

HCMV infects nearly 80% of the world's population and causes significant morbidity and mortality. The current antiviral agents used to treat HCMV infections are prone to resistance and can be toxic to patients, and there is no current vaccine against HCMV available. The data in this report will lead to a better understanding of how essential HCMV envelope glycoproteins function during infection of biologically important cell types and will have significant implications for understanding HCMV pathogenesis for developing new therapeutics.
KEYWORDS: HCMV entry, HCMV tropism, pentamer, trimer
ABSTRACT
Human cytomegalovirus (HCMV) infects a wide variety of human cell types by different entry pathways that involve distinct envelope glycoprotein complexes that include gH/gL, a trimer complex consisting of gHgL/gO, and a pentamer complex consisting of gH/gL/UL128/UL130/UL131. We characterized the effects of soluble forms of these proteins on HCMV entry. Soluble trimer and pentamer blocked entry of HCMV into epithelial and endothelial cells, whereas soluble gH/gL did not. Trimer inhibited HCMV entry into fibroblast cells, but pentamer and gH/gL did not. Both trimer and pentamer bound to the surfaces of fibroblasts and epithelial cells, whereas gH/gL did not bind to either cell type. Cell surface binding of trimer and pentamer did not involve heparin sulfate moieties. The ability of soluble trimer to block entry of HCMV into epithelial cells did not involve platelet-derived growth factor PDGFRα, which has been reported as a trimer receptor for fibroblasts. Soluble trimer reduced the amount of virus particles that could be adsorbed onto the surface of epithelial cells, whereas soluble pentamer had no effect on virus adsorption. However, soluble pentamer reduced the ability of virus particles to exit from early endosomes into the cytoplasm and then travel to the nucleus. These studies support a model in which both the trimer and pentamer are required for HCMV entry into epithelial and endothelial cells, with trimer interacting with cell surface receptors other than PDGFR and pentamer acting later in the entry pathway to promote egress from endosomes.
IMPORTANCE HCMV infects nearly 80% of the world's population and causes significant morbidity and mortality. The current antiviral agents used to treat HCMV infections are prone to resistance and can be toxic to patients, and there is no current vaccine against HCMV available. The data in this report will lead to a better understanding of how essential HCMV envelope glycoproteins function during infection of biologically important cell types and will have significant implications for understanding HCMV pathogenesis for developing new therapeutics.
INTRODUCTION
Human cytomegalovirus (HCMV) is a ubiquitous betaherpesvirus that establishes a lifelong persistent infection. Infection of a host with a functioning immune system typically results in a mild fever or mononucleosis-type disease. In contrast, infection of a host with a compromised immune system can result in severe pneumonia, gastrointestinal disease, hepatitis, retinitis, and encephalitis (1, 2). HCMV infection can be particularly devastating to newborns and is the leading viral cause of birth defects with 5 to 10% of congenitally infected children developing serious neurologic defects, including hearing loss, mental retardation, and cerebral palsy (3, 4). HCMV is also a major problem in transplant patients due to immunosuppression and can exacerbate graft-versus-host disease and increase transplant vascular sclerosis (5).
All human herpesviruses require the highly conserved envelope glycoproteins gB and gH/gL in order to infect cells. The glycoprotein gB is thought to function as the virus fusogen that is somehow triggered by upstream events involving gH/gL complexes (6–8). HCMV encodes distinct gH/gL molecules that are required for entry, including gH/gL/gO, denoted as the trimer, and gH/gL/UL128/UL130/UL131, known as the pentamer (9, 10). Genetic studies clearly demonstrated that the pentamer is required for entry into epithelial and endothelial cells and is dispensable for entry into fibroblasts (11–13). In contrast, the trimer is thought to be sufficient for entry into fibroblasts. We have previously shown, using interference assays in which these proteins were expressed in cells before infection, that the trimer can interfere with entry into fibroblasts, whereas the pentamer interfered with entry into epithelial cells (14, 15). These studies supported a model in which the trimer and pentamer recognize distinct receptors on fibroblast and epithelial cells, respectively. A previous report also showed that a gO-null virus not only was unable to infect fibroblasts but, importantly, was also unable to infect epithelial cells (16).
The structure of the pentamer was recently solved, showing that HCMV gH/gL resembles Epstein-Barr virus gH/gL with proteins UL128 to UL131 assembled onto gL (17). The structure of the trimer has not yet been reported. Biochemical studies showed that gO and UL128 compete with one another to form disulfide bonds with gL cysteine-144 so that one of two mutually exclusive complexes is formed, the gH/gL/gO trimer or the gH/gL/UL128-131 pentamer (18). Besides these two complexes, a stable complex of gB with gH/gL has been coprecipitated from gB- and gH/gL-expressing cells, from HCMV-infected cells, and from solubilized extracellular particles (19). The best evidence for how gH/gL proteins function comes from studies showing that trimer interacts with the platelet-derived growth factor (PDGFRα) in a process required for entry into fibroblasts (20, 21, 21, 22). To date no cellular proteins have been identified that interact with the pentamer. However, CD147 can enhance entry of pentamer-expressing HCMV, but not HCMV lacking pentamer, into epithelial and endothelial cells, apparently acting as a chaperone to increase cell surface or endosomal localization of pentamer receptors (23). The epidermal growth factor receptor (EGFR) was described as an entry mediator for HCMV entry (24). Subsequent reports found no role for EGFR in HCMV entry into fibroblasts or epithelial and endothelial cells, although some studies suggest that EGFR signaling can increase macropinocytosis into certain cells or prepare cells for early stages of HCMV replication (19, 20, 25, 26).
Importantly, the mechanisms by which gH/gL, trimer, and pentamer complexes function during virus entry and trigger gB for membrane fusion remain unclear. In this report, we constructed and purified soluble forms of gH/gL, trimer, and pentamer. Using these soluble proteins, we showed that the soluble trimer can block entry into fibroblast cells, whereas both the trimer and pentamer block entry into epithelial and endothelial cells. Our studies support a model in which trimer acts at cell surfaces to increase HCMV binding to cells, while pentamer acts later, likely in endosomes, to allow the virus to gain access to the cytoplasm and nucleus.
RESULTS
Expression, purification, and characterization of HCMV gH/gL complexes.
We used 293 HEK-6E cells together with 6E cell-optimized vectors to transiently express the HCMV wild-type (wt) gH/gL, gH/gLC144S mutant, pentamer, and trimer proteins (27). We typically obtain final yields of 1 to 2 mg/liter of gH/gLC144S, ∼1 mg/liter of trimer, and 2 to 4 mg/liter of pentamer after purification. Notably, protein yields of wt gH/gL were less than that of gHgLC144S. SDS-PAGE analysis indicates that each protein component of these three complexes was present in the purified preparations and migrated on SDS-PAGE as reported previously (Fig. 1A) (18, 20). Western blot analysis using anti-UL128, anti-UL130, and anti-UL131 rabbit serum further confirmed the presence of UL128, UL130, and UL131 in the pentamer (data not shown). The trimer eluted with an apparent molecular weight (MW) of 380 kDa on a Superdex 200 gel filtration column, consistent with the observations of Kabanova et al. (20) (Fig. 1B). The gO subunit is heavily glycosylated, and deglycosylation of the trimer with PNGase F resulted in a shift in the apparent gO MW from ∼100 kDa to ∼55 kDa (https://www.ohsu.edu/johnson-lab-supplement). The pentamer eluted with an apparent molecular weight of 300 kDa on the Superdex 200. Since gH/gL has been previously reported to form a dimer of gH/gL heterodimers through gL cysteine 144, we replaced cysteine 144 with serine to preclude the ability of dimeric forms of gH/gL (dimers of gH/gL heterodimers) to assemble (18). We expressed both wild-type gH/gL and the gHgLC144S mutant. Gel filtration analysis of the gH/gLC144S mutant indicated that gH/gL is still able to form dissociable dimers of gH/gL molecules at higher concentrations (Fig. 1C). Expression of wt gH/gL resulted in a heterogeneous population consisting of both gH/gL and dimers of gH/gL molecules (https://www.ohsu.edu/johnson-lab-supplement). Binding studies with the HCMV gH/gL proteins showed that the trimer formed stable complexes with PDGFRα and pentamer bound to scFv constructs from monoclonal antibodies (MAbs) 13H11 (gH specific), 8J16 (UL128 to UL131A specific), and 8I21 (pentamer specific; data not shown).
