.1016/j.jacc.2009.08.091. [DOI] [PubMed] [Google Scholar]
- 111.Yesilbursa D, Serdar A, Senturk T, Serdar Z, Sağ S, Cordan J. Effect of N-acetylcysteine on oxidative stress and ventricular function in patients with myocardial infarction. Heart Vessels. 2006;21:33–37. doi: 10.1007/s00380-005-0854-4. [DOI] [PubMed] [Google Scholar]
- 112.Abe M, Takiguchi Y, Ichimaru S, Tsuchiya K, Wada K. Comparison of the protective effect of N-acetylcysteine by different treatments on rat myocardial ischemia-reperfusion injury. J Pharmacol Sci. 2008;106:571–577. doi: 10.1254/jphs.fp0071664. [DOI] [PubMed] [Google Scholar]
- 113.Pasupathy S, Tavella R, Grover S, Raman B, Procter NEK, Du YT, Mahadavan G, Stafford I, Heresztyn T, Holmes A, et al. Early Use of N-acetylcysteine With Nitrate Therapy in Patients Undergoing Primary Percutaneous Coronary Intervention for ST-Segment-Elevation Myocardial Infarction Reduces Myocardial Infarct Size (the NACIAM Trial [N-acetylcysteine in Acute Myocardial Infarction]) Circulation. 2017;136:894–903. doi: 10.1161/CIRCULATIONAHA.117.027575. [DOI] [PubMed] [Google Scholar]
- 114.Kolnagou A, Kleanthous M, Kontoghiorghes GJ. Reduction of body iron stores to normal range levels in thalassaemia by using a deferiprone/deferoxamine combination and their maintenance thereafter by deferiprone monotherapy. Eur J Haematol. 2010;85:430–438. doi: 10.1111/j.1600-0609.2010.01499.x. [DOI] [PubMed] [Google Scholar]
- 115.Freeman AP, Giles RW, Berdoukas VA, Walsh WF, Choy D, Murray PC. Early left ventricular dysfunction and chelation therapy in thalassemia major. Ann Intern Med. 1983;99:450–454. doi: 10.7326/0003-4819-99-4-450. [DOI] [PubMed] [Google Scholar]
- 116.Reddy BR, Kloner RA, Przyklenk K. Early treatment with deferoxamine limits myocardial ischemic/reperfusion injury. Free Radic Biol Med. 1989;7:45–52. doi: 10.1016/0891-5849(89)90099-3. [DOI] [PubMed] [Google Scholar]
- 117.Lesnefsky EJ, Repine JE, Horwitz LD. Deferoxamine pretreatment reduces canine infarct size and oxidative injury. J Pharmacol Exp Ther. 1990;253:1103–1109. [PubMed] [Google Scholar]
- 118.Williams RE, Zweier JL, Flaherty JT. Treatment with deferoxamine during ischemia improves functional and metabolic recovery and reduces reperfusion-induced oxygen radical generation in rabbit hearts. Circulation. 1991;83:1006–1014. doi: 10.1161/01.cir.83.3.1006. [DOI] [PubMed] [Google Scholar]
- 119.Paraskevaidis IA, Iliodromitis EK, Vlahakos D, Tsiapras DP, Nikolaidis A, Marathias A, Michalis A, Kremastinos DT. Deferoxamine infusion during coronary artery bypass grafting ameliorates lipid peroxidation and protects the myocardium against reperfusion injury: immediate and long-term significance. Eur Heart J. 2005;26:263–270. doi: 10.1093/eurheartj/ehi028. [DOI] [PubMed] [Google Scholar]
- 120.Chan W, Taylor AJ, Ellims AH, Lefkovits L, Wong C, Kingwell BA, Natoli A, Croft KD, Mori T, Kaye DM, et al. Effect of iron chelation on myocardial infarct size and oxidative stress in ST-elevation-myocardial infarction. Circ Cardiovasc Interv. 2012;5:270–278. doi: 10.1161/CIRCINTERVENTIONS.111.966226. [DOI] [PubMed] [Google Scholar]
- 121.Karahaliou A, Katsouras C, Koulouras V, Nikas D, Niokou D, Papadopoulos G, Nakos G, Sideris D, Michalis L. Ventricular arrhythmias and antioxidative medication: experimental study. Hellenic J Cardiol. 2008;49:320–328. [PubMed] [Google Scholar]