

Abstract
AIM
To investigate the hypothesis that cardiomyocyte-specific loss of the electrogenic NBCe1 Na+-HCO3- cotransporter is cardioprotective during in vivo ischemia-reperfusion (IR) injury.
METHODS
An NBCe1 (Slc4a4 gene) conditional knockout mouse (KO) model was prepared by gene targeting. Cardiovascular performance of wildtype (WT) and cardiac-specific NBCe1 KO mice was analyzed by intraventricular pressure measurements, and changes in cardiac gene expression were determined by RNA Seq analysis. Response to in vivo IR injury was analyzed after 30 min occlusion of the left anterior descending artery followed by 3 h of reperfusion.
RESULTS
Loss of NBCe1 in cardiac myocytes did not impair cardiac contractility or relaxation under basal conditions or in response to β-adrenergic stimulation, and caused only limited changes in gene expression patterns, such as those for electrical excitability. However, following ischemia and reperfusion, KO heart sections exhibited significantly fewer apoptotic nuclei than WT sections.
CONCLUSION
These studies indicate that cardiac-specific loss of NBCe1 does not impair cardiovascular performance, causes only minimal changes in gene expression patterns, and protects against IR injury in vivo .
Keywords: Deep sequencing, Ischemic, Apoptosis, Slc4a4, NBCe1
Core tip: The NBCe1 Na+-HCO3- cotransporter and NHE1 Na+/H+ exchanger both mediate Na+-loading and intracellular pH regulation in cardiomyocytes. Inhibition of NHE1 protects against ischemia-reperfusion (IR) injury, and evidence suggests that loss of NBCe1 activity could also be cardioprotective. We have developed a conditional NBCe1 knockout mouse model and have used it to determine the effects of NBCe1 ablation in cardiac muscle. These studies demonstrate that loss of NBCe1 does not impair cardiac performance. However, cardiomyocyte apoptosis following IR injury in vivo is much lower in hearts that lack NBCe1, thus indicating that loss of NBCe1 is cardioprotective.
INTRODUCTION
Regulation of intracellular pH (pHi) in cardiac myocytes is critical for cardiac function[1-4]. Cardiomyocytes express a sarcolemmal Na+/H+ exchanger (NHE1), two Na+-HCO3- cotransporters, and a number of Na+-independent electroneutral Cl-/HCO3- exchangers[1,2,5]. The Cl-/HCO3- exchangers extrude HCO3- in exchange for Cl- and therefore serve as acid-loading mechanisms[1,2]. In addition, they facilitate Na+-loading by operating in concert with the Na+-dependent acid extruders[6] and have the potential to contribute to CO2 disposal[7]. The sarcolemmal Na+/H+ exchanger and Na+-HCO3- cotransporters are activated by intracellular acidification and serve to alkalinize the cell by extruding H+ or bringing in HCO3[1,2]. Because their acid-base transport activities are coupled with uptake of Na+, these transporters stimulate Ca2+-loading via effects on the Na+/Ca2+ exchanger[4], which in turn can affect contractility[8].
The effects of NHE1 activity on Na+- and Ca2+-loading and on ischemia-reperfusion (IR) injury in heart, using both inhibitors[9-13] and a gene-targeted mouse model[14], are well established. The cardiac functions of the Na+-HCO3- cotransporters and their relevance to disease processes are less understood. Two Na+/HCO3- cotransporters, one electrogenic (NBCe1) and one electroneutral (NBCn1), are expressed in mammalian hearts. Based on RNA seq data[5,7], NBCe1 mRNA expression in mouse heart is about double that of NBCn1 and data available in the EMBL-EBI Expression Atlas (https://www.ebi.ac.uk/gxa/home) shows that this is also the case in the human heart. NBCe1 has been localized to the lateral sarcolemma, intercalated disc, and t-tubules of cardiomyocytes[1], and it has been suggested that the presence of NBCe1 in the t-tubule may contribute to electrical events involved in excitation-contraction coupling[1]. Inhibition of NBCe1 has been shown to reduce infarct size and improve ventricular function during reperfusion of the isolated rodent heart[15,16], and the expression of NBCe1 is elevated in hearts of human patients with heart failure[15]. In a rat model of pressure overload hypertrophy, both NBCe1 and NBCn1 mRNAs were elevated[17]. These observations suggest that increased Na+-HCO3- cotransport activity may be a contributing factor during pathological conditions like heart failure, myocardial infarction, and ischemic injury.
Mice with a targeted global disruption of the Slc4a4 gene exhibit severe metabolic acidosis, absorptive and secretory defects, growth retardation, hyperaldosteronism, splenomegaly, and abnormal dentition, and they die before weaning[18]. Because of the severe phenotype it was not possible to examine the cardiac functions of NBCe1 using the global KO. The studies performed to assess cardio-protective effects of inhibiting NBCe1 in the heart have been performed using an anti-NBCe1 antibody[15,16] or the compound S0859, reported to be a common inhibitor of all NBCs[16]. To better understand the functions of NBCe1 in heart, we generated cardiomyocyte-specific conditional NBCe1 KO mice. The cardiovascular functions of WT and KO mice were analyzed in vivo under basal conditions and in response to β-adrenergic stimulation. To determine if the loss of NBCe1 is protective against IR injury in vivo, WT and NBCe1 KO mice were subjected to ligation of the left anterior descending artery followed by a period of reperfusion.
MATERIALS AND METHODS
Generation of a conditional NBCe1 knockout mouse model
Design and construction of the targeting vector, gene targeting of embryonic stem (ES) cells, and subsequent steps needed to generate mice carrying a floxed allele of NBCe1 were performed by the Animal Models Core Facility of the University of Cincinnati. The targeting construct was prepared with LoxP sites flanking coding exon 12 for transcripts ENSMUST00000148750 and ENSMUST00000156238 of the Slc4a4 gene, both of which have an upstream non-coding exon. It should be noted that there are multiple transcripts for the Slc4a4 gene and that the numbering of exons differs in some transcripts. However, because the targeted exon is an essential exon in all transcripts, the model can be used for conditional deletion of Slc4a4 transcripts in any tissue. The targeted exon is 134 nucleotides in length, begins with the codons for the amino acid sequence GVLESFLGT and ends with the codons for the amino acid sequence FERLLFNFS. Because it also contains two nucleotides of the next codon, deletion of this exon causes any transcripts that might be produced to go out of frame, thus eliminating all codons following that for amino acid 499, which include sequences that encode the transmembrane domains necessary for ion transport.
To prepare the targeting construct, LoxP sites were inserted at the ends of a 1.2 kb genomic DNA fragment containing exon 12. The proximal LoxP site was 709 nucleotides upstream of exon 12, and the distal LoxP site was 393 nucleotides downstream of exon 12. A neomycin resistance gene (neo), which allowed for positive selection of ES cells, was inserted just inside the distal LoxP site and was flanked by flippase recognition target (FRT) sites, which allowed its removal at a later step. The 5’ arm of the targeting construct was a 3.4 kb fragment from sequences in intron 11 that immediately preceded the insertion site of the proximal LoxP site, and the 3’ arm was a 2.3 kb fragment from intron 12 that immediately followed the insertion site of the distal LoxP site. Each of the genomic fragments used to prepare the construct were amplified by polymerase chain reaction (PCR) from mouse genomic DNA. A thymidine kinase gene was included after the 3’ arm in order to allow negative selection of ES cells. The targeting construct was electroporated into ES cells derived from 129/SvJ mice and targeted ES cells were identified by long-range PCR and used to generate chimeric mice. When germline transmission of the targeted allele was achieved, the mice were bred with C57Bl6 mice expressing FLP recombinase to remove the Neo gene. For cardiac myocyte-specific deletion of Slc4a4 exon 12, the mice were bred with a C57Bl6 mouse carrying the Cre recombinase gene driven by the β-myosin promoter, which is an effective strategy for Cre-mediated recombination beginning during embryonic development in cardiac myocytes[19].
