

MERS-CoV represents a high public health threat. Because CoV nsp1 is a major viral virulence factor, uncovering the biological functions of MERS-CoV nsp1 could contribute to our understanding of MERS-CoV pathogenicity and spur development of medical countermeasures. Expressed MERS-CoV nsp1 suppresses host gene expression, but its biological functions for virus replication and effects on host gene expression in infected cells are largely unexplored. We found that nsp1 suppressed host gene expression in infected cells. Our data further demonstrated that nsp1, which was not detected in virus particles, promoted virus assembly or budding in a 293-derived cell line, leading to efficient virus replication. These data suggest that nsp1 plays an important role in MERS-CoV replication and possibly affects virus-induced diseases by promoting virus particle production in infected hosts. Our data, which uncovered an unexpected novel biological function of nsp1 in virus replication, contribute to further understanding of the MERS-CoV replication strategies.
KEYWORDS: MERS coronavirus, nsp1, virus assembly/budding
ABSTRACT
Middle East respiratory syndrome coronavirus (MERS-CoV) nsp1 suppresses host gene expression in expressed cells by inhibiting translation and inducing endonucleolytic cleavage of host mRNAs, the latter of which leads to mRNA decay. We examined the biological functions of nsp1 in infected cells and its role in virus replication by using wild-type MERS-CoV and two mutant viruses with specific mutations in the nsp1; one mutant lacked both biological functions, while the other lacked the RNA cleavage function but retained the translation inhibition function. In Vero cells, all three viruses replicated efficiently with similar replication kinetics, while wild-type virus induced stronger host translational suppression and host mRNA degradation than the mutants, demonstrating that nsp1 suppressed host gene expression in infected cells. The mutant viruses replicated less efficiently than wild-type virus in Huh-7 cells, HeLa-derived cells, and 293-derived cells, the latter two of which stably expressed a viral receptor protein. In 293-derived cells, the three viruses accumulated similar levels of nsp1 and major viral structural proteins and did not induce IFN-β and IFN-λ mRNAs; however, both mutants were unable to generate intracellular virus particles as efficiently as wild-type virus, leading to inefficient production of infectious viruses. These data strongly suggest that the endonucleolytic RNA cleavage function of the nsp1 promoted MERS-CoV assembly and/or budding in a 293-derived cell line. MERS-CoV nsp1 represents the first CoV gene 1 protein that plays an important role in virus assembly/budding and is the first identified viral protein whose RNA cleavage-inducing function promotes virus assembly/budding.
IMPORTANCE MERS-CoV represents a high public health threat. Because CoV nsp1 is a major viral virulence factor, uncovering the biological functions of MERS-CoV nsp1 could contribute to our understanding of MERS-CoV pathogenicity and spur development of medical countermeasures. Expressed MERS-CoV nsp1 suppresses host gene expression, but its biological functions for virus replication and effects on host gene expression in infected cells are largely unexplored. We found that nsp1 suppressed host gene expression in infected cells. Our data further demonstrated that nsp1, which was not detected in virus particles, promoted virus assembly or budding in a 293-derived cell line, leading to efficient virus replication. These data suggest that nsp1 plays an important role in MERS-CoV replication and possibly affects virus-induced diseases by promoting virus particle production in infected hosts. Our data, which uncovered an unexpected novel biological function of nsp1 in virus replication, contribute to further understanding of the MERS-CoV replication strategies.
INTRODUCTION
Middle East respiratory syndrome (MERS) is a viral respiratory illness caused by MERS coronavirus (MERS-CoV), which was first identified in Saudi Arabia in 2012 (1). MERS outbreaks continue with increasing geographical distribution (2), and the mortality rate of MERS is approximately 36% (http://www.who.int/emergencies/mers-cov/en/). MERS-CoV represents a high public health threat, yet no vaccine or specific treatment for MERS is currently available.
CoVs belong to the order Nidovirales in the family Coronaviridae, and are currently classified into four genera, alpha, beta, gamma, and delta CoVs. CoV is an enveloped virus carrying a large single-stranded, nonsegmented RNA with the 5′ end capped and the 3′ end polyadenylated (3–5). Replication of MERS-CoV, a beta CoV, starts with binding of the virus particle to a receptor, dipeptidyl peptidase 4 (6), which is also called CD26. After virus-host membrane fusion (7), the viral genomic RNA is released into the cytoplasm and undergoes translation of partially overlapping two large precursor polyproteins from gene 1, which encompasses the 5′ two-thirds of the genome. These precursor polyproteins are proteolytically processed by two virally encoded proteinases to generate 16 mature proteins, nonstructural protein 1 (nsp1) to nsp16 (8). All of these gene 1 proteins, except for nsp1 (9) and nsp2 (10), are considered to be essential for CoV RNA synthesis (11). MERS-CoV replication results in accumulation of eight viral mRNAs, including mRNA 1, the intracellular forms of viral genome, and subgenomic mRNAs 2 to 8 (12, 13); these viral mRNAs form the 3′-coterminal nested structure, and all carry the same leader sequence of ∼70 nucleotides at the 5′ end (14–16). Viral structural proteins (S, E, M, and N proteins) and four accessory proteins (3, 4a, 4b, and 5 proteins) are translated from these subgenomic mRNAs. MERS-CoV accessory proteins are not essential for virus replication, and yet they affect viral pathogenicity (17–19). Accumulation of viral proteins and mRNA 1 leads to the assembly of virus particles and budding of virus particles at endoplasmic reticulum Golgi intermediate compartment (ERGIC) membranes (20–22), followed by subsequent release of the virus from the cells. CoV M protein plays a central role in virus assembly (23–29). In many CoVs, including MERS-CoV, E protein, a low-abundance protein in the virus particle, is essential for production of infectious virus particles (23, 30–32), while severe acute respiratory syndrome CoV (SARS-CoV) mutant lacking E protein is viable but attenuated in growth (33).
Among the four CoV genera, only alpha and beta CoVs encode nsp1 (34). In contrast to nsp3 to nsp16 that play essential roles in exert viral RNA synthesis, nsp1 shares low amino acid homology among CoVs (35–39), and the sizes of beta CoV nsp1 and alpha CoV nsp1 differ; the former and the latter were ∼28 and ∼9 kDa, respectively. Nonetheless, structural analysis suggests that CoV nsp1 has a common origin (36) and that the nsp1s of alpha and beta CoVs share a biological function to inhibit host gene expression. Past studies suggest that mechanisms of host gene expression suppression induced by nsp1 of each CoV species may differ (39–43). Among CoV nsp1s, mechanisms of nsp1-induced host gene suppression have been well characterized in severe respiratory syndrome CoV (SARS-CoV) nsp1. SARS-CoV nsp1 is a cytoplasmic protein that binds to the 40S ribosomal subunit (40, 41) and inactivates its translation function, which leads to translation inhibition. The SARS-CoV nsp1-40S ribosome complex also induces endonucleolytic cleavage of host mRNAs. Host 5′-3′ exonuclease, Xrn 1, further degrades host mRNAs that undergo the nsp1-induced RNA cleavage (44). Although nsp1 suppresses translation of SARS-CoV mRNAs, it does not induce endonucleolytic cleavage of SARS-CoV mRNAs (45). Like SARS-CoV nsp1, expressed MERS-CoV nsp1 suppresses translation and induces endonucleolytic RNA cleavage of host mRNA, leading to host mRNA decay (43). However, unlike SARS-CoV nsp1, MERS-CoV nsp1 localized in both the cytoplasm and nucleus, does not bind to 40S ribosomes, and targets host mRNAs of the nuclear origin, but not mRNAs of cytoplasmic origin (43). Several lines of evidence point toward the strong possibility that nsp1 is a major virulence factor of CoVs. SARS-CoV nsp1 suppresses the host innate immune functions by inhibiting interferon (IFN) expression (46) and host antiviral signaling pathways in infected cells (47). The nsp1 of porcine epidemic diarrhea virus suppresses type II IFN (48). The contribution of nsp1 in CoV pathogenesis has been demonstrated for mouse hepatitis virus and SARS-CoV (49–51).
Although it has been considered that nsp1 is not essential for CoV RNA synthesis (9), the biological roles of nsp1 in CoV replication are not well understood (39–41, 46, 52, 53). Our present study demonstrated that, as in expressed cells, MERS-CoV nsp1 suppressed host gene expression in infected cells. Unexpectedly, our studies revealed that the RNA cleavage function of the MERS-CoV nsp1 promoted virus assembly or budding in a 293-derived cell line. To our knowledge, MERS-CoV nsp1 is the first recognized CoV gene 1 protein that plays an important role in the production of infectious virus particles. Furthermore, MERS-CoV nsp1 is the first viral protein whose RNA cleavage-inducing function promoted the assembly/budding of virus particles.
RESULTS
Generation of MERS-CoV nsp1 mutants lacking host gene suppression functions.
Toward understanding the roles of MERS-CoV nsp1 in host gene expression and virus replication, we aimed to generate a MERS-CoV nsp1 mutant that lacks both host mRNA cleavage and host mRNA translation inhibition functions. Because the alanine substitutions of two charged amino acid residues, K164 and H165, of the 180-amino-acid-long SARS-CoV nsp1 abolish translation inhibition function and the endonucleolytic RNA cleavage function (46), we hypothesized that alanine substitution of a charged amino acid residue(s) near the C-terminal region of MERS-CoV nsp1 (193 amino acids) would also disrupt the MERS-CoV nsp1's host gene suppression functions. Since alignment of amino acid sequences of MERS-CoV nsp1 and SARS-CoV nsp1 showed that K181 of MERS-CoV nsp1 corresponded to K164 of SARS-CoV nsp1, we hypothesized that K181A mutation in MERS-CoV nsp1 would disrupt the host gene suppression functions and constructed a T7 plasmid that expressed transcripts encoding MERS-CoV nsp1 with K181A mutation (MERS-CoV nsp1-mt).
To investigate the biological functions of MERS-CoV nsp1-mt, we independently transfected 293 cells with capped and polyadenylated RNA transcripts encoding chloramphenicol acetyltransferase (CAT), SARS-CoV nsp1, wild-type MERS-CoV nsp1 (MERS-CoV nsp1-WT), MERS-CoV nsp1-mt, and MERS-CoV nsp1 mutant carrying R125A and K126A mutation (MERS-CoV nsp1-CD), the latter of which lacks the endonucleolytic RNA cleavage activity but retains the translation suppression function (43). All encoded proteins carried a C-terminal myc tag. The cells were radiolabeled with Tran35S-label from 8.5 to 9.5 h after transfection, and cell extracts were subjected to SDS-PAGE analysis. Consistent with our previous reports (43, 46), expression of SARS-CoV nsp1, MERS-CoV nsp1-WT, and MERS-CoV nsp1-CD suppressed host protein synthesis (Fig. 1A, top two panels). In contrast, MERS-CoV nsp1-mt protein expression did not inhibit host protein synthesis. We also confirmed the expression of CAT, SARS-CoV nsp1, MERS-CoV nsp1-WT, MERS-CoV nsp1-CD, and MERS-CoV nsp1-mt (Fig. 1A, bottom two panels).
FIG 1.