FIG 1.
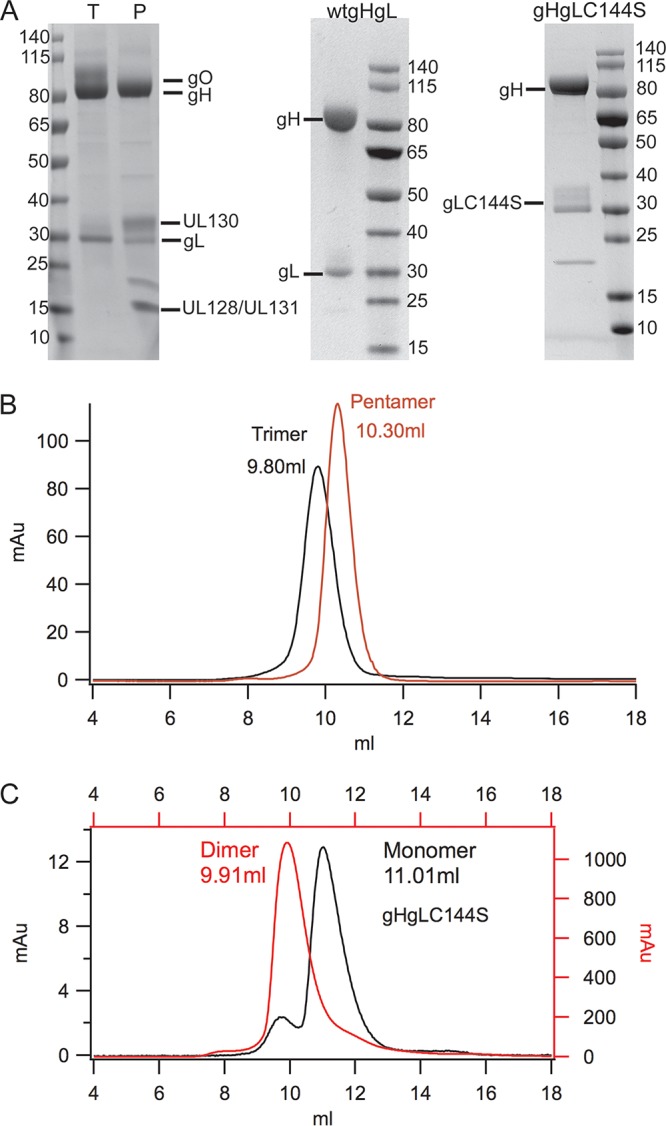
Expression and purification of different HCMV gH/gL, pentamer, and trimer complexes. (A) SDS-PAGE analysis of purified pentamer, trimer, gH/gL, and gH/gLC144S complexes under reducing conditions. P, pentamer complex; T, trimer complex. The ∼20-kDa band in the pentamer prep is likely a cleavage product of gL based on Western blot analysis. (B and C) Superdex 200 size exclusion chromatography profiles of pentamer, trimer, and gH/gLC144S at two concentrations.
Inhibition of virus entry by soluble gH/gL proteins.
To assess the effects of soluble gH/gL, trimer, and pentamer complexes on HCMV entry, ARPE-19 epithelial cells were incubated with various concentrations of these proteins for 1 h at 4°C. Cell monolayers were then incubated for 1 h at 4°C with BADrUL131, a virus derived from HCMV strain AD169 that was repaired for the UL131 gene so as to express the pentamer and also expresses a green fluorescent protein (GFP) reporter gene (13). The cells were then shifted to 37°C for an additional 2 h to allow virus entry. After 24 h, GFP expression was assessed by fluorescence microscopy, and the GFP-positive cells were quantified. The results of these experiments showed dose-dependent inhibition of virus entry when cells were incubated with soluble pentamer and trimer (Fig. 2A). GFP expression by HCMV BADrUL131 was reduced by ∼50% with 25 μg/ml of soluble trimer and ∼50% with 25 μg/ml of the soluble pentamer (Fig. 2A). In all experiments with ARPE-19 cells, lower concentrations of trimer were more efficient at inhibiting virus entry than the pentamer. In contrast, soluble gH/gL did not inhibit HCMV infection of epithelial cells (Fig. 2A). Similarly, we did not see any inhibition of HCMV entry with soluble gH/gL C144S on epithelial cells (data not shown). Experiments performed with human umbilical vascular endothelial cells (HUVECs) showed inhibition of HCMV entry with soluble trimer (Fig. 2B). There was a 50% reduction in GFP expression with ∼6 μg/ml of soluble trimer and complete inhibition with 25 μg/ml. Pentamer inhibited HCMV entry much less effectively, requiring as much as 50 μg/ml to reduce HCMV entry by ∼50% (Fig. 2B). Similar to epithelial cells, soluble gH/gL or soluble gH/gL C144S did not inhibit HCMV entry into HUVECs.
FIG 2.

Inhibition of HCMV entry into cells by soluble protein complexes. (A and B) ARPE-19 epithelial cells or HUVEC monolayers were incubated in culture medium supplemented with increasing concentrations of soluble protein complexes for 1 h at 4°C. The cells were then incubated with HCMV BADrUL131 virus particles, 10 IU per cell, and allowed to incubate for an additional 1 h at 4°C. The cells were then shifted to 37°C and allowed to incubate for an additional 2 h; then, the virus inoculum was removed, and the cells were washed and then incubated in fresh growth medium at 37°C for 24 h. (C) Fibroblast cell monolayers were incubated in culture medium supplemented with increasing concentrations of soluble protein complexes for 1 h at 4°C. The cells were then incubated with HCMV BADrUL131 (or AD169, green diamonds) virus particles at 1 IU per cell and allowed to incubate for an additional 1 h at 4°C. The cells were then shifted to 37°C and allowed to incubate for an additional 2 h; then, the virus inoculum was removed and the cells were washed and then incubated in fresh growth medium at 37°C for 24 h. (D) ARPE-19 or fibroblast cell monolayers were incubated with soluble proteins as described for panels A to C and then challenged with HSV-1 F-BAC VP26GFP virus particles at 1 PFU per cell. The level of virus entry was determined by assessing GFP expression by fluorescence microscopy and comparing soluble-protein-treated samples to no-protein control samples and is displayed as percent infection relative to no-protein samples. The data were determined from three separate wells for each condition.
Experiments with fibroblasts involved two viruses, HCMV BADrUL131 and an HCMV AD169 derivative that expresses GFP but not the pentamer. Trimer inhibited infection of both the BADrUL131 and AD169 virus strains with fibroblasts (Fig. 2C). Soluble trimer was much more efficient at inhibiting HCMV entry into fibroblasts than into epithelial and endothelial cells, with complete inhibition of entry observed with 6 μg/ml and 12 μg/ml for AD169 and BADrUL131, respectively. In contrast, soluble pentamer and soluble gH/gL molecules were unable to inhibit entry of fibroblast cells (Fig. 2C).
To test for the specificity of the effects of these soluble proteins, entry assays were performed with herpes simplex virus 1 (HSV-1) on ARPE-19 cells and fibroblasts. Neither soluble trimer nor pentamer reduced entry of HSV-1 into epithelial and fibroblast cells (Fig. 2D). We concluded that both soluble trimer and pentamer block entry into epithelial and endothelial cells, while gH/gL does not. Moreover, trimer but not pentamer or gH/gL blocked entry into fibroblast cells.
Binding of soluble gH/gL proteins to cell surfaces.