All procedures using animals conformed to guidelines published by the National Institutes of Health (Guide for the Care and Use of Laboratory Animals) and were approved by the Institutional Animal Care and Use Committee at the University of Cincinnati. Mice were used in this study, as it is the best mammalian model for preparing genetic modifications and appropriate techniques are available for analysis of cardiovascular performance and response to in vivo IR injury. All appropriate measures were taken to minimize pain or discomfort. Mice were maintained in a specific pathogen free, temperature controlled barrier facility, with a 12 h light-dark cycle, and access to food and water ad libitum.
Genotype analysis
PCR genotyping of mice carrying the floxed allele was performed using DNA from tail biopsies and the following primers: Forward primer: 5’-TGGTGGCTTAAATTGCAAATGGC-3’; Reverse primer: 5’-CATAACCCACTAAGTCCAGTACG-3’. These primers flank the proximal LoxP site and yield a 176-base pair PCR product for the wild-type Slc4a4 allele, and a 223-base pair PCR product for the floxed allele, with the increase in size being due to the LoxP site. An additional PCR reaction was performed to determine the presence or absence of the Cre transgene.
Quantitative PCR analysis of the degree of knockdown of functional NBCe1 mRNA
To determine the degree of knockdown of NBCe1 mRNA in KO (Slc4a4flx/flx(Cre)) relative to WT (Slc4a4flx/flx) hearts, quantitative reverse transcriptase-PCR analysis (qRT-PCR) was performed using an ABI 7300 Real Time PCR system as described previously[7]. cDNA was prepared from mRNA isolated from whole heart of 4 mo old male mice (n = 6 for each genotype) and was PCR amplified using a forward primer (5’-TTCAGGCTCTCTCTGCGATT-3’) from coding exon 11 and reverse primer from coding exon 12 (5’-CTCAAGATGGTAAGCGGTTGA-3’).
Analysis of left ventricular function and blood pressure
Cardiovascular performance was determined using intraventricular and intra-arterial pressure measurements as described previously[20,21]. The mice (2-3 mo old; n = 4 male and 4 female Slc4a4flx/flx and 8 Slc4a4flx/flx(Cre)) were anesthetized with a mixture of ketamine and inactin, surgically instrumented, and their body temperatures maintained using a thermally controlled surgical stage. A high fidelity pressure transducer (Millar Instruments, Houston, TX) was introduced into the left ventricle through the right carotid artery to measure left ventricular pressure, and blood pressure was recorded via a fluid filled catheter in the right femoral artery. A catheter in the right femoral vein was used for infusion of dobutamine, a β-adrenergic agonist.
RNA Seq analysis
Total RNA was isolated from whole hearts of 4 mo old male Slc4a4flx/flx and Slc4a4flx/flx(Cre) mice (n = 6 of each genotype). RNA Seq analysis was performed in the University of Cincinnati Genomics and Sequencing Core using the Illumina HiSeq 1000 platform, and sequence reads were aligned to the reference mouse genome using TopHat aligner[22]. Statistical analysis was performed using the negative-binomial model of read counts as carried out in the DeSeq Bioconductor package[23]. mRNA expression data are presented as Reads Per Kilobase per Million mapped reads (RPKM), which normalizes for the size of the mRNA and allows direct comparisons of transcript abundance.
Gene Ontology analysis
For Gene Ontology (GO) analyses, we used the GOrilla program[24] (http://cbl-gorilla.cs.technion.ac.il/). As discussed previously[7,25], two analysis options are available: (1) Two Unranked Lists, in which a target list of genes with a specific range of P values is compared against the list of all genes analyzed; and (2) a Single Rank List, with the entire gene set ranked according to P values, thereby avoiding the use of an arbitrary cutoff of P values. Both analyses were performed. However, because of the low number of genes with highly significant changes, we relied primarily on the Two List analysis, in which significance and enrichment are calculated based on the number of genes in each GO category that appear in the target list and background list. Enrichment is defined as = (b/n)/(B/N), where b is the number of genes in the intersection, n is the number of genes in the top of the user’s input list or in the target set when appropriate, B is the total number of genes associated with a specific GO term, and N is the total number of genes. The program calculates statistical probabilities using the hypergeometric distribution[24]. In addition to a P value, the program provides an FDR q-value, which considers the total number of GO categories and corrects for the false discovery rate (FDR).
In vivo myocardial ischemia-reperfusion injury
Surgery to induce IR injury was performed on 4-5 mo old male mice (n = 4 for each genotype) as described previously[26]. Mice were anesthetized with ketamine-inactin and were mechanically ventilated using a rodent ventilator (Model 845, Harvard Apparatus) connected to an endotracheal tube. The heart was exposed by a left side limited thoracotomy and a 6-0 silk suture was passed beneath the left anterior descending artery (LAD) with a tapered needle[26]. A loose double knot was made with the suture and a 5 mm long piece of PE-10 tubing was placed through the loop. The loop was tightened around the artery and tubing to occlude blood flow. After 30 min, the knot was untied and the tubing was removed. The chest cavity was closed with continuous 4-0 silk sutures and the heart was reperfused for 3 h. The mice were euthanized and the chest cavity was opened to isolate the heart. The aorta was cannulated and perfused initially with PBS containing Heparin (10 units Heparin per ml) followed by a modified Krebs-Henseleit buffer (pH 7.4) containing 110 mmol/L NaCl, 16 mmol/L KCl, 16 mmol/L MgCl2, 1.2 mmol/L CaCl2, and 10 mmol/L NaHCO3. Finally, the isolated heart was perfused with 10% buffered formalin and left in 10% buffered formalin for 48 h. The left ventricle was embedded in paraffin and sectioned, with great care taken to identify the exact location of the sections relative to the LAD occlusion site.
TUNEL assay
Cardiomyocyte death by apoptosis in hearts from mice subjected to IR injury (described above) was analyzed using the In Situ Cell Death Detection Kit, TMR red from Roche. Sections that were carefully oriented relative to the occlusion site were deparaffinized, hydrated and pretreated with proteinase K (20 μg/mL in 10 mmol/L tris/HCl, pH 7.4) for 30 min at 37 °C. The sections were then incubated with the TUNEL mixture for 60 min at 37 °C. The sections were rinsed with PBS and counterstained with 1:200 dilution of α-Sarcomeric actin antibody (Sigma, A2172) for 60 min at 37 °C. The section was then rinsed and stained with 1:200 dilution of Fluorescein (FITC) anti-mouse secondary antibody (Jackson Immunoresearch, 115-095-146) for 60 min at 37 °C. The sections were mounted with ProLong gold antifade mountant with DAPI from Life technologies. Images were taken under 40 × objective on a Olympus BX41 microscope equipped with a digital camera and MagnaFireTM software.
Statistical analysis
Values are presented as means ± SEM. Individual comparisons were made using a two-tailed Student’s t-test, and a P value < 0.05 was considered significant. For group comparisons, a mixed factor analysis of variance with repeated measures on the second factor was used. Statistical analyses of RNA Seq data and GO data are described in the respective sections above. Statistical methods were reviewed by coauthor Dr. Mario Medvedovic, Director of the Division of Biostatistics and Bioinformatics in the Department of Environmental Health.
RESULTS
Generation of a mouse model with cardiomyocyte-specific deletion of NBCe1 mRNA
A targeting construct in which coding exon 12 of the Slc4a4 gene was flanked with LoxP sites was prepared (Figure 1) and electroporated into ES cells, which were then subjected to a positive-negative selection procedure. Cells carrying the floxed allele were injected into blastocyts and used to generate chimeric animals. After achieving germline transmission of the targeted allele, the neo gene was deleted by breeding the mice with a transgenic mouse expressing Flip recombinase.
Figure 1.
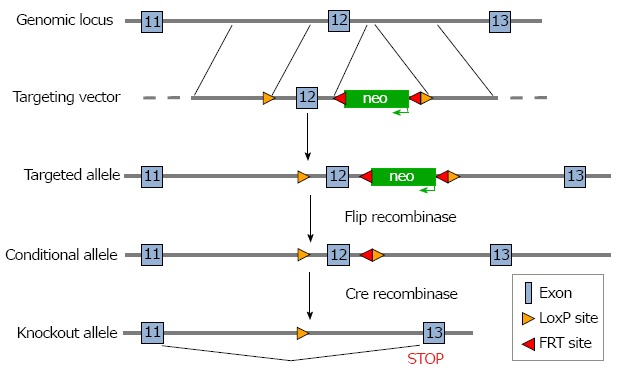
Targeting strategy for generation of Slc4a4 cardiac myocyte conditional KO mice. The targeting construct contained the neomycin resistance gene (neo) to allow selection of ES cells after homologous recombination. The neo gene was flanked with flippase recognition target (FRT) sites, which allowed removal of neo when mice carrying the targeted allele were bred with transgenic mice expressing Flip recombinase. After confirming the deletion of neo, mice carrying the floxed allele were bred with mice expressing Cre recombinase controlled by the β-MHC promoter, which mediates the deletion of exon 12 of Slc4a4 in cardiac myocytes.