Characterization of the loss-of-function mutant, MERS-CoV nsp1-mt, in expressed cells. (A) 293 cells were transfected with 2 μg of capped and polyadenylated RNA transcripts encoding CAT, SARS-CoV nsp1, MERS-CoV nsp1-WT, MERS-CoV nsp1-CD, or MERS-CoV nsp1-mt, all of which carried a C-terminal myc epitope tag, radiolabeled with Tran35S-label from 8.5 to 9.5 h posttransfection. Lysates were resolved using SDS–12% PAGE, followed by autoradiography (top panel), colloidal Coomassie blue staining (middle panel), and Western blot analysis with anti-myc and tubulin antibodies (bottom two panels). (B) 293 cells were transfected with RNA transcripts as described in panel A. Intracellular RNAs were extracted at 9 h posttransfection and subjected to Northern blot analysis using a probe for GAPDH mRNA (top). The 28S and 18S rRNAs were detected by ethidium bromide staining (bottom). (C) A schematic diagram of Ren-EMCV-FF is shown at the top of the panel. 293 cells were cotransfected with a plasmid encoding Ren-EMCV-FF and the plasmid expressing CAT, SARS-CoV nsp1, MERS-CoV nsp1-WT, MERS-CoV nsp1-CD, or MERS-CoV nsp1-mt protein; all nsp1s carried the C-terminal myc tag. At 24 h posttransfection, intracellular RNAs were extracted and subjected to Northern blot analysis using an RNA probe that binds to the rLuc gene (second panel). Arrowhead, full-length Ren-EMCV-FF; arrow, cleaved RNA fragment. The 28S and 18S rRNAs were detected by ethidium bromide staining (third panel). Cell extracts, prepared at 24 h posttransfection, were used for Western blot analysis using anti-myc and tubulin antibodies (fourth and fifth panels).
Next, we tested the effect of MERS-CoV nsp1-mt expression on abundance of a host mRNA. First, 293 cells were transfected with the RNA transcripts as described above. Intracellular RNAs were extracted at 9 h posttransfection and subjected to Northern blot analysis using a probe detecting glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA (Fig. 1B). Reduction of GAPDH mRNA abundance occurred in cells expressing SARS-CoV nsp1 or MERS-CoV nsp1-WT, but not in those expressing MERS-CoV nsp1-CD or CAT (43). MERS-CoV nsp1-mt expression also did not induce reduction in the abundance of GAPDH mRNA, suggesting that MERS-CoV-mt did not induce the endonucleolytic RNA cleavage to GAPDH mRNA and subsequent mRNA degradation.
To establish that MERS-CoV nsp1-mt lacks the endonucleolytic RNA cleavage function, 293 cells were transfected with a plasmid encoding CAT, MERS-CoV nsp1-WT, MERS-CoV nsp1-CD, or MERS-CoV nsp1-mt, together with a plasmid encoding a bicistronic reporter mRNA (Ren-EMCV-FF RNA) carrying the encephalomyocarditis virus internal ribosomal entry sites (EMCV IRES) between the upstream Renilla luciferase (rLuc) gene and the downstream firefly luciferase (fLuc) gene (Fig. 1C, top panel); all expressed proteins carried a C-terminal myc tag. SARS-CoV nsp1 and MERS-CoV nsp1-WT served as positive controls as they induce endonucleolytic RNA cleavage within the EMCV IRES region of Ren-EMCV-FF RNA (40, 43, 45), while CAT and MERS-CoV nsp1-CD served as negative controls. Intracellular RNAs were extracted at 24 h posttransfection and subjected to Northern blot analysis using rLuc probe. Expression of MERS-CoV nsp1-WT and SARS-CoV nsp1 induced endonucleolytic cleavage of Ren-EMCV-FF RNA, generating a fast migrating RNA fragment (Fig. 1C, second panel; see arrowhead) and reduction in the amounts of the full-length Ren-EMCV-FF RNA (Fig. 1C, second panel; see arrow). Consistent with our previous report (43), SARS-CoV nsp1 was more active than MERS-CoV nsp1-WT for inducing RNA cleavage. The RNA fragment was absent in cells expressing the MERS-CoV nsp1-mt, demonstrating that the MERS-CoV nsp1-mt lacked the endonucleolytic RNA cleavage activity. Western blat analysis confirmed expression of SARS-CoV nsp1, MERS-CoV nsp1-WT, MERS-CoV nsp1-CD, and MERS-CoV nsp1-mt in transfected cells (Fig. 1C, fourth panel). Consistent with our previous report (43), SARS-CoV nsp1 and MERS-CoV nsp1-WT accumulated poorly in expressed cells; these nsp1s probably targeted their own template mRNAs for degradation, leading to poor protein accumulation.
MERS-CoV nsp1-CD, which is deficient for the endonucleolytic RNA cleavage function (43), suppressed host translation (Fig. 1A), demonstrating that MERS-CoV nsp1-CD retained its translational suppression function. The absence of host translation inhibition in cells expressing MERS-CoV nsp1-mt demonstrated that MERS-CoV nsp1-mt lost both the RNA cleavage function and the translation suppression function.
Replication of MERS-CoV mutants encoding mutant nsp1 in Vero cells.
To explore the role of nsp1 in virus replication and host gene expression, we rescued MERS-CoV-WT encoding MERS-CoV nsp1-WT, MERS-CoV-CD carrying MERS-CoV nsp1-CD, and MERS-CoV-mt carrying MERS-CoV nsp1-mt by using a reverse-genetics system (54). All three viruses replicated efficiently with similar replication kinetics in Vero cells (Fig. 2A). Also, all of the viruses accumulated similar levels of viral structural proteins, S, M, and N, nsp1, and virus-specific mRNAs at each indicated time point (Fig. 2B and C).
FIG 2.

Growth kinetics of MERS-CoV-WT, -CD, and -mt and accumulation of viral proteins and RNA in infected Vero cells. (A) Vero cells were infected with MERS-CoV-WT (WT), MERS-CoV-CD (CD), or MERS-CoV-mt (mt) at an MOI of 0.01 (left panel) or 3 (right panel). Culture supernatants were collected at the indicated times, and virus titers were determined by plaque assay in Vero cells. The results represent the averages of three independent experiments. Each bar represents the mean (± the standard deviation) for three samples. (B) Vero cells were infected with each of the three viruses at an MOI of 3. At indicated times postinfection, total proteins were extracted and Western blot analysis was performed to detect the S, M, N, nsp1, and tubulin by using anti-MERS-CoV S, M, N, nsp1, and tubulin antibody, respectively. (C) Vero cells were infected with each of the three viruses at an MOI of 3. At the indicated times, total RNAs were extracted. The viral mRNAs were detected by Northern blotting using a probe that binds to the 3′ end of the MERS-CoV genome. The 28S and 18S rRNAs were detected by ethidium bromide staining.
Next, we examined the effects of nsp1 for host mRNA stability and host protein synthesis in infected Vero cells. The abundance of host GAPDH mRNA was lower in MERS-CoV-WT-infected cells than in MERS-CoV-CD- and MERS-CoV-mt-infected cells (Fig. 3A). Replication of MERS-CoV-WT, but not the two mutant viruses, in the presence of actinomycin D (ActD), also resulted in reduced GAPDH mRNA levels (Fig. 3B, right panel), demonstrating that nsp1 induced efficient degradation of preexisting GAPDH mRNA in infected cells. Metabolic radiolabeling experiments showed that replication of MERS-CoV-WT, as well as the two mutant viruses, induced an inhibition of host protein synthesis. (Fig. 3C). Although the extent of host translation inhibition induced by these viruses was modest at 24 h postinfection (p.i.), a stronger inhibition of host translation was observed in MERS-CoV-WT-infected cells than in those infected with the mutant viruses at 32 h p.i., suggesting that the strong inhibition of host gene expression was due to a combined effect of the nsp1-mediated RNA cleavage and the translation suppression function. Taken together, these data established that nsp1 suppressed host gene expression by inducing host mRNA decay and inhibiting host translation in infected cells.
FIG 3.
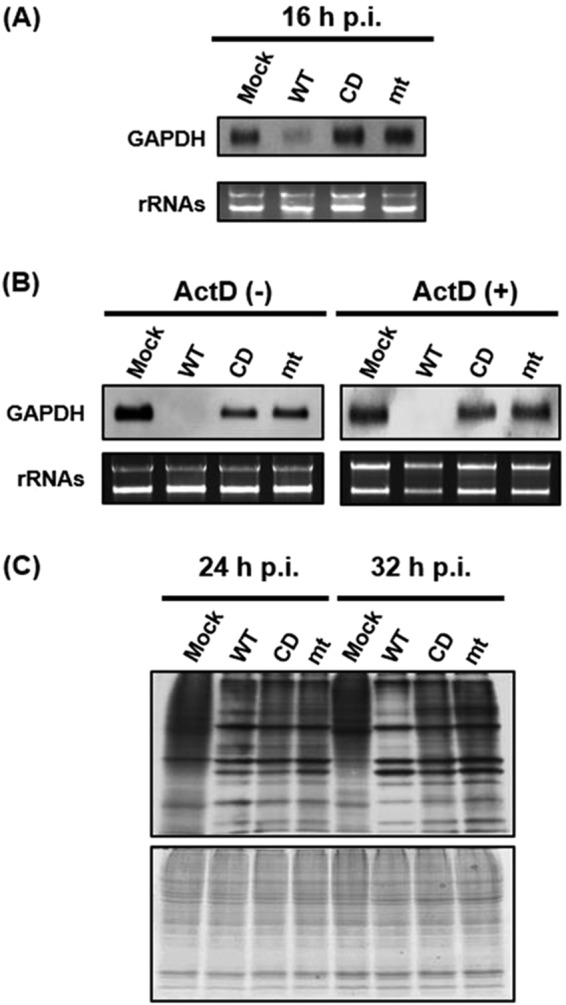
Effect of replication of MERS-CoV-WT, MERS-CoV-CD, and MERS-CoV-mt on abundance of host endogenous mRNA and host protein synthesis in Vero cells. (A and B) Vero cells were either mock infected (Mock) or infected with MERS-CoV-WT (WT), MERS-CoV-CD (CD), or MERS-CoV-mt (mt) at an MOI of 3. At 16 h p.i. (A) or 24 h p.i. (B, left panel), intracellular RNAs were extracted. For testing host mRNA decay in infected cells, ActD was added to the culture at 1 h p.i., and intracellular RNAs were extracted at 24 h p.i. (B, right panel). The abundance of GAPDH mRNA was determined using Northern blot analysis (top panel). The 28S and 18S rRNAs were detected by ethidium bromide staining (bottom panel). (C) Vero cells were either mock infected (Mock) or infected with MERS-CoV-WT (WT), MERS-CoV-CD (CD), or MERS-CoV-mt (mt) at an MOI of 3. The cells were radiolabeled for 1 h with Tran35S-label, and cell lysates were prepared at the indicated times postinfection. Cell lysates were subjected to SDS-PAGE analysis, followed by autoradiography (top panel) and colloidal Coomassie blue staining (bottom panel).
Replication of MERS-CoV-WT and the mutant viruses in various cell lines.
We subsequently examined replication kinetics of the three viruses in various cell lines. All of the three viruses replicated efficiently with similar replication kinetics in Calu-3 cells, a human airway epithelial cell line (55), regardless of the multiplicity of infections (MOIs) (Fig. 4A). The three viruses replicated efficiently and similarly at an MOI of 3 in Huh-7 cells, a well-differentiated hepatocyte-derived cellular carcinoma cell line (56), except that the titer of MERS-CoV-WT was statistically ∼10-fold higher at peak titers than those of the mutants at 32 h p.i. (Fig. 4B). In contrast, the two mutant viruses replicated ∼2 logs less efficiently than MERS-CoV-WT in Huh-7 cells at an MOI of 0.01 (Fig. 4B). The titers of MERS-CoV-WT were statistically higher than those of the mutant viruses from 24 to 48 h p.i. at an MOI of 3 in 293 cells stably expressing human CD26 (293/CD26 cells) (Fig. 4C). Likewise, both mutant viruses replicated less efficiently than MERS-CoV-WT in 293/CD26 cells at an MOI of 0.01. MERS-CoV-WT also replicated to statistically higher titers than the two mutants throughout the infection at an MOI of 3 in HeLa cells stably expressing CD26 (HeLa/CD26 cells), while both mutants showed similar titers and replication kinetics. At an MOI of 0.01 in HeLa/CD26 cells, MERS-CoV-WT replicated to higher titers than the mutant viruses after 12 h p.i. Taken together, these data suggested that nsp1 promoted virus replication in a cell type-dependent manner.
