

HIV is most commonly transmitted sexually, and semen is the primary vector. Despite progress in studies of HIV pathogenesis and the success of combination antiretroviral therapy in controlling viral replication, current therapy cannot completely control sexual transmission. Thus, there is a need to identify effective methods of controlling HIV replication and transmission. Recently, it was shown that human semen contains exosomes that protect against HIV infection in vitro. In this study, we identified a mechanism by which semen exosomes inhibited HIV-1 RNA expression. We found that semen exosomes inhibit recruitment of transcription factors NF-κB and Sp1, as well as RNA Pol II, to the promoter region in the 5′ long terminal repeat (LTR) of HIV-1. The HIV-1 early protein transcriptional activator (Tat) was a target of semen exosomes, and semen exosomes inhibited the binding and recruitment of Tat to the HIV-1 LTR.
KEYWORDS: exosomes, HIV-1, transcription, Tat, NF-κB, Sp1, RNA Pol II, LTR, promoter, semen, long terminal repeat
ABSTRACT
Exosomes play various roles in host responses to cancer and infective agents, and semen exosomes (SE) inhibit HIV-1 infection and transmission, although the mechanism(s) by which this occurs is unclear. Here, we show that SE block HIV-1 proviral transcription at multiple transcriptional checkpoints, including transcription factor recruitment to the long terminal repeat (LTR), transcription initiation, and elongation. Biochemical and functional studies show that SE inhibit HIV-1 LTR-driven viral gene expression and virus replication. Through partitioning of the HIV-1 RNA, we found that SE reduced the optimal expression of various viral RNA species. Chromatin immunoprecipitation–real-time quantitative PCR (ChIP–RT-qPCR) and electrophoretic mobility shift assay (EMSA) analysis of infected cells identified the human transcription factors NF-κB and Sp1, as well as RNA polymerase (Pol) II and the viral protein transcriptional activator (Tat), as targets of SE. Of interest, SE inhibited HIV-1 LTR activation mediated by HIV-1 or Tat, but not by the mitogen phorbol myristate acetate (PMA) or tumor necrosis factor alpha (TNF-α). SE inhibited the DNA binding activities of NF-κB and Sp1 and blocked the recruitment of these transcription factors and Pol II to the HIV-1 LTR promoter. Importantly, SE directly blocked NF-κB, Sp1, and Pol II binding to the LTR and inhibited the interactions of Tat/NF-κB and Tat/Sp1, suggesting that SE-mediated inhibition of the functional quadripartite complex NF-κB–Sp1–Pol II–Tat may be a novel mechanism of proviral transcription repression. These data provide a novel molecular basis for SE-mediated inhibition of HIV-1 and identify Tat as a potential target of SE.
IMPORTANCE HIV is most commonly transmitted sexually, and semen is the primary vector. Despite progress in studies of HIV pathogenesis and the success of combination antiretroviral therapy in controlling viral replication, current therapy cannot completely control sexual transmission. Thus, there is a need to identify effective methods of controlling HIV replication and transmission. Recently, it was shown that human semen contains exosomes that protect against HIV infection in vitro. In this study, we identified a mechanism by which semen exosomes inhibited HIV-1 RNA expression. We found that semen exosomes inhibit recruitment of transcription factors NF-κB and Sp1, as well as RNA Pol II, to the promoter region in the 5′ long terminal repeat (LTR) of HIV-1. The HIV-1 early protein transcriptional activator (Tat) was a target of semen exosomes, and semen exosomes inhibited the binding and recruitment of Tat to the HIV-1 LTR.
INTRODUCTION
The HIV-1 long terminal repeat (LTR) is responsible for transcriptional regulation of proviral DNA. The LTR is divided into four functional regions, namely, the modulatory element, enhancer, promoter, and transcriptional activator (Tat)-activating region (TAR). TAR forms a stable RNA stem-loop that binds and recruits the HIV-encoded transactivator Tat, which activates HIV-1 transcription by increasing host polymerase (Pol) II processivity via interactions with the cellular target positive transcription elongation factor b (P-TEFb) (1, 2). Although HIV-1 proviral transcription is initiated by host Pol II, in the absence of Tat, Pol II is unable to transcribe the complete viral template, and only short viral transcripts are generated (3, 4). Tat transactivates the HIV-1 LTR promoter and plays a role in HIV-1 reverse transcription (5, 6).