To determine whether the soluble gH/gL proteins bound to cell surfaces, ARPE-19 and fibroblast monolayers were incubated with soluble gH/gL, trimer, and pentamer complexes at 100 μg/ml at 4°C for 1 h, and then the cells were washed and fixed with phosphate-buffered saline (PBS) containing 4% paraformaldehyde. gH/gL complexes were detected by immunofluorescent staining using anti-gH MAb 14-4b, which recognizes gH/gL alone or when associated with gO or UL128-131 (28). Detection of this antibody with a fluorophore-labeled secondary antibody produced evidence of trimer and pentamer complexes binding to cell surfaces. Both trimer and pentamer bound equally well to ARPE-19 cells, whereas we were unable to detect surface binding of gH/gL (Fig. 3A to C and G). Similarly, trimer and pentamer complexes bound to fibroblasts at similar levels and again no soluble gH/gL was detected on fibroblast cell surfaces (Fig. 3D to F and H). To confirm that the inability of soluble gH/gL to bind to cells was not due to changes in the native conformation of gH/gL, we performed immunoprecipitations of soluble gH/gL, trimer, and pentamer complexes using the conformational-specific gH MAb 14-4b (29). Western blot analysis of these immunoprecipitations showed that 14-4b MAbs could precipitate gH/gL as well as trimer- and pentamer-associated gH/gL, indicating that the soluble gH/gL was structurally intact (Fig. 3I).
FIG 3.

Binding of soluble protein complexes to cell surfaces. ARPE-19 cells (A to C) or fibroblast cells (D to F) seeded on glass coverslips were incubated with different soluble protein complexes (100 μg/ml) for 1 h at 4°C. The cells were then washed once in cold PBS and then fixed with 4% paraformaldehyde and processed for immunofluorescence microscopy. Protein complexes were detected using anti-gH MAb 14-4b followed by Dylight-594 goat anti-mouse secondary antibody. The cells were then permeabilized, counterstained with DAPI (4′,6-diamidino-2-phenylindole), and then mounted onto glass slides with Fluoromount-G. Images were captured by a 60× objective with the Deltavision Core DV Widefield Deconvolution system. (G and H) Histograms showing the quantification of soluble protein binding to cell surfaces for conditions represented in panels A to C (G) and D to F (H). The data are displayed as mean gray values (arbitrary units) as analyzed by ImageJ. Data were collected from three separate images for each condition. (I) Immunoprecipitation of soluble gH/gL protein complexes followed by Western blot analysis. The top of the panel indicates whether the conformational specific anti-gH MAb 14-4b was included (plus) or omitted (minus) in the immunoprecipitation, and the bottom of the panel indicates which protein complex was used for the immunoprecipitation. The membrane was probed with MAb AP-86, which is specific for the gH polypeptide.
Effects of soluble glycoproteins on binding of HCMV to cells.
To examine the effects of the gH/gL proteins on binding of HCMV virus particles to cells, ARPE-19 cells were incubated with soluble proteins using 100 μg/ml at 4°C for 1 h followed by incubation with HCMV TB40E UL32-GFP particles for an additional 1 h at 4°C. This recombinant HCMV expresses a GFP-tagged form of the UL32 gene product, pp150, a capsid-associated tegument protein, and is derived from TB40E, which expresses the pentamer (30). After incubation, the cells were washed and fixed and the presence of HCMV was detected by fluorescence microscopy. In the absence of any soluble protein, GFP-positive virus particles were detected on ARPE-19 epithelial cells (Fig. 4A). TB40E UL32-GFP binding to ARPE-19 cells was reduced by 80% by preincubation with soluble trimer (Fig. 4B and D). In contrast, preincubation of ARPE-19 cells with pentamer had only minimal effects on surface binding of HCMV particles (Fig. 4C and D). These experiments were extended to fibroblasts incubated with the soluble proteins and then either with HCMV TB40E UL32-GFP or with the related HCMV TB40F UL32-GFP, which expresses UL32-GFP but not the pentamer (26, 30). Again, preincubation with soluble trimer reduced the number of TB40F UL32-GFP particles bound to fibroblast cell surfaces by 90% (Fig. 4E, F, and H). Similarly, soluble trimer reduced the number of TB40E UL32-GFP particles bound to fibroblast cell surfaces by 93% (Fig. 4I, J, and L). In contrast, preincubation of fibroblasts with soluble pentamer did not drastically affect binding of either TB40E UL32-GFP or TB40F UL32-GFP to fibroblast surfaces (Fig. 4E, G, H, I, J, and L). Therefore, trimer substantially reduced the binding of HCMV to both epithelial cells and fibroblasts, but gH/gL or pentamer did not.
FIG 4.

Effects of soluble protein on adsorption of HCMV virus particles onto cells. (A to C) ARPE-19 cells on glass coverslips were either left untreated or incubated with soluble trimer or pentamer complexes (100 μg/ml) for 1 h at 4°C followed by 1 h of incubation with HCMV TB40 E-UL32-GFP virus particles at 4°C. (D) Histograms showing the quantification of HCMV virus particles binding to cell surfaces for the conditions represented in panels A to C. (E to G) Fibroblast cell monolayers were either not treated or incubated with soluble trimer or pentamer complexes (100 μg/ml) for 1 h at 4°C followed by 1 h of incubation with HCMV TB40 F-UL32-GFP. (H) Histograms showing the quantification of HCMV virus particles binding to cell surfaces for the conditions represented in panels E to G. (I to K) Fibroblast cell monolayers were either not treated or incubated with soluble trimer or pentamer complexes (100 μg/ml) for 1 h at 4°C followed by 1 h of incubation with HCMV TB40 E-UL32-GFP virus particles at 4°C. (L) Histograms showing the quantification of HCMV virus particles binding to cell surfaces for the conditions represented in panels I to K. In all cases, the cells were washed once with cold PBS and then fixed with 4% paraformaldehyde. The cells were then permeabilized, counterstained with DAPI (4′,6-diamidino-2-phenylindole), and mounted onto glass slides with Fluoromount-G. Images were captured by a 60× objective with the Deltavision Core DV Widefield Deconvolution system. (D, H, and L) The data in the histograms are displayed as mean gray values (arbitrary units) relative to control conditions as analyzed by ImageJ. Data were collected from three separate images for each condition.
Role of heparan sulfate GAGs in binding of gH/gL proteins to cells.
Previously, it was reported that heparan sulfate glycosaminoglycans (GAGs) play an important role in the initial attachment of HCMV onto fibroblasts (31). Virus entry is effectively blocked with heparin, and treatment of fibroblasts with heparinase to remove cell surface heparan GAGs prevented virus entry of cells. HCMV glycoproteins gB and gM have both been shown to bind heparan GAGs (32, 33). To test whether the binding of trimer and pentamer complexes to cells was related to the effects of surface heparan sulfate GAGs, we treated ARPE-19 cell monolayers with heparinase followed by incubation with HCMV. HCMV TB40E-UL32-GFP failed to bind to ARPE-19 cell surfaces treated with heparinase (Fig. 5A and B). Heparinase treatment also inhibited virus entry and IE-1 expression after cells were incubated with BADrUL131 (Fig. 5C and D). In contrast, binding of soluble trimer and pentamer to cell surfaces of ARPE-19 cells was minimally affected by heparinase treatment and showed levels of 85% compared to no heparinase treatment (Fig. 5E, F, G, H, O, and P). Similarly, heparinase treatment of fibroblasts blocked virus binding and expression of IE-1 (Fig. 5I to L). However, heparinase treatment did not reduce binding of trimer to fibroblasts (Fig. 5M, N, and Q). We concluded that binding of trimer and pentamer to cells is not dependent upon heparan sulfate GAGs, and it is unlikely that trimer and pentamer act to block HCMV entry by obstructing heparan sulfate GAGs.
FIG 5.

