

Abstract
Chemotherapy is routinely used in cancer treatment to eliminate primary and metastatic tumor cells. However, tumors often display or develop resistance to chemotherapy. Mechanisms of chemoresistance can be either tumor cell autonomous or mediated by the tumor surrounding non-malignant cells, also known as stromal cells, which include fibroblasts, immune cells, and cells from the vasculature. Therapies targeting cancer cells have shown limited effectiveness in tumors characterized by a rich tumor stroma. Tumor-associated macrophages (TAMs) and cancer-associated fibroblasts (CAFs) are the most abundant non-cancerous cells in the tumor stroma and have emerged as key players in cancer progression, metastasis and resistance to therapies. This review describes the recent advances in our understanding of how CAFs and TAMs confer chemoresistance to tumor cells and discusses the therapeutic opportunities of combining anti-tumor with anti-stromal therapies. The continued elucidation of the mechanisms by which TAMs and CAFs mediate resistance to therapies will allow the development of improved combination treatments for cancer patients.
Keywords: macrophages, fibroblasts, tumor stroma, tumor microenvironment, chemoresistance, therapy resistance
Introduction
The treatment of cancer with chemical substances, known as chemotherapy, is routinely used for cancer treatment because as it circulates throughout the body it targets not only the primary tumor site but also tumor cells that have spread to other organs which are usually missed with surgical intervention or radiotherapy treatment (Eguchi et al., 2008).
The birth of chemotherapy came after the first world war, using nitrogen mustard as an anti-cancer agent in non-Hodgkin’s lymphoma (Gilman, 1963). This agent was non-specific and showed limited effectiveness as patients experienced relapse after a few weeks. However, this discovery triggered investigation into the drug’s mechanism of action leading to the development of other alkylating agents (Haddow et al., 1948). Targeted chemotherapy was developed in the late 1980s after the elucidation of some of the signaling pathways aberrantly regulated in tumors. Targeted chemotherapy included pharmacological targeting of the cell cycle regulating proteins, growth factors and angiogenesis mediators (Hanahan and Weinberg, 2000; Chabner and Roberts, 2005).
Since its beginning, chemotherapy has provided a plethora of benefits for many cancer patients (Klastersky and Paesmans, 2001; Benedetti-Panici et al., 2003; Gebski et al., 2007). Chemotherapeutic agents given before surgery as ‘neoadjuvant’ therapy can be used to reduce the tumor mass before surgical resection. This has many benefits as the reduction of the tumor size decreases the level of invasiveness required for resection and often improves the distinction between healthy and neoplastic tissue during resection (Hayes and Schott, 2015). Adjuvant administration of chemotherapy occurs post-surgery with the purpose of minimizing the chance of recurrence. Adjuvant therapy is effective in two ways: firstly, against micro or macro-metastasis which are already seeded but were not detectable at the time of surgery, and secondly, against micro-metastasis created as a by-product of surgery due to tissue regeneration promoting cytokine storms released after invasive surgery (Hayes and Schott, 2015).
Despite the development of targeted agents with improved toxicity profiles, in some cancers chemotherapeutic agents only provide a minimal improvement of overall survival (Burris et al., 1997; Marquette and Nabell, 2012). The reduced effectiveness of chemotherapy in patients is due to tumor resistance mechanisms, which can be either tumor cell autonomous and/or mediated by the tumor surrounding non-malignant cells present in the tumor microenvironment (TME) (Joyce and Pollard, 2009; De Palma and Lewis, 2013; Mielgo and Schmid, 2013; Zheng, 2017). Tumor cell autonomous mechanisms of drug resistance have been extensively reviewed before (Zahreddine and Borden, 2013; Housman et al., 2014; Zheng, 2017) so the focus of this review is on the emerging TME-mediated mechanisms of tumor resistance to chemotherapy with a main focus on chemoresistance mechanisms mediated by tumor-associated macrophages (TAMs) and fibroblasts.
The TME describes the complete tumor milieu including the malignant tumor cells and the surrounding tumor stroma. The tumor stroma consists of non-malignant cells including immune cells (macrophages, neutrophils, and T cells), fibroblasts, cells from the vasculature (pericytes and endothelial cells) and extracellular matrix (ECM) proteins (Hanahan and Weinberg, 2011). Accumulating evidence shows that the tumor stroma develops and interacts with the tumor cells, participating in bi-directional tumor-stroma signaling which supports tumor progression, metastasis and resistance to therapy (Hanahan and Coussens, 2012; Quail and Joyce, 2013). The most abundant non-cancerous cell types present in the tumor stroma are TAMs and cancer-associated fibroblasts (CAFs). This review will discuss the various mechanisms, discovered to date, by which TAMs and CAFs support tumor chemoresistance, the controversies and current gaps in this research field and the potential future perspectives.
Origin of Macrophages and Fibroblasts
Macrophages
Tissue resident macrophages are a diverse population of cells which perform tissue-specific functions in tissue homeostasis, repair, immunity and angiogenesis (Davies et al., 2013a). Macrophages can originate from three independent sources. Embryonic macrophage populations have been mapped back to two sources: fetal liver-derived monocytes or precursor cells found in the yolk sac (Yona et al., 2013; Mass et al., 2016). In adult tissue, macrophage populations differentiate from hematopoietic stem cells in the bone marrow (Orkin and Zon, 2008).
Once established in adult tissue, macrophages maintain their population via self-renewal in the steady state but increase their rate of proliferation in response to stimuli such as interleukin 4 (IL-4) and colony stimulating factor 1 (CSF-1) (Jenkins et al., 2011, 2013; Davies et al., 2013a). During inflammation, bone marrow-derived monocytes are recruited into the tissue and mature into macrophage populations which act alongside tissue resident macrophages (Shi and Pamer, 2011). These converted monocytes display cell surface markers associated with resident macrophages increasing their responsiveness to IL-4 and IL-3 (Yona et al., 2013; Dal-Secco et al., 2015).
Bone-marrow derived macrophages (BM-DMs) and tissue resident macrophages appear to intermingle and work together to resolve inflammation and promote tissue repair. However, it is currently undetermined if BM-DMs play the exact same role as tissue resident macrophages (Davies et al., 2013b). Bone marrow transplant studies have shown that BM-DMs and tissue resident macrophages share similar characteristics (van de Laar et al., 2016). These similarities have been further confirmed by transcriptome analysis of lung alveolar resident macrophages which revealed different genes expressed in BM-DMs compared to tissue resident macrophages (Gibbings et al., 2015).
Fibroblasts
Fibroblasts are of mesenchymal origin and dependent on their tissue of origin have a distinct transcriptional profile (Chang et al., 2002). Fibroblasts have never been identified in embryonic tissue but are hypothesized to arise during the epithelial-to-mesenchymal transition (EMT) of the epiblast during gastrulation with the generation of mesoderm tissue (Kalluri, 2016). Virchow (1858) identified cells in adult tissue that, produced collagen, were resistant to apoptosis, and reverted to quiescence upon the completion of tissue development. These cells were later called fibroblasts (Virchow, 1858). Due to the inability to identify fibroblasts in embryonic tissue it remains unknown whether the majority of activated fibroblasts originate from fibrocytes or mesenchymal stem cells (MSCs) in adult tissue (Kalluri, 2016).
Stellate cells are found in the pancreas, liver, lung, and kidney and although stellate cells are similar to fibroblasts, they display some distinctly different functions such as vitamin A storage as retinol droplets in their cytoplasm which is required for cellular homeostasis (Keane et al., 2005; Liu, 2006; Erkan et al., 2010). Quiescent stellate cells usually constitute <10% of the organ where they reside and are found in perivascular and peri-parenchymal regions (Wake and Sato, 1993; Apte et al., 1998; Bachem et al., 1998). Like fibroblasts, the origin of stellate cells is still debated. Neuroectoderm is suggested as a potential origin of pancreatic stellate cells (PaSCs) and hepatic stellate cells (hStCs) (Friedman, 2000). Lineage tracing studies have shown that hStCs can originate from mesoderm in mice, however, lineage tracing studies are currently lacking for PaSCs (Asahina et al., 2009, 2011).
Activated fibroblasts (also known as myofibroblasts) can originate from several different cell types that include quiescent fibroblasts from normal parenchyma, endothelial cells, MSCs, and stellate cells (LeBleu et al., 2013; Kalluri, 2016). For example, the origin of activated fibroblasts which support ductal outgrowth has been disputed in breast tissue. Ucar et al. (2010), reported that miR-212/132 expression in stromal fibroblasts is required to support ductal outgrowth. However, in another study, targeted deletion of miR-212 and miR-132 in embryonic stem cells did not show an effect in ductal outgrowth, instead, this study claims that Hic1 expression in stromal cells is required for mammary ductal outgrowth (Kayo et al., 2014). These contradictory results suggest that further studies aiming to understand the role of fibroblasts in mammary gland development are required (Ucar et al., 2014). The heterogeneous origins of myofibroblasts may play a role in generating populations with different phenotypes and functions. Recent studies have described heterogeneous populations of activated fibroblasts present in pancreatic and breast tumors (Ohlund et al., 2017; Costa et al., 2018) and understanding the functions of these different fibroblast populations in cancer is currently an intensive field of research.
Physiological Functions of Macrophages and Fibroblasts
Macrophages represent a heterogeneous population of cells that are highly plastic and adapt to their surroundings to perform a variety of functions in tissue homeostasis, repair, and immunity (Wynn et al., 2013). Macrophages respond to tissue-derived or external stimuli adapting their phenotype and function accordingly (Biswas and Mantovani, 2010). A spectrum of different subsets of macrophages with diverse phenotypes and functions co-exist in tissues and the macrophage subsets at the extremes of this spectrum are known as M1 (or classically activated) and M2 (or alternatively activated) macrophages (Murray and Wynn, 2011; Mills, 2012). Macrophages can be polarized into M1-like or M2-like macrophages and their polarization depends on the stimulating cytokine and the length of exposure (Gordon and Martinez, 2010). However, the nomenclature and understanding of macrophage subtypes and functions is still evolving.
M1-like macrophages are generated in response to interferon gamma (INFγ) and lipopolysaccharide (LPS) stimulation, factors produced by infiltrating bacteria and pathogens. M1-like macrophages are pro-inflammatory and secrete factors to promote inflammation, microbicidal activity and immunostimulation, such as cytokines IL-12, IL-6, IL-1β, tumor-necrosis factor alpha (TNFα) as well as reactive oxygen species (ROS) and nitric oxide (NO) (Gordon and Martinez, 2010; Biswas et al., 2013) (Figure 1).
FIGURE 1.