FIG 4.

Replication kinetics of MERS-CoV-WT, MERS-CoV-CD, and MERS-CoV-mt in various cell lines. Calu-3 cells (A), Huh7-cells (B), 293/CD26 cells (C), and HeLa/CD26 cells (D) were infected with MERS-CoV-WT (WT), MERS-CoV-CD (CD), or MERS-CoV-mt (mt) at an MOI of 0.01 (left panels) or an MOI of 3 (right panels). Culture supernatants were collected at the indicated times, and virus titers were determined by plaque assay in Vero cells. The results represent the averages of three independent experiments. Each bar represents the mean (± the standard deviation) for three samples. Asterisks represent statically significant differences between the titers of MERS-CoV-WT and mutant viruses (P < 0.05).
Replication of the three viruses does not induce IFN-β and IFN-λ mRNAs.
To determine the mechanisms of nsp1-mediated promotion of virus replication, we used 293/CD26 cells for subsequent analyses. We first tested the possibility that the mutant nsp1 did not suppress the host innate immune responses, thereby promoting production of type I IFN and/or III IFN and leading to inhibition of virus replication. To this end, we examined induction of IFN-β and IFN-λ mRNAs in infected 293/CD26 cells (Fig. 5). Sendai virus (SeV) infection resulted in efficient induction of IFN-β and IFN-λ mRNAs, whereas mock-infected cells did not. None of the three viruses efficiently induced IFN-β and IFN-λ mRNAs from 8 h to 32 h p.i., suggesting that inefficient replication of MERS-CoV-mt and MERS-CoV-CD was not due to induction of type I and III IFNs and that the host gene suppression functions of MERS-CoV nsp1 did not play a significant role in inhibiting the induction of IFN-β and IFN-λ mRNAs in 293/CD26 cells.
FIG 5.
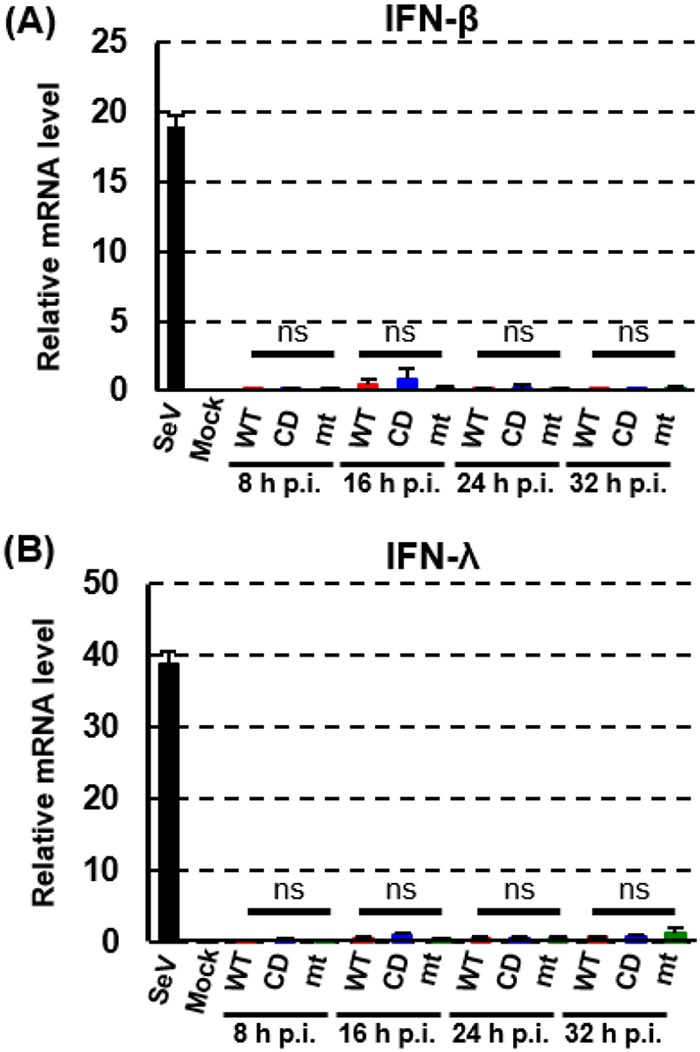
IFN-β and IFN-λ mRNA expression in infected 293/CD26 cells. 293/CD26 cells were either mock infected (M) or infected with MERS-CoV-WT (WT), MERS-CoV-CD (CD), or MERS-CoV-mt at an MOI of 3. SeV infection (100 HA units) was inoculated as a positive control. Total intracellular RNAs were extracted at the indicated times, and the amounts of endogenous IFN-β and -λ mRNA mRNAs were determined by qRT-PCR analysis. Expression levels of the genes were normalized to levels of 18S rRNA. Each bar represents the mean (± the standard deviation) for three wells.
Accumulation of viral proteins and mRNAs in infected 293/CD26 cells.
To discern whether inefficient replication of mutant viruses in 293/CD26 cells was due to poor accumulation of viral structural proteins, we examined the abundance of major viral structural proteins, including S, M, and N proteins. No substantial differences in the accumulation of these structural proteins were noted among cells infected with the three viruses (Fig. 6A). The three viruses also accumulated similar levels of nsp1 (Fig. 6A).
FIG 6.
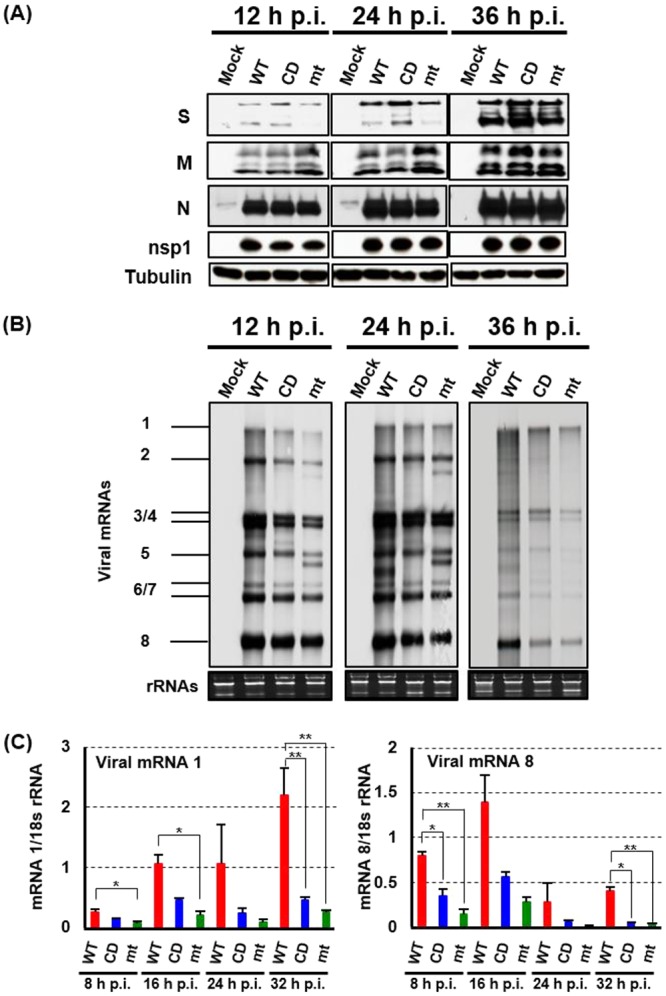
Accumulation of viral proteins and RNA in infected 293/CD26 cells. (A) 293/CD26 cells were either mock infected (Mock) or infected with MERS-CoV-WT (WT), MERS-CoV-CD (CD), or MERS-CoV-mt (mt) at an MOI of 3. At indicated times postinfection, total proteins were extracted (A) or total RNAs were extracted (B and C). (A) Western blot analysis was performed to detect the S, M, N, nsp1, and tubulin. (B) The viral mRNAs were detected by Northern blotting using a probe that binds to the 3′ end of the MERS-CoV genome, and the 28S and 18S rRNAs were detected by ethidium bromide staining. (C) The amounts of genomic RNA and subgenomic mRNA 8 were quantified by qRT-PCR, and expression levels were normalized to levels of 18s rRNA. Each bar represents the mean (± the standard deviation) for three wells.
Northern blot analysis showed that accumulation of viral mRNAs were marginally higher in MERS-CoV-WT-infected cells than in cells infected with the mutant viruses (Fig. 6B). In addition to eight mRNA species, we noted the presence of two additional virus-specific RNA bands; one migrated faster than mRNA 2 and the other migrated faster than mRNA 5 in MERS-CoV-mt-infected cells. We also detected another virus-specific RNA band that migrated between mRNA 5 and mRNAs 6/7 in MERS-CoV-WT-infected cells at 24 h p.i. The origins of these viral RNAs are currently unclear, yet they may represent defective RNAs or subgenomic mRNAs. qRT-PCR analyses revealed that mRNA 1 of MERS-CoV-WT accumulated higher abundance than that of MERS-CoV-mt at 8 h, 16 h, and 32 h p.i. and that of MERS-CoV-CD at 32 h p.i. In addition, the amount of mRNA 8 of MERS-CoV-WT was higher than those of the mutant viruses at 8 and 32 h p.i. (Fig. 6C). These studies showed that there was a trend that MERS-CoV-WT accumulated higher levels of viral mRNAs than the mutant viruses.
Analyses of host gene expression in infected 293/CD26 cells.
The effects of nsp1 for host mRNA stability and host protein synthesis in infected 293/CD26 cells were examined next. Replication of both mutant viruses did not affect abundance of GAPDH mRNA, while the abundance of GAPDH mRNA was substantially reduced in MERS-CoV-WT-infected cells (Fig. 7A), suggesting that MERS-CoV nsp1, but not MERS-CoV nsp1-CD and MERS-CoV nsp1-mt, induced degradation of GADPH mRNA in infected cells. Metabolic radiolabeling experiments showed that replication of all three viruses induced host protein synthesis inhibition at 24 h p.i. (Fig. 7B). Because MERS-CoV nsp1-mt were deficient for the translation inhibition and mRNA cleavage functions (Fig. 1), host translational suppression in MERS-CoV-mt-infected cells was independent from the nsp1 function. MERS-CoV-WT and MERS-CoV-CD induced slightly stronger host translational suppression than MERS-CoV-mt at 24 h p.i., suggesting that the translation suppression function of the nsp1 modestly contributed to host translation suppression in 293/CD26 cells.
FIG 7.