Binding of soluble proteins to cell surfaces is not affected by heparinase treatment. ARPE-19 or fibroblast cells seeded on glass coverslips were incubated in buffered Opti-MEM at 30°C and either left untreated (no heparinase) or treated with heparinase enzyme and incubated at 30°C for 2 h. The cells were then subjected to one of the following conditions. For panels A, B, I, and J, ARPE-19 (A, B) and fibroblast (I, J) cells were incubated with HCMV TB40E-UL32-GFP virus particles at 4°C for 1 h and then fixed and analyzed by deconvolution microscopy as described for Fig. 4. For panels C, D, K, and L, ARPE-19 (C, D) or fibroblast (K, L) cells were incubated with HCMV BADrUL131 for 2 h and then washed and allowed to incubate for an additional 24 h, at which time the cells were fixed and analyzed by immunofluorescence microscopy to detect the HCMV GFP expression. For panels E, F, M, and N, cells were incubated with soluble trimer complexes (100 μg/ml). For panels G and H, cells were incubated with soluble pentamer complexes (100 μg/ml) at 4°C for 1 h. The cells were then washed once in cold PBS and then fixed with 4% paraformaldehyde and processed for immunofluorescence microscopy using anti-gH MAb 14-4b as described for Fig. 3. (O to Q) Histograms showing the quantification of HCMV virus particles binding to cell surfaces for the conditions represented in panels E and F, G and H, and M and N, respectively. The data are displayed as mean gray values (arbitrary units) relative to control conditions as analyzed by ImageJ. Data were collected from three separate images for each condition.
Binding of trimer to epithelial cells is not related to expression of the platelet growth factor receptor.
There have been multiple reports that PDGFRα promotes HCMV entry into fibroblasts and trimer binds directly to PDGFRα to promote this entry (20–22, 34). Given our observations that trimer binds to epithelial and endothelial cells, it was important to determine whether this binding to these cells involved PDGFRα. For these experiments, we used an ARPE-19 KD cell line that we previously described, which expresses a short hairpin RNA (shRNA) targeting PDGFRα (19, 35). Western blot analysis showed that PDGFRα expression was silenced in ARPE-19 KD compared to normal APRE-19 cells (Fig. 6A). However, binding of soluble trimer to ARPE-19 KD cells was similar to that seen with normal ARPE-19 cells (Fig. 6B and C). This is consistent with the previous report that silencing of PDGFRα in ARPE-19 cells did not reduce HCMV entry into ARPE-19 cells and indicated that the binding of soluble trimer to ARPE-19 cells does not involve PDGFRα (19). We note that in contrast to this report, Wu et al. were unable to detect PDGFRα expression in ARPE-19 cells by Western blotting (22). We acknowledge that detecting PDGFRα expression in ARPE-19 is difficult and in our hands, we have been able to detect PDGFRα from ARPE-19 cells only by using the anti-PDGFRα antibody described in this and in previous reports (19, 35).
FIG 6.

Binding of soluble trimer to ARPE-19 cells is not affected by silencing PDGFRα. (A) Cell lysates from normal ARPE-19 cells or ARPE-19 KD cells expressing an shRNA targeting PDGFRα were analyzed by Western blotting using anti-PDGFRα polyclonal antibody 3164S (R & D Systems) according to the manufacturer's instructions. The membrane was also probed with a monoclonal antibody to β-actin (Sigma AC-74) to serve as a loading control. (B) Normal ARPE-19 cells or ARPE-19 KD cells were incubated with soluble trimer complexes (100 μg/ml) for 1 h at 4°C. The cells were then washed once in cold PBS and then fixed with 4% paraformaldehyde and processed for immunofluorescence microscopy. Protein complexes were detected using anti-gH MAb 14-4b followed by Dylight-594 goat anti-mouse secondary antibody. The cells were counterstained with DAPI (4′,6-diamidino-2-phenylindole), and the coverslips were mounted onto glass slides with Fluoromount-G. Images were captured by a 60× objective using the Deltavision Core DV Widefield Deconvolution system. (C) Histograms showing the quantification of HCMV soluble trimer binding to cell surfaces of ARPE-19 and ARPE-19 KD cells. The data are displayed as mean gray values (arbitrary units) relative to control conditions as analyzed by ImageJ. Data were collected from three separate images for each condition.
Pentamer blocks virus entry by inhibiting translocation of virus DNA to the nucleus.
To better understand the mechanism by which soluble pentamer blocked HCMV infection of ARPE-19 cells, experiments were performed in epithelial cells that expressed a red fluorescent protein (RFP)-tagged early endosome marker, EEA1. These cells were then incubated with soluble pentamer complexes at 4°C for 1 h followed by incubation with TB40E-UL32-GFP at 4°C for 1 h. Following these incubations, the cells were warmed to 37°C for 2 h and then fixed and processed for analysis by deconvolution microscopy. Images of cells incubated with no protein were compared to images of pentamer-treated cells to determine whether virus particles internalized into EEA1 labeled endosomes in the presence of pentamer. Analysis of these images showed no obvious differences in the localization of TB40E-UL32-GFP to EEA-1+ endosomes when pentamer was present (Fig. 7A). This supports the conclusion that pentamer does not block HCMV internalization into EEA1+ endosomes. We initially attempted to follow fluorescent HCMV particles as they departed from EEA1+ endosomes but found that this was an inefficient process and too difficult to characterize by immunofluorescence. However, we could follow the HCMV DNA from endosomes into the nucleus by using quantitative PCR. For these experiments, control or pentamer-treated cells were infected with BADrUL131 in the presence of foscarnet and ganciclovir to prevent replication of virus DNA. Cells were incubated for 24 h and then suspended in nucleus isolation buffer, consisting of 15 mM Tris-HCl (pH 7.5), 60 mM KCl, 15 mM NaCl, 5 mM MgCl,1 mM CaCl2, 250 mM sucrose, and 0.5% NP-40, and the nuclei were pelleted. DNA from isolated nuclei was extracted, and viral DNA was quantified by PCR. The results of these experiments showed that epithelial cells treated with soluble pentamer had 85% less HCMV DNA in the nucleus than did untreated control cells (Fig. 7B). These results indicated that the soluble form of the pentamer is able to block HCMV entry by competing with virus-associated pentamer, which under normal conditions functions to promote virion trafficking from early endosomes to the nucleus during the entry pathway.
FIG 7.
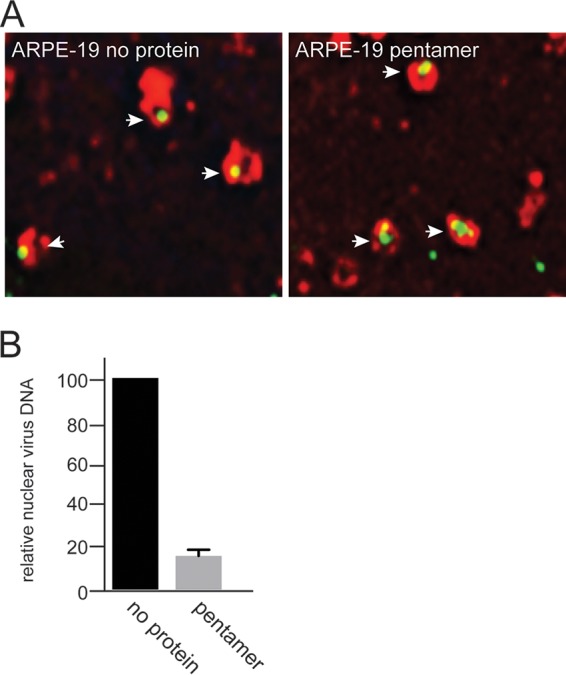
Soluble pentamer inhibits HCMV entry at a step after entering endosomes. (A) ARPE-19 cells on glass coverslips were transduced with a BacMam vector expressing an EEA-1-RFP fusion protein for 24 h. The cells were then either left untreated or incubated with soluble pentamer complexes (100 μg/ml) for 1 h at 4°C followed by 1 h of incubation with HCMV TB40 E-UL32-GFP virus particles at 4°C. The cells were shifted to 37°C for 2 h and then fixed and processed for deconvolution microscopy. Images were captured by a 60× objective using the Deltavision Core DV Widefield Deconvolution system. (B) ARPE-19 cells were either left untreated (no protein) or treated with soluble pentamer (100 μg/ml) for 1 h at 4°C followed by incubation with HCMV virus BADrUL131 for 1 h at 4°C. The cells were then shifted to 37°C and allowed to incubate for an additional 2 h, and then the virus inoculum was removed and the cells were washed and then incubated in fresh growth medium that was supplemented with 100 μM foscarnet and ganciclovir at 37°C for 24 h. After 24 h, the cellular nuclei were isolated and the nuclear DNA was extracted and used for quantitative PCR. Quantitative PCR was performed with probes specific for a region in the HCMV gB open reading frame. Input DNA was controlled with probes that annealed to a region of the GAPDH open reading frame.