Macrophage polarization. Bone marrow derived monocytes or tissue derived monocytes can be polarized toward either an M1 or M2 phenotype. Classical activation toward M1 polarization occurs in response to interferon gamma (IFNγ) and lipopolysaccharide (LPS) leading to a Th1 response associated with bacteria and viruses as well as possessing anti-tumorigenic properties. Alternative activation toward an M2 phenotype is triggered in response to toll-like receptors (TLRs), immune complexes, IL-4, IL-13, IL-10, and glucocorticoids. M2 macrophages lead to a Th2 response and exhibit anti-parasitic behavior. In cancer, M2-like macrophages promote tumor progression.
In contrast, M2-like macrophages are polarized by IL-4 and IL-13 produced by invading parasites and release anti-inflammatory cytokines IL-10, arginase I and transforming growth factor beta (TGF-β), as well as vascular endothelial growth factor (VEGF), promoting the remodeling of their surrounding tissue. Concurrently, macrophages upregulate expression of scavenging receptors while downregulating receptors and markers associated with antigen presentation (Biswas and Mantovani, 2010; Mantovani and Sica, 2010) (Figure 1).
Tissue resident macrophages play a variety of roles in a tissue context-dependent manner. Largely, they participate in functions usually associated with an M2 phenotype including mediating resolution of inflammation, maintaining tissue homeostasis via the removal of debris, supporting angiogenesis and partaking in immune surveillance (Davies et al., 2013a).
Angiogenesis occurs as part of homeostasis throughout life and is tightly regulated by macrophages (Fantin et al., 2010; Outtz et al., 2011). In mouse embryos, microglia (central-nervous system specific macrophages) migrate to the brain and assist in developmental angiogenesis (Arnold and Betsholtz, 2013). In the central nervous system, macrophages promote endothelial tip cell fusion by acting as a chaperone for endothelial cells in vascular development (Fantin et al., 2010). However, it appears that the actions undertaken by macrophages are tissue-dependent as, conversely, macrophages mediate the regression of blood vessels in the developing retina (Lobov et al., 2005; Fantin et al., 2010).
Another homeostatic function of macrophages is the removal of apoptotic and excess cell debris. This function is extremely important in the regulation of hematopoiesis in which macrophages phagocytose excess erythrocytes and neutrophils (Gordy et al., 2011; Klei et al., 2017). When this process was interrupted in mice they suffered severe neutrophilia, splenomegaly, extramedullary hematopoiesis and decreased body weight (Gordy et al., 2011). Macrophages also regulate immune responses through the ingestion of apoptotic cells preventing leakage of cell-death related factors which could promote inflammation (Savill et al., 2002).
In the event of injury or infection, pro-inflammatory macrophages are recruited to the afflicted area and secrete factors including IL-1β, NO, and TNFα as a defense mechanism to kill any invading pathogens (Murray and Wynn, 2011). The release of these factors can also result in secondary damage to host tissue. To limit the impact of this damage, macrophages either undergo apoptosis or reprogram toward an anti-inflammatory M2-like phenotype (Murray and Wynn, 2011). However, when this process goes awry, and macrophages maintain their pro-inflammatory functions, chronic inflammation occurs and becomes the basis of some auto-immune diseases such as Crohn’s disease, rheumatoid arthritis and autoimmune hepatitis (Sindrilaru et al., 2011; Navegantes et al., 2017). Along with mediating the immunity side of wound healing, macrophages alter their secretory phenotype after inflammation subsides, to promote tissue regeneration. To promote the closure of the wound, macrophages attract and activate fibroblasts through the secretion of TGF-β (Khalil et al., 1989; Murray and Wynn, 2011).
In healthy tissue, fibroblasts and stellate cells exist in a quiescent state within the ECM making few cell-cell or cell-basement membrane connections. They are usually found as single cells, elongated and spindle-like in morphology situated in the interstitial space between the functional tissues of adult organs (Tarin and Croft, 1969). Quiescent fibroblasts and stellate cells produce very little ECM components such as collagen 1 and fibronectin and secrete a few factors including pigment epithelium-derived factor (PEDF) and thrombospondin-2, although their actual role while quiescent is yet to be fully elucidated (Tarin and Croft, 1969; Pollina et al., 2008). Specific markers for fully quiescent fibroblasts are not yet known, however, fibroblasts specific protein-1 positive (FSP1+) cells are often considered as quiescent (Strutz et al., 1995) (Figure 2).
FIGURE 2.

Fibroblast activation. Quiescent fibroblasts produce few extracellular matrix (ECM) components such as fibronectin and collagen type 1 (Col1a1). They express fibroblasts specific protein-1 (FSP-1), actin and vimentin and secrete pigment epithelium-derived factor (PEDF) and thrombospondin-2 (THBS2). When stimulated with transforming growth factor beta (TGF-β), reactive oxygen species (ROS) or hypoxia, quiescent cells become activated increasing their contractility, proliferation and secretion. Activated myofibroblasts produce larger volumes of fibronectin and collagen along with tenascin-c and secreted protein acidic and rich in cysteine (SPARC). Increased secretion includes IL-6, tissue inhibitor of metalloproteinase (TIMPs), TGF-β, vascular endothelial growth factor (VEGF), epidermal growth factor (EGF), and CXCL10. Upregulated receptors/markers include alpha smooth muscle actin (αSMA), platelet derived growth factor receptor alpha/beta (PDGFRα/β), fibroblast activation protein (FAP), discoidin domain-containing receptor 2 (DDR2), desmin, and vimentin.
The activation of fibroblasts and stellate cells is triggered in response to stress factors produced during tissue stress and damage, including TGF-β and ROS (Kalluri and Zeisberg, 2006). Activated fibroblasts acquire smooth muscle-like properties with increased contractility, motility, proliferation and a stellate morphology, and are known as myofibroblasts (Sappino et al., 1988; Ronnovjessen and Petersen, 1993). Upon activation, stellate cells also acquire a myoblastic phenotype but lose their cytoplasmic retinol lipid droplets (Blaner et al., 2009). Common markers of myofibroblasts include alpha-smooth muscle actin (αSMA), platelet derived growth factor receptor beta (PDGFRβ), PDGFRα, fibroblast activated protein (FAP), vimentin, desmin, Fibronectin Extra-domain A (EDA-FN) and discoidin domain-containing receptor 2 (DDR2) (Ronnovjessen and Petersen, 1993; Sugimoto et al., 2006; Quail and Joyce, 2013; Kalluri, 2016; Jiang et al., 2017) (Figure 2).
Myofibroblasts classically function in acute wound healing, becoming ‘reversibly’ activated and depositing the ECM proteins, collagens and fibronectin to close the wound (Dvorak et al., 1986; Darby and Hewitson, 2007). Myofibroblasts also modulate ECM consistency secreting matrix metalloproteases (MMPs) and tissue inhibitor of metalloproteinase (TIMPs) (Tampe and Zeisberg, 2014). Activated myofibroblasts also possess an altered secretory phenotype producing factors such as TGF-β, VEGF, C-X-C motif chemokine ligand 10 (CXCL10), CXCL12, IL-6, and epidermal growth factor (EGF) to promote proliferation and mediate recruitment of other cell types to the damaged tissue (Dvorak et al., 1986) (Figure 2).
Chronic activation of fibroblasts and stellate cells occurs in response to prolonged afflictions including toxins or auto-immune disorders. This results in chronic tissue fibrosis with myofibroblasts continuing to aberrantly perform their wound healing functions without resolution. These myofibroblasts become fibrosis-associated fibroblasts (FAFs), are irreversibly activated and exhibit enhanced proliferation and survival (Rock et al., 2011; Zeisberg and Zeisberg, 2013; Kalluri, 2016).
Tumor-Associated Macrophages (TAMs) and Cancer-Associated Fibroblasts (CAFs)
Macrophages and fibroblasts are the two most abundant non-cancerous cells in tumors. Tumors become infiltrated with BM-DMs that are attracted to the tumor via the secretion of damage associated molecular patterns (DAMPs) and specific macrophage chemoattractants CSF-1 and chemokine C-C motif ligand 2 (CCL2). M1-like macrophages derived from the bone marrow and tissue resident macrophages are recruited to and activated in the tumor site in response to antigen presentation and inflammatory responses (Zhu et al., 2017). Once the tumor is established, tumor cells secrete cytokines IL-4, IL-10, IL-13 and lactic acid, and along with the presence of CD4+ Th2 cells, cause the polarization of TAMs toward an M2-like phenotype. The M2 TAMs no longer serve to destroy the tumor but rather support cancer growth, metastasis and resistance to therapies (Gocheva et al., 2010; Qian and Pollard, 2010; Ruffell et al., 2012; Colegio et al., 2014). M2 TAMs support tumor progression by directly stimulating the growth of cancer cells through the production of growth factors, including EGF, TNFα, IL-6 (Grivennikov et al., 2010).
Solid tumors can undergo periods of hypoxia as its growing size limits the disposal of waste products and nutrient delivery becomes limited, triggering the angiogenic switch (Bergers and Benjamin, 2003; Hanahan and Weinberg, 2011). The activation of the angiogenic switch in tumors triggers dysregulated angiogenesis resulting in leaky vasculature with abnormal branching and enlarged diameter (Bergers and Benjamin, 2003). Macrophages are a source of VEGF and are known to support angiogenesis under normal physiological conditions (Fantin et al., 2010; Outtz et al., 2011). However, tumors depleted of myeloid-derived VEGF have a normalized vasculature with increased pericyte coverage and reduced vessel length and this accelerates tumor progression (Stockmann et al., 2008). Conversely, another study showed that hypoxia-related TAMs possess reduced mTOR activation, and that stimulation of mTOR activity in TAMs resulted in normalized vasculature with decreased vessel leakiness, hypoxia and metastasis (Wenes et al., 2016). TAMs are attracted to areas of tumor hypoxia through the release of Semaphorin 3A by cancer cells and TAMs promote angiogenesis via the phosphorylation of VEGF-receptor on endothelial cells (Casazza et al., 2013). CSF-1 stimulation has been shown to upregulate TIE2 expression on macrophages (Forget et al., 2014). Once inside the tumor, TIE2+ macrophages bind to angiopoietin-2 (Ang-2) expressed by endothelial cells and stimulate the growth of blood vessels promoting tumor growth and metastasis (De Palma et al., 2005; Mazzieri et al., 2011).
Metastatic spread of tumor cells to distant organs involves a multi-step process that requires local tissue invasion, intravasation, circulation through the blood stream, extravasation and successful colonization of the distant organ by the cancer cells (Hanahan and Coussens, 2012; Massague and Obenauf, 2016). Macrophages play a role in each of these stages of the metastatic cascade. Macrophages help tumor cell invasion into the basement membrane (Condeelis and Pollard, 2006; Wyckoff et al., 2007). In the PyMT breast cancer mouse model, CSF-1 produced by tumor cells and EGF secreted by TAMs results in the migration of both macrophages and cancer cells along collagen fibers and intravasation into the blood vessels (Goswami et al., 2005; Wyckoff et al., 2007). This phenomenon was also seen in glioblastoma, resulting in enhanced cancer cell invasion (Coniglio et al., 2012). TAMs can also promote tumor cell migration and invasion through the secretion of MMPs, secreted protein acidic and rich in cysteine (SPARC) and cathepsins which degrade and remodel the ECM (Bergers et al., 2000; Gocheva et al., 2006) as well as through the secretion of TGF-β which promotes EMT of tumor cells and increased tumor cell migration (Bonde et al., 2012).