To delete Slc4a4 in cardiac myocytes, the Slc4a4flx/flx mouse was crossed with a transgenic mouse expressing Cre recombinase driven by the β-myosin heavy chain (β-MHC) promoter[19] to obtain Slc4a4flx/+(Cre) and Slc4a4flx/+ mice. Further breeding was performed to obtain Slc4a4flx/flx(Cre) (KO) and Slc4a4flx/flx (WT) mice, which were mated to obtain experimental pairs. This allowed mice of both genotypes being used for experiments to be housed in the same cages, with sibling pairs often used for experiments. Male and female KO mice appeared normal, and WT and KO mice were indistinguishable in body weight, appearance, and behavior. Also, there was no significant difference in the heart weight: body weight ratios (WT: 4.38 ± 0.15 mg/g; KO: 4.56 ± 0.11 mg/g; n = 4 male and 4 female of each genotype at 4 mo of age).
Genotypes were determined by PCR analysis of tail DNA, which yielded a 176-base pair product for the WT allele and a 223-base pair product for the floxed allele (Figure 2A); a separate reaction was performed to test for the presence or absence of the Cre gene. The knockdown of Slc4a4 mRNA in the heart was confirmed by qRT-PCR analysis of whole heart mRNA using primers from coding exons 11 and 12. This allowed a quantitative assessment of the percentage of NBCe1 transcripts that lack coding exon 12. Loss of this exon causes the sequences following codon 499, which encode most of the transmembrane domains, to be out of frame. As shown in Figure 2B, the amount of NBCe1 mRNA containing exon 12 was reduced by approximately 70% in whole hearts of Slc4a4flx/flx(Cre) mice relative to those of Slc4a4flx/flx mice. It should be noted that expression of NBCe1 is not restricted to cardiac myocytes, and has been shown to be expressed in both vascular and nerve tissues[27]. Thus, the reduction in functional NBCe1 mRNA in cardiac myocytes is likely to be greater than that indicated by qRT-PCR analysis of whole heart mRNA.
Figure 2.

Polymerase chain reaction genotyping and quantitative reverse transcriptase-polymerase chain reaction of NBCe1 mRNA. A: PCR analysis of DNA from tail biopsies, using primers flanking the proximal LoxP site, showing bands for the alleles in Slc4a4+/+, Slc4a4flx/+ and Slc4a4flx/flx mice. The larger size of the band for the floxed allele is due to the presence of the LoxP site; B: Quantitative RT-PCR analysis of cDNA prepared from whole heart RNA (n = 6 for each genotype) showing a reduction of Slc4a4 mRNA in hearts of KO (Slc4a4flx/flx(Cre)) relative to WT (Slc4a4flx/flx without Cre) mice. PCR: Polymerase chain reaction.
Cardiovascular performance in NBCe1 KO and WT mice under basal conditions and in response to β-adrenergic stimulation
Cardiovascular performance in vivo was analyzed under basal conditions and in response to β-adrenergic stimulation using dobutamine. Anesthetized and surgically instrumented mice were analyzed using a pressure transducer in the left ventricle to measure intraventricular pressures and a catheter in the right femoral artery to measure blood pressure and heart rate. There was no difference in heart rate (HR), systolic ventricular pressure, or mean arterial pressure (Figure 3A, 3B and 3C) between WT (Slc4a4flx/flx) and KO (Slc4a4flx/flx(Cre)) mice under basal conditions or in response to β-adrenergic stimulation. Left ventricular contractility (+dP/dT and +dP/dt40) and relaxation (-dP/dt) (Figure 3D, 3E and 3F) were essentially the same in WT and KO mice, indicating normal cardiac function in NBCe1-deficient mice.
Figure 3.

Cardiovascular performance in WT and KO mice. Intraventricular and intra-arterial pressure measurements were recorded using transducers in the left ventricle and right femoral artery of anesthetized 2-3 mo old WT (Slc4a4flx/flx) and KO (Slc4a4flx/flx(Cre)) mice under both basal conditions and in response to β-adrenergic stimulation (intravenous infusion of increasing doses of dobutamine). A: Heart rate; B: Systolic left ventricular pressure; C: Mean arterial pressure; D: Positive dP/dt in mm Hg/sec; E: Positive dP/dt at 40 mm Hg; F: Negative dP/dt in mm Hg/sec is shown for WT and KO mice. n = 8 WT (4 female, 4 male) and 8 KO (4 female, 4 male) mice.
RNA sequencing of mRNA from NBCe1 KO and WT hearts
To identify patterns of differential gene expression changes in response to the loss of NBCe1 in cardiac myocytes, we performed RNA Seq analysis using mRNA from hearts of WT and KO mice. Between WT and KO hearts, there were 452 differentially expressed genes with P < 0.05. However, only a few of these genes had a highly significant FDR value. Many of the genes that fell within the range of P < 0.05 were expressed at very low levels or were absent in some samples. The genes that were expressed at low levels included sixteen major urinary proteins (Mups) and six cytochrome P450s, which were sharply induced in the KO. After removal of those genes with very low expression, the remaining 347 genes were subjected to GO analyses.
Because it has been proposed that the electrogenic activity of NBCe1 and its expression in t-tubules is likely to affect electrical activity of cardiac myocytes[1], we anticipated that there might be changes in genes involved in membrane excitability and cardiac conduction. In addition, because it has been suggested that NBCe1 activity can serve as a major mechanism for Na+-loading, with subsequent effects on Ca2+-handling and contractility[4,8], we were interested in whether the loss of NBCe1 might affect expression of genes encoding myofibrillar proteins, Ca2+-handling proteins, and proteins that affect Na+-loading.
As shown in Table 1, changes in Biological Function GO categories for cellular ion homeostasis, including subcategories for intracellular pH and cation homeostasis, were indicated, but the changes were modest and their statistical significance was poor (low FDR). Two Biological Function GO categories relating to apoptosis were identified, and their statistical significance was poor. However, when GO analysis was run using the single rank option for GOrilla GO analysis (see materials and methods), additional genes were identified for GO:0043652 (Engulfment of Apoptotic Cell) and an acceptable FDR value for this GO category was attained. There were no significant Molecular Function GO categories, but among Cellular Component GO categories there were a number dealing with membrane compartments (Table 1). These included GO categories for the endoplasmic and sarcoplasmic reticulum, t-tubules, and sarcolemma.
Table 1.
Enriched Gene Ontology categories in NBCe1 cardiac conditional KO mice
| GO category | P-value | Q-value | Enrichment | (N, B, n, b) |
| GO categories dealing with ion homeostasis | ||||
| GO:0006873 Cellular ion homeostasis | 1.13E-4 | 5.68E-1 | 2.45 | (21545, 579, 334, 22) |
| GO:0051453 Regulation of intracellular pH | 1.92E-4 | 5.80E-1 | 5.86 | (21545, 77, 334, 7) |
| GO:0030003 Cellular cation homeostasis | 2.12E-4 | 4.58E-1 | 2.4 | (21545, 564, 334, 21) |
| GO categories dealing with apoptosis | ||||
| GO:0043652 Engulfment of apoptotic cell | 4.09E-4 | 4.42E-1 | 19.35 | (21545, 10, 334, 3) |
| GO:1901030 Mitochondrial membrane permeabilization involved in apoptotic signaling | 4.09E-4 | 4.12E-1 | 19.35 | (21545, 10, 334, 3) |
| GO categories dealing with membrane compartments | ||||
| GO:0005783 Endoplasmic reticulum | 2.82E-6 | 1.06E-3 | 2.03 | (21545, 1493, 334, 47) |
| GO:0030315 T-tubule | 3.45E-5 | 7.18E-3 | 7.65 | (21545, 59, 334, 7) |
| GO:0016529 Sarcoplasmic reticulum | 3.00E-4 | 3.30E-2 | 6.56 | (21545, 59, 334, 6) |
| GO:0042383 Sarcolemma | 4.84E-4 | 4.77E-2 | 4.41 | (21545, 117, 334, 8) |
GO categories were identified using the Two unranked list option of the GOrilla Gene Ontology program[24]. N is the total number of genes, B is the total number of genes associated with a specific GO term, n is the number of genes in the target set, b is the number of genes in the intersection. Enrichment = (b/n)/(B/N). Differential mRNA expression data were generated by RNA Seq analysis of hearts from 6 adult male mice for the NBCe1 KO (Slc4a4flx/flx(Cre)) and NBCe1 WT (Slc4a4flx/flx) genotypes.