DISCUSSION
Our previous models describing how HCMV trimer and pentamer function during virus entry were largely based on interference studies that proposed that trimer bound to fibroblast-specific receptors whereas pentamer bound to epithelial and endothelial cell-specific receptors. However, an HCMV gO-null virus was unable to enter any cell type and exhibited highly reduced amounts of gH/gL and increased quantities of pentamer in the virus envelope (16). In addition, altering the levels of trimer present in HCMV strain Merlin influenced virus entry into both fibroblast and epithelial cells (36). In this report, we showed that both soluble trimer and soluble pentamer were able to block HCMV entry into epithelial and endothelial cells. In fact, the trimer inhibited HCMV entry into epithelial and endothelial cells at lower concentrations than those required to produce inhibition with the pentamer. Our observation that both soluble trimer and pentamer blocked virus entry into epithelial and endothelial provides the best direct evidence that both trimer and pentamer play important roles in virus entry into epithelial and endothelial cells. These data would also suggest that HCMV entry into epithelial and endothelial cells is a multistep process and likely involves more than a single cellular factor, e.g., putative trimer and pentamer receptors.
Inhibition assays with soluble pentamer did not block entry of pentamer-expressing HCMV viruses into fibroblast cells, which is consistent with the pentamer being dispensable for HCMV entry into fibroblast cells. However, it was interesting that we observed robust binding of soluble pentamer to the surface of fibroblast cells. Whether the soluble pentamer is binding the same molecules on fibroblasts and epithelial cells is unclear. It was also somewhat surprising that we were unable to inhibit HCMV entry into either fibroblasts or epithelial cells with soluble gH/gL alone and we were unable to detect any soluble gH/gL that bound to the surface of these cell types as well. These results could argue against a role for gH/gL during entry into these cell types; however, more importantly, these results indicate that the gO and UL128, UL130, and UL131 components of the trimer and pentamer complexes, respectively, are facilitating the binding of these complexes to saturable receptors on the cell surface. This is consistent with previous results showing that gO facilitates binding of the trimer to PDGFRα molecules (19, 20). Alternatively, it is formally possible that association of gO or UL128 to 131 onto gH/gL may induce conformational changes in gH/gL that expose protein surfaces involved in protein-protein interactions with receptors.
The role of heparan sulfate heparin sulfate GAGs in binding of these glycoproteins to cells was characterized by using heparinase to remove the GAGs. Like other herpesviruses, HCMV adsorbs onto cells by interacting with very numerous GAGs that constitute the extracellular matrix (22, 31). Heparinase treatment of fibroblasts and epithelial cells blocked HCMV adsorption onto cells and virus entry. However, the same heparinase treatment did not reduce cell surface binding of trimer or pentamer. We concluded that the binding of trimer and pentamer does not involve binding to heparan sulfate GAGs. Moreover, gH/gL did not bind to any cell surfaces, suggesting that gH/gL does not bind heparan sulfate GAGs. This is different from observations with Kaposi's sarcoma herpesvirus gH/gL, which binds heparan sulfate GAGs (37).
The HCMV trimer binds with relatively high affinity to PDGFRα, and this interaction promotes HCMV entry into fibroblasts (20–22). It is likely that our soluble trimer binds PDGFRα on the surfaces of fibroblasts and blocks entry as reported in these previous papers. In contrast, it appears unlikely that the trimer blocks HCMV entry into epithelial or endothelial cells by binding to PDGFRα. First, ARPE-19 cells express dramatically less PDGFRα than do fibroblast cells, yet the cell surface binding of the trimer to epithelial cells was similar to that with fibroblasts (22). Second, silencing of the already lower levels of endogenous expression of PDGFRα in APRE-19 cells did not reduce trimer binding to APRE-19 cells. Third, we and others have shown that shRNA silencing of PDGFRα in epithelial and endothelial cells did not affect the ability of HCMV to enter either cell type (19–21). It is possible that soluble trimer bound to the small quantities of PDGFRα on ARPE-19 cells, but this does not influence virus entry. Together, these observations suggest that the trimer binds to a molecule or molecules other than PDGFRα on ARPE-19 and endothelial cells that are required for entry.
The requirement for both trimer and pentamer for HCMV entry into epithelial and endothelial cells suggests a model in which HCMV enters these cells by a multistep pathway. Treatment of fibroblasts and epithelial cells with soluble trimer reduced the number of HCMV-UL32-GFP particles that attached to cell surfaces. This suggests that trimer may act at an early stage of entry, such as by mediating virus attachment onto cell surface receptors. In contrast, soluble pentamer did not reduce the number of HCMV particles that bound onto epithelial cell surfaces. Thus, it appears that the pentamer acts downstream of the trimer at a step in the entry pathway following virus attachment to cells. Supporting this were our observations that pentamer did not reduce entry of HCMV-UL32-GFP particles into EEA-1-positive endosomes. Thus, soluble pentamer did not block the internalization of HCMV particles into early endosomes that follows attachment. However, treatment of cells with soluble pentamer reduced the amount of virus DNA that reached the nucleus from these endosomes compared to what was seen in untreated cells. This suggests that HCMV was blocked in egress from endosomes and reaching the nucleus. It seems unlikely that soluble pentamer could act to inhibit cytoplasmic events, as it would be unlikely that pentamer reaches the cytoplasm. This model fits with observations that HCMV pentamer-expressing viruses enter APRE-19 cells by macropinocytosis followed by low-pH-mediated fusion between the virion enveloped and endosomal membranes (19, 38). HCMV virions lacking the pentamer do not reach the cytoplasm, and it appears that soluble pentamer also produces the same effect, suggesting that the pentamer functions in some capacity to promote entry fusion within endosomes.
How these sequential effects of trimer and pentamer that promote HCMV entry into epithelial and endothelial cells relate to gB, the viral fusion protein, remains unclear. We have shown previously that HCMV gB and gH/gL are all that is required to promote cell-cell fusion of epithelial cells and fibroblasts (39). Moreover, gB and gH/gL can be coprecipitated from solubilized virions, suggesting that preformed gB and gH/gL complexes may be present in the virion envelope (22, 28). However, clearly HCMV entry into epithelial and endothelial cells requires more than just gB and gH/gL; trimer and pentamer are also needed. Obviously, virus entry differs substantially from cell-cell fusion in that the virion envelope must be brought into apposition with different cellular membranes (cell surface and endosomal). Our working hypothesis of how HCMV enters epithelial cells suggests that trimer binding to some cell surface proteins leads to surface attachment and leads to macropinocytosis and that pentamer binding to other cellular proteins in endosomes triggers gB for membrane fusion.
MATERIALS AND METHODS
Cells and viruses.