As outlined earlier, fibroblasts are activated in response to tissue damage. After resolution of the insult, fibroblasts will reprogram back to quiescence or undergo apoptosis (Tomasek et al., 2002). However, tumors are referred to as “wounds that do not heal” (Dvorak et al., 1986). Persistent activation signals, in the context of cancer, maintain fibroblasts in a chronically activated state triggering a desmoplastic reaction and generating a dense fibrotic stroma which envelopes the tumor mass. Fibroblast activation signals are tumor-specific and determine the phenotype and function of the resulting myofibroblast. In the TME, a myofibroblast will exert a pro- or anti-tumorigenic response depending upon which chemokines/cytokines it encounters (Tampe and Zeisberg, 2014). TGF-β is a common activating factor released by tumors which increases the expression of PDGF receptors on activated PaSCs (Apte et al., 1999). Sonic hedgehog (Shh) signaling in PDAC tumors has been reported to promote fibroblast activation and fibrosis in the pancreas (Bailey et al., 2008; Yauch et al., 2008). Other common factors involved in CAF activation include fibroblast growth factor (FGF), platelet derived growth factor (PDGF), and monocyte chemotactic protein (MCP1) (Kalluri and Zeisberg, 2006; Marsh et al., 2013).
Concurrent with their activated state, CAFs express an altered secretory phenotype, compared to quiescent fibroblasts, including ECM proteins and ECM modulating factors such as tenascin C, periostin, SPARC and EDA-FN; and tumor promoting factors such as nuclear factor-kB (NF-kB), IL-8, prostaglandin E2 (PGE2), connective tissue growth factor (CTGF) and CXCL7 (Kalluri, 2003; Hanahan and Coussens, 2012).
Recent advances in the field of CAF research has shown that different subsets of CAF populations with different functions co-exist within tumors (Costea et al., 2013; Brechbuhl et al., 2017; Ohlund et al., 2017; Costa et al., 2018). For example, in PDAC a specific subset of CAFs expressing high levels of αSMA but low levels of IL-6 was found in the fibrotic area juxtaposed to cancer cells and was called the myofibroblast CAF subset (myCAFs) (Ohlund et al., 2017). A different subset of CAFs, expressing low levels of αSMA but high levels of IL-6 was found at the periphery of the tumor and was termed the inflammatory CAF subset (iCAFs) (Ohlund et al., 2017). Ohlund et al. (2017) showed that the proximity of the myofibroblasts to the PDAC tumor cells, and the concentration of tumor-secreted factors alters the phenotype of the CAFs and the proteins they secrete. Another recent study performed with luminal A, human epidermal growth factor receptor 2+ (HER2+) and triple negative breast cancer (TNBC) patient samples revealed the co-existence of four different CAF subsets in breast tumors (Costa et al., 2018). TNBC samples predominantly had two types of myofibroblast-like CAFs; CAF-S1 and CAF-S4 identified by their high expression of αSMA. However, only CAF-S1 defined as CD29Med, FAPHi, FSP1Low-Hi, aSMAHi, PDGFRbMed-Hi, and CAV1Low; showed an immunosuppressive role by attracting T lymphocytes and promoting their survival and differentiation into immunosuppressive T regulatory cells (Costa et al., 2018). Thus, CAFs, like TAMs, are a heterogeneous population of cells and uncovering the different CAF populations and their functions in cancer is currently an important area of research.
Tumor-associated macrophages and CAFs take part in a complex interplay and can regulate each other’s functions. For example, cancer cells and myofibroblasts are known sources of VEGF which promotes the accumulation of immune cells including macrophages at the site of fibrosis (Fukumura et al., 1998). VEGF-dependent recruitment and activation of macrophages promotes tumorigenesis, angiogenesis and invasion in skin cancer (Linde et al., 2012). Reciprocally, in liver metastasis of pancreatic cancer, macrophages recruited to the metastatic liver secrete granulin and activate resident quiescent hStCs which subsequently produce periostin supporting the growth of metastatic cancer cells in the liver (Nielsen et al., 2016).
Mechanisms of Chemotherapy Resistance Driven by TAMs and CAFs
Chemotherapy is used as a treatment in many different cancer types and is used either alone or in combination with surgical resection or radiation. Chemotherapy targets tumor cells at both the primary tumor site and the metastatic site. However, a common problem encountered with the treatment of many tumors is an acquired resistance to chemotherapeutic agents. Chemoresistance can be mediated by tumor cell-autonomous mechanisms, including changes in tumor cell epigenetics, drug inactivation, EMT, activation of alternative survival and proliferative pathways, and/or selection of drug-resistant cancer cell clones (Housman et al., 2014). However, many solid tumors such as breast cancer and PDAC have a rich stroma which contains, as described before, a plethora of non-malignant cell types that influence cancer progression and response to therapy in various ways. In fact, these non-malignant stromal cells are not simple bystanders but engage in bi-directional tumor-stroma signaling which can result in impaired therapeutic efficacy. For instance, the attraction of TAMs in a MCF-7 breast cancer xenograft model, via CSF-1 signaling, reduces the efficacy of a combination treatment with cyclophosphamide, methotrexate and 5-fluorouracil (CMF) (Paulus et al., 2006) (Figure 3A). The presence of TAMs in the genetic MMTV-PyMT mouse model of breast cancer makes tumors more resistant to paclitaxel therapy (DeNardo et al., 2011). Another study revealed TAM-derived cathepsins B and S as responsible for mediating chemoresistance to taxol in the MMTV-PyMT mouse model (Shree et al., 2011) (Figure 3B). In a subcutaneous mouse model of colorectal cancer, IL-6 released by TAMs mediates chemoresistance to 5-FU via activation of the IL-6R/STAT3 signaling axis (Yin et al., 2017).
FIGURE 3.
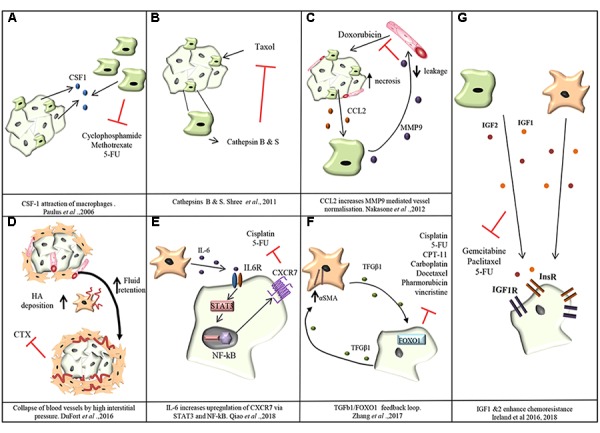
Mechanisms of chemoresistance mediated by TAMs and CAFs. (A) Cancer cells attract TAMs via CSF-1. TAMs confer resistance of MCF-7 breast cancer cells toward cyclophosphamide, methotrexate and 5-fluorouracil (5-FU; Paulus et al., 2006). (B) Cathepsins B and S secreted by TAMS mediate resistance of breast cancer cells to taxol in MMTV-PyMT mouse model (Shree et al., 2011). (C) In the MMTV-PyMT transgenic mouse model, cancer cell necrosis caused by doxorubicin treatment causes cancer cells to release the monocyte chemoattractant CCL2. Recruited TAMs produce MMP-9 which causes leakiness of blood vessels and reduction in doxorubicin delivery (Nakasone et al., 2012). (D) In PDAC, CAFs increase deposition of hyaluronan (HA) creating an increase in fluid retention and subsequently interstitial pressure in the tumor rises causing the collapse of blood vessels and limiting the delivery of chemotherapeutic agents (DuFort et al., 2016). (E) CAF secreted IL-6 stimulates the upregulation of CXCR7 through STAT3/NF-kB signaling promoting resistance of esophageal squamous cell carcinoma cells against cisplatin and 5-FU (Qiao et al., 2018). (F) CAF-derived TGF-β upregulates FOXO1 expression in esophageal squamous cell carcinoma cells triggering reciprocal TGF-β secretion which in turn increases the levels of αSMA expression in CAFs and resistance to cisplatin, taxol, irinotecan (CPT-11), 5-FU, carboplatin, docetaxel, pharmorubicin, and vincristine (Zhang et al., 2017). (G) TAM and CAF derived IGF-1 and IGF-2 activate insulin and IGF-1 receptor signaling on tumor cells conferring resistance of pancreatic and breast tumors to gemcitabine and paclitaxel (Ireland et al., 2016, 2018).
Tumor-associated macrophages can also regulate the delivery of chemotherapy to tumor cells. In the MMTV-PyMT transgenic breast cancer mouse model, doxorubicin administration causes necrosis of cancer cells with the release of CCL2, a chemokine that attracts monocytes/macrophages. MMP-9 secretion by the recruited myeloid cells was shown to decrease vasculature leakiness and to impair doxorubicin delivery into the tumors (Nakasone et al., 2012). In fact, MMP-9 null mice showed an improved response to Doxorubicin that correlated with increased vascular leakage (Nakasone et al., 2012) (Figure 3C). Conversely, in a Lewis lung carcinoma subcutaneous isograft model, myeloid derived VEGF promotes resistance to cyclophosphamide treatment by promoting the formation of abnormal vessels with reduced pericyte coverage, tortuosity, and vessel density (Stockmann et al., 2008).
Cancer-associated fibroblasts also play a role in tumor chemoresistance. In fact, a dense fibrotic stroma correlates with a poor response to neoadjuvant treatment with 5-fluorouracil, epirubicin and cyclophosphamide (FEC) in breast cancer and with gemcitabine in PDAC (Farmer et al., 2009; Olive et al., 2009; Pandol et al., 2009). One way fibrosis promotes chemoresistance in PDAC is through CAF secretion of hyaluronan, generating high interstitial pressure within the tumor, causing the collapse of blood vessels supplying the tumor mass and impairing drug delivery (DuFort et al., 2016) (Figure 3D).
In esophageal squamous cell carcinoma, CXCR7 expression is upregulated in tumor cells through STAT3/NF-kB signaling stimulated by CAF-derived IL-6, ultimately promoting resistance against cisplatin and 5-fluorouracil (Qiao et al., 2018) (Figure 3E). IL-6 has pleiotropic effects in the TME and also mediates chemoresistance by promoting EMT of cancer cells (Shintani et al., 2016). TGF-β secretion by CAFs was shown to confer resistance of esophageal squamous cell carcinoma against cisplatin, taxol, irinotecan (CPT-11), 5-fluorouracil (5-FU), carboplatin, docetaxel, pharmorubicin, and vincristine (Zhang et al., 2017) (Figure 3F).