Tat is located in the nucleus and cytoplasm (7). Nuclear Tat is required for transactivation of the HIV-1 LTR promoter, resulting in transcription of high levels of viral mRNA by Pol II (8). Nuclear Tat binds the HIV-1 TAR (9), promoting transcriptional initiation and elongation through interaction with cellular transacting factors and cofactors, including TFIID (10, 11), C/EBPb (12), cyclin T1/CDK9 (13, 14), E2F-2 (15), histone acetyltransferases p300/CBP and P/CAF (16), Sp1 (17, 18), and NF-κB (19). The LTR contains key binding sites for cellular transcription factors, including Sp1 and NF-κB, that are involved in HIV-1 transcription. Through the induction of IKK activity and proteasomal degradation of IκB-a, Tat increases NF-κB p65 transcriptional activity (20). NF-κB is bound to IκB proteins in the cytoplasm. Following release from IκB, NF-κB translocates to the nucleus, where it associates with the HIV-1 LTR (21). NF-κB interacts with two NF-κB binding sites that cooperatively interact with three proximal Sp1 binding sites to stimulate LTR transcriptional activity (22, 23). Binding of these factors results in cooperation with TATA binding protein and associated factors to drive transcription. Further, these factors recruit P-TEFb and are involved in chromatin remodeling to regulate transcription (21). Thus, NF-κB is critical for HIV promoter activity. The p65 subunit is primarily responsible for LTR transactivation, and induction of NF-κB results in NF-κB-dependent HIV-1 gene expression. However, binding of NF-κB alone without Sp1 is insufficient to induce HIV-1 gene expression (4, 19, 23–25).
Factors that impair or block Tat-mediated processes reduce provirus transcription and possibly inhibit viral replication. We previously demonstrated that semen exosomes (SE), a subset of extracellular vesicles, inhibit HIV-1 infection of various cell types and inhibit HIV-1 transmission (26–28). In addition to the protection provided by the female mucosa, SE may contribute to the low risk of HIV-1 infection per heterosexual sexual act (0.04 to 0.08%) by interfering with HIV replication (29). SE inhibit HIV-1 infection and transmission in part due to their ability to impair viral reverse transcriptase function and to repress viral RNA gene expression (26–28). Since reverse transcription and viral gene expression require Tat activity, we hypothesized that SE interfere with Tat-dependent transcriptional events. Furthermore, we hypothesized that Tat protein is targeted by SE.
We examined potential mechanisms by which SE inhibit HIV-1 provirus transcription and characterized the binding of HIV-1 Tat, cellular transcription factors, and the recruitment of Pol II to the HIV-1 LTR in the presence and absence of SE. Our results indicate that, in addition to damping NF-κB activation, SE inhibit NF-κB and Sp1 binding/recruitment and interaction with HIV-1, impair Pol II recruitment to the HIV-1 promoter, and specifically block Tat protein-mediated LTR activation. These findings provide new insights into how SE control HIV-1 transcription and provirus replication. The findings also reveal a novel function for exosomes: interaction with host transcription factors.
RESULTS
Three classes of HIV-1 transcripts are susceptible to inhibition by SE.