Primary human neonatal dermal fibroblasts (NHDFs) were obtained from Invitrogen and grown in Dulbecco's modified Eagle medium (DMEM) with 10% fetal bovine serum (FBS). Human umbilical cord vascular endothelial cells (HUVECs) were a kind gift from Ashlee Moses at the Vaccine and Gene Therapy Institute in Portland, OR, and were maintained in Medium-200 plus low serum growth supplement (Invitrogen). Human retinal pigmented epithelial (ARPE-19) cells were obtained from ATCC and grown in DMEM/F12 plus 10% FBS. ARPE-19 KD cells were previously described (19, 35). All cells were maintained at 37°C with 5% CO2. The HCMV BADrUL131 (kindly provided by Tom Shenk, Princeton University) is a derivative of AD169 that has had the UL131 gene repaired to allow for expression of pentamer and also carries a GFP reporter gene under the control of the simian virus 40 (SV40) promoter. AD169-GFP is a bacterial artificial chromosome (BAC)-derived AD169 virus that has a GFP marker gene inserted into the UL21.5 locus (40). HCMV TB40E UL32-GFP (kindly provided by Andrew Yurochko at the Louisiana State University Health Sciences Center, Shreveport) is a derivative of the endotheliotropic strain TB40E, which has a GFP reporter gene fused to the tegument phosphoprotein (pp150) encoded by the UL32 open reading frame, and TB40F UL32-GFP (kindly provided by Christian Sinzger at the Institute of Virology, University of Ulm, Ulm, Germany) is a derivative of TB40F that lacks the pentamer but expresses UL32-GFP (30, 41). HCMV stocks were produced from NHDFs grown in roller bottles, and viral particles were concentrated from culture supernatants by centrifugation through a cushion of 20% sorbitol in PBS at 80,000 × g for 1 h. Pellets were suspended in DMEM plus 10% FBS and frozen at −70°C. Because HCMV does not plaque very well, titers of HCMV stocks were calculated by determining the number of infectious units per ml (IU ml−1) by serial diluting virus stocks, adding the dilutions onto NHDF monolayers, and then staining for the HCMV immediate early gene IE-86 after 24 with anti-IE-86 rabbit polyclonal serum 6658. HSV-1 F-VP26-GFP has been described previously and was propagated, and its titers were determined on Vero cells. For TB40E UL32-GFP and TB40F UL32-GFP, cells on glass coverslips were incubated in 1 ml of infected cell culture supernatant.
Expression and purification of HCMV pentamer, trimer, wt gH/gL, and gHgLC144S.
Initial attempts to obtain pentamer using two recombinant baculoviruses containing gH/gL and UL128/UL130/UL131A did not yield sufficient amounts of pentamer. We therefore cloned the insect codon-optimized gH (AD169, amino acids [aa] 27 to 718), gL (AD169), UL128 (AD169), UL130 (AD169), and UL131A (Merlin) into mammalian cell expression vectors (pYD7 vector; National Research Council [NRC], Canada) via Gibson assembly. Each HCMV protein is fused to the vascular endothelial growth factor (VEGF) signal sequence derived from the pTTVH8G vector (NRC). At the C terminus of gH, there is a thrombin protease cleavage site followed by a Strep-II tag and a His8 purification tag. Mammalian codon-optimized full-length TR strain gO (TRgO) was cloned into the pTT5 vector (NRC) and contains a C-terminal Strep-tag. Equal amounts of the vectors were mixed and then transiently cotransfected into HEK293-EBNA1-6E cells (HEK-6E; NRC) according to NRC protocols using linear polyethylenimine (PEI) at a 1:3 plasmid-to-PEI ratio. Six days after the transfection, the cell culture was harvested and spun at 8,500 relative centrifugal force (rcf) for 30 min. Supernatants were passed through a 0.45-μm filter and then loaded onto His60 Superflow resin (Clontech) preequilibrated in buffer A (300 mM NaCl, 20 mM Tris-HCl [pH 8.0], and 5 mM imidazole). The resin was washed with 10 resin bed volumes of buffer A and eluted with 5 resin bed volumes of buffer B (300 mM NaCl, 20 mM Tris-HCl [pH 8.0], and 300 mM imidazole). The elution was concentrated and loaded onto a Superdex 200 10/300 GL (S200; GE) column preequilibrated with a running buffer consisting of 300 mM NaCl and 20 mM HEPES (pH 7.5). gHgLC144S and trimer always eluted as a symmetric peak on the S200 column, and the peak fractions were collected for further experiments. Expression of wt gH/gL consisted of both gH/gL and dimeric forms of gH/gL (dimers of gH/gL heterodimers) and were collected in separate fractions (https://www.ohsu.edu/johnson-lab-supplement). The pentamer variably eluted as an asymmetric peak with a lagging shoulder, which might indicate incomplete pentamer assembly. We therefore collected the first 2 or 3 fractions (0.5 ml/fraction) of the peak for further studies. SDS-PAGE analysis was done with precast 4 to 20% gradient ExpressPlus gels (GenScript) using PageRuler (26616; Thermo Fisher) as protein molecular weight standards.
Soluble protein, virus binding, and virus entry assays.
For protein binding assays, cell monolayers seeded in culture dishes or on glass cover slides were incubated with soluble protein complexes in Opti-MEM without FBS and at 4°C for 1 h with gentle rocking. The cells were then washed once with Opti-MEM without FBS and then fixed with PBS containing 4% paraformaldehyde. For virus binding assays, the cells were incubated with soluble protein complexes as just described. After 1 h of incubation at 4°C, virus was added to the cells, and then the mixture was incubated at 4°C for an additional hour. The cells were washed once with Opti-MEM without FBS and then fixed with PBS containing 4% paraformaldehyde. For virus entry assays, cells were incubated with soluble protein complexes as described above followed by incubating cells with virus inoculum at 4°C for an additional 1 h. The cells were then shifted to 37°C and allowed to incubate for another 2 h. The cells were then washed once with Opti-MEM without FBS, and then normal growth medium was added and the cells were incubated for 24 h at 37°C. For all experiments, ARPE-19 cells were infected with HCMV using 10 IU per cell and fibroblasts were infected with 1 IU per cell. HSV-1 was used at 1 PFU per cell. Virus entry was measured by assessing GFP expression by fluorescence microscopy.
Fluorescence microscopy and immunostaining.
Cells seeded on glass coverslips were treated with soluble proteins and virus as described above. For fluorescent staining, cells on coverslips were fixed with PBS containing 4% formaldehyde for 10 min at room temperature. If necessary, cells were permeabilized with immunofluorescence (IF) buffer (0.5% Triton X-100, 0.5% deoxycholate, 2% bovine serum albumin, and 0.05% sodium azide in PBS) for 30 min. To detect soluble gH/gL proteins, cells were incubated with anti-gH MAb 14-4b (0.5 μg/ml) for 1 h at room temperature. The cells were then washed extensively with IF buffer and incubated with Dylight goat anti-mouse 594 secondary (0.5 μg/ml) antibody for 1 h. Cells were washed several times with IF buffer, and then the nuclei were counterstained with DAPI (4′,6-diamidino-2-phenylindole) for 10 min and the coverslips were mounted on glass slides with Fluoromount-G (Southern Biotech). To detect EEA1-positive endosomes, ARPE-19 epithelial cells on glass slides were transduced with a CellLight BacMam (Invitrogen) expressing a red-fluorescent-tagged early endosome marker, EEA-1. Images were captured and analyzed using a Deltavision Core DV Widefield Deconvolution system in the OHSU Advanced Light Microscopy Core. Quantification of soluble protein or virus particles binding to cell surfaces was performed by analyzing three separate grayscale images for each sample using ImageJ software and is displayed in mean gray values.
Cell fractionation and quantitative PCR.