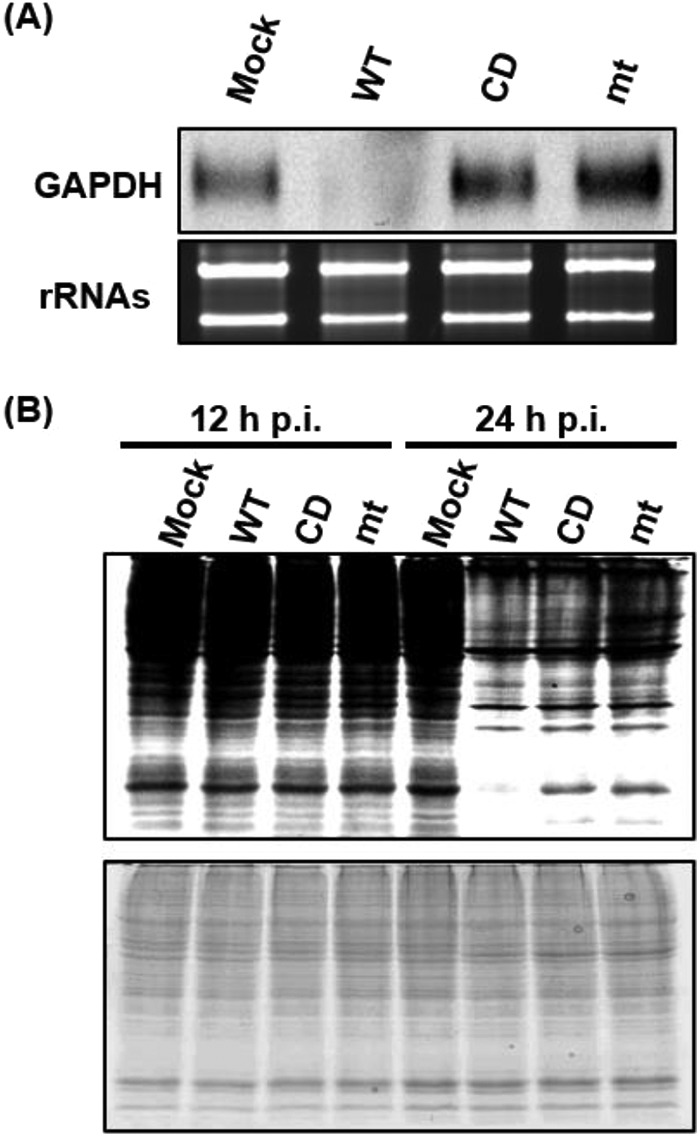
Effects of virus replication on GAPDH mRNA level and host protein synthesis in 293/CD26 cells. 293/CD26 cells were either mock-infected (Mock) or infected with MERS-CoV-WT (WT), MERS-CoV-CD (CD), or MERS-CoV-mt (mt) at an MOI of 3. (A) The abundance of GAPDH mRNA at 24 h p.i. was determined using Northern blot analysis. The 28S and 18S rRNAs were detected by ethidium bromide staining. (B) Cells were radiolabeled for 1 h with Tran35S-label, and cell lysates were prepared at the indicated times p.i. Cell lysates were subjected to SDS-PAGE analysis, followed by autoradiography (top panel) and colloidal Coomassie blue staining (bottom panel).
Titers of cell-associated virus and abundances of released virus particles among the three viruses.
MERS-CoV-WT replicated to higher titers than two mutant viruses in 293/CD26 cells (Fig. 4C), whereas accumulation of intracellular viral structural proteins were comparable among the three viruses (Fig. 6A). One possible interpretation of these data is that the mutant viruses were able to undergo assembly and budding of infectious virus particles as efficient as MERS-CoV-WT, yet the mutant viruses were unable to efficiently release infectious viruses from infected cells. If this is the case, the titers of the cell-associated viruses would be similar among the three viruses. We found that the titers of cell-associated MERS-CoV-WT were higher than those of the cell-associated mutant viruses at 24, 36, and 48 h p.i (Fig. 8A). Also, the titers of cell-associated MERS-CoV-CD were higher than those of cell-associated MERS-CoV-mt at 36 and 48 h p.i. (Fig. 8A). Low titers of cell-free and the cell-associated viruses (Fig. 4C and 8A) in mutant viruses suggested that the release of infectious viruses was not selectively inhibited in the mutant viruses. Rather, these data implied that the low titers of cell-free viruses in the mutant virus-infected cells were due to the accumulation of low titers of cell-associated viruses.
FIG 8.
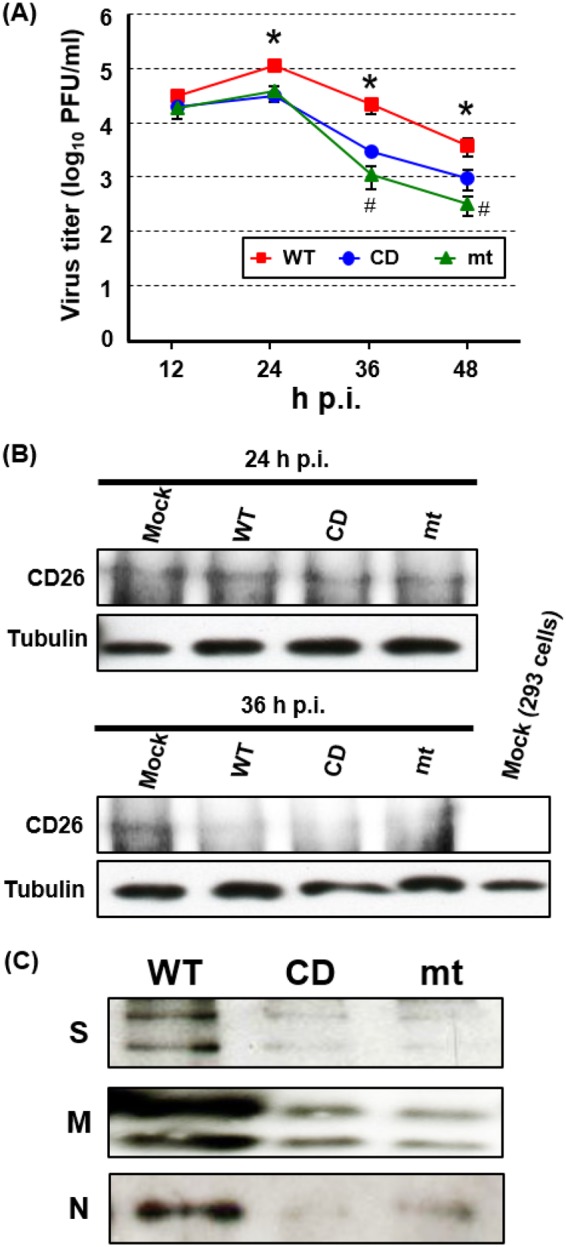
MERS-CoV-CD and MERS-CoV-mt undergo inefficient virus assembly in 293/CD26 cells. 293/CD26 cells were infected with MERS-CoV-WT (WT), MERS-CoV-CD (CD), or MERS-CoV-mt (mt) at an MOI of 3. (A) At the indicated time points postinfection, the titers of cell-associated viruses were determined by plaque assay. The results represent the averages of three independent experiments. Each bar represents the mean (± the standard deviation) for three samples. Asterisks represent statically significant differences between the titers of MERS-CoV-WT and mutant viruses (P < 0.05). Hash marks represent statically significant differences between the titers of MERS-CoV-CD and MERS-CoV-mt (P < 0.05). (B) At the indicated times postinfection, the total proteins were extracted and subjected to Western blot analysis using anti-human CD26 or tubulin antibody. Lysate of mock-infected 293 cells was included as a negative control in the far right lane of bottom panels. (C) At 36 h p.i., supernatants were collected and subjected to 60Co irradiation. The purified viruses were pelleted, dissolved in the same volume of sample buffer, and subjected to Western blot analysis by using anti-S, -M, and -N protein antibodies.
Because soluble CD26 binds to MERS-CoV particles and neutralizes virus infectivity (57), expressed CD26 might have bound to intracellular virus particles in the mutant virus-infected cells, leading to neutralization of the cell-associated virus particles and/or preventing virus release. In contrast, MERS-CoV nsp1-WT expression might have efficiently suppressed CD26 expression in MERS-CoV-WT-infected cells, preventing the putative binding of CD26 to intracellular virus particles. To test a likelihood of this possibility, we examined the abundance of CD26 in infected cells (Fig. 8B). Although replication of the three viruses did not affect the levels of CD26 at 24 h p.i., it caused a reduction in CD26 abundance at 36 h p.i. There were no substantial differences in the amounts of CD26 among MERS-CoV-WT-infected cells and mutant virus-infected cells at both time points. These data showed that CD26 expression levels did not play significant roles in low titers of cell-associated and cell-free viruses in the mutant viruses.
Although infection with the mutant viruses produced low titers of infectious viruses, it is possible that high levels of noninfectious viruses could have been released into the supernatant from the cells infected with the mutant viruses. To test this possibility, we examined the amount of virus particles, including both infectious and noninfectious, that are released from infected cells. We harvested culture fluid from infected 293/CD26 cells, inactivated the released viruses by 60Co irradiation, purified the virus particles by sucrose gradient centrifugation, and estimated the amounts of the released viruses by Western blotting using antibodies detecting S, M, and N proteins. Substantially stronger signals of S, M, and N proteins were observed in the purified virus particles obtained from MERS-CoV-WT-infected cells showed than in those obtained from mutant virus-infected cells (Fig. 8C), suggesting that the amounts of virus particles, including both infectious and noninfectious virus particles, released from the mutant virus-infected cells were lower than those released from MERS-CoV-WT-infected cells. Since virus inactivation by gamma irradiation is believed to be mainly caused by radiolytic cleavage or cross-linking of genetic material (58–62), we did not examine the amount of viral genomic RNA in the purified 60Co-irradiated virus particles.
Transmission electron microscopic analysis of infected 293/CD26 cells.
To further understand the mechanism of inefficient replication of mutant viruses, we performed transmission electron microscopic analysis of infected 293/CD26 cells (Fig. 9). We observed the accumulation of intracellular virus particles, with the expected average size, within intracellular vesicles (Fig. 9A to C). We also noted the presence of particles, whose sizes were similar to virus particles, outside these vesicles, and yet the identity of these particles was unclear. Because CoV undergoes assembly and budding of virus particles at ERGIC membranes (20–22) and then follows the secretory pathway for egress (63), these vesicles containing virus particles most probably represented those in the secretory pathway. Counting the number of intracellular virus particles in an arbitrarily selected 30 vesicles for each virus showed the presence of statistically lower numbers of virus particles within these vesicles inside the cells infected with the mutant viruses versus MERS-CoV-WT-infected cells (Fig. 9D). However, the number of virus particles within these virus-containing vesicles between MERS-CoV-CD-infected cells and MERS-CoV-mt-infected cells showed no statistical difference. These data suggested that the mutant viruses were less efficient at the production of virus particles, including virus assembly and/or virus budding, than MERS-CoV-WT. Taken together, these data support a notion that the mutant viruses were able to accumulate viral structural proteins as efficiently as MERS-CoV-WT in 293/CD26 cells (Fig. 6A), whereas they were inefficient for assembly or budding of virus particles (Fig. 9). This resulted in low titers of cell-associated viruses (Fig. 8A) and the release of a low number of virus particles (Fig. 8C), including infectious viruses (Fig. 4C).
FIG 9.

Transmission electron microscopy of 293/CD26 cells infected with MERS-CoV-WT, -CD, or -mt. Ultrastructure analyses of 293/CD26 cells infected with MERS-CoV-WT (A), -CD (B), or -mt (C). Arrowheads indicate vesicles containing virus particles. Scale bars, 0.5 μm. (D) Numbers of virus particles in randomly selected 30 vesicles for each virus sample. Each dot represent the number of virus particles in each vesicle. Asterisks represent statically significant differences in virus titers (P < 0.05). ns, not significant.