The effect of SE on cell-associated HIV RNA expression was determined by infecting SUPT1 cells in the presence and absence of SE prior to RNA extraction, DNase treatment, reverse transcription, and real-time quantitative PCR (RT-qPCR) with HIV gene-specific primers (Fig. 1A and Table 1). SE inhibited the accumulation of multiply spliced Tat-Rev (Fig. 1B), singly spliced Vif and Vpr (Fig. 1C and D), and unspliced U5-Gag and Gag-Pol (Fig. 1E and F) HIV-1 RNAs. The extent of SE-mediated inhibition observed in SUPT1 cells was similar to that in a different cell line, TZM-bl cells (Fig. 1G to K), indicating that the inhibition is cell type independent. Since the presence of SE produced drastic changes in the steady-state levels of the different RNA species, we examined the levels of particle-associated RNA. Using a similar technique for assaying cell-associated RNA expression, we found that SE inhibit particle-associated viral RNA (Fig. 1L). Copy number analysis of particle-associated viral RNA confirmed a reduction in RNA levels in the presence of SE (Fig. 1M). Given the reductions in all classes of viral RNA by SE, we evaluated the effect of SE on the levels of viral protein. Analysis of intracellular p24 protein revealed no significant alteration (Fig. 1N and O). Similarly, progeny p24 levels were unchanged in the presence of SE (Fig. 1P). Previously, a similar disconnect between RNA expression and HIV-1 protein levels was described in which pr55Gag (the precursor to p24) protein synthesis could be rescued in the presence of HIV-1 RNA defects and de novo RNA synthesis was dispensable for synthesis and processing of HIV-1 Gag (30, 31). Normalizing for p24 protein, viral particles produced by cells incubated in SE contained reduced RNA (Fig. 1L, M, and P). The infectivity of these viral particles was significantly reduced (Fig. 1Q), suggesting that nascent HIV-1 particles produced in the presence of SE are less infectious. Thus, alteration of RNA levels may be a contributing mechanism for SE-related inhibition of HIV-1 infection, and changes in HIV-1 RNA levels do not have to be accompanied by reduced protein levels.
FIG 1.

Effects of SE on viral RNA species produced within HIV-1-infected cells. Vehicle (PBS) or semen exosomes (100 μg/ml) were incubated with HIV-1 NL4.3 (8 reverse transcriptase units [RTU]/ml) at 37°C for 1 h before infection. qRT-PCR analysis of HIV-1 RNA species transcript levels in cells was performed 24 h after SE or vehicle treatment and HIV-1 infection. (A) Schematic of HIV-1 genome. The arrows represent primer pairs used for qRT-PCR analysis. US, unspliced; SS, singly spliced; MS, multiply spliced. (B to F) HIV-1 RNA from SUPT1 cells representing multiply spliced Tat-Rev mRNA, singly spliced Vif and Vpr mRNA, and unspliced U5-Gag and Gag-Pol mRNA expression, respectively. qRT-PCR analysis of HIV-1 RNA species transcript levels in TZM-bl cells was performed 24 h after SE or vehicle treatment and HIV-1 infection. Fold Δ, fold change. (G to K) HIV-1 RNA from TZM-bl cells representing multiply spliced Tat-Rev mRNA, singly spliced Vif and Vpr mRNA, and unspliced U5-Gag and Gag-Pol mRNA expression, respectively. (L) Progeny viral RNA was extracted from infected SUPT1 supernatants, and qRT-PCR analysis was used to analyze Gag-Pol mRNA expression. (M) Droplet digital PCR analysis was used to measure progeny Gag-Pol RNA copy numbers. The copy numbers were normalized to GAPDH. Vehicle was set to 1 as a reference. (N) Cell-associated Gag protein was measured 24 h after infection by cell permeabilization and flow cytometry analysis of anti-p24 or isotype control staining. Shown is one representative experiment. (O) Median fluorescence intensity (MFI) was quantified from flow cytometry analysis of intracellular p24. Vehicle was set to 1 as a reference. (P) Progeny virus produced in the presence and absence of SE was analyzed for p24 content by ELISA. (Q) Naive TZM-bl indicator cells were infected with supernatants from infected SUPT1 cells for 24 h, and infectivity was measured by reporter expression and presented as relative light units (RLU). Vehicle was set to 100% as a reference. PCR analysis was normalized to GAPDH, and vehicle was set to 1 as a reference for all experiments. Significance was determined by Student's t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant. The error bars indicate standard errors of the mean of the results of triplicate experiments.
TABLE 1.