Genes for channels, pumps, and transporters that exhibited differential expression are shown in Table 2. Among K+ channels, modest upregulation of Kcnj2 and Kcnj11 (both in the t-tubule GO category) was observed along with higher upregulation of Kcna4 (Kv1.4; 1.50-fold increase), which is expressed in t-tubules and sarcolemma[28], and Kcnk2 (TREK-1; 1.65x), which is expressed in intercalated discs[29]. Among Ca2+ channels, Cacna1s (Cav1.1; L-type α1S) was upregulated and Cacnb1 (a beta subunit) was downregulated. Both Ca2+ channel subunits are in the t-tubule. However, they are major components of the skeletal muscle Ca2+ channel and are expressed at much lower levels in heart. The only other gene for a Ca2+-handling protein that was changed was Atp2a1, the skeletal muscle sarcoplasmic reticulum (SR) Ca2+ pump, which is expressed at very low levels in heart.
Table 2.
Ion channel and transporter genes altered in NBCe1-null hearts
| Gene symbol | Description | Fold change | P value |
| Kcnj2 | Inwardly-rectifying K+ channel J2 | 1.18 | 0.02 |
| Kcnj11 | Inwardly-rectifying K+ channel J11 | 1.14 | 0.03 |
| Kcnk2 | K+ channel K2; TREK-1 | 1.65 | 0 |
| Kcna4 | K+ channel, shaker-related 4; Kv1.4 | 1.50 | 0.05 |
| Cacna1s | L type Ca2+ channel, alpha 1S subunit | 1.44 | 0 |
| Cacnb1 | Ca2+ channel, beta 1 subunit | 0.51 | 0 |
| Atp2a1 | Skeletal muscle SR Ca2+ pump | 1.82 | 0.04 |
| Slc9a1 | Sarcolemmal Na+/H+ Exchanger | 1.18 | 0.05 |
| Slc9a9 | Organellar Na+/H+ Exchanger | 1.44 | 0.03 |
| Slc16a3 | Monocarboxylic acid transporter, | 1.92 | 0.04 |
| Slc1a5 | Neutral amino acid transporter | 0.81 | 0.04 |
| Slc7a1 | Cationic amino acid transporter, y+ system | 1.24 | 0 |
| Slc15a2 | Proton/peptide transporter | 1.87 | 0.01 |
| Slc15a1 | Oligopeptide transporter | 0.56 | 0.03 |
| Slc40a1 | Iron transporter; ferroportin | 0.86 | 0.02 |
| Slc39a13 | Zinc transporter | 1.23 | 0.03 |
| Slc36a1 | Proton/amino acid symporter | 0.83 | 0.05 |
Differential mRNA expression is presented as fold-changes in NBCe1-null hearts (Slc4a4flx/flx(Cre)) relative to WT (Slc4a4flx/flx) hearts.
Slc9a1 mRNA, encoding the NHE1 Na+/H+ exchanger, was slightly upregulated, and Slc9a9, an organellar Na+/H+ exchanger, was also upregulated. Other H+-coupled transporters with altered expression (Table 2) were transporters for peptides (Slc15a1, Slc15a2), amino acids (Slc36a1), and lactate (Slc16a3). Additional transporters included two amino acid transporters (Slc1a5 and Slc7a1), an iron transporter (Slc40a1), and a zinc transporter (Slc39a13). The expression of NBCn1 (Slc4a7), the only other Na+-HCO3- cotransporter expressed in heart, was not significantly changed (RPKM values: 6.67 ± 0.42 in WT; 6.99 ± 0.15 in KO).
Differentially expressed genes for proteins involved in apoptosis are shown in Table 3. Some of these were identified because of their inclusion in the apoptosis GO categories shown in Table 1 (Xkr8, Rac3, Thbs1, Zfp13, Sh3glb1, and Siva1). Others were identified based on literature searches and a P value ≤ 0.01 (Msrb3, Xlrl, Ier5, Hint1, and Stmn1).
Table 3.
Differential expression of apoptosis genes in NBCe1-null hearts
| Gene symbol | Description | Fold change | P value |
| Xkr8 | X Kell Blood Group Precursor Related 8 | 0.61 | 0.015 |
| Rac3 | RAS-Related C3 Botulinum Substrate 3 | 0.36 | 0.016 |
| Thbs1 | Thrombospondin 1 | 0.79 | 0.020 |
| Zfp13 | Zinc Finger Protein 13 | 0.46 | 0.023 |
| Sh3glb1 | SH3-Domain GRB2-like B1 (Endophilin) | 1.13 | 0.024 |
| Siva1 | SIVA1, Apoptosis-Inducing Factor | 0.66 | 0.030 |
| Msrb3 | Methionine Sulfoxide Reductase B3 | 1.19 | 0.002 |
| Xlrl | Xlr-like | 1.77 | 0.003 |
| Ier5 | Immediate Early Response 5 | 0.80 | 0.005 |
| Hint1 | Histidine Triad Nucleotide Binding Protein 1 | 0.84 | 0.010 |
| Stmn1 | Stathmin 1 | 1.37 | 0.010 |
Differential mRNA expression is presented as fold-changes in NBCe1-null hearts (Slc4a4flx/flx(Cre)) relative to WT (Slc4a4flx/flx) hearts. The first six genes on this list were identified based on inclusion in the two apoptosis GO categories identified in Table 1.
Reduced apoptosis in NBCe1 KO hearts in response to in vivo ischemia-reperfusion
To determine whether ablation of NBCe1 would provide protection against IR injury, WT and KO hearts were subjected to 30 min of ischemia by temporary ligation of the left anterior descending (LAD) artery, followed by 3 h of reperfusion. TUNEL staining was performed to detect apoptotic cell death and to quantify apoptotic nuclei in WT and KO heart sections. Apoptotic nuclei were detected primarily in the left ventricular free wall in both WT and KO heart sections (Figure 4A). However, there were significantly fewer apoptotic nuclei (Figure 4B) in KO (40.6 ± 6.4 apoptotic nuclei per mm2) than in WT (142.0 ± 13.6 apoptotic nuclei per mm2) heart sections.
Figure 4.
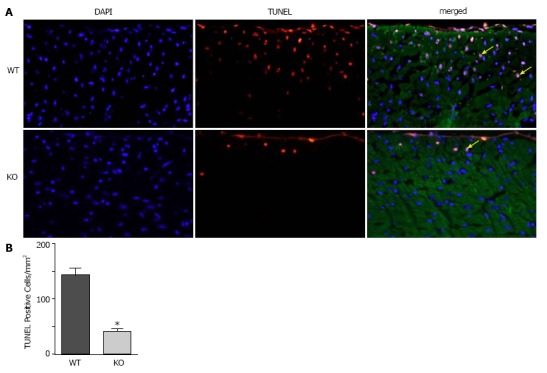
TUNEL staining to quantify apoptosis in WT and KO heart sections after ischemia and reperfusion. Mice were subjected to 30 min of ischemia by occlusion of the LAD, followed by 3 h of reperfusion, and heart sections were processed to analyze apoptosis. A: Representative images from WT and KO heart sections, taken from a region approximately 1 mm distal to the occlusion site, showing nuclei stained with DAPI (blue), TUNEL (red) and sarcomeric actin (green). Yellow arrows point to apoptotic nuclei (pink); B: Quantification of apoptotic nuclei. n = 4 mice of each genotype, P ≤ 0.05.