We recently showed that TAMs and CAFs are the main sources of Insulin-like growth factors 1 and 2 (IGF-1, IGF-2) in pancreatic and breast tumors, and that IGF signaling mediates resistance of murine pancreatic and breast tumors to gemcitabine and paclitaxel (Figure 3G) (Ireland et al., 2016, 2018). Importantly, we found that 72% of PDAC patients and 87% of patients with invasive breast cancer have the IGF signaling pathway activated in their tumors, and this correlates with an increased number of TAMs and CAFs (Ireland et al., 2016, 2018). Similarly, IGF1 was also shown to be secreted by TAMs in glioblastoma multiforme and to mediate resistance to a CSF-1R small molecule inhibitor through activation of PI3K signaling (Quail et al., 2016).
Targeting TAMs and CAFs in Cancer
Currently, approaches are being undertaken to block macrophage recruitment to the tumor site, to repolarize TAMs back into an M1-like anti-tumorigenic phenotype, and to target specific tumorigenic functions of TAMs. Preventing recruitment of macrophages to the tumor site has been achieved through targeting macrophage chemoattractants such as CSF-1 and CCL2 or their corresponding receptors: CSF-1 receptor (CSF-1R) and C-C chemokine receptor type 2 (CCR2). Anti-CSF-1R agents have been shown to be effective against recruitment of M2-like macrophages in breast cancer models, and anti-CSF1R inhibitors used in combination with paclitaxel decreased tumor growth and pulmonary metastasis (DeNardo et al., 2011). CSF-1R and CCR2 antagonists have been reported to prevent infiltration of TAMs into the tumor mass increasing response to gemcitabine treatment in mouse models of PDAC (Mitchem et al., 2013). CCL2 inhibition in combination with docetaxel has shown increased efficacy, compared to docetaxel treatment alone, resulting in decreased tumor growth and metastatic spread in prostate cancer (Loberg et al., 2007). This combination has also shown promise in lung cancer, breast cancer metastasis, and PDAC (Lu and Kang, 2009; Fridlender et al., 2011; Kalbasi et al., 2017). Due to these successes CSF-1, CCL2, and CSF-1R targeting agents are being investigated in clinical trials in combination with chemotherapy in a range of solid tumors (Table 1). However, the targeting of chemokines and cytokines has limitations due to their redundant and promiscuous nature. In fact, chemokines and cytokines can often bind to more than one receptor, and at the same time different cytokines/chemokines can bind to the same receptor and activate the same signaling pathway (O’Shea and Murray, 2008; Turner et al., 2014). In addition, to add more complexity, certain cytokine receptors are expressed by several cell types and as a result, inhibiting the cytokine/receptor affects all cell populations expressing the receptor. This is the case with CSF-1R which is not exclusively expressed by M2-like macrophages but is also expressed by M1-like macrophages, neutrophils, myeloid-derived suppressor cells (MDSCs) and dendritic cells (DCs; Cannarile et al., 2017).
Table 1.
Summary of combination treatments of chemotherapy and stromal targeting agents.
| Molecular target | Treatment combination | Cancer type | Clinical trial | Outcome | Reference |
|---|---|---|---|---|---|
| CSF1R | Pexidartinib (PLX3397) (αCSF-lR) + eribulin | Metastatic breast cancer | Phase 1/2 NCT01596751 | Ongoing | |
| Pexidartinib (PLX3397 αCSF-lR) + paclitaxel | Solid tumors | Phase 1 NCT01525602 | ORR: 4/23 (17%) CBR: 14/23 (61%) | Rugo et al., 2014 | |
| CSF1 | MCS110 (αCSFl) + carboplatin plus gemcitabine | Triple negative breast cancer | Phase 2 NCT02435680 | Ongoing | |
| CCL2 | CNTO888 (αCCL2) + DOXIL®/Caelyx® doxorubicin HC1 liposome injection CNTO888 + gemcitabine CNTO888 + paclitaxel and carboplatin CNTO888 + docetaxel | Solid tumors | Phase 1 NCT01204996 | Hematological complications in >93% | |
| CNTO888 + docetaxel | Metastatic resistant prostate cancer | Phase 2 NTC00992186 | 34% maintained stable disease | ||
| CD40 | Dacetuzumab + bortezomib | Relapsed or refractory multiple myeloma | Phase 1 NCT00664898 | Completed results not posted | |
| Dacetuzumab + R-ICE (rituximab, etoposide, carboplatin, ifosfamide) | Diffuse large B cell lymphoma | Phase IIb NCT00529503 | Terminated | ||
| Smo | LDE225 (αSmo) + temozolomide | Medulloblastoma | Phase 3 NCT01708174 | ORR: 18.8% | Kieran et al., 2013 |
| IGF | BI 836845 + enzalutamide | Castration-resistant Prostatic neoplasms | Phase 1 NCT02204072 | Ongoing | |
| BI 836845 + everolimus + exemestane | HR+/HER2- advanced breast cancer | Phase 1 NCT02123823 | Ongoing | ||
| MEDI-573 + aromatase inhibitor | HER-2 negative metastatic breast cancer | Phase 2 NCT01446159 | Ongoing |
Repolarizing macrophages back into an M1-like tumoricidal phenotype appears an attractive approach as the M2 TAMs are already present in the tumor and repolarization could therefore provide an effective strategy to restore the tumoricidal function of macrophages and prevent cancer progression. This has been investigated using an anti-CD40 antibody in combination with gemcitabine in a genetic KPC (Kras LSL.G12D/+; p53R172H/+; PdxCretg/+) PDAC mouse model and in PDAC patients (Beatty et al., 2011). The administration of an agonist CD40 antibody repolarized TAMs back into an M1-like phenotype leading to an increased response to gemcitabine and reduced tumor burden (Beatty et al., 2011). A phase 1 clinical trial has recently been completed for the use of Dacetuzumab (human anti-CD40 mAb) + Bortezomib chemotherapy in patients with relapsed or refractory multiple myeloma, however, results have yet to be published (Table 1).
Since TAMs can act as a double edge sword in cancer, with M1-like TAMs exerting anti-tumorigenic functions and M2-like TAMs exerting pro-tumorigenic functions, targeting TAMs pro-tumorigenic functions seems a more promising approach compared to ablation therapies targeting all TAMs. As previously mentioned, TAMs are known to facilitate the intravasation of tumor cells and promote angiogenesis (Wyckoff et al., 2007). Therefore, targeting TAMs role in pathological angiogenesis is an attractive therapeutic opportunity. In MMTV-PyMT mammary carcinomas and RIP1-Tag2 pancreatic insulinomas, an Ang-2 neutralizing antibody administration did not reduce the recruitment of Tie-2+ TAMs but instead, prevented their binding to Ang-2 on activated endothelial cells subsequently decreasing angiogenesis and tumor progression (Mazzieri et al., 2011). CSF-1R inhibition increased the efficacy of anti-VEGFR-2 anti-angiogenic therapy in a mouse model of Lewis lung carcinoma (Priceman et al., 2010). M2 TAMs produce IL-10 at the tumor site leading to resistance of breast cancer to paclitaxel treatment (Yang et al., 2015) and this resistance can be abrogated with the administration of an IL-10 neutralizing antibody (Yang et al., 2015) (Figure 4).
FIGURE 4.

Therapeutic strategies to overcome chemoresistance mediated by TAMs and CAFs. CAFs: Reprogramming activated CAFs back toward a quiescent phenotype by anti-Smoothened (Smo), anti-sonic hedgehog (Shh), all-trans retinoic acid and calcipotriol (vitamin D analog) while fibroblast growth factor 2 (FGF2) targeting agents prevents resistance of tumor cells to anti-estrogens in breast cancer. TAMs: Repolarizing M2 macrophages back to an M1-like phenotype can be mediated by a CD40 agonist. Prevention of macrophage recruitment to tumor sites is currently being achieved by targeting the colony-stimulating factor 1 (CSF-1) and C-C motif chemokine 2 (CCL2) signaling axis. Anti-angiopoietin-2 (Ang-2) antibodies prevent TAM interaction with blood vessels. IL-10 produced by TAMs promotes chemoresistance which can be abrogated by treatment with anti-IL-10 antibodies. TAMs and CAFs secrete insulin-like growth factor 1 and 2 (IGF1 and IGF2) which makes pancreatic and breast tumors chemoresistant and more metastatic. Treatment of tumors with anti-IGF blocking antibodies increases the response of pancreatic and breast tumors to chemotherapy and decreases tumor growth and metastasis.
It is currently unclear whether CAFs play a supportive or restrictive role in tumor progression. Based on the correlation between a large desmoplastic reaction and poor patient outcome it was hypothesized that ablation of the myofibroblasts would improve therapy response and decrease tumor growth. Shh is overexpressed by neoplastic PDAC cells (Thayer et al., 2003), stimulating Gli activity in surrounding fibroblasts and triggering their activation (Tian et al., 2009). Therefore, Shh became a target to inhibit fibroblast activation and Shh inhibition initially showed promising results in a pre-clinical PDAC mouse models. Shh inhibition reduced fibrosis and increased tumor vascularization, improving the delivery of gemcitabine to PDAC tumors (Olive et al., 2009). However, a clinical trial of Saridegib, a Shh inhibitor, with gemcitabine, in metastatic PDAC patients, failed at phase II as patients had reduced survival (Madden, 2012). Further investigation into fibroblast function in PDAC in longer-term experiments with mouse PDAC models showed that fibroblast ablation using smoothened inhibitor or genetic depletion of Shh or αSMA+ myofibroblasts, in fact showed that the stroma restrained tumor growth and metastasis (Oezdemir et al., 2014; Rhim et al., 2014). These conflicting results, combined with the emerging evidence that different CAF populations co-exist in tumors, suggest that different CAF populations may have different and possibly opposing effects in cancer progression (Ohlund et al., 2017; Costa et al., 2018). Despite these results, a phase 3 trial in medulloblastoma, using an oral sonidegib (smoothened inhibitor) in combination with temozolomide showed promising results with an objective response rate of 18.8% (Table 1).
One approach which warrants further investigation is the reprogramming of the activated CAFs back into their quiescent state. This approach has seen some success in PDAC mouse models using Calcipotriol (vitamin D analog) which reverts myofibroblasts to quiescence, reducing the desmoplastic reaction which in turn improves gemcitabine delivery (Sherman et al., 2014). In 3D models and genetic mouse models of PDAC the use of all-trans retinoic acid to restore the quiescence of stellate cells increased vascularity, resulting in increased response to gemcitabine and reduced tumor growth (Carapuca et al., 2016). In estrogen receptor positive breast cancer, CAF-derived FGF-2 promotes resistance to anti-estrogens which is abrogated with administration of an FGF-2 neutralizing antibody (Shee et al., 2018) (Figure 4).