Primer sequences
| Gene | Primer sequence |
Reference | |
|---|---|---|---|
| Forward | Reverse | ||
| GAPDH | 5′-CCCCTTCATTGACCTCAACTACA-3′ | 5′-CGCTCCTGGAGGATGGTGAT-3′ | 55 |
| Gag-Pol | 5′-TTCTTCAGAGCAGACCAGAGC-3′ | 5′-GCTGCCAAAGAGTGATCTGA-3′ | 56 |
| Tat-Rev (M669-LA23) | 5′-GTGTGCCCGTCTGTTGTGTGACTCTGGTAAC-3′ | 5′-GCCTATTCTGCTATGTGACACCC-3′ | 57 |
| Vif | 5′-GGGAGCCACACAATGAATG-3′ | 5′-CCAGGGCTCTAGTCTAGG ATC-3′ | 58 |
| Vpr | 5′-ACTTACGGGGATACTTGGGCAG-3′ | 5′-CTCCATTTCTTGCTCTCCTCTGTC-3′ | 56 |
| U5-Gag | 5′-TGTGTGCCCGTCTGTTGTGTGA-3′ | 5′-GAGTCCTGCGTCGAGAGAGCT-3′ | 58 |
| NF-κB p65 | 5′-TCTCATCCCATCTTTGACAATC-3′ | 5′-TGAAATACACCTCAATGTCCTCTTTC-3′ | 59 |
| Sp1 | 5′-TTGAAAAAGGAGTTGGTGGC-3′ | 5′-TGCTGGTTCTGTAAGTTGGG-3′ | 60 |
| Pre-GAPDH | 5′-CCACCAACTGCTTAGCACC-3′ | 5′-CTCCCCACCTTGAAAGGAAAT-3′ | 61 |
| HIV-1 LTR | 5′-TGGAAAATCTCTAGCAGTGGC-3′ | 5′-GAGTCCTGCGTCGAGAGATCT-3′ | 62 |
| ELK1 | 5′-CTGACCCCATCCCTGCTTCCTA-3′ | 5′-GAAGTGAATGCTAGGAGGCAGCG-3′ | 63 |
| MAZ | 5′-GCTTCTCCCGGCCGGAT-3′ | 5′-GAAAGCTGCCTCACATTTCTCACATTTG-3′ | 64 |
SE do not alter the intracellular distribution of viral transcripts.
The intracellular distribution of HIV-1 RNA in infected cells was examined using cellular fractionation (Fig. 2A). Efficiency of cell fractionation was verified by measuring cytoplasmic GAPDH (glyceraldehyde-3-phosphate dehydrogenase) and nuclear PCNA (Fig. 2B). Quantification of total viral genomic RNA (gRNA), as well as cytoplasmic and nuclear gRNA, revealed significant but similar decreases in viral gRNA in the different cellular compartments (Fig. 2C) that could not be attributed to reduced cell viability (Fig. 2D). The RNA encapsidation efficiency was evaluated as the ratio of progeny virion-associated and cytoplasmic gRNA levels (32). Incubation of cells with SE did not reduce the relative amount of packaged viral gRNA (Fig. 2E). There was no preferential localization of viral RNA in either of the compartments, indicating that SE-mediated reduction in total viral RNA was not due to impairment of nucleocytoplasmic shuttling or of the encapsidation machinery.
FIG 2.
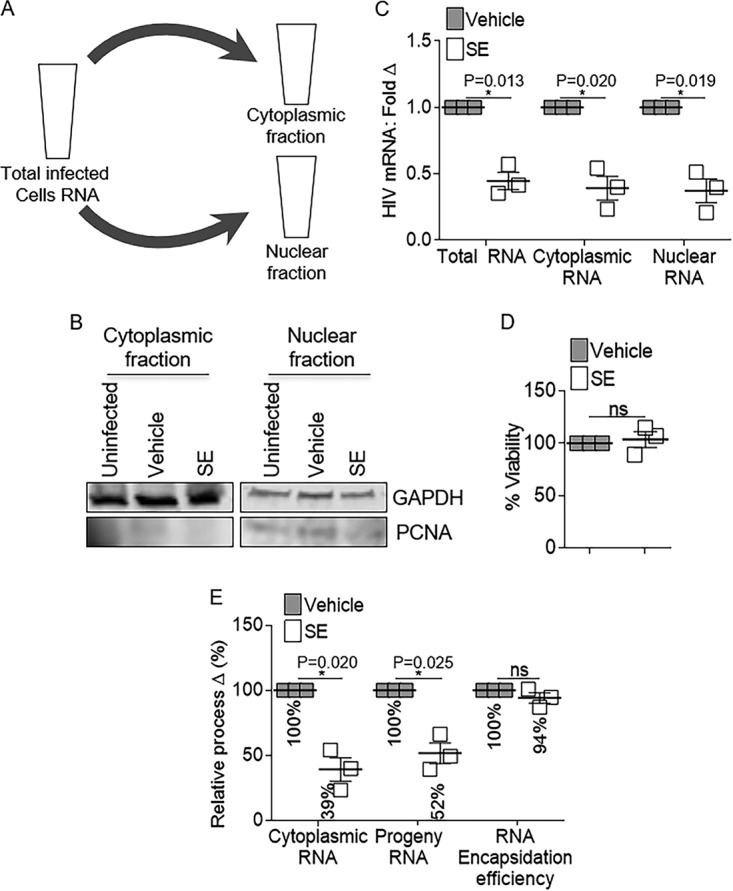
Semen exosome-mediated reduction in HIV RNA expression is operative in host cytoplasmic and nuclear subcellular compartments. Vehicle PBS or semen exosomes (100 μg/ml) were incubated with HIV-1 NL4.3 (8 RTU/ml) at 37°C for 1 h before infection of SUPT1 cells. (A and B) Total cellular RNA and RNA from cells fractionated into cytoplasmic and nuclear compartments. Fractionation was evaluated by Western blot analysis of cytoplasmic GAPDH and nuclear PCNA. Shown is one representative experiment. (C) Total, nuclear, and cytoplasmic RNAs were analyzed