As our studies revealed the importance of nsp1 for production of virus particles, we also explored the possibility that MERS-CoV nsp1 promotes virus assembly/budding by incorporating itself into virus particles. Western blot analysis of purified MERS-CoV-WT, MERS-CoV-CD, and MERS-CoV-mt using anti-nsp1 antibody did not show the presence of nsp1 in the purified virus particles (data not shown), suggesting that MERS-CoV nsp1 was not associated with virus particles or was not a major viral protein in the virus particles.
DISCUSSION
The present study explored the biological significance of MERS-CoV nsp1 in virus replication. By characterizing MERS-CoV nsp1-WT and MERS-CoV nsp-1-CD, the latter of which lacked the endonucleolytic RNA cleavage function, our previous study showed that expressed MERS-CoV nsp1 suppresses host gene expression by inducing endonucleolytic cleavage of host mRNAs and inhibiting translation, the latter of which is independent from the former function (43). SARS-CoV nsp1 also suppresses host gene expression by inducing endonucleolytic cleavage of mRNAs and inhibiting translation, yet existing data imply that MERS-CoV nsp1 and SARS-CoV nsp1 exert these functions by different mechanisms (43). Namely, SARS-CoV nsp1, a cytoplasmic protein (41), binds to 40S ribosomal subunits, inactivates translational function of the 40S ribosomes (40), and induces degradation of host nucleus-derived mRNAs and cytoplasmically synthesized mRNAs (43). In contrast, MERS-CoV nsp1 localizes in both the cytoplasm and the nucleus (43), does not bind to 40S ribosomes, and induces the degradation of mRNAs of nuclear origin but not those of cytoplasmic origin (43). The present study revealed that MERS-CoV nsp1-mt with K181A mutation lost the RNA cleavage and translation inhibition functions (Fig. 1). The K181A mutation corresponded to one of the K164A and H165A mutations introduced in SARS-CoV nsp1-mt, which also lacks both RNA cleavage and translation inhibition functions (46). These data suggest the importance of the C-terminal regions of SARS-CoV nsp1 and MERS-CoV nsp1 for the biological functions. Because SARS-CoV nsp1-mt is deficient for binding to 40S ribosomes (40), SARS-CoV nsp1 probably interacts with the 40S ribosome through its C-terminal region. MERS-CoV nsp1's selective biological effects toward nucleus-derived mRNAs led us to hypothesize that MERS-CoV nsp1 targets nucleus-derived mRNAs, by binding to one of the mRNA-binding proteins that form the host mRNP complex, and inhibits the expression of host genes (43). If this hypothesis is correct, disruption of MERS-CoV nsp1's functions by the K181A mutation imply that the MERS-CoV nsp1 accesses the host mRNP complex through its C-terminal region. It is conceivable that both MERS-CoV nsp1 and SARS-CoV nsp1 access target host protein/factors through their C-terminal regions.
Although MERS-CoV-WT, MERS-CoV-CD, and MERS-CoV-mt replicated efficiently with similar growth kinetics in Vero and Calu-3 cells, the two mutant viruses replicated less efficiently than MERS-CoV-WT in 293/CD26 cells, Huh-7 cells, and HeLa/CD26 cells (Fig. 4). We explored whether induction of type I and/or III IFNs caused inefficient replication of the mutant viruses in 293/CD26 cells, which were competent for induction of IFN-β and IFN-λ mRNAs by SeV infection (Fig. 5). Replication of the three viruses did not induce high levels of IFN-β and IFN-λ mRNAs (Fig. 5), demonstrating that inefficient replication of the two mutant viruses was not due to induction of the type I and III IFNs. It has been reported that MERS-CoV mutant lacking all accessory genes induced higher levels of IFN-β, IFN-λ1, and IFN-λ3 mRNAs than wt MERS-CoV in Calu-3 2B4 cells (19). Accordingly, it is possible that other viral proteins, including the accessory proteins, suppressed the induction of IFN-β and IFN-λ mRNAs in MERS-CoV-CD-infected 293/CD26 cells and MERS-CoV-mt-infected 293/CD26 cells. In contrast to MERS-CoV, SARS-CoV carrying biological inactive nsp1 induced high levels of IFN-β mRNA and type I IFN in infected cells (46). These data suggest that MERS-CoV and SARS-CoV use different strategies to suppress induction of innate immune responses.
We observed that nsp1-induced changes in translational activities differed between infected Vero cells (Fig. 3) and infected 293/CD26 cells (Fig. 7). For each cell line, the levels of nsp1 accumulation were similar among the three viruses (Fig. 2B and 6A), suggesting that the differences in the functions of nsp1, but not their expression levels, affected translational activities. MERS-CoV-WT inhibited translation at 32 h p.i. in Vero cells, while the mutant viruses induced modest and similar levels of translational inhibition (Fig. 3C), demonstrating that MERS-CoV-nsp1, particularly its RNA cleavage function, played a significant role in translational suppression in Vero cells. The moderate level of host translation inhibition in MERS-CoV-mt-infected Vero cells (Fig. 3C) could be due to the induction of the cellular stress response to virus infection, which is independent of the mode of translation inhibition induced by nsp1. In contrast to Vero cells, nsp1 did not play a significant role in virus-induced translation suppression in 293/CD26 cells, since the three viruses, including MERS-CoV-mt, efficiently inhibited translation at 24 h p.i. (Fig. 7B). These data suggest a cell line-specific effect of MERS-CoV nsp1 on host gene expression.
There was a trend of increased viral mRNA accumulation in MERS-CoV-WT-infected 293/CD26 cells than in mutant virus-infected 293/CD26 cells (Fig. 6B and C), yet the differences in the amounts of viral mRNAs did not determine the amount of viral proteins (Fig. 6A). As translational activities in 293/CD26 cells infected with MERS-CoV-WT, -CD, or -mt were similar and lower than that of mock-infected 293/CD26 cells at 24 h p.i. (Fig. 7B), low translational activities might have served as a bottle neck, which allowed translation of only a fraction of viral mRNAs in MERS-CoV-WT-infected 293/CD26 cells, resulting in similar amounts of viral protein accumulation in the three viruses.
We explored the mechanism of inefficient replication of MERS-CoV-CD and MERS-CoV-mt in 293/CD26 cells. Low titers of cell-associated and cell-free viruses in mutant virus-infected cells (Fig. 8 and 4C) were not due to inefficient accumulations of major viral structural proteins and nsp1 (Fig. 6). The data showing similar levels of CD26 expression in 293/CD26 cells infected with the three viruses (Fig. 8B) did not support a possibility that MERS-CoV-WT, but not the mutant viruses, inhibited CD26 expression and prevented interaction of CD26 with intracellular virus particles, which might have induced neutralization of intracellular virus particles and/or inhibition of virus release. Electron microscopic analysis showed less efficient intracellular virus particle accumulation in the virus-containing vesicles in mutant virus-infected cells than in MERS-CoV-WT-infected cells (Fig. 9). These data strongly suggested that low levels of virus particle accumulation, which was probably due to inefficient virus assembly/budding, in mutant virus-infected cells caused low titers of cell-associated viruses (Fig. 8A) and cell-free viruses (Fig. 8B). The data that MERS-CoV-CD, expressing MERS-CoV nsp1-CD lacking the endonucleolytic RNA cleavage function and retaining the translation inhibition function, was inefficient for accumulation of intracellular virus particles strongly suggested that the RNA cleavage function of the nsp1 was required for efficient assembly/budding of MERS-CoV particles. To our knowledge, this is the first demonstration that a CoV gene 1 protein affects efficiency of virus assembly.
Several different mechanisms are conceivable for inefficient assembly/budding of the two mutant viruses in 293/CD26 cells. One possible mechanism may be that low accumulation of E protein, which is known to be important for assembly of many CoVs (23, 30, 64), might have occurred in mutant virus-infected cells and prevented efficient virus assembly. The absence of appropriate anti-E protein antibodies prevented us from directly examining this possibility. However, it seems illogical that the loss of the RNA cleavage function of the MERS-CoV nsp1, which did not severely affect the accumulation of S, M, and N proteins (Fig. 6A) and mRNA 6 encoding E protein (Fig. 6B), selectively suppressed E protein expression in mutant virus-infected 293/CD26 cells. Furthermore, similar translational activities in MERS-CoV-WT- and mutant virus-infected 293/CD26 cells (Fig. 7B) did not support a possibility of selective inhibition of E protein accumulation in the mutant virus-infected 293/CD26 cells. We suspect that E protein accumulation was not low in mutant virus-infected 293/CD26 cells.
Another possible mechanism for inefficient assembly of the two mutant viruses could be due to lower levels of mRNA 1 accumulation in the mutant virus-infected 293/CD26 cells (Fig. 6B and C). CoV-like particles are produced from cells expressing viral structural proteins in the absence of mRNA 1 (23), yet it is unknown whether mRNA 1 affects efficiency of CoV particle assembly. There was a trend of higher accumulation of mRNA 1 in MERS-CoV-WT-infected 293/CD26 cells than in mutant virus-infected 293/CD26 cells (Fig. 6C). If mRNA 1 promotes the assembly of CoV particles, reduced amounts of mRNA 1 would have caused inefficient virus assembly in mutant virus-infected 293/CD26 cells. Another possibility of the inefficient assembly/budding of the two mutant viruses would be that MERS-CoV-nsp1-WT, but not MERS-CoV-nsp1-CD and MERS-CoV-nsp1-mt, suppressed expression of a host protein that restricts assembly/budding of MERS-CoV particles. Although host translation was inhibited to similar levels between MERS-CoV-WT-infected 293/CD26 cells and MERS-CoV-CD-infected 293/CD26 cells (Fig. 7B), the RNA cleavage function of the MERS-CoV nsp1-WT might have induced efficient degradation of the mRNA encoding this putative virus assembly/budding restriction protein, preventing the accumulation of this putative protein and promoting virus assembly/budding in MERS-CoV-WT-infected 293/CD26 cells. In contrast, due to lack of the RNA cleavage function in MERS-CoV-nsp1-CD and MERS-CoV-nsp1-mt, this putative host protein might have been expressed abundantly in mutant virus-infected 293/CD26 cells, preventing efficient virus assembly. If this possibility is the case, a plausible reason for the efficient replication of mutant viruses in Vero and Calu-3 cells may be that these cells express the putative virus restriction protein at low levels, the amounts of which are not sufficient for inhibiting MERS-CoV assembly/budding. Tetherin has been known as a host restriction factor capable of impeding the release of multiple viruses, including CoV (65–69). Because tetherin primarily prevents release of viruses from the cells, but does not affect virus assembly/budding, this putative virus-assembly restriction protein may not be tetherin.
Viral proteins that inhibit host gene expression, including nsp1 of CoV (49–51), are often major virulence factors (70–79). Accordingly, it is likely that MERS-CoV nsp1 also plays a critical role in MERS-CoV pathogenesis. Because MERS-CoV nsp1-CD and MERS-CoV nsp1-mt negatively affected the efficient production of infectious viruses in several human cell lines, MERS-CoV-CD and MERS-CoV-mt could exhibit a reduced virulence in infected hosts, compared to MERS-CoV-WT, at least partly due to the inefficient production of infectious viruses. Further studies are warranted, including a detailed characterization of the replication and virulence of MERS-CoV-WT, MERS-CoV-CD, and MERS-CoV-mt in animal models (80–83), to clarify the role of nsp1 in MERS-CoV pathogenicity.