As previously mentioned TAMs and CAFs act as stromal sources of IGF 1 and 2 in PDAC, and invasive breast cancer (Ireland et al., 2016, 2018) and this makes tumors resistant to chemotherapy and more metastatic. Blockade of IGF1 receptor signaling in PDAC, using IGF-1R inhibitors, has failed in the clinic (Guha, 2013; King et al., 2014; Gradishar et al., 2016) but appears to be more effective in certain tumor types such as glioblastoma (Quail et al., 2016). In PDAC and invasive breast cancer mouse models, we have shown that both Insulin and IGF1 receptors are activated, and the use of IGF1/IGF2 ligand blocking antibodies, which inhibit IGF-1 and IGF-2 signaling through both IGF-1 and insulin receptors, increases response to chemotherapy and reduces tumor growth and metastasis (Ireland et al., 2016, 2018). These studies suggest that inhibition of signaling through both Insulin and IGF1 receptors by blocking IGF 1 and 2 ligands may be more effective compared to IGF1R inhibitors in certain cancer types which have both receptors activated, such as pancreatic and breast cancer (Figure 4). IGF1/IGF2 blocking antibodies are currently being tested in phase I and II clinical trials in patients with castration resistant prostate cancer and metastatic breast cancer patients in combination with chemotherapy (Table 1).
Future Perspectives
Macrophages and fibroblasts are key regulators of tissue homeostasis, repair, angiogenesis and immunity. In tumors, cancer cells, macrophages and fibroblasts co-exist, co-evolve and continuously interact with each other. Tumor cells “hijack” macrophages and fibroblasts to support their own growth and expansion. Specifically, tumors exploit the natural plasticity of macrophages polarizing them into M2-like pro-tumorigenic TAMs that, support tumor growth in numerous ways, as described in this review. The same phenomenon is observed with respect to fibroblast function. Under normal physiological conditions fibroblasts facilitate wound repair by promoting cell growth, migration and ECM deposition. Tumor cells stimulate fibroblast activation and, reciprocally, activated fibroblasts support cancer cell survival, proliferation and resistance to therapies. However, recent findings have shown that different CAF populations with different and possibly even opposite functions co-exist in tumors.
Ablation therapies that eliminate macrophage recruitment to the tumor site have shown some promising results (DeNardo et al., 2011; Mitchem et al., 2013). However, this approach has some limitations, including the lack of specificity for different macrophage subsets and the redundancy of macrophage chemo-attractants. Inhibition of CAFs activation in PDAC patients actually resulted in enhanced tumor progression (Madden, 2012) and CAF ablation therapies in mouse tumor models resulted in increased tumor growth and metastasis (Oezdemir et al., 2014; Rhim et al., 2014). These findings suggest that further investigation into the role of different CAF subtypes is required to design therapies that specifically target defined CAF subtypes and/or functions that support cancer progression. Therapies that specifically target the pro-tumorigenic functions of TAMs and CAFs could lead to a more specific and effective anti-tumor response. To develop specific anti-stroma therapies that only target the pro-tumorigenic functions of TAMs and CAFs, while sparing their anti-tumorigenic functions, we first need to gain a better understanding of the complex composition and function of the tumor stroma.
While TAMs and CAFs are the most abundant stromal cell types in tumors, and as described in this review affect resistance to chemotherapy using a plethora of mechanisms, other stromal/immune cells present in the TME, including MDSCs, DCs, and T cells can also affect the response of tumors to therapies (for reviews/articles on this topic see Castells et al., 2012; Palucka et al., 2013; Son et al., 2017; Weber et al., 2018).
While some key stroma-derived signaling molecules have already been identified, the complex tumor-stroma interactions and the dynamic evolution of these interactions during tumor progression and in response to treatment need to be fully elucidated in order to develop effective anti-cancer therapies with a durable effect.
Author Contributions
LI and AM co-wrote this review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. AM and LI are supported by the Wellcome Trust and the Royal Society (grant no. 102521/Z/13/Z) and North West Cancer Research.
References
- Apte M. V., Haber P. S., Applegate T. L., Norton I. D., Mccaughan G. W., Korsten M. A., et al. (1998). Periacinar stellate shaped cells in rat pancreas: identification, isolation, and culture. Gut 43 128–133. 10.1136/gut.43.1.128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apte M. V., Haber P. S., Darby S. J., Rodgers S. C., Mccaughan G. W., Korsten M. A., et al. (1999). Pancreatic stellate cells are activated by proinflammatory cytokines: implications for pancreatic fibrogenesis. Gut 44 534–541. 10.1136/gut.44.4.534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold T., Betsholtz C. (2013). The importance of microglia in the development of the vasculature in the central nervous system. Vasc. Cell 5:4. 10.1186/2045-824X-5-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asahina K., Tsai S. Y., Li P., Ishii M., Maxson R. E., Sucov H. M., et al. (2009). Mesenchymal origin of hepatic stellate cells, submesothelial cells, and perivascular mesenchymal cells during mouse liver development. Hepatology 49 998–1011. 10.1002/hep.22721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asahina K., Zhou B., Pu W. T., Tsukamoto H. (2011). Septum transversum-derived mesothelium gives rise to hepatic stellate cells and perivascular mesenchymal cells in developing mouse liver. Hepatology 53 983–995. 10.1002/hep.24119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachem M. G., Schneider E., Gross H., Weidenbach H., Schmid R. M., Menke A., et al. (1998). Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology 115 421–432. 10.1016/S0016-5085(98)70209-4 [DOI] [PubMed] [Google Scholar]
- Bailey J. M., Swanson B. J., Hamada T., Eggers J. P., Singh P. K., Caffery T., et al. (2008). Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin. Cancer Res. 14 5995–6004. 10.1158/1078-0432.CCR-08-0291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty G. L., Chiorean E. G., Fishman M. P., Saboury B., Teitelbaum U. R., Sun W. J., et al. (2011). Cd40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 331 1612–1616. 10.1126/science.1198443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedetti-Panici P., Bermudez A., Blake P., Cardenas J., Chang T. C., Chiara S., et al. (2003). Neoadjuvant chemotherapy for locally advanced cervical cancer: a systematic review and meta-analysis of individual patient data from 21 randomised trials. Eur. J. Cancer 39 2470–2486. 10.1016/S0959-8049(03)00425-8 [DOI] [PubMed] [Google Scholar]
- Bergers G., Benjamin L. E. (2003). Tumorigenesis and the angiogenic switch. Nat. Rev. Cancer 3 401–410. 10.1038/nrc1093 [DOI] [PubMed] [Google Scholar]
- Bergers G., Brekken R., Mcmahon G., Vu T. H., Itoh T., Tamaki K., et al. (2000). Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell Biol. 2 737–744. 10.1038/35036374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas S. K., Allavena P., Mantovani A. (2013). Tumor-associated macrophages: functional diversity, clinical significance, and open questions. Semin. Immunopathol. 35 585–600. 10.1007/s00281-013-0367-7 [DOI] [PubMed] [Google Scholar]
- Biswas S. K., Mantovani A. (2010). Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat. Immunol. 11 889–896. 10.1038/ni.1937 [DOI] [PubMed] [Google Scholar]
- Blaner W. S., O’byrne S. M., Wongsiriroj N., Kluwe J., D’ambrosio D. M., Jiang H. F., et al. (2009). Hepatic stellate cell lipid droplets: a specialized lipid droplet for retinoid storage. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 1791 467–473. 10.1016/j.bbalip.2008.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonde A. K., Tischler V., Kumar S., Soltermann A., Schwendener R. A. (2012). Intratumoral macrophages contribute to epithelial-mesenchymal transition in solid tumors. BMC Cancer 12:35. 10.1186/1471-2407-12-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brechbuhl H. M., Finlay-Schultz J., Yamamoto T. M., Gillen A. E., Cittelly D. M., Tan A. C., et al. (2017). Fibroblast subtypes regulate responsiveness of luminal breast cancer to estrogen. Clin. Cancer Res. 23 1710–1721. 10.1158/1078-0432.CCR-15-2851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burris H. A., Moore M. J., Andersen J., Green M. R., Rothenberg M. L., Madiano M. R., et al. (1997). Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J. Clin. Oncol. 15 2403–2413. 10.1200/JCO.1997.15.6.2403 [DOI] [PubMed] [Google Scholar]
- Cannarile M. A., Weisser M., Jacob W., Jegg A. M., Ries C. H., Ruttinger D. (2017). Colony-stimulating factor 1 receptor (Csf1R) inhibitors in cancer therapy. J. Immunother. Cancer 5:4. 10.1186/s40425-017-0257-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carapuca E. F., Gemenetzidis E., Feig C., Bapiro T. E., Williams M. D., Wilson A. S., et al. (2016). Anti-stromal treatment together with chemotherapy targets multiple signalling pathways in pancreatic adenocarcinoma. J. Pathol. 239 286–296. 10.1002/path.4727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casazza A., Laoui D., Wenes M., Rizzolio S., Bassani N., Mambretti M., et al. (2013). Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell 24 695–709. 10.1016/j.ccr.2013.11.007 [DOI] [PubMed] [Google Scholar]
- Castells M., Thibault B., Delord J. P., Couderc B. (2012). Implication of tumor microenvironment in chemoresistance: tumor-associated stromal cells protect tumor cells from cell death. Int. J. Mol. Sci. 13 9545–9571. 10.3390/ijms13089545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabner B. A., Roberts T. G. (2005). Timeline – Chemotherapy and the war on cancer. Nat. Rev. Cancer 5 65–72. 10.1038/nrc1529 [DOI] [PubMed] [Google Scholar]
- Chang H. Y., Chi J. T., Dudoit S., Bondre C., Van De Rijn M., Botstein D., et al. (2002). Diversity, topographic differentiation, and positional memory in human fibroblasts. Proc. Natl. Acad. Sci. U.S.A. 99 12877–12882. 10.1073/pnas.162488599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colegio O. R., Chu N. Q., Szabo A. L., Chu T., Rhebergen A. M., Jairam V., et al. (2014). Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513 559–563. 10.1038/nature13490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condeelis J., Pollard J. W. (2006). Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell 124 263–266. 10.1016/j.cell.2006.01.007 [DOI] [PubMed] [Google Scholar]