Soluble protein inhibition assays on ARPE-19 cells were performed as described above, except that cells were also treated with 100 μM foscarnet and 100 μM ganciclovir to prevent replication of input virus DNA. To isolate nuclei, cells were scraped from tissue culture dishes and collected by centrifugation at 1,000 × g. The cells were then suspended in approximately 10 times the volume of the cell pellet of nuclei isolation buffer (15 mM Tris-HCl-7.5, 60 mM KCl, 15 mM NaCl, 5 mM MgCl,1 mM CaCl2, 250 mM sucrose, and 0.5% NP-40). The cells were gently pipetted several times and incubated on ice for 5 min. The nuclei were then pelleted by centrifugation at 600 rcf for 5 min at 4°C. The cytosolic fraction was removed, and the nuclei were washed with nucleus isolation buffer without NP-40. DNA was extracted from the nucleus fractions using the Gentra Puregene Tissue kit. Quantitative PCR was performed with the following primer and probes specific for HCMV gB and the cellular GAPDH (glyceraldehyde-3-phosphate dehydrogenase) gene: gB forward primer, AGGTCTTCAAGGAACTCAGCAAGA; gB reverse primer, CGGCAATCGGTTTGTTGTAAA; gB probe, 56-FAM/AACCCGTCA/ZEN/GCCATTCTCTCGGC/3IABkFQ; GAPDH forward primer, GAAGGTGAAGGTCGGAGTC; GAPDH reverse primer, GAAGATGGTGATGGGATTTC; GAPDH probe, 56-FAM/CAAGCTTCC/ZEN/CGTTCTCAGCC/3IABkFQ. Amplification was performed with TaqMan Fast Advanced master mix (Applied Biosystems) according to the manufacturer's instructions using an Applied Biosystems Step-One plus thermocycler.
Heparinase treatment of cells.
Cell monolayers were incubated in Opti-MEM with no FBS that was supplemented with 1× NEB heparinase buffer and 4 units each of heparinase I, II, and III (NEB). The cells were then incubated for 2 h at 30°C, after which the enzyme and medium were removed and the cell monolayers washed, and then the heparinase-treated cells were used for binding and entry assays as described above.
Immunoprecipitation, SDS-PAGE, and Western blotting.
Cell extracts were prepared by scraping cells into 1 ml PBS, centrifuging the cells at 800 × g for 2 min and then lysing the cell pellet in protein extraction buffer (10 mM Tris-HCl [pH 7.4], 5 mM EDTA, 150 mM NaCl, 1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride) and then centrifuged for 5 min at 13,000 × g. The solubilized fraction was diluted in 2× SDS gel loading buffer, boiled for 5 min, and then samples were loaded onto SDS-polyacrylamide gel and separated by electrophoresis. For immunoprecipitation of soluble gH/gL complexes, 0.5 μg of purified gH/gL, trimer, or pentamer was diluted in 1 ml PBS and incubated with 0.5 μg of MAb 14-4b for 1 h at 4°C followed by 1 h with protein A agarose. The protein A agarose was then washed 3 times with PBS supplemented with 0.5% NP-40, and the protein was eluted from the protein A agarose by addition of SDS gel loading buffer and prepared for electrophoresis as described above.
Proteins were electrophoretically transferred to Immobilon membranes (Millipore) in a buffer containing 25 mM Tris, 192 mM glycine, and 20% methanol. Membranes were blocked in TBST (Tris-buffered saline with 0.1% Tween 20 and 5% FBS) for 30 min and then probed with anti-PDGFRα rabbit polyclonal antibody 31614 (Cell Signaling Technologies), anti-beta actin polyclonal (Sigma-Aldrich), or anti-gH MAb AP86 at 1:1,000 dilution in TBST and allowed to incubate at 4°C overnight. The blots were then washed three times with TBST and then incubated for 1 h with horseradish peroxidase (HRP)-conjugated goat anti-mouse-HRP IgG (Jackson) diluted 1:1,000 in TBST. The blots were washed with TBST, and proteins were visualized by incubating membranes in ECL chemiluminescent detection reagent (Pierce) and then exposing the membranes in an Image-Quant LAS 4000 detection system (General Electric).
ACKNOWLEDGMENTS
We are very grateful to Aurelie Snyder and Stefanie Kaech Petrie of the OHSU Advanced Microscopy Core for technical assistance with fluorescence microscopy. We also thank Sunwen Chou for providing foscarnet and ganciclovir.
Funding for this research was provided by National Institutes of Health grants R01AI081517 (D.C.J.) and R21AI133192 (T.S.J.). The project was also supported by the Stanford Child Health Research Institute. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Britt W. 2006. Human cytomegalovirus infections and mechanisms of disease, p 1–28. In Reddehase MJ. (ed), Cytomegaloviruses: molecular biology and immunology. Caister Academic Press, Poole, United Kingdom. [Google Scholar]
- 2.Britt W. 2008. Manifestations of human cytomegalovirus infection: proposed mechanisms of acute and chronic disease. Curr Top Microbiol Immunol 325:417–470. [DOI] [PubMed] [Google Scholar]
- 3.Alford CA, Britt WJ. 1990. Cytomegalovirus, p 1981–2010. In Fields BN, Knipe DM, Howley PM, Chanock RM, Monath TP, Melnick JL, Roizman B, Straus SE (ed), Fields virology. Raven Press Ltd, New York, NY. [Google Scholar]
- 4.Pass RF. 2001. Cytomegalovirus, p 2675–2706. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MD, Roizman B, Straus SE (ed), Fields virology, 4th ed Lippincott Willliams & Davis, Philadelphia, PA. [Google Scholar]
- 5.Streblow DN, Orloff SL, Nelson JA. 2007. Acceleration of allograft failure by cytomegalovirus. Curr Opin Immunol 19:577–582. doi: 10.1016/j.coi.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sathiyamoorthy K, Chen J, Longnecker R, Jardetzky TS. 2017. The COMPLEXity in herpesvirus entry. Curr Opin Virol 24:97–104. doi: 10.1016/j.coviro.2017.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krummenacher C, Carfí A, Eisenberg RJ, Cohen GH. 2013. Entry of herpesviruses into cells: the enigma variations. Adv Exp Med Biol 790:178–195. doi: 10.1007/978-1-4614-7651-1_10. [DOI] [PubMed] [Google Scholar]
- 8.Vanarsdall AL, Johnson DC. 2012. Human cytomegalovirus entry into cells. Curr Opin Virol 2:37–42. doi: 10.1016/j.coviro.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huber MT, Compton T. 1998. The human cytomegalovirus UL74 gene encodes the third component of the glycoprotein H-glycoprotein L-containing envelope complex. J Virol 72:8191–8197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang D, Shenk T. 2005. Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tropism. Proc Natl Acad Sci U S A 102:18153–18158. doi: 10.1073/pnas.0509201102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adler B, Scrivano L, Ruzcics Z, Rupp B, Sinzger C, Koszinowski U. 2006. Role of human cytomegalovirus UL131A in cell type-specific virus entry and release. J Gen Virol 87:2451–2460. doi: 10.1099/vir.0.81921-0. [DOI] [PubMed] [Google Scholar]
- 12.Hahn G, Revello MG, Patrone M, Percivalle E, Campanini G, Sarasini A, Wagner M, Gallina A, Milanesi G, Koszinowski U, Baldanti F, Gerna G. 2004. Human cytomegalovirus UL131-128 genes are indispensable for virus growth in endothelial cells and virus transfer to leukocytes. J Virol 78:10023–10033. doi: 10.1128/JVI.78.18.10023-10033.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang D, Shenk T. 2005. Human cytomegalovirus UL131 open reading frame is required for epithelial cell tropism. J Virol 79:10330–10338. doi: 10.1128/JVI.79.16.10330-10338.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vanarsdall AL, Chase MC, Johnson DC. 2011. Human cytomegalovirus glycoprotein gO complexes with gH/gL, promoting interference with viral entry into human fibroblasts but not entry into epithelial cells. J Virol 85:11638–11645. doi: 10.1128/JVI.05659-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ryckman BJ, Chase MC, Johnson DC. 2008. HCMV gH/gL/UL128-131 interferes with virus entry into epithelial cells: evidence for cell type-specific receptors. Proc Natl Acad Sci U S A 105:14118–14123. doi: 10.1073/pnas.0804365105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wille PT, Knoche AJ, Nelson JA, Jarvis MA, Johnson DC. 2010. A human cytomegalovirus gO-null mutant fails to incorporate gH/gL into the virion envelope and is unable to enter fibroblasts and epithelial and endothelial cells. J Virol 84:2585–2596. doi: 10.1128/JVI.02249-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chandramouli S, Malito E, Nguyen T, Luisi K, Donnarumma D, Xing Y, Norais N, Yu D, Carfi A. 2017. Structural basis for potent antibody-mediated neutralization of human cytomegalovirus. Sci Immunol 2(12):eaan1457. doi: 10.1126/sciimmunol.aan1457. [DOI] [PubMed] [Google Scholar]