Herpes simplex virus 1 and 2 virion host shutoff protein, Kaposi's sarcoma-associated herpesvirus SOX, and influenza A virus PA-X are virus-encoded RNases that induce endonucleolytic cleavage of host mRNAs, leading to host mRNA degradation (84, 85). These viral endonucleases contribute to evasion of host antiviral responses, including IFN response and stress granule formation, and contribute to viral pathogenesis (70–79), while roles of these virus-encoded endonucleases in production of virus particle have not been explored. To our knowledge, MERS-CoV nsp1 represents the first viral protein whose RNA cleavage-inducing function promotes virus assembly/budding.
MATERIALS AND METHODS
Cells.
Vero cells (ATCC number CCL-81), Calu-3 cells, and Huh-7 cells were maintained in minimum essential medium supplemented with 10% fetal calf serum (FBS), GlutaMAX (Gibco) supplemented with 10% FBS, and Dulbecco modified Eagle medium supplemented plus 10% FBS, respectively. HeLa/CD26 cells were generated as described in a previous report (43). Briefly, HeLa cells were transfected with pCAGGS-CD26-BlasticidinR and grown in selection medium containing blasticidin (10 μg/ml) for 2 weeks. 293/CD26 cells were generated by transfecting pCAGGS-CD26-BlasticidinR into 293 cells (ATCC) and subsequent incubation of the transfected cells in the presence of blasticidin (12 μg/ml). Stable expression of human CD26 in 293/CD26 and HeLa/CD26 cells was confirmed by Western blotting with anti-human DPP4 antibody (R&D Systems).
Viruses.
MERS-CoV-WT, MERS-CoV-CD, and MERS-CoV-mt were rescued by using reverse-genetics system as reported previously (54). All virus strains were passaged once in Vero cells and used for infection studies. The presence of the expected mutation and absence of other mutations in nsp1 was confirmed prior to use of these viruses. For virus growth analysis, Vero, Calu-3, Huh-7, HeLa/CD26, and 293/CD26 cells were infected with MERS-CoV-WT, -CD, or -mt at an MOI of 3 or 0.01. After virus adsorption for 1 h at 37°C, the cells were washed twice with phosphate-buffered saline (PBS) and incubated with the appropriate medium. The culture fluid was collected at indicated time points, and the infectious virus titers were determined by plaque assay on Vero cells. All experiments with infectious MERS-CoV were performed in an approved biosafety level 3 laboratory at The University of Texas Medical Branch at Galveston. Cantell strain of SeV was obtained from Charles River Laboratory (Wilmington, MA) and was used to infect cells at 100 hemagglutination (HA) unit/ml. Viral stocks were prepared in Vero cells and stored at −80°C.
Plasmids.
pCAGGA-based expression plasmids, pCAGGS-CAT, -SARS-CoV nsp1, -MERS-CoV nsp1-WT, and -MERS-CoV nsp1-CD, all of which carried a C-terminal myc tag, were described previously (43). pCAGGS-MERS-CoV nsp1-mt, expressing a C-terminal myc-tagged MERS-CoV nsp1 carrying a Lys-to-Ala substitution at position 181, was generated from pCAGGS-MERS-CoV nsp1 by using a recombinant PCR-based method. Sequence analysis of the plasmid confirmed the presence of the expected nsp1 sequence. pRL-EMCV-FL expressing a bicistronic reporter mRNA carrying the EMCV IRES between the upstream rLuc gene and the downstream fLuc gene was used (43).
Northern blot analysis.
Subconfluent 293 cells were transfected with a plasmid encoding CAT, SARS-CoV nsp1, MERS-CoV nsp1-WT, -CD, or -mt, together with pRL-EMCV-FL. At 24 h posttransfection, total RNAs were extracted and subjected to Northern blot analysis with an rLuc probe and a GAPDH probe. Northern blot analysis was performed as described previously (43). Vero and 293/CD26 cells were infected with MERS-CoV-WT, MERS-CoV-CD, or MERS-CoV-mt at an MOI of 3. At the indicated times postinfection, the total RNAs were extracted and subjected to Northern blot analysis with GAPDH probe. To detect MERS-CoV mRNAs, a digoxigenin-labeled random-primed probe corresponding to nucleotides 29084 to 29608 of the MERS-CoV genome were used.
Metabolic radiolabeling of intracellular proteins.
Subconfluent 293 cells were transfected with in vitro-synthesized capped and polyadenylated RNA transcripts encoding CAT, SARS-CoV nsp1, MERS-CoV nsp1-WT, MERS-CoV nsp1-CD, or MERS-CoV nsp1-mt. All encoded proteins carried the C-terminal myc tag. After incubation in methionine-deficient medium for 30 min, the cells were metabolically labeled with 20 μCi of Tran35S-label (1,000 Ci/mmol; Perkin-Elmer)/ml from 8.5 to 9.5 h posttransfection. Infected Vero cells were radiolabeled with 75 μCi of Tran35S-label/ml for 1 h at 16 or 24 h p.i. The cell extracts were prepared by lysing the cells in SDS-PAGE sample buffer. Cell lysates were subjected to SDS-PAGE analysis, followed by autoradiography and colloidal Coomassie blue staining.
Western blot analysis.
Antibodies for MERS-CoV proteins were generated by immunizing rabbits with the following synthetic peptides: NDITNTNLSRGRGRNPKPR for anti-MERS-CoV N protein peptide antibody, DDRTEVPQLVNANQYSPCVSIVC for anti-MERS-CoV S protein peptide antibody, and CDYDRLPNEVTVAK for anti-MERS-CoV M protein peptide antibody. Anti-MERS-CoV nsp1 antibody was generated by immunizing rabbits with purified C-terminal His-tagged MERS-CoV nsp1. Vero and 293/CD26 cells were infected with MERS-CoV-WT, MERS-CoV-CD, or MERS-CoV-mt at an MOI of 3. At the indicated time points, whole-cell lysates were prepared and subjected to Western blot analysis as described previously (43). Anti-myc antibody (Millipore), anti-tubulin antibody (Calbiochem), or antibodies against each of MERS-CoV protein described above were used as primary antibodies. Goat anti-mouse immunoglobulin G–horseradish peroxidase or goat anti-rabbit immunoglobulin G–horseradish peroxidase (Santa Cruz) were used as secondary antibodies.
Total RNA extraction and qRT-PCR.
Total cellular RNAs were extracted from virus-infected cells by using TRIzol LS reagent (Invitrogen) and Direct-zol RNA MiniPrep (Zymo Research), according to the instruction manuals. cDNAs were synthesized using SuperScript III reverse transcriptase (Invitrogen) and random primers (Invitrogen). To specifically detect MERS-CoV genomic or subgenomic RNAs, cDNAs were synthesized by using a MERS-CoV gene-specific primer (5′-TTTTTTTTCTAATCAGTGTTAACATCAATCATTGG-3′). qRT-PCR was performed using a Bio-Rad CFX96 real-time PCR apparatus and SYBR green Master mix (Bio-Rad). The PCR conditions were as follows: preincubation at 95°C for 30 s and amplification with 40 cycles of 95°C for 15 s and 60°C for 20 s. The purity of the amplified PCR products was confirmed by the dissociation melting curves obtained after each reaction. The primers used for human IFN-β mRNA were 5′-AAGGCCAAGGAGTACAGTC-3′ (forward) and 5′-ATCTTCAGTTTVGGAGGTAA-3′ (reverse), the primers used for IFN-λ mRNA were 5′-CGCCTTGGAAGAGTCACTCA-3′ (forward) and 5′-GAAGCCTCAGGTCCCAATTC-3′ (reverse), the primers used for 18S rRNA were 5′-CCGGTACAGTGAAACTGCGAATG-3′ (forward) and 5′-GTTATCCAAGTAGGAGAGGAGCGAG-3′ (reverse), the primers used for MERS-CoV genomic RNA/mRNA 1 were 5′-AATACACGGTTTCGTCCGGTG-3′ (forward) and 5′-ACCACAGAGTGGCACAGTTAG-3′ (reverse), and the primers used for MERS-CoV subgenomic RNA 8 were 5′-CTCGTTCTCTTGCAGAACTTTG-3′ (forward) and 5′-TGCCCAGGTGGAAAGGT-3′ (reverse). The relative expression level of each gene mRNA were normalized to 18S rRNA levels. All of the assays were performed in triplicate, and the results are expressed as means ± the standard deviations.
Titration of intracellular infectious particles in infected 293/CD26 cells.
At the indicated times postinfection, infected 293/CD26 cells were washed two times in PBS and pelleted by centrifugation at 3,000 rpm for 10 min. The pelleted cells were suspended in growth medium and lysed by three freeze-thaw cycles. After centrifugation at 3,000 rpm for 10 min, supernatant was collected and subjected to plaque assays using Vero cells.
Purification of released virus particles.
After centrifugation at 1,500 × g for 10 min at 4°C, supernatants from 293/CD26 cells infected with MERS-CoV-WT, -CD or –mt were irradiated with 2 × 106 rads from a Gammacell 60Co source (model 109A; J. L. Shepherd and Associates, San Fernando, CA) to completely inactivate viruses. Inactivation of virus infectivity was confirmed by blind passages on the samples in Vero cells two times. The inactivated samples were applied onto a discontinuous sucrose gradient consisted of 20, 30, 50, and 60% sucrose in NTE buffer (100 mM NaCl, 10 mM Tris-HCl [pH 7.5], 1 mM EDTA) and subjected to centrifugation at 26,000 rpm for 3 h in an SW28 rotor. The virus particles in the interface of 50 to 30% fraction were collected, diluted with NTE buffer, applied onto a discontinuous sucrose gradient, and centrifuged at 26,000 rpm for 18 h in an SW28 rotor. After collecting the purified MERS-CoV particle in the interface of 50 to 30% sucrose, MERS-CoV particles were pelleted by centrifugation at 38,000 rpm for 2 h using a Beckman SW41 rotor. The purified virus particles in the pellets were dissolved in the same amount of 1× SDS sample buffer and subjected to Western blot analysis.
Electron microscopic analysis.
Monolayer 293/CD26 cells were infected with MERS-CoV-WT, -CD, or -mt at an MOI of 3. At 36 h p.i., the cells were washed with PBS and fixed with 4% formaldehyde and 0.1% glutaraldehyde in 0.05 M cacodylate buffer (pH 7.3), to which 0.03% picric acid and 0.03% CaCl2 were added. The monolayers were washed in 0.1 M cacodylate buffer, and the cells were scraped off and processed further as a pellet. The pellets were postfixed in 1% OsO4 in 0.1 M cacodylate buffer (pH 7.3) for 1 h, washed with distilled water, and en bloc stained with 2% aqueous uranyl acetate for 20 min at 60°C. The pellets were dehydrated in ethanol, processed through propylene oxide, and embedded in Poly/Bed 812 (Polysciences). Ultrathin sections were cut on Leica EM UC7 ultramicrotome (Leica Microsystems, Buffalo Grove, IL), stained with lead citrate, and examined with a CM-100 electron microscope at 60 kV.
Statistical analysis.
One-way analysis of variance with Tukey's multiple-comparison test was conducted to determine statistical significance. A P value of <0.05 was considered statistically significant.
ACKNOWLEDGMENTS
We thank Boyd Yount, Rachel Graham, and Amy Sims in the Baric lab for technical advice regarding reverse genetics system of MERS-CoV. We also thank Julie Wen and Zhixia Ding for suggestions regarding the electron microscopic analysis.
This study was supported by Public Health Service grants AI99107 and AI114657 to S.M. and AI108197 and AI110700 to R.S.B. from the National Institutes of Health and a grant from the Institute for Human Infections and Immunity at The University of Texas Medical Branch to S.M. K.N. was supported by the James W. McLaughlin fellowship fund.