- Coniglio S. J., Eugenin E., Dobrenis K., Stanley E. R., West B. L., Symons M. H., et al. (2012). Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (Egfr) and colony stimulating factor 1 receptor (Csf-1R) signaling. Mol. Med. 18 519–527. 10.2119/molmed.2011.00217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa A., Kieffer Y., Scholer-Dahirel A., Pelon F., Bourachot B., Cardon M., et al. (2018). Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell 33 463.e10–479.e10. 10.1016/j.ccell.2018.01.011 [DOI] [PubMed] [Google Scholar]
- Costea D. E., Hills A., Osman A. H., Thurlow J., Kalna G., Huang X. H., et al. (2013). Identification of two distinct carcinoma-associated fibroblast subtypes with differential tumor-promoting abilities in oral squamous cell carcinoma. Cancer Res. 73 3888–3901. 10.1158/0008-5472.CAN-12-4150 [DOI] [PubMed] [Google Scholar]
- Dal-Secco D., Wang J., Zeng Z. T., Kolaczkowska E., Wong C. H. Y., Petri B., et al. (2015). A dynamic spectrum of monocytes arising from the in situ reprogramming of Ccr2( + ) monocytes at a site of sterile injury. J. Exp. Med. 212 447–456. 10.1084/jem.20141539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darby I. A., Hewitson T. D. (2007). Fibroblast differentiation in wound healing and fibrosis. Int. Rev. Cytol. 257 257 143–179. 10.1016/S0074-7696(07)57004-X [DOI] [PubMed] [Google Scholar]
- Davies L. C., Jenkins S. J., Allen J. E., Taylor P. R. (2013a). Tissue-resident macrophages. Nat. Immunol. 14 986–995. 10.1038/ni.2705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies L. C., Rosas M., Jenkins S. J., Liao C. T., Scurr M. J., Brombacher F., et al. (2013b). Distinct bone marrow-derived and tissue-resident macrophage lineages proliferate at key stages during inflammation. Nat. Commun. 4:1886. 10.1038/ncomms2877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Palma M., Lewis C. E. (2013). Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell 23 277–286. 10.1016/j.ccr.2013.02.013 [DOI] [PubMed] [Google Scholar]
- De Palma M., Venneri M. A., Galli R., Sergi L. S., Politi L. S., Sampaolesi M., et al. (2005). Tie2 identifies a hematopoietic monocytes required for tumor lineage of proangiogenic vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell 8 211–226. 10.1016/j.ccr.2005.08.002 [DOI] [PubMed] [Google Scholar]
- DeNardo D. G., Brennan D. J., Rexhepaj E., Ruffell B., Shiao S. L., Madden S. F., et al. (2011). Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 1 54–67. 10.1158/2159-8274.CD-10-0028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuFort C. C., Delgiorno K. E., Hingorani S. R. (2016). Mounting pressure in the microenvironment: fluids, solids, and cells in pancreatic ductal adenocarcinoma. Gastroenterology, 150 1545.e2–1557.e2. 10.1053/j.gastro.2016.03.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvorak H. F., Flier J., Frank H. (1986). Tumors – Wounds that do not heal – Similarities between tumor stroma generation and wound-healing. N. Engl. J. Med. 315 1650–1659. 10.1056/NEJM198612253152606 [DOI] [PubMed] [Google Scholar]
- Eguchi T., Kodera Y., Nakanishi H., Yokoyama H., Ohashi N., Ito Y., et al. (2008). The effect of chemotherapy against micrometastases and isolated tumor cells in lymph nodes: an in vivo study. In Vivo 22707–712. [PubMed] [Google Scholar]
- Erkan M., Weis N., Pan Z., Schwager C., Samkharadze T., Jiang X., et al. (2010). Organ-, inflammation- and cancer specific transcriptional fingerprints of pancreatic and hepatic stellate cells. Mol. Cancer 9:88. 10.1186/1476-4598-9-88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantin A., Vieira J. M., Gestri G., Denti L., Schwarz Q., Prykhozhij S., et al. (2010). Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of Vegf-mediated endothelial tip cell induction. Blood 116829–840. 10.1182/blood-2009-12-257832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer P., Bonnefoi H., Anderle P., Cameron D., Wirapati P., Becette V., et al. (2009). A stroma-related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer (vol 15, pg 68, 2009). Nat. Med. 15 220–220. 10.1038/nm0209-220a [DOI] [PubMed] [Google Scholar]
- Forget M. A., Voorhees J. L., Cole S. L., Dakhlallah D., Patterson I. L., Gross A. C., et al. (2014). Macrophage colony-stimulating factor augments Tie2-expressing monocyte differentiation, angiogenic function, and recruitment in a mouse model of breast cancer. PLoS One 9:e98623. 10.1371/journal.pone.0098623 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Fridlender Z. G., Kapoor V., Buchlis G., Cheng G., Sun J., Wang L. C. S., et al. (2011). Monocyte chemoattractant protein-1 blockade inhibits lung cancer tumor growth by altering macrophage phenotype and activating Cd8( + ) cells. Am. J. Respir. Cell Mol. Biol. 44 230–237. 10.1165/rcmb.2010-0080OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman S. L. (2000). Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J. Biol. Chem. 275 2247–2250. 10.1074/jbc.275.4.2247 [DOI] [PubMed] [Google Scholar]
- Fukumura D., Xavier R., Sugiura T., Chen Y., Park E. C., Lu N. F., et al. (1998). Tumor induction of Vegf promoter activity in stromal cells. Cell 94 715–725. 10.1016/S0092-8674(00)81731-6 [DOI] [PubMed] [Google Scholar]
- Gebski V., Burmeister B., Smithers B. M., Foo K., Zalcberg J., Simes J., et al. (2007). Survival benefits from neoadjuvant chemoradiotherapy or chemotherapy in oesophageal carcinoma: a meta-analysis. Lancet Oncol. 8 226–234. 10.1016/S1470-2045(07)70039-6 [DOI] [PubMed] [Google Scholar]
- Gibbings S. L., Goyal R., Desch A. N., Leach S. M., Prabagar M., Atif S. M., et al. (2015). Transcriptome analysis highlights the conserved difference between embryonic and postnatal-derived alveolar macrophages. Blood 126 1357–1366. 10.1182/blood-2015-01-624809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman A. (1963). The initial clinical trial of nitrogen mustard. Am. J. Surg. 105 574–578. 10.1016/0002-9610(63)90232-0 [DOI] [PubMed] [Google Scholar]
- Gocheva V., Wang H. W., Gadea B. B., Shree T., Hunter K. E., Garfall A. L., et al. (2010). Il-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev. 24 241–255. 10.1101/gad.1874010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gocheva V., Zeng W., Ke D. X., Klimstra D., Reinheckel T., Peters C., et al. (2006). Distinct roles for cysteine cathepsin genes in multistage tumorigenesis. Genes Dev. 20 543–556. 10.1101/gad.1407406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S., Martinez F. O. (2010). Alternative activation of macrophages: mechanism and functions. Immunity 32 593–604. 10.1016/j.immuni.2010.05.007 [DOI] [PubMed] [Google Scholar]
- Gordy C., Pua H., Sempowski G. D., He Y. W. (2011). Regulation of steady-state neutrophil homeostasis by macrophages. Blood 117 618–629. 10.1182/blood-2010-01-265959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami S., Sahai E., Wyckoff J. B., Cammer N., Cox D., Pixley F. J., et al. (2005). Macrophages promote the invasion of breast carcinoma cells via a colony-stimulating factor-1/epidermal growth factor paracrine loop. Cancer Res. 65 5278–5283. 10.1158/0008-5472.CAN-04-1853 [DOI] [PubMed] [Google Scholar]
- Gradishar W. J., Yardley D. A., Layman R., Sparano J. A., Chuang E., Northfelt D. W., et al. (2016). Clinical and translational results of a phase Ii, randomized trial of an anti-Igf-1R (cixutumumab) in women with breast cancer that progressed on endocrine therapy. Clin. Cancer Res. 22 301–309. 10.1158/1078-0432.CCR-15-0588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov S. I., Greten F. R., Karin M. (2010). Immunity, inflammation, and cancer. Cell 140 883–899. 10.1016/j.cell.2010.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guha M. (2013). Anticancer Igf1R classes take more knocks. Nat. Rev. Drug Discov. 12:250. 10.1038/nrd3992 [DOI] [PubMed] [Google Scholar]
- Haddow A., Kon G. A. R., Ross W. C. J. (1948). Effects upon tumours of various haloalkylarylamines. Nature 162 824–825. 10.1038/162824a0 [DOI] [PubMed] [Google Scholar]
- Hanahan D., Coussens L. M. (2012). Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 21 309–322. 10.1016/j.ccr.2012.02.022 [DOI] [PubMed] [Google Scholar]
- Hanahan D., Weinberg R. A. (2000). The hallmarks of cancer. Cell 100 57–70. 10.1016/S0092-8674(00)81683-9 [DOI] [PubMed] [Google Scholar]
- Hanahan D., Weinberg R. A. (2011). Hallmarks of cancer: the next generation. Cell 144 646–674. 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- Hayes D. F., Schott A. F. (2015). Neoadjuvant chemotherapy: what are the benefits for the patient and for the investigator? J. Natl. Cancer Inst. Monogr. 2015 36–39. 10.1093/jncimonographs/lgv004 [DOI] [PubMed] [Google Scholar]
- Housman G., Byler S., Heerboth S., Lapinska K., Longacre M., Snyder N., et al. (2014). Drug resistance in cancer: an overview. Cancers 6 1769–1792. 10.3390/cancers6031769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ireland L., Santos A., Ahmed M. S., Rainer C., Nielsen S. R., Quaranta V., et al. (2016). Chemoresistance in pancreatic cancer is driven by stroma-derived insulin-like growth factors. Cancer Res. 76 6851–6863. 10.1158/0008-5472.CAN-16-1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ireland L., Santos A., Campbell F., Figueiredo C., Hammond D., Ellies L. G., et al. (2018). Blockade of insulin-like growth factors increases efficacy of paclitaxel in metastatic breast cancer. Oncogene 37 2022–2036. 10.1038/s41388-017-0115-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins S. J., Ruckerl D., Cook P. C., Jones L. H., Finkelman F. D., Van Rooijen N., et al. (2011). Local macrophage proliferation, rather than recruitment from the blood, is a signature of T(H)2 inflammation. Science 332 1284–1288. 10.1126/science.1204351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins S. J., Ruckerl D., Thomas G. D., Hewitson J. P., Duncan S., Brombacher F., et al. (2013). Il-4 directly signals tissue-resident macrophages to proliferate beyond homeostatic levels controlled by Csf-1. J. Exp. Med. 210 2477–2491. 10.1084/jem.20121999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H., Hegde S., Denardo D. G. (2017). Tumor-associated fibrosis as a regulator of tumor immunity and response to immunotherapy. Cancer Immunol. Immunother. 66 1037–1048. 10.1007/s00262-017-2003-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce J. A., Pollard J. W. (2009). Microenvironmental regulation of metastasis. Nat. Rev. Cancer 9 239–252. 10.1038/nrc2618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalbasi A., Komar C., Tooker G. M., Liu M. G., Lee J. W., Gladney W. L., et al. (2017). Tumor-derived Ccl2 mediates resistance to radiotherapy in pancreatic ductal adenocarcinoma. Clin. Cancer Res. 23 137–148. 10.1158/1078-0432.CCR-16-0870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R. (2003). Basement membranes: structure, assembly and role in tumour angiogenesis. Nat. Rev. Cancer 3 422–433. 10.1038/nrc1094 [DOI] [PubMed] [Google Scholar]