DISCUSSION
On the basis of functional studies and expression data it is clear that NBCe1 serves as a major uptake mechanism for both Na+ and HCO3- in cardiac myocytes[1,4]. Na+-loading can affect both cardiac contractility and response to IR injury[4,8,15,16], and there is evidence that HCO3- has a major effect on contractility, both in isolated myocytes[5] and in the isolated work-performing heart[30]. Also, because NBCe1 is electrogenic and expressed in t-tubules, lateral sarcolemma, and intercalated discs[1], it has the potential to affect membrane potential and electrical activity of the myocyte. To begin testing the cardiovascular functions of NBCe1 we developed a mouse model carrying a floxed allele for NBCe1 and analyzed the effects of cardiomyocyte-specific ablation on cardiovascular performance and in vivo IR injury. In addition, we performed RNA Seq analysis to determine whether remodeling of cardiac systems occurs at the level of mRNA expression patterns in response to the loss of NBCe1.
The loss of NBCe1 did not impair cardiovascular performance, and heart weights did not differ between the two genotypes. NBCe1 mutant mice exhibited normal heart rates, blood pressures, and basal contraction and relaxation, and when they were treated with a β-adrenergic agonist, they exhibited normal increases in contraction and relaxation. The lack of any apparent detrimental effects supports the notion that inhibition of cardiac NBCe1 activity could be used for cardioprotection. These results also suggest that uptake of HCO3- by NBCe1 is not required for the stimulation of contraction observed in isolated myocytes and hearts in the presence of HCO3-[5,30], although it is possible that the high rate of HCO3- production from metabolically produced CO2 in vivo prevents any deficit in intracellular HCO3- levels. Alternatively, reduced HCO3--efflux via Cl-/HCO3- exchange or increased uptake via NBCn1 may be sufficient to maintain intracellular HCO3- homeostasis in vivo when NBCe1 was ablated. Also, the NHE1 Na+/H+ exchanger was slightly upregulated at the mRNA level and provides a powerful alternative mechanism of Na+-loading and pHi regulation. It should be noted that genetic ablation of NHE1 also caused no impairment of cardiovascular performance[31], and it had beneficial effects on cardiac energy metabolism, including increased glucose utilization and metabolic flexibility[31]. Interestingly, NBCe1 activity is involved in acute stimulation of glycolysis in astrocytes in response to membrane depolarization[32]. Thus, one could speculate that the increased glucose utilization observed in hearts of NHE1-null mice[31] might be due in part to a compensatory increase in NBCe1 activity.
Because they have opposing activities with respect to HCO3- fluxes, it is of interest to compare the results of ablating NBCe1, the predominant HCO3--uptake mechanism in heart, with the results of ablating the AE3 (Slc4a3) Cl-/HCO3- exchanger. Both the very high mRNA expression levels of AE3[7] and its predominant role in recovery of myocytes from an alkaline load[33] demonstrate that it is the predominant HCO3--extrusion mechanism in the heart. In humans, heterozygous mutations in AE3 contribute to heart disease[34]. In mice, the loss of AE3 caused a mild impairment of force-frequency responses[35] and exacerbated heart disease caused by other genetic defects[21,36]. However, AE3-null mice exhibited normal cardiac contraction and relaxation under both basal conditions and after β-adrenergic stimulation[33]. Nevertheless, RNA Seq analysis indicated that AE3-null hearts undergo major remodeling of cardiac gene expression patterns involved in hypoxia responses and angiogenesis, energy metabolism, sarcomere function, and membrane excitability and electrical conduction[7].
Unlike the results of the AE3 RNA Seq study, RNA Seq analysis of NBCe1-null hearts revealed little evidence of cardiac remodeling at the mRNA level. For example, altered expression of genes involved in sarcomere function were very prominent in AE3 null-hearts[7], but no such changes were observed in NBCe1-null hearts, which is consistent with the normal contractility and β-adrenergic responses. Similarly, changes in energy metabolism genes were extensive in AE3-null hearts, but were not observed in NBCe1-null hearts, despite the known stimulatory effect of NBCe1 activity on glycolysis in astrocytes[32]. We also observed no changes in hypoxia response and angiogenesis genes, which were prominent in AE3-null hearts. However, there were several interesting changes related to membrane excitability.
One of the predictions by other investigators[1] was that the electrogenic activity of NBCe1 in the sarcolemma, t-tubules, and intercalated discs, with inward transport of one Na+ and two HCO3- ions during depolarization, would affect membrane excitability. The changes in genes involved in membrane excitability and electrical conduction were extensive in AE3-null hearts, and included voltage-sensitive Na+, K+, and Ca2+ channels, and gap junction proteins involved in electrical coupling between myocytes. Few such changes were observed in NBCe1-null hearts. However, a number of K+ channel genes were upregulated. These included Kcna4, encoding Kv1.4, and Kcnk2, encoding TREK-1. Kv1.4 is co-expressed with NBCe1 in t-tubules and the sarcolemma[28]. It mediates transient outward currents during the initial phase of depolarization[37] and might therefore counteract the lengthening of the action potential expected with a reduction in NBCe1 activity[38]. TREK-1 is expressed in intercalated discs[29] and plays an important role in regulating excitability of the sinoatrial node[39]. Thus, increased expression and activity of TREK-1 has the potential to counteract the electrical effects of the loss of NBCe1 activity in intercalated discs.
Although there were changes in two voltagesensitive Ca2+ channel subunits in t-tubules (Cacna1s, an α subunit expressed at low levels; Cacnb1, a β subunit, expressed at very low levels), both are primarily skeletal muscle isoforms. In contrast, Cacna1c, which encodes the major L-type Ca2+ channel in cardiac muscle (Cav1.2), was not changed (RPKM values: 26.8 ± 0.8 in WT; 26.2 ± 0.6 in KO). The only other gene for a Ca2+-handling protein that was changed was Atp2a1, the skeletal muscle sarcoplasmic reticulum Ca2+ pump. However, Atp2a1 is expressed at exceedingly low levels in heart (< 1 RPKM in WT), whereas Atp2a2, the cardiac Ca2+ pump was expressed at very high levels and was not changed (RPKM values: 2185 ± 47 in WT; 2183 ± 23 in KO). The major Na+/Ca2+ exchanger (NCX1, Slc8a1) also was not significantly changed (RPKM values: 25.0 ± 0.6 in WT; 27.3 ± 0.7 in KO). These data provide little support for remodeling of Ca2+-handling systems at the mRNA level.
A major objective was to determine whether genetic ablation of NBCe1 is cardioprotective during IR injury. The role of NHE1 in cardiac IR injury has been extensively studied using both inhibitors and genetic approaches[9-14], and it is generally accepted that the mechanism of protection involves reductions in Na+-loading, Ca2+-loading, and the rate of pHi recovery during reperfusion. Since NBCe1, like NHE1, mediates both Na+-loading and recovery from an acid load[40], a number of investigators have noted that NBCe1 activity could potentially contribute to IR injury. In fact, two groups have shown that inhibition of NBCe1 during reperfusion of mouse or rat hearts that were subjected to ischemia provides some protection against IR injury[15,16]. These results using isolated heart preparations support a role for NBCe1 in IR injury that is similar to that of NHE1 and indicate that a reduction in its activity can be cardioprotective.
Previous studies have shown that apoptotic cell death occurs mainly during reperfusion of ischemic tissue[41-43]. An increase in intracellular Ca2+ results in opening of the mitochondrial permeability transition pore[44], which then leads to apoptosis. In the current study, after WT and KO mice were subjected to 30 min of LAD occlusion and 3 h of reperfusion, apoptotic cell death and apoptotic nuclei were detected and quantified in heart sections taken from just below the point of occlusion. The significant reduction in the numbers of apoptotic nuclei in NBCe1 KO hearts following cardiac IR injury in vivo indicates that reduced NBCe1 activity is cardioprotective. Thus, both the studies using isolated hearts[15,16] and the current studies showing reduced apoptosis following in vivo IR injury support the hypothesis that a reduction in NBCe1 activity is cardioprotective. Given the severe effects of global NBCe1 ablation[18], only partial inhibition of NBCe1 is likely to be acceptable as part of a treatment strategy, however, it is possible that an NBCe1 inhibitor could be used in combination with other treatments.