REFERENCES
- 1.Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. 2012. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 367:1814–1820. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- 2.Hui DS, Azhar EI, Kim YJ, Memish ZA, Oh MD, Zumla A. 2018. Middle East respiratory syndrome coronavirus: risk factors and determinants of primary, household, and nosocomial transmission. Lancet Infect Dis 18:e217–e227. doi: 10.1016/S1473-3099(18)30127-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee HJ, Shieh CK, Gorbalenya AE, Koonin EV, La Monica N, Tuler J, Bagdzhadzhyan A, Lai MM. 1991. The complete sequence (22 kilobases) of murine coronavirus gene 1 encoding the putative proteases and RNA polymerase. Virology 180:567–582. doi: 10.1016/0042-6822(91)90071-I. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lomniczi B. 1977. Biological properties of avian coronavirus RNA. J Gen Virol 36:531–533. doi: 10.1099/0022-1317-36-3-531. [DOI] [PubMed] [Google Scholar]
- 5.Lomniczi B, Kennedy I. 1977. Genome of infectious bronchitis virus. J Virol 24:99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raj VS, Mou H, Smits SL, Dekkers DH, Muller MA, Dijkman R, Muth D, Demmers JA, Zaki A, Fouchier RA, Thiel V, Drosten C, Rottier PJ, Osterhaus AD, Bosch BJ, Haagmans BL. 2013. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 495:251–254. doi: 10.1038/nature12005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burkard C, Verheije MH, Wicht O, van Kasteren SI, van Kuppeveld FJ, Haagmans BL, Pelkmans L, Rottier PJ, Bosch BJ, de Haan CA. 2014. Coronavirus cell entry occurs through the endo-/lysosomal pathway in a proteolysis-dependent manner. PLoS Pathog 10:e1004502. doi: 10.1371/journal.ppat.1004502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Snijder EJ, Decroly E, Ziebuhr J. 2016. The nonstructural proteins directing coronavirus RNA synthesis and processing. Adv Virus Res 96:59–126. doi: 10.1016/bs.aivir.2016.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hurst-Hess KR, Kuo L, Masters PS. 2015. Dissection of amino-terminal functional domains of murine coronavirus nonstructural protein 3. J Virol 89:6033–6047. doi: 10.1128/JVI.00197-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Graham RL, Sims AC, Brockway SM, Baric RS, Denison MR. 2005. The nsp2 replicase proteins of murine hepatitis virus and severe acute respiratory syndrome coronavirus are dispensable for viral replication. J Virol 79:13399–13411. doi: 10.1128/JVI.79.21.13399-13411.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neuman BW, Chamberlain P, Bowden F, Joseph J. 2014. Atlas of coronavirus replicase structure. Virus Res 194:49–66. doi: 10.1016/j.virusres.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Masters PS. 2006. The molecular biology of coronaviruses. Adv Virus Res 66:193–292. doi: 10.1016/S0065-3527(06)66005-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sawicki SG, Sawicki DL, Siddell SG. 2007. A contemporary view of coronavirus transcription. J Virol 81:20–29. doi: 10.1128/JVI.01358-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lai MM, Baric RS, Brayton PR, Stohlman SA. 1984. Characterization of leader RNA sequences on the virion and mRNAs of mouse hepatitis virus, a cytoplasmic RNA virus. Proc Natl Acad Sci U S A 81:3626–3630. doi: 10.1073/pnas.81.12.3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lai MM, Patton CD, Baric RS, Stohlman SA. 1983. Presence of leader sequences in the mRNA of mouse hepatitis virus. J Virol 46:1027–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lai MM, Patton CD, Stohlman SA. 1982. Replication of mouse hepatitis virus: negative-stranded RNA and replicative form RNA are of genome length. J Virol 44:487–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu DX, Fung TS, Chong KK, Shukla A, Hilgenfeld R. 2014. Accessory proteins of SARS-CoV and other coronaviruses. Antiviral Res 109:97–109. doi: 10.1016/j.antiviral.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Narayanan K, Huang C, Makino S. 2008. SARS coronavirus accessory proteins. Virus Res 133:113–121. doi: 10.1016/j.virusres.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Menachery VD, Mitchell HD, Cockrell AS, Gralinski LE, Yount BL Jr, Graham RL, McAnarney ET, Douglas MG, Scobey T, Beall A, Dinnon K III, Kocher JF, Hale AE, Stratton KG, Waters KM, Baric RS. 2017. MERS-CoV accessory ORFs play key role for infection and pathogenesis. mBio 8:e00665-17. doi: 10.1128/mBio.00665-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stertz S, Reichelt M, Spiegel M, Kuri T, Martinez-Sobrido L, Garcia-Sastre A, Weber F, Kochs G. 2007. The intracellular sites of early replication and budding of SARS-coronavirus. Virology 361:304–315. doi: 10.1016/j.virol.2006.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tooze J, Tooze S, Warren G. 1984. Replication of coronavirus MHV-A59 in sac- cells: determination of the first site of budding of progeny virions. Eur J Cell Biol 33:281–293. [PubMed] [Google Scholar]
- 22.Klumperman J, Locker JK, Meijer A, Horzinek MC, Geuze HJ, Rottier PJ. 1994. Coronavirus M proteins accumulate in the Golgi complex beyond the site of virion budding. J Virol 68:6523–6534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vennema H, Godeke GJ, Rossen JW, Voorhout WF, Horzinek MC, Opstelten DJ, Rottier PJ. 1996. Nucleocapsid-independent assembly of coronavirus-like particles by coexpression of viral envelope protein genes. EMBO J 15:2020–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Haan CA, Smeets M, Vernooij F, Vennema H, Rottier PJ. 1999. Mapping of the coronavirus membrane protein domains involved in interaction with the spike protein. J Virol 73:7441–7452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Escors D, Ortego J, Laude H, Enjuanes L. 2001. The membrane M protein carboxy terminus binds to transmissible gastroenteritis coronavirus core and contributes to core stability. J Virol 75:1312–1324. doi: 10.1128/JVI.75.3.1312-1324.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Escors D, Camafeita E, Ortego J, Laude H, Enjuanes L. 2001. Organization of two transmissible gastroenteritis coronavirus membrane protein topologies within the virion and core. J Virol 75:12228–12240. doi: 10.1128/JVI.75.24.12228-12240.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuo L, Masters PS. 2002. Genetic evidence for a structural interaction between the carboxy termini of the membrane and nucleocapsid proteins of mouse hepatitis virus. J Virol 76:4987–4999. doi: 10.1128/JVI.76.10.4987-4999.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nguyen VP, Hogue BG. 1997. Protein interactions during coronavirus assembly. J Virol 71:9278–9284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Opstelten DJ, Raamsman MJ, Wolfs K, Horzinek MC, Rottier PJ. 1995. Envelope glycoprotein interactions in coronavirus assembly. J Cell Biol 131:339–349. doi: 10.1083/jcb.131.2.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Corse E, Machamer CE. 2000. Infectious bronchitis virus E protein is targeted to the Golgi complex and directs release of virus-like particles. J Virol 74:4319–4326. doi: 10.1128/JVI.74.9.4319-4326.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bos EC, Luytjes W, van der Meulen HV, Koerten HK, Spaan WJ. 1996. The production of recombinant infectious DI-particles of a murine coronavirus in the absence of helper virus. Virology 218:52–60. doi: 10.1006/viro.1996.0165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fischer F, Stegen CF, Masters PS, Samsonoff WA. 1998. Analysis of constructed E gene mutants of mouse hepatitis virus confirms a pivotal role for E protein in coronavirus assembly. J Virol 72:7885–7894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.DeDiego ML, Alvarez E, Almazan F, Rejas MT, Lamirande E, Roberts A, Shieh WJ, Zaki SR, Subbarao K, Enjuanes L. 2007. A severe acute respiratory syndrome coronavirus that lacks the E gene is attenuated in vitro and in vivo. J Virol 81:1701–1713. doi: 10.1128/JVI.01467-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Narayanan K, Ramirez SI, Lokugamage KG, Makino S. 2015. Coronavirus nonstructural protein 1: common and distinct functions in the regulation of host and viral gene expression. Virus Res 202:89–100. doi: 10.1016/j.virusres.2014.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Connor RF, Roper RL. 2007. Unique SARS-CoV protein nsp1: bioinformatics, biochemistry, and potential effects on virulence. Trends Microbiol 15:51–53. doi: 10.1016/j.tim.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jansson AM. 2013. Structure of alphacoronavirus transmissible gastroenteritis virus nsp1 has implications for coronavirus nsp1 function and evolution. J Virol 87:2949–2955. doi: 10.1128/JVI.03163-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Snijder EJ, Bredenbeek PJ, Dobbe JC, Thiel V, Ziebuhr J, Poon LL, Guan Y, Rozanov M, Spaan WJ, Gorbalenya AE. 2003. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J Mol Biol 331:991–1004. doi: 10.1016/S0022-2836(03)00865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thiel V, Ivanov KA, Putics A, Hertzig T, Schelle B, Bayer S, Weissbrich B, Snijder EJ, Rabenau H, Doerr HW, Gorbalenya AE, Ziebuhr J. 2003. Mechanisms and enzymes involved in SARS coronavirus genome expression. J Gen Virol 84:2305–2315. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- 39.Tohya Y, Narayanan K, Kamitani W, Huang C, Lokugamage K, Makino S. 2009. Suppression of host gene expression by nsp1 proteins of group 2 bat coronaviruses. J Virol 83:5282–5288. doi: 10.1128/JVI.02485-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kamitani W, Huang C, Narayanan K, Lokugamage KG, Makino S. 2009. A two-pronged strategy to suppress host protein synthesis by SARS coronavirus Nsp1 protein. Nat Struct Mol Biol 16:1134–1140. doi: 10.1038/nsmb.