- Kalluri R. (2016). The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 16 582–598. 10.1038/nrc.2016.73 [DOI] [PubMed] [Google Scholar]
- Kalluri R., Zeisberg M. (2006). Fibroblasts in cancer. Nat. Rev. Cancer 6 392–401. 10.1038/nrc1877 [DOI] [PubMed] [Google Scholar]
- Kayo H., Kiga K., Fukuda-Yuzawa Y., Hedlund S., Murakami K., De La Rosa-Velazquez I. A., et al. (2014). miR-212 and miR-132 are dispensable for mouse mammary gland development. Nat. Genet. 46 802–804. 10.1038/ng.2990 [DOI] [PubMed] [Google Scholar]
- Keane M. P., Strieter R. M., Belperio J. A. (2005). Mechanisms and mediators of pulmonary fibrosis. Crit. Rev. Immunol. 25 429–463. 10.1615/CritRevImmunol.v25.i6.10 [DOI] [PubMed] [Google Scholar]
- Khalil N., Bereznay O., Sporn M., Greenberg A. H. (1989). Macrophage production of transforming growth factor-beta and fibroblast collagen-synthesis in chronic pulmonary inflammation. J. Exp. Med. 170 727–737. 10.1084/jem.170.3.727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieran M., Hargrave D., Wen P. Y., Goldman S., Amakye D., Patton M., et al. (2013). A phase 3, multicenter, open-label, randomized, controlled study of the efficacy and safety of oral sonidegib (Lde225) versus temozolomide in patients with hedgehog pathway-activated relapsed medulloblastoma. Neuro-Oncol. 15 115–115. [Google Scholar]
- King H., Aleksic T., Haluska P., Macaulay V. M. (2014). Can we unlock the potential of Igf-1R inhibition in cancer therapy? Cancer Treat. Rev. 40 1096–1105. 10.1016/j.ctrv.2014.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klastersky J., Paesmans M. (2001). Response to chemotherapy, quality of life benefits and survival in advanced non-small cell lung cancer: review of literature results. Lung Cancer 34 S95–S101. 10.1016/S0169-5002(01)00383-X [DOI] [PubMed] [Google Scholar]
- Klei T. R. L., Meinderts S. M., Van Den Berg T. K., Van Bruggen R. (2017). From the cradle to the grave: the role of macrophages in erythropoiesis and erythrophagocytosis. Front. Immunol. 8:73. 10.3389/fimmu.2017.00073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBleu V. S., Taduri G., O’connell J., Teng Y. Q., Cooke V. G., Woda C., et al. (2013). Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 19 1047–1054. 10.1038/nm.3218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linde N., Lederle W., Depner S., Van Rooijen N., Gutschalk C. M., Mueller M. M. (2012). Vascular endothelial growth factor-induced skin carcinogenesis depends on recruitment and alternative activation of macrophages. J. Pathol. 227 17–28. 10.1002/path.3989 [DOI] [PubMed] [Google Scholar]
- Liu Y. H. (2006). Renal fibrosis: new insights into the pathogenesis and therapeutics. Kid. Int. 69 213–217. 10.1038/sj.ki.5000054 [DOI] [PubMed] [Google Scholar]
- Loberg R. D., Ying C., Craig M., Day L. L., Sargent E., Neeley C., et al. (2007). Targeting Ccl2 with systemic delivery of neutralizing antibodies induces prostate cancer tumor regression in vivo. Cancer Res. 67 9417–9424. 10.1158/0008-5472.CAN-07-1286 [DOI] [PubMed] [Google Scholar]
- Lobov I. B., Rao S., Carroll T. J., Vallance J. E., Ito M., Ondr J. K., et al. (2005). Wnt7b mediates macrophage-induced programmed cell death in patterning of the vasculature. Nature 437 417–421. 10.1038/nature03928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X., Kang Y. B. (2009). Chemokine (C-C Motif) ligand 2 engages Ccr2( + ) stromal cells of monocytic origin to promote breast cancer metastasis to lung and bone. J. Biol. Chem. 284 29087–29096. 10.1074/jbc.M109.035899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madden J. I. (2012). Infinity Reports Update from Phase 2 Study of Saridegib Plus Gemcitabine in Patients with Metastatic Pancreatic Cancer. Available at: https://www.businesswire.com/news/home/20120127005146/en/Infinity-Reports-Update-Phase-2-Study-Saridegib [Google Scholar]
- Mantovani A., Sica A. (2010). Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr. Opin. Immunol. 22 231–237. 10.1016/j.coi.2010.01.009 [DOI] [PubMed] [Google Scholar]
- Marquette C., Nabell L. (2012). Chemotherapy-resistant metastatic breast cancer. Curr. Treat. Options. Oncol. 13 263–275. 10.1007/s11864-012-0184-6 [DOI] [PubMed] [Google Scholar]
- Marsh T., Pietras K., Mcallister S. S. (2013). Fibroblasts as architects of cancer pathogenesis. Biochim. Biophys. Acta-Mol. Basis Dis. 1832 1070–1078. 10.1016/j.bbadis.2012.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mass E., Ballesteros I., Farlik M., Halbritter F., Gunther P., Crozet L., et al. (2016). Specification of tissue-resident macrophages during organogenesis. Science 353:aaf4238. 10.1126/science.aaf4238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J., Obenauf A. C. (2016). Metastatic colonization by circulating tumour cells. Nature 529 298–306. 10.1038/nature17038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzieri R., Pucci F., Moi D., Zonari E., Ranghetti A., Berti A., et al. (2011). Targeting the ang2/Tie2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer Cell 19 512–526. 10.1016/j.ccr.2011.02.005 [DOI] [PubMed] [Google Scholar]
- Mielgo A., Schmid M. C. (2013). Impact of tumour associated macrophages in pancreatic cancer. BMB Rep. 46 131–138. 10.5483/BMBRep.2013.46.3.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills C. D. (2012). M1 and M2 macrophages: oracles of health and disease. Crit. Rev. Immunol. 32 463–488. 10.1615/CritRevImmunol.v32.i6.10 [DOI] [PubMed] [Google Scholar]
- Mitchem J. B., Brennan D. J., Knolhoff B. L., Belt B. A., Zhu Y., Sanford D. E., et al. (2013). Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 73 1128–1141. 10.1158/0008-5472.CAN-12-2731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray P. J., Wynn T. A. (2011). Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 11 723–737. 10.1038/nri3073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakasone E. S., Askautrud H. A., Kees T., Park J. H., Plaks V., Ewald A. J., et al. (2012). Imaging tumor-stroma interactions during chemotherapy reveals contributions of the microenvironment to resistance. Cancer Cell 21 488–503. 10.1016/j.ccr.2012.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navegantes K. C., Gomes R. D., Pereira P. A. T., Czaikoski P. G., Azevedo C. H. M., Monteiro M. C. (2017). Immune modulation of some autoimmune diseases: the critical role of macrophages and neutrophils in the innate and adaptive immunity. J. Trans. Med. 15:36. 10.1186/s12967-017-1141-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen S. R., Quaranta V., Linford A., Emeagi P., Rainer C., Santos A., et al. (2016). Macrophage-secreted granulin supports pancreatic cancer metastasis by inducing liver fibrosis. Nat. Cell Biol. 18 549–560. 10.1038/ncb3340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oezdemir B. C., Pentcheva-Hoang T., Carstens J. L., Zheng X., Wu C.-C., Simpson T. R., et al. (2014). Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 25 719–734. 10.1016/j.ccr.2014.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlund D., Handly-Santana A., Biffi G., Elyada E., Almeida A. S., Ponz-Sarvise M., et al. (2017). Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 214 579–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive K. P., Jacobetz M. A., Davidson C. J., Gopinathan A., Mcintyre D., Honess D., et al. (2009). Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 324 1457–1461. 10.1126/science.1171362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orkin S. H., Zon L. I. (2008). Hematopoiesis: an evolving paradigm for stem cell biology. Cell 132 631–644. 10.1016/j.cell.2008.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Shea J. J., Murray P. J. (2008). Cytokine signaling modules in inflammatory responses. Immunity 28 477–487. 10.1016/j.immuni.2008.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Outtz H. H., Tattersall I. W., Kofler N. M., Steinbach N., Kitajewski J. (2011). Notch1 controls macrophage recruitment and Notch signaling is activated at sites of endothelial cell anastomosis during retinal angiogenesis in mice. Blood 118 3436–3439. 10.1182/blood-2010-12-327015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palucka K., Coussens L. M., O’shaughnessy J. (2013). Dendritic cells, inflammation, and breast cancer. Cancer J. 19 511–516. 10.1097/PPO.0000000000000007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandol S., Edderkaoui M., Gukovsky I., Lugea A., Gukovskaya A. (2009). Desmoplasia of pancreatic ductal adenocarcinoma. Clin. Gastroenterol. Hepatol. 7 S44–S47. 10.1016/j.cgh.2009.07.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulus P., Stanley E. R., Schafer R., Abraham D., Aharinejad S. (2006). Colony-stimulating factor-1 antibody reverses chemoresistance in human Mcf-7 breast cancer xenografts. Cancer Res. 66 4349–4356. 10.1158/0008-5472.CAN-05-3523 [DOI] [PubMed] [Google Scholar]
- Pollina E. A., Legesse-Miller A., Haley E. M., Goodpaster T., Randolph-Habecker J., Coller H. A. (2008). Regulating the angiogenic balance in tissues – A potential role for the proliferative state of fibroblasts. Cell Cycle 7 2056–2070. 10.4161/cc.7.13.6240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priceman S. J., Sung J. L., Shaposhnik Z., Burton J. B., Torres-Collado A. X., Moughon D. L., et al. (2010). Targeting distinct tumor-infiltrating myeloid cells by inhibiting Csf-1 receptor: combating tumor evasion of antiangiogenic therapy. Blood 115 1461–1471. 10.1182/blood-2009-08-237412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian B. Z., Pollard J. W. (2010). Macrophage diversity enhances tumor progression and metastasis. Cell 141 39–51. 10.1016/j.cell.2010.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao Y., Zhang C., Li A., Wang D., Luo Z., Ping Y., et al. (2018). Il6 derived from cancer-associated fibroblasts promotes chemoresistance via Cxcr7 in esophageal squamous cell carcinoma. Oncogene 37 873–883. 10.1038/onc.2017.387 [DOI] [PubMed] [Google Scholar]