- 18.Ciferri C, Chandramouli S, Donnarumma D, Nikitin PA, Cianfrocco MA, Gerrein R, Feire AL, Barnett SW, Lilja AE, Rappuoli R, Norais N, Settembre EC, Carfi A. 2015. Structural and biochemical studies of HCMV gH/gL/gO and Pentamer reveal mutually exclusive cell entry complexes. Proc Natl Acad Sci U S A 112:1767–1772. doi: 10.1073/pnas.1424818112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vanarsdall AL, Wisner TW, Lei H, Kazlauskas A, Johnson DC. 2012. PDGF receptor-alpha does not promote HCMV entry into epithelial and endothelial cells but increased quantities stimulate entry by an abnormal pathway. PLoS Pathog 8:e1002905. doi: 10.1371/journal.ppat.1002905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kabanova A, Marcandalli J, Zhou T, Bianchi S, Baxa U, Tsybovsky Y, Lilleri D, Silacci-Fregni C, Foglierini M, Fernandez-Rodriguez BM, Druz A, Zhang B, Geiger R, Pagani M, Sallusto F, Kwong PD, Corti D, Lanzavecchia A, Perez L. 2016. Platelet-derived growth factor-α receptor is the cellular receptor for human cytomegalovirus gHgLgO trimer. Nat Microbiol 1:16082. doi: 10.1038/nmicrobiol.2016.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stegmann C, Hochdorfer D, Lieber D, Subramanian N, Stöhr D, Laib Sampaio K, Sinzger C. 2017. A derivative of platelet-derived growth factor receptor alpha binds to the trimer of human cytomegalovirus and inhibits entry into fibroblasts and endothelial cells. PLoS Pathog 13:e1006273. doi: 10.1371/journal.ppat.1006273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu Y, Prager A, Boos S, Resch M, Brizic I, Mach M, Wildner S, Scrivano L, Adler B. 2017. Human cytomegalovirus glycoprotein complex gH/gL/gO uses PDGFR-α as a key for entry. PLoS Pathog 13:e1006281. doi: 10.1371/journal.ppat.1006281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vanarsdall AL, Pritchard SR, Wisner TW, Liu J, Jardetzky TS, Johnson DC. 2018. CD147 promotes entry of pentamer-expressing human cytomegalovirus into epithelial and endothelial cells. mBio 9(3):e00781-. doi: 10.1128/mBio.00781-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang X, Huong SM, Chiu ML, Raab-Traub N, Huang ES. 2003. Epidermal growth factor receptor is a cellular receptor for human cytomegalovirus. Nature 424:456–461. doi: 10.1038/nature01818. [DOI] [PubMed] [Google Scholar]
- 25.Isaacson MK, Compton T. 2009. Human cytomegalovirus glycoprotein B is required for virus entry and cell-to-cell spread but not for virion attachment, assembly, or egress. J Virol 83:3891–3903. doi: 10.1128/JVI.01251-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chan G, Nogalski MT, Yurochko AD. 2009. Activation of EGFR on monocytes is required for human cytomegalovirus entry and mediates cellular motility. Proc Natl Acad Sci U S A 106:22369–22374. doi: 10.1073/pnas.0908787106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Durocher Y, Perret S, Kamen A. 2002. High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res 30:E9. doi: 10.1093/nar/30.2.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vanarsdall AL, Howard PW, Wisner TW, Johnson DC. 2016. Human cytomegalovirus gH/gL forms a stable complex with the fusion protein gB in virions. PLoS Pathog 12:e1005564. doi: 10.1371/journal.ppat.1005564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bogner E, Reschke M, Reis B, Reis E, Britt W, Radsak K. 1992. Recognition of compartmentalized intracellular analogs of glycoprotein H of human cytomegalovirus. Arch Virol 126:67–80. doi: 10.1007/BF01309685. [DOI] [PubMed] [Google Scholar]
- 30.Sampaio KL, Cavignac Y, Stierhof YD, Sinzger C. 2005. Human cytomegalovirus labeled with green fluorescent protein for live analysis of intracellular particle movements. J Virol 79:2754–2767. doi: 10.1128/JVI.79.5.2754-2767.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Compton T, Nowlin DM, Cooper NR. 1993. Initiation of human cytomegalovirus infection requires initial interaction with cell surface heparan sulfate. Virology 193:834–841. doi: 10.1006/viro.1993.1192. [DOI] [PubMed] [Google Scholar]
- 32.Carlson C, Britt WJ, Compton T. 1997. Expression, purification, and characterization of a soluble form of human cytomegalovirus glycoprotein B. Virology 239:198–205. doi: 10.1006/viro.1997.8892. [DOI] [PubMed] [Google Scholar]
- 33.Kari B, Gehrz R. 1993. Structure, composition and heparin binding properties of a human cytomegalovirus glycoprotein complex designated gC-II. J Gen Virol 74:255–264. doi: 10.1099/0022-1317-74-2-255. [DOI] [PubMed] [Google Scholar]
- 34.Soroceanu L, Akhavan A, Cobbs CS. 2008. Platelet-derived growth factor-alpha receptor activation is required for human cytomegalovirus infection. Nature 455:391–395. doi: 10.1038/nature07209. [DOI] [PubMed] [Google Scholar]
- 35.Lei H, Rheaume MA, Velez G, Mukai S, Kazlauskas A. 2011. Expression of PDGFRalpha is a determinant of the PVR potential of ARPE19 cells. Invest Ophthalmol Vis Sci 52:5016–5021. doi: 10.1167/iovs.11-7442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou M, Lanchy JM, Ryckman BJ. 2015. Human cytomegalovirus gH/gL/gO promotes the fusion step of entry into all cell types, whereas gH/gL/UL128-131 broadens virus tropism through a distinct mechanism. J Virol 89:8999–9009. doi: 10.1128/JVI.01325-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hahn A, Birkmann A, Wies E, Dorer D, Mahr K, Sturzl M, Titgemeyer F, Neipel F. 2009. Kaposi's sarcoma-associated herpesvirus gH/gL: glycoprotein export and interaction with cellular receptors. J Virol 83:396–407. doi: 10.1128/JVI.01170-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ryckman BJ, Jarvis MA, Drummond DD, Nelson JA, Johnson DC. 2006. Human cytomegalovirus entry into epithelial and endothelial cells depends on genes UL128 to UL150 and occurs by endocytosis and low-pH fusion. J Virol 80:710–722. doi: 10.1128/JVI.80.2.710-722.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vanarsdall AL, Ryckman BJ, Chase MC, Johnson DC. 2008. Human cytomegalovirus glycoproteins gB and gH/gL mediate epithelial cell-cell fusion when expressed either in cis or in trans. J Virol 82:11837–11850. doi: 10.1128/JVI.01623-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang D, Bresnahan W, Shenk T. 2004. Human cytomegalovirus encodes a highly specific RANTES decoy receptor. Proc Natl Acad Sci U S A 101:16642–16647. doi: 10.1073/pnas.0407233101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim JH, Collins-McMillen D, Buehler JC, Goodrum FD, Yurochko AD. 2017. Human cytomegalovirus requires epidermal growth factor receptor signaling to enter and initiate the early steps in the establishment of latency in CD34+ human progenitor cells. J Virol 91:e01206-16. doi: 10.1128/JVI.01206-16. [DOI] [PMC free article] [PubMed] [Google Scholar]