As discussed above, the primary mechanism of NBCe1-mediated cardioprotection is likely to be the reductions in Na+-loading, Ca2+-loading, and the rate of pHi recovery during reperfusion. However, because a genetic ablation strategy was employed, rather than acute inhibition of NBCe1, it is possible that the long-term absence of NBCe1 has elicited remodeling that contributes to cardioprotection. This possibility is supported by the observation that genetic ablation of NHE1 reduces oxidative stress in the heart, high-fat diet-induced myocardial stress[31], and fatty liver disease[45], which are clear indications of long-term remodeling. A potentially interesting set of changes observed in the RNA Seq data were those involving genes with functions in apoptosis. The q-values for the apoptosis GO categories (Table 1) were not highly significant and these findings must therefore be considered with caution. Nevertheless, the observation provides suggestive evidence that ablation of the NBCe1 gene using the β-myosin promoter, which would be largely complete by birth, may cause changes in gene expression that tend to protect against apoptosis. For example, Siva1 (apoptosis-inducing factor) normally interacts with and inhibits Stathmin, thus promoting apoptosis[46,47]. Thus, down-regulation of Siva1 (0.66-fold, RPKM: 4.8 ± 0.3 in WT; 3.2 ± 0.2 in KO) and upregulation of Stathmin (1.37-fold, RPKM: 9.6 ± 0.5 in WT; 13.2 ± 1.2 in KO) would be expected to reduce apoptosis. Another interesting example is Xlrl, which was upregulated (1.77-fold, RPKM: 1.96 ± 0.34 in WT; 3.49 ± 0.17 in KO). Little is known about this gene, but Xlrl is almost identical to Xlr (95% amino acid identity and no gaps over their entire length of 208 amino acids), which protects against apoptosis[48]. Xlr was also upregulated (1.36-fold), but its levels of expression were lower.
The current study has a number of limitations. First, our mice were on a mixed 129/Svj and C57Bl6 background, rather than on a highly inbred background. The lack of a highly inbred background was likely responsible for the relatively high variability in our RNA Seq data compared to the very low variability that we observed when performing RNA Seq analysis using mRNA from hearts of AE3-null and WT mice on a highly inbred background[7]. With much lower variability, it would have been possible to detect more subtle patterns of changes in gene expression. However, it was clear from the data that there were no extensive changes in gene expression patterns for sarcomeric or metabolic genes, and that the changes in ion channel genes were not extensive.
A second limitation is that the 30 min period of ischemia and 3 h of reperfusion were chosen to maximize our ability to detect apoptotic cell death, which plays a major role in ischemic injury. However, this protocol is not optimal for analysis of necrotic cell death, for detecting differences in cardiac function between WT and KO mice, or for detecting markers of myocardial infarction such as serum troponin levels. For such studies, a longer period of ischemia or a myocardial infarction model, with permanent occlusion of the LAD and long-term follow-up, would be necessary. In myocardial infarction studies it would also be useful to determine whether NBCe1 heterozygosity could provide some degree of cardioprotection, since any cardiac therapy using complete pharmacological inhibition of NBCe1 would have a severe impact on kidney function. Even limited cardioprotection caused by partial inhibition could be useful, however, as it might be possible to combine partial inhibition of NBCe1 with partial inhibition of the NHE1 Na+/H+ exchanger, which appear to act via similar mechanisms (see introduction).
An additional potential limitation was that our WT controls were Slc4a4flx/flx mice rather than Slc4a4+/+(Cre+) mice. A study published after we had performed our RNA Seq analysis reported that prolonged Cre expression driven by the α-myosin heavy chain promoter can be cardiotoxic[49]. However, we used a Cre transgene driven by the β-myosin promoter[19], which is expressed at much lower levels than the α-myosin promoter after birth. As an indication of cardiotoxicity, long-term expression of Cre via the α-myosin promoter caused approximately 7-fold increase in expression of atrial natriuretic factor mRNA and approximately 2.5-fold increase in expression of β-myosin mRNA[48]. However, these genes were not changed in the NBCe1 KO carrying Cre driven by the β-myosin promoter. Of greater relevance, 6 mo old mice expressing Cre via the α-myosin promoter exhibited an approximately 3-fold increase in apoptosis. In our own studies, however, only the NBCe1 cardiac-specific KO mice were carrying the Cre transgene, and they exhibited significantly less apoptosis than the Slc4a4flox/flox controls. These results clearly indicated a cardioprotective effect of NBCe1 ablation, despite the presence of Cre.
In summary, we have developed a conditional knockout mouse model for Slc4a4 and have used it to test the effects of cardiac-specific ablation of NBCe1 in heart. The results show that loss of NBCe1 does not impair cardiovascular performance and that it significantly reduces the incidence of apoptosis following in vivo IR injury. These results are consistent with the results of previous studies using the isolated heart[15,16] and support the view that inhibition of NBCe1 during reperfusion has the potential to serve as a component of a cardioprotective treatment strategy.
ARTICLE HIGHLIGHTS
Research background
There is a strong rationale for the hypothesis that inhibition of NBCe1-mediated Na+-HCO3- cotransport activity protects against cardiac ischemia-reperfusion injury. This suggests that inhibition of NBCe1 could become part of a cardioprotective strategy.
Research motivation
Previous studies have been performed using the isolated heart, but there are no in vivo studies supporting the hypothesis that loss of NBCe1 activity is cardioprotective. Such studies are critical if NBCe1 inhibition is to be developed as a therapeutic strategy.
Research objectives
The objective of this study was to test whether loss of NBCe1 in the heart would protect against cardiac ischemia-reperfusion injury in vivo.
Research methods
Gene targeting was used to develop a conditional knockout mouse model in which the NBCe1 gene was ablated in cardiomyocytes. Hemodynamic measurements were performed to assess the effects of cardiac-specific NBCe1 ablation on cardiovascular performance, RNA Seq analysis was used to study changes in the cardiac transcriptome, and histological techniques were used to analyze cardiomyocyte apoptosis in response to ischemia-reperfusion injury.
Research results
NBCe1 ablation did not impair cardiovascular performance and caused only limited changes in the cardiac transcriptome. However, it caused a significant reduction in apoptosis following in vivo cardiac ischemia-reperfusion injury.
Research conclusions
Loss of NBCe1 in heart does not cause any apparent adverse effects, but does have a cardioprotective effect following ischemia and reperfusion in vivo.
Research perspectives
Future studies should focus on whether NBCe1 ablation is cardioprotective following myocardial infarction and whether partial inhibition of NBCe1 can be combined with other treatments that reduce Na+ and Ca2+ loading.
ACKNOWLEDGEMENTS
We thank Jeffery D Molkentin for providing the mouse model with the Cre transgene driven by the β-myosin promoter, Michelle L Nieman for help with the cardiovascular physiology experiments, and Glenn Doerman for preparing the figures.
Footnotes
Institutional review board statement: Because human subjects or tissues were not used in this study, approval from the Institutional review board was not required. Ethical issues relating to the animal protocol were reviewed and approved by the Institutional Animal Care and Use Committee of the University of Cincinnati.
Institutional animal care and use committee statement: All procedures involving animals were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Cincinnati (protocol number: 15-07-27-01).
Conflict-of-interest statement: The authors have no conflict of interest related to this manuscript.
ARRIVE guidelines statement: The authors have read the ARRIVE guidelines, and the manuscript was prepared according these guidelines.
Manuscript source: Invited manuscript
Peer-review started: April 7, 2018
First decision: June 5, 2018
Article in press: July 17, 2018
Specialty type: Cardiac and cardiovascular systems
Country of origin: United States
Peer-review report classification
Grade A (Excellent): A, A, A
Grade B (Very good): 0
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
P- Reviewer: Anan R, Montecucco F, Salvadori M, Ueda H S- Editor: Ji FF L- Editor: Filipodia E- Editor: Wu YXJ
Contributor Information
Kanimozhi Vairamani, Division of Oncology, Cancer and Blood Diseases Institute, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229-3026, United States.