- Quail D. F., Bowman R. L., Akkari L., Quick M. L., Schuhmacher A. J., Huse J. T., et al. (2016). The tumor microenvironment underlies acquired resistance to Csf-1R inhibition in gliomas. Science 352:aad3018. 10.1126/science.aad3018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail D. F., Joyce J. A. (2013). Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 19 1423–1437. 10.1038/nm.3394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhim A. D., Oberstein P. E., Thomas D. H., Mirek E. T., Palermo C. F., Sastra S. A., et al. (2014). Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 25 735–747. 10.1016/j.ccr.2014.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock J. R., Barkauskas C. E., Cronce M. J., Xue Y., Harris J. R., Liang J. R., et al. (2011). Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc. Natl. Acad. Sci. U.S.A. 108 E1475–E1483. 10.1073/pnas.1117988108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronnovjessen L., Petersen O. W. (1993). Induction of alpha-smooth muscle actin by transforming growth-factor-beta-1 in quiescent human breast gland fibroblasts – Implications for myofibroblast generation in breast neoplasia. Labor. Investig. 68 696–707. [PubMed] [Google Scholar]
- Ruffell B., Affara N. I., Coussens L. M. (2012). Differential macrophage programming in the tumor microenvironment. Trends Immunol. 33 119–126. 10.1016/j.it.2011.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rugo H. S., Sharma N., Reebel L., Rodal M. B., Peck A., West B. L., et al. (2014). Phase Ib study of Plx3397, a Csf1R inhibitor, and paclitaxel in patients with advanced solid tumors. Ann. Oncol. 25 S3127–S3127. 10.1200/jco.2014.32.15_suppl.tps3127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sappino A. P., Skalli O., Jackson B., Schurch W., Gabbiani G. (1988). Smooth-muscle differentiation in stromal cells of malignant and non-malignant breast tissues. Int. J. Cancer 41 707–712. 10.1002/ijc.2910410512 [DOI] [PubMed] [Google Scholar]
- Savill J., Dransfield I., Gregory C., Haslett C. (2002). A blast from the past: clearance of apoptotic cells regulates immune responses. Nat. Rev. Immunol. 2 965–975. 10.1038/nri957 [DOI] [PubMed] [Google Scholar]
- Shee K., Yang W., Hinds J. W., Hampsch R. A., Varn F. S., Traphagen N. A., et al. (2018). Therapeutically targeting tumor microenvironment–mediated drug resistance in estrogen receptor–positive breast cancer. J. Exp. Med. 215 895–910. 10.1084/jem.20171818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman M. H., Yu R. T., Engle D. D., Ding N., Atkins A. R., Tiriac H., et al. (2014). Vitamin d receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 159 80–93. 10.1016/j.cell.2014.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C., Pamer E. G. (2011). Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 11 762–774. 10.1038/nri3070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintani Y., Fujiwara A., Kimura T., Kawamura T., Funaki S., Minami M., et al. (2016). Il-6 secreted from cancer-associated fibroblasts mediates chemoresistance in nsclc by increasing epithelial-mesenchymal transition signaling. J. Thor. Oncol. 11 1482–1492. 10.1016/j.jtho.2016.05.025 [DOI] [PubMed] [Google Scholar]
- Shree T., Olson O. C., Elie B. T., Kester J. C., Garfall A. L., Simpson K., et al. (2011). Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes Dev. 25 2465–2479. 10.1101/gad.180331.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sindrilaru A., Peters T., Wieschalka S., Baican C., Baican A., Peter H., et al. (2011). An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J. Clin. Investig. 121 985–997. 10.1172/JCI44490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son B., Lee S., Youn H., Kim E., Kim W., Youn B. (2017). The role of tumor microenvironment in therapeutic resistance. Oncotarget 8 3933–3945. 10.18632/oncotarget.13907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockmann C., Doedens A., Weidemann A., Zhang N., Takeda N., Greenberg J. I., et al. (2008). Deletion of vascular endothelial growth factor in myeloid cells accelerates tumorigenesis. Nature 456 814–818. 10.1038/nature07445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strutz F., Okada H., Lo C. W., Danoff T., Carone R. L., Tomaszewski J. E., et al. (1995). Identification and characterization of a fibroblast marker - Fsp1. J. Cell Biol. 130 393–405. 10.1083/jcb.130.2.393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto H., Mundel T. M., Kieran M. W., Kalluri R. (2006). Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol. Ther. 5 1640–1646. 10.4161/cbt.5.12.3354 [DOI] [PubMed] [Google Scholar]
- Tampe B., Zeisberg M. (2014). Contribution of genetics and epigenetics to progression of kidney fibrosis. Nephrol. Dialysis Transpl. 29 72–79. 10.1093/ndt/gft025 [DOI] [PubMed] [Google Scholar]
- Tarin D., Croft C. B. (1969). Ultrastructural features of wound healing in mouse skin. J. Anat. 105 189–190. [PubMed] [Google Scholar]
- Thayer S. P., Di Magliano M. P., Heiser P. W., Nielsen C. M., Roberts D. J., Lauwers G. Y., et al. (2003). Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 425 851–856. 10.1038/nature02009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian H., Callahan C. A., Dupree K. J., Darbonne W. C., Ahn C. P., Scales S. J., et al. (2009). Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc. Natl. Acad. Sci. U.S.A. 106 4254–4259. 10.1073/pnas.0813203106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasek J. J., Gabbiani G., Hinz B., Chaponnier C., Brown R. A. (2002). Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 3 349–363. 10.1038/nrm809 [DOI] [PubMed] [Google Scholar]
- Turner M. D., Nedjai B., Hurst T., Pennington D. J. (2014). Cytokines and chemokines: at the crossroads of cell signalling and inflammatory disease. Biochim. Biophys. Acta-Mol. Cell Res. 1843 2563–2582. 10.1016/j.bbamcr.2014.05.014 [DOI] [PubMed] [Google Scholar]
- Ucar A., Erikci E., Ucar O., Chowdhury K. (2014). miR-212 and miR-132 are dispensable for mouse mammary gland development reply. Nat. Genet. 46 804–805. 10.1038/ng.3032 [DOI] [PubMed] [Google Scholar]
- Ucar A., Vafaizadeh V., Jarry H., Fiedler J., Klemmt P. A. B., Thum T., et al. (2010). miR-212 and miR-132 are required for epithelial stromal interactions necessary for mouse mammary gland development. Nat. Genet. 42 1101–1108. 10.1038/ng.709 [DOI] [PubMed] [Google Scholar]
- van de Laar L., Saelens W., De Prijck S., Martens L., Scott C. L., Van Isterdael G., et al. (2016). Yolk sac macrophages, fetal liver, and adult monocytes can colonize an empty niche and develop into functional tissue-resident macrophages. Immunity 44 755–768. 10.1016/j.immuni.2016.02.017 [DOI] [PubMed] [Google Scholar]
- Virchow R. (1858). Die Cellularpathologie in ihrer Begrundung auf Physiologische und Pathologische Gewebelehre. Berlin: August Hirschwald. [PubMed] [Google Scholar]
- Wake K., Sato T. (1993). Intralobular heterogeneity of perisinusoidal stellate cells in porcine liver. Cell Tissue Res. 273 227–237. 10.1007/BF00312824 [DOI] [PubMed] [Google Scholar]
- Weber R., Fleming V., Hu X. Y., Nagibin V., Groth C., Altevogt P., et al. (2018). Myeloid-derived suppressor cells hinder the anti-cancer activity of immune checkpoint inhibitors. Front. Immunol. 9:1310. 10.3389/fimmu.2018.01310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenes M., Shang M., Di Matteo M., Goveia J., Martin-Perez R., Serneels J., et al. (2016). Macrophage metabolism controls tumor blood vessel morphogenesis and metastasis. Cell Metab. 24 701–715. 10.1016/j.cmet.2016.09.008 [DOI] [PubMed] [Google Scholar]
- Wyckoff J. B., Wang Y., Lin E. Y., Li J. F., Goswami S., Stanley E. R., et al. (2007). Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. 67 2649–2656. 10.1158/0008-5472.CAN-06-1823 [DOI] [PubMed] [Google Scholar]
- Wynn T. A., Chawla A., Pollard J. W. (2013). Macrophage biology in development, homeostasis and disease. Nature 496 445–455. 10.1038/nature12034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C. X., He L. Y., He P. Q., Liu Y. W., Wang W. J., He Y. Q., et al. (2015). Increased drug resistance in breast cancer by tumor-associated macrophages through Il-10/Stat3/bcl-2 signaling pathway. Med. Oncol. 32:352. 10.1007/s12032-014-0352-6 [DOI] [PubMed] [Google Scholar]
- Yauch R. L., Gould S. E., Scales S. J., Tang T., Tian H., Ahn C. P., et al. (2008). A paracrine requirement for hedgehog signalling in cancer. Nature 455 406–410. 10.1038/nature07275 [DOI] [PubMed] [Google Scholar]
- Yin Y., Yao S., Hu Y., Feng Y., Li M., Bian Z., et al. (2017). The immune-microenvironment confers chemoresistance of colorectal cancer through macrophage-derived Il6. Clin. Cancer Res. 23 7375–7387. 10.1158/1078-0432.CCR-17-1283 [DOI] [PubMed] [Google Scholar]
- Yona S., Kim K. W., Wolf Y., Mildner A., Varol D., Breker M., et al. (2013). Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38 79–91. 10.1016/j.immuni.2012.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahreddine H., Borden K. L. B. (2013). Mechanisms and insights into drug resistance in cancer. Front. Pharmacol. 4:28 10.3389/fphar.2013.00028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisberg E. M., Zeisberg M. (2013). The role of promoter hypermethylation in fibroblast activation and fibrogenesis. J. Pathol. 229 264–273. 10.1002/path.4120 [DOI] [PubMed] [Google Scholar]
- Zhang H. F., Xie C. H., Yue J., Jiang Z. Z., Zhou R. J., Xie R. F., et al. (2017). Cancer-associated fibroblasts mediated chemoresistance by a Foxo1/Tgf1 signaling loop in esophageal squamous cell carcinoma. Mol. Carcinog. 56 1150–1163. 10.1002/mc.22581 [DOI] [PubMed] [Google Scholar]
- Zheng H. C. (2017). The molecular mechanisms of chemoresistance in cancers. Oncotarget 8 59950–59964. 10.18632/oncotarget.19048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y., Herndon J. M., Sojka D. K., Kim K. W., Knolhoff B. L., Zuo C., et al. (2017). Tissue-resident macrophages in pancreatic ductal adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity 47 323–338. 10.1016/j.immuni.2017.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]