by qRT-PCR for viral Gag-Pol mRNA expression. GAPDH and pre-GAPDH were used for normalization of total/cytoplasmic and nuclear RNA, respectively. Vehicle was set as a reference for RNA analysis from each cellular compartment. (D) Cell viability was measured by 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay. Vehicle was used for normalization and set to 100%. (E) Cellular cytoplasmic RNA and progeny viral RNA were analyzed by qRT-PCR for Gag-Pol expression. GAPDH was used for normalization. Vehicle was set as a reference. Encapsidation efficiency was mathematically determined as the ratio of progeny and cytoplasmic Gag-Pol expression. Vehicle was used for normalization and set to 100%. *, P < 0.05; ns, not significant. The error bars indicate standard errors of the mean of the results of triplicate experiments.
SE have minimal off-target effects on human RNA.
The potential for SE to affect the integrity of host cell total RNA and/or the expression of host mRNAs was evaluated in cells infected in the presence and absence of SE. Although SE decreased viral gRNA in SUPT1 cells (Fig. 3A), SE did not affect the level of host GAPDH mRNA (Fig. 3B). SE also did not affect host GAPDH mRNA levels in TZM-bl cells (Fig. 3B). In order to determine if SE induced more subtle effects on host RNA, cellular genes that have more variable expression during HIV-1 infection than the GAPDH gene were analyzed in the presence or absence of SE with and without HIV-1 infection (33). SE did not significantly alter mRNA levels of the transcriptional regulators ELK1 and MAZ in infected and uninfected SUPT1 and TZM-bl cells (Fig. 3C and D). SUPT1 RNA integrity was assessed using an Agilent bioanalyzer. All the cells analyzed (uninfected, vehicle [HIV-1 alone], and SE [HIVSE]) contained similar RNA concentrations (ranging from an average of 88 ng/μl to 127 ng/μl) and RNA integrity (ranging from an average of 5.97 to 6.23) (Fig. 3E and F). Similar results were observed when we examined the levels of 28S and 18S rRNAs using nondenaturing agarose gel electrophoresis in TZM-bl cells (Fig. 3G). The ratios of host 28S and 18S rRNAs were 2.7, 2.5, and 2.4 for uninfected, vehicle (HIV alone), and SE (HIVSE), respectively (Fig. 3H), indicating that SE-mediated downregulation of HIV-1 mRNA expression is not due to a reduction in host RNA integrity. The results also suggest that SE can inhibit HIV-1 expression with minimal regulatory effects on human mRNA production, in general, but it is possible that SE may affect specific host genes that are yet to be discovered.
FIG 3.

Semen exosomes do not alter the integrity of host RNA. Vehicle (PBS) or semen exosomes (100 μg/ml) were