Vikram Prasad, Department of Pediatrics, Cincinnati Children’s Hospital Medical Center, University of Cincinnati, Cincinnati, OH 45229-3039, United States.
Yigang Wang, Department of Pathology, University of Cincinnati, College of Medicine, Cincinnati, OH 45267-0529, United States.
Wei Huang, Department of Pathology, University of Cincinnati, College of Medicine, Cincinnati, OH 45267-0529, United States.
Yinhua Chen, Division of Developmental Biology, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229-3039, United States.
Mario Medvedovic, Department of Environmental Health, University of Cincinnati, College of Medicine, Cincinnati, OH 45267-0056, United States.
John N Lorenz, Department of Pharmacology and Systems Physiology, University of Cincinnati, College of Medicine, Cincinnati, OH 45267-0575, United States.
Gary E Shull, Department of Molecular Genetics, Biochemistry and Microbiology, University of Cincinnati, College of Medicine, Cincinnati, OH 45267-0524, United States. shullge@ucmail.uc.edu.
References
- 1.Garciarena CD, Ma YL, Swietach P, Huc L, Vaughan-Jones RD. Sarcolemmal localisation of Na+/H+ exchange and Na+-HCO3- co-transport influences the spatial regulation of intracellular pH in rat ventricular myocytes. J Physiol. 2013;591:2287–2306. doi: 10.1113/jphysiol.2012.249664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vaughan-Jones RD, Spitzer KW, Swietach P. Intracellular pH regulation in heart. J Mol Cell Cardiol. 2009;46:318–331. doi: 10.1016/j.yjmcc.2008.10.024. [DOI] [PubMed] [Google Scholar]
- 3.Lagadic-Gossmann D, Buckler KJ, Vaughan-Jones RD. Role of bicarbonate in pH recovery from intracellular acidosis in the guinea-pig ventricular myocyte. J Physiol. 1992;458:361–384. doi: 10.1113/jphysiol.1992.sp019422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garciarena CD, Youm JB, Swietach P, Vaughan-Jones RD. H+-activated Na+ influx in the ventricular myocyte couples Ca²+-signalling to intracellular pH. J Mol Cell Cardiol. 2013;61:51–59. doi: 10.1016/j.yjmcc.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 5.Wang HS, Chen Y, Vairamani K, Shull GE. Critical role of bicarbonate and bicarbonate transporters in cardiac function. World J Biol Chem. 2014;5:334–345. doi: 10.4331/wjbc.v5.i3.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alvarez BV, Fujinaga J, Casey JR. Molecular basis for angiotensin II-induced increase of chloride/bicarbonate exchange in the myocardium. Circ Res. 2001;89:1246–1253. doi: 10.1161/hh2401.101907. [DOI] [PubMed] [Google Scholar]
- 7.Vairamani K, Wang HS, Medvedovic M, Lorenz JN, Shull GE. RNA SEQ Analysis Indicates that the AE3 Cl-/HCO3- Exchanger Contributes to Active Transport-Mediated CO2 Disposal in Heart. Sci Rep. 2017;7:7264. doi: 10.1038/s41598-017-07585-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shen X, Cannell MB, Ward ML. Effect of SR load and pH regulatory mechanisms on stretch-dependent Ca(2+) entry during the slow force response. J Mol Cell Cardiol. 2013;63:37–46. doi: 10.1016/j.yjmcc.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 9.Moffat MP, Karmazyn M. Protective effects of the potent Na/H exchange inhibitor methylisobutyl amiloride against post-ischemic contractile dysfunction in rat and guinea-pig hearts. J Mol Cell Cardiol. 1993;25:959–971. doi: 10.1006/jmcc.1993.1108. [DOI] [PubMed] [Google Scholar]
- 10.Scholz W, Albus U, Lang HJ, Linz W, Martorana PA, Englert HC, Schölkens BA. Hoe 694, a new Na+/H+ exchange inhibitor and its effects in cardiac ischaemia. Br J Pharmacol. 1993;109:562–568. doi: 10.1111/j.1476-5381.1993.tb13607.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hendrikx M, Mubagwa K, Verdonck F, Overloop K, Van Hecke P, Vanstapel F, Van Lommel A, Verbeken E, Lauweryns J, Flameng W. New Na(+)-H+ exchange inhibitor HOE 694 improves postischemic function and high-energy phosphate resynthesis and reduces Ca2+ overload in isolated perfused rabbit heart. Circulation. 1994;89:2787–2798. doi: 10.1161/01.cir.89.6.2787. [DOI] [PubMed] [Google Scholar]
- 12.Hartmann M, Decking UK. Blocking Na(+)-H+ exchange by cariporide reduces Na(+)-overload in ischemia and is cardioprotective. J Mol Cell Cardiol. 1999;31:1985–1995. doi: 10.1006/jmcc.1999.1029. [DOI] [PubMed] [Google Scholar]
- 13.Strömer H, de Groot MC, Horn M, Faul C, Leupold A, Morgan JP, Scholz W, Neubauer S. Na(+)/H(+) exchange inhibition with HOE642 improves postischemic recovery due to attenuation of Ca(2+) overload and prolonged acidosis on reperfusion. Circulation. 2000;101:2749–2755. doi: 10.1161/01.cir.101.23.2749. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, Meyer JW, Ashraf M, Shull GE. Mice with a null mutation in the NHE1 Na+-H+ exchanger are resistant to cardiac ischemia-reperfusion injury. Circ Res. 2003;93:776–782. doi: 10.1161/01.RES.0000094746.24774.DC. [DOI] [PubMed] [Google Scholar]
- 15.Khandoudi N, Albadine J, Robert P, Krief S, Berrebi-Bertrand I, Martin X, Bevensee MO, Boron WF, Bril A. Inhibition of the cardiac electrogenic sodium bicarbonate cotransporter reduces ischemic injury. Cardiovasc Res. 2001;52:387–396. doi: 10.1016/s0008-6363(01)00430-8. [DOI] [PubMed] [Google Scholar]
- 16.Fantinelli JC, Orlowski A, Aiello EA, Mosca SM. The electrogenic cardiac sodium bicarbonate co-transporter (NBCe1) contributes to the reperfusion injury. Cardiovasc Pathol. 2014;23:224–230. doi: 10.1016/j.carpath.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 17.Yamamoto T, Shirayama T, Sakatani T, Takahashi T, Tanaka H, Takamatsu T, Spitzer KW, Matsubara H. Enhanced activity of ventricular Na+-HCO3- cotransport in pressure overload hypertrophy. Am J Physiol Heart Circ Physiol. 2007;293:H1254–H1264. doi: 10.1152/ajpheart.00964.2006. [DOI] [PubMed] [Google Scholar]
- 18.Gawenis LR, Bradford EM, Prasad V, Lorenz JN, Simpson JE, Clarke LL, Woo AL, Grisham C, Sanford LP, Doetschman T, et al. Colonic anion secretory defects and metabolic acidosis in mice lacking the NBC1 Na+/HCO3- cotransporter. J Biol Chem. 2007;282:9042–9052. doi: 10.1074/jbc.M607041200. [DOI] [PubMed] [Google Scholar]
- 19.Parsons SA, Millay DP, Wilkins BJ, Bueno OF, Tsika GL, Neilson JR, Liberatore CM, Yutzey KE, Crabtree GR, Tsika RW, et al. Genetic loss of calcineurin blocks mechanical overload-induced skeletal muscle fiber type switching but not hypertrophy. J Biol Chem. 2004;279:26192–26200. doi: 10.1074/jbc.M313800200. [DOI] [PubMed] [Google Scholar]
- 20.Lorenz JN, Robbins J. Measurement of intraventricular pressure and cardiac performance in the intact closed-chest anesthetized mouse. Am J Physiol. 1997;272:H1137–H1146. doi: 10.1152/ajpheart.1997.272.3.H1137. [DOI] [PubMed] [Google Scholar]