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kamitani W, Narayanan K, Huang C, Lokugamage K, Ikegami T, Ito N, Kubo H, Makino S. 2006. Severe acute respiratory syndrome coronavirus nsp1 protein suppresses host gene expression by promoting host mRNA degradation. Proc Natl Acad Sci U S A 103:12885–12890. doi: 10.1073/pnas.0603144103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang C, Lokugamage KG, Rozovics JM, Narayanan K, Semler BL, Makino S. 2011. Alphacoronavirus transmissible gastroenteritis virus nsp1 protein suppresses protein translation in mammalian cells and in cell-free HeLa cell extracts but not in rabbit reticulocyte lysate. J Virol 85:638–643. doi: 10.1128/JVI.01806-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lokugamage KG, Narayanan K, Nakagawa K, Terasaki K, Ramirez SI, Tseng CT, Makino S. 2015. Middle East respiratory syndrome coronavirus nsp1 inhibits host gene expression by selectively targeting mRNAs transcribed in the nucleus while sparing mRNAs of cytoplasmic origin. J Virol 89:10970–10981. doi: 10.1128/JVI.01352-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gaglia MM, Covarrubias S, Wong W, Glaunsinger BA. 2012. A common strategy for host RNA degradation by divergent viruses. J Virol 86:9527–9530. doi: 10.1128/JVI.01230-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang C, Lokugamage KG, Rozovics JM, Narayanan K, Semler BL, Makino S. 2011. SARS coronavirus nsp1 protein induces template-dependent endonucleolytic cleavage of mRNAs: viral mRNAs are resistant to nsp1-induced RNA cleavage. PLoS Pathog 7:e1002433. doi: 10.1371/journal.ppat.1002433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Narayanan K, Huang C, Lokugamage K, Kamitani W, Ikegami T, Tseng CT, Makino S. 2008. Severe acute respiratory syndrome coronavirus nsp1 suppresses host gene expression, including that of type I interferon, in infected cells. J Virol 82:4471–4479. doi: 10.1128/JVI.02472-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wathelet MG, Orr M, Frieman MB, Baric RS. 2007. Severe acute respiratory syndrome coronavirus evades antiviral signaling: role of nsp1 and rational design of an attenuated strain. J Virol 81:11620–11633. doi: 10.1128/JVI.00702-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Q, Ke H, Blikslager A, Fujita T, Yoo D. 2018. Type III interferon restriction by porcine epidemic diarrhea virus and the role of viral protein nsp1 in IRF1 signaling. J Virol 92:e01677-17. doi: 10.1128/JVI.01677-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zust R, Cervantes-Barragan L, Kuri T, Blakqori G, Weber F, Ludewig B, Thiel V. 2007. Coronavirus nonstructural protein 1 is a major pathogenicity factor: implications for the rational design of coronavirus vaccines. PLoS Pathog 3:e109. doi: 10.1371/journal.ppat.0030109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang R, Li Y, Cowley TJ, Steinbrenner AD, Phillips JM, Yount BL, Baric RS, Weiss SR. 2015. The nsp1, nsp13, and M proteins contribute to the hepatotropism of murine coronavirus JHM.WU. J Virol 89:3598–3609. doi: 10.1128/JVI.03535-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jimenez-Guardeno JM, Regla-Nava JA, Nieto-Torres JL, DeDiego ML, Castano-Rodriguez C, Fernandez-Delgado R, Perlman S, Enjuanes L. 2015. Identification of the mechanisms causing reversion to virulence in an attenuated SARS-CoV for the design of a genetically stable vaccine. PLoS Pathog 11:e1005215. doi: 10.1371/journal.ppat.1005215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tanaka T, Kamitani W, DeDiego ML, Enjuanes L, Matsuura Y. 2012. Severe acute respiratory syndrome coronavirus nsp1 facilitates efficient propagation in cells through a specific translational shutoff of host mRNA. J Virol 86:11128–11137. doi: 10.1128/JVI.01700-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Terada Y, Kawachi K, Matsuura Y, Kamitani W. 2017. MERS coronavirus nsp1 participates in an efficient propagation through a specific interaction with viral RNA. Virology 511:95–105. doi: 10.1016/j.virol.2017.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Scobey T, Yount BL, Sims AC, Donaldson EF, Agnihothram SS, Menachery VD, Graham RL, Swanstrom J, Bove PF, Kim JD, Grego S, Randell SH, Baric RS. 2013. Reverse genetics with a full-length infectious cDNA of the Middle East respiratory syndrome coronavirus. Proc Natl Acad Sci U S A 110:16157–16162. doi: 10.1073/pnas.1311542110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tseng CT, Tseng J, Perrone L, Worthy M, Popov V, Peters CJ. 2005. Apical entry and release of severe acute respiratory syndrome-associated coronavirus in polarized Calu-3 lung epithelial cells. J Virol 79:9470–9479. doi: 10.1128/JVI.79.15.9470-9479.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nakabayashi H, Taketa K, Miyano K, Yamane T, Sato J. 1982. Growth of human hepatoma cells lines with differentiated functions in chemically defined medium. Cancer Res 42:3858–3863. [PubMed] [Google Scholar]
- 57.Mou H, Raj VS, van Kuppeveld FJ, Rottier PJ, Haagmans BL, Bosch BJ. 2013. The receptor binding domain of the new Middle East respiratory syndrome coronavirus maps to a 231-residue region in the spike protein that efficiently elicits neutralizing antibodies. J Virol 87:9379–9383. doi: 10.1128/JVI.01277-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hume AJ, Ames J, Rennick LJ, Duprex WP, Marzi A, Tonkiss J, Muhlberger E. 2016. Inactivation of RNA viruses by gamma irradiation: a study on mitigating factors. Viruses 8:E204. doi: 10.3390/v8070204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lomax ME, Folkes LK, O'Neill P. 2013. Biological consequences of radiation-induced DNA damage: relevance to radiotherapy. Clin Oncol 25:578–585. doi: 10.1016/j.clon.2013.06.007. [DOI] [PubMed] [Google Scholar]
- 60.Ohshima H, Iida Y, Matsuda A, Kuwabara M. 1996. Damage induced by hydroxyl radicals generated in the hydration layer of gamma-irradiated frozen aqueous solution of DNA. J Radiat Res 37:199–207. doi: 10.1269/jrr.37.199. [DOI] [PubMed] [Google Scholar]
- 61.Summers WC, Szybalski W. 1967. Gamma-irradiation of deoxyribonucleic acid in dilute solutions. II. Molecular mechanisms responsible for inactivation of phage, its transfecting DNA, and of bacterial transforming activity. J Mol Biol 26:227–235. [DOI] [PubMed] [Google Scholar]
- 62.Ward RL. 1980. Mechanisms of poliovirus inactivation by the direct and indirect effects of ionizing radiation. Radiat Res 83:330–344. doi: 10.2307/3575284. [DOI] [PubMed] [Google Scholar]
- 63.Ruch TR, Machamer CE. 2012. The coronavirus E protein: assembly and beyond. Viruses 4:363–382. doi: 10.3390/v4030363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Baudoux P, Carrat C, Besnardeau L, Charley B, Laude H. 1998. Coronavirus pseudoparticles formed with recombinant M and E proteins induce alpha interferon synthesis by leukocytes. J Virol 72:8636–8643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang SM, Huang KJ, Wang CT. 2014. BST2/CD317 counteracts human coronavirus 229E productive infection by tethering virions at the cell surface. Virology 449:287–296. doi: 10.1016/j.virol.2013.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Neil SJ, Zang T, Bieniasz PD. 2008. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 451:425–430. doi: 10.1038/nature06553. [DOI] [PubMed] [Google Scholar]
- 67.Kaletsky RL, Francica JR, Agrawal-Gamse C, Bates P. 2009. Tetherin-mediated restriction of filovirus budding is antagonized by the Ebola glycoprotein. Proc Natl Acad Sci U S A 106:2886–2891. doi: 10.1073/pnas.0811014106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mansouri M, Viswanathan K, Douglas JL, Hines J, Gustin J, Moses AV, Fruh K. 2009. Molecular mechanism of BST2/tetherin downregulation by K5/MIR2 of Kaposi's sarcoma-associated herpesvirus. J Virol 83:9672–9681. doi: 10.1128/JVI.00597-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jouvenet N, Neil SJ, Zhadina M, Zang T, Kratovac Z, Lee Y, McNatt M, Hatziioannou T, Bieniasz PD. 2009. Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. J Virol 83:1837–1844. doi: 10.1128/JVI.02211-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hayashi T, MacDonald LA, Takimoto T. 2015. Influenza A virus protein PA-X contributes to viral growth and suppression of the host antiviral and immune responses. J Virol 89:6442–6452. doi: 10.1128/JVI.00319-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xu G, Zhang X, Liu Q, Bing G, Hu Z, Sun H, Xiong X, Jiang M, He Q, Wang Y, Pu J, Guo X, Yang H, Liu J, Sun Y. 2017. PA-X protein contributes to virulence of triple-reassortant H1N2 influenza virus by suppressing early immune responses in swine. Virology 508:45–53. doi: 10.1016/j.virol.2017.05.002. [DOI] [PubMed] [Google Scholar]
- 72.Lee J, Yu H, Li Y, Ma J, Lang Y, Duff M, Henningson J, Liu Q, Li Y, Nagy A, Bawa B, Li Z, Tong G, Richt JA, Ma W. 2017. Impacts of different expressions of PA-X protein on 2009 pandemic H1N1 virus replication, pathogenicity and host immune responses. Virology 504:25–35. doi: 10.1016/j.virol.2017.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tigges MA, Leng S, Johnson DC, Burke RL. 1996. Human herpes simplex virus (HSV)-specific CD8+ CTL clones recognize HSV-2-infected fibroblasts after treatment with IFN-gamma or when virion host shutoff functions are disabled. J Immunol 156:3901–3910. [PubMed] [Google Scholar]
- 74.Pasieka TJ, Lu B, Crosby SD, Wylie KM, Morrison LA, Alexander DE, Menachery VD, Leib DA. 2008. Herpes simplex virus virion host shutoff attenuates establishment of the antiviral state. J Virol 82:5527–5535. doi: 10.1128/JVI.02047-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Murphy JA, Duerst RJ, Smith TJ, Morrison LA. 2003. Herpes simplex virus type 2 virion host shutoff protein regulates alpha/beta interferon but not adaptive immune responses during primary infection in vivo. J Virol 77:9337–9345. doi: 10.1128/JVI.77.17.9337-9345.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Finnen RL, Zhu M, Li J, Romo D, Banfield BW. 2016. Herpes simplex virus 2 virion host shutoff endoribonuclease activity is required to disrupt stress granule formation. J Virol 90:7943–7955. doi: 10.1128/JVI.00947-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dauber B, Pelletier J, Smiley JR. 2011. The herpes simplex virus 1 Vhs protein enhances translation of viral true late mRNAs and virus production in a cell type-dependent manner. J Virol 85:5363–5373. doi: 10.1128/JVI.00115-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Finnen RL, Hay TJ, Dauber B, Smiley JR, Banfield BW. 2014. The herpes simplex virus 2 virion-associated ribonuclease Vhs interferes with stress granule formation. J Virol 88:12727–12739. doi: 10.1128/JVI.01554-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dauber B, Poon D, Dos Santos T, Duguay BA, Mehta N, Saffran HA, Smiley JR. 2016. The herpes simplex virus virion host shutoff protein enhances translation of viral true late mRNAs independently of suppressing protein kinase R and stress granule formation. J Virol 90:6049–6057. doi: 10.1128/JVI.03180-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Agrawal AS, Garron T, Tao X, Peng BH, Wakamiya M, Chan TS, Couch RB, Tseng CT. 2015. Generation of a transgenic mouse model of Middle East respiratory syndrome coronavirus infection and disease. J Virol 89:3659–3670. doi: 10.1128/JVI.03427-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Falzarano D, de Wit E, Rasmussen AL, Feldmann F, Okumura A, Scott DP, Brining D, Bushmaker T, Martellaro C, Baseler L, Benecke AG, Katze MG, Munster VJ, Feldmann H. 2013. Treatment with interferon-α2b and ribavirin improves outcome in MERS-CoV-infected rhesus macaques. Nat Med 19:1313–1317. doi: 10.1038/nm.3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Falzarano D, de Wit E, Feldmann F, Rasmussen AL, Okumura A, Peng X, Thomas MJ, van Doremalen N, Haddock E, Nagy L, LaCasse R, Liu T, Zhu J, McLellan JS, Scott DP, Katze MG, Feldmann H, Munster VJ. 2014. Infection with MERS-CoV causes lethal pneumonia in the common marmoset. PLoS Pathog 10:e1004250. doi: 10.1371/journal.ppat.1004250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cockrell AS, Yount BL, Scobey T, Jensen K, Douglas M, Beall A, Tang XC, Marasco WA, Heise MT, Baric RS. 2016. A mouse model for MERS coronavirus-induced acute respiratory distress syndrome. Nat Microbiol 2:16226. doi: 10.1038/nmicrobiol.2016.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Abernathy E, Glaunsinger B. 2015. Emerging roles for RNA degradation in viral replication and antiviral defense. Virology 479-480:600–608. doi: 10.1016/j.virol.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Read GS. 2013. Virus-encoded endonucleases: expected and novel functions. Wiley Interdiscip Rev RNA 4:693–708. doi: 10.1002/wcs.1258. [DOI] [PubMed] [Google Scholar]