- 21.Al Moamen NJ, Prasad V, Bodi I, Miller ML, Neiman ML, Lasko VM, Alper SL, Wieczorek DF, Lorenz JN, Shull GE. Loss of the AE3 anion exchanger in a hypertrophic cardiomyopathy model causes rapid decompensation and heart failure. J Mol Cell Cardiol. 2011;50:137–146. doi: 10.1016/j.yjmcc.2010.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics. 2009;10:48. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bradford EM, Vairamani K, Shull GE. Differential expression of pancreatic protein and chemosensing receptor mRNAs in NKCC1-null intestine. World J Gastrointest Pathophysiol. 2016;7:138–149. doi: 10.4291/wjgp.v7.i1.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu Z, Alloush J, Beck E, Weisleder N. A murine model of myocardial ischemia-reperfusion injury through ligation of the left anterior descending artery. J Vis Exp. 2014:(86). doi: 10.3791/51329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.GTEx Consortium. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348:648–660. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O’Connell KM, Whitesell JD, Tamkun MM. Localization and mobility of the delayed-rectifer K+ channel Kv2.1 in adult cardiomyocytes. Am J Physiol Heart Circ Physiol. 2008;294:H229–H237. doi: 10.1152/ajpheart.01038.2007. [DOI] [PubMed] [Google Scholar]
- 29.Hund TJ, Snyder JS, Wu X, Glynn P, Koval OM, Onal B, Leymaster ND, Unudurthi SD, Curran J, Camardo C, et al. β(IV)-Spectrin regulates TREK-1 membrane targeting in the heart. Cardiovasc Res. 2014;102:166–175. doi: 10.1093/cvr/cvu008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fülöp L, Szigeti G, Magyar J, Szentandrássy N, Ivanics T, Miklós Z, Ligeti L, Kovács A, Szénási G, Csernoch L, et al. Differences in electrophysiological and contractile properties of mammalian cardiac tissues bathed in bicarbonate - and HEPES-buffered solutions. Acta Physiol Scand. 2003;178:11–18. doi: 10.1046/j.1365-201X.2003.01114.x. [DOI] [PubMed] [Google Scholar]
- 31.Prasad V, Lorenz JN, Miller ML, Vairamani K, Nieman ML, Wang Y, Shull GE. Loss of NHE1 activity leads to reduced oxidative stress in heart and mitigates high-fat diet-induced myocardial stress. J Mol Cell Cardiol. 2013;65:33–42. doi: 10.1016/j.yjmcc.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ruminot I, Gutiérrez R, Peña-Münzenmayer G, Añazco C, Sotelo-Hitschfeld T, Lerchundi R, Niemeyer MI, Shull GE, Barros LF. NBCe1 mediates the acute stimulation of astrocytic glycolysis by extracellular K+ J Neurosci. 2011;31:14264–14271. doi: 10.1523/JNEUROSCI.2310-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sowah D, Brown BF, Quon A, Alvarez BV, Casey JR. Resistance to cardiomyocyte hypertrophy in ae3-/- mice, deficient in the AE3 Cl-/HCO3- exchanger. BMC Cardiovasc Disord. 2014;14:89. doi: 10.1186/1471-2261-14-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thorsen K, Dam VS, Kjaer-Sorensen K, Pedersen LN, Skeberdis VA, Jurevičius J, Treinys R, Petersen IMBS, Nielsen MS, Oxvig C, et al. Loss-of-activity-mutation in the cardiac chloride-bicarbonate exchanger AE3 causes short QT syndrome. Nat Commun. 2017;8:1696. doi: 10.1038/s41467-017-01630-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prasad V, Lorenz JN, Lasko VM, Nieman ML, Al Moamen NJ, Shull GE. Loss of the AE3 Cl(-)/HCO(-) 3 exchanger in mice affects rate-dependent inotropy and stress-related AKT signaling in heart. Front Physiol. 2013;4:399. doi: 10.3389/fphys.2013.00399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prasad V, Bodi I, Meyer JW, Wang Y, Ashraf M, Engle SJ, Doetschman T, Sisco K, Nieman ML, Miller ML, et al. Impaired cardiac contractility in mice lacking both the AE3 Cl-/HCO3- exchanger and the NKCC1 Na+-K+-2Cl- cotransporter: effects on Ca2+ handling and protein phosphatases. J Biol Chem. 2008;283:31303–31314. doi: 10.1074/jbc.M803706200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmitt N, Grunnet M, Olesen SP. Cardiac potassium channel subtypes: new roles in repolarization and arrhythmia. Physiol Rev. 2014;94:609–653. doi: 10.1152/physrev.00022.2013. [DOI] [PubMed] [Google Scholar]
- 38.Aiello EA, De Giusti VC. Regulation of the cardiac sodium/bicarbonate cotransporter by angiotensin II: potential Contribution to structural, ionic and electrophysiological myocardial remodelling. Curr Cardiol Rev. 2013;9:24–32. doi: 10.2174/157340313805076340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Unudurthi SD, Wu X, Qian L, Amari F, Onal B, Li N, Makara MA, Smith SA, Snyder J, Fedorov VV, et al. Two-Pore K+ Channel TREK-1 Regulates Sinoatrial Node Membrane Excitability. J Am Heart Assoc. 2016;5:e002865. doi: 10.1161/JAHA.115.002865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vaughan-Jones RD, Villafuerte FC, Swietach P, Yamamoto T, Rossini A, Spitzer KW. pH-Regulated Na(+) influx into the mammalian ventricular myocyte: the relative role of Na(+)-H(+) exchange and Na(+)-HCO Co-transport. J Cardiovasc Electrophysiol. 2006;17 Suppl 1:S134–S140. doi: 10.1111/j.1540-8167.2006.00394.x. [DOI] [PubMed] [Google Scholar]
- 41.Gottlieb RA, Burleson KO, Kloner RA, Babior BM, Engler RL. Reperfusion injury induces apoptosis in rabbit cardiomyocytes. J Clin Invest. 1994;94:1621–1628. doi: 10.1172/JCI117504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takashi E, Ashraf M. Pathologic assessment of myocardial cell necrosis and apoptosis after ischemia and reperfusion with molecular and morphological markers. J Mol Cell Cardiol. 2000;32:209–224. doi: 10.1006/jmcc.1999.1067. [DOI] [PubMed] [Google Scholar]
- 43.Freude B, Masters TN, Robicsek F, Fokin A, Kostin S, Zimmermann R, Ullmann C, Lorenz-Meyer S, Schaper J. Apoptosis is initiated by myocardial ischemia and executed during reperfusion. J Mol Cell Cardiol. 2000;32:197–208. doi: 10.1006/jmcc.1999.1066. [DOI] [PubMed] [Google Scholar]
- 44.Halestrap AP, Clarke SJ, Javadov SA. Mitochondrial permeability transition pore opening during myocardial reperfusion--a target for cardioprotection. Cardiovasc Res. 2004;61:372–385. doi: 10.1016/S0008-6363(03)00533-9. [DOI] [PubMed] [Google Scholar]
- 45.Prasad V, Chirra S, Kohli R, Shull GE. NHE1 deficiency in liver: implications for non-alcoholic fatty liver disease. Biochem Biophys Res Commun. 2014;450:1027–1031. doi: 10.1016/j.bbrc.2014.06.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li N, Jiang P, Du W, Wu Z, Li C, Qiao M, Yang X, Wu M. Siva1 suppresses epithelial-mesenchymal transition and metastasis of tumor cells by inhibiting stathmin and stabilizing microtubules. Proc Natl Acad Sci USA. 2011;108:12851–12856. doi: 10.1073/pnas.1017372108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ma Y, Liu T, Song X, Tian Y, Wei Y, Wang J, Li X, Yang X. Siva 1 inhibits proliferation, migration and invasion by phosphorylating Stathmin in ovarian cancer cells. Oncol Lett. 2017;14:1512–1518. doi: 10.3892/ol.2017.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kang TH, Noh KH, Kim JH, Bae HC, Lin KY, Monie A, Pai SI, Hung CF, Wu TC, Kim TW. Ectopic expression of X-linked lymphocyte-regulated protein pM1 renders tumor cells resistant to antitumor immunity. Cancer Res. 2010;70:3062–3070. doi: 10.1158/0008-5472.CAN-09-3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pugach EK, Richmond PA, Azofeifa JG, Dowell RD, Leinwand LA. Prolonged Cre expression driven by the α-myosin heavy chain promoter can be cardiotoxic. J Mol Cell Cardiol. 2015;86:54–61. doi: 10.1016/j.yjmcc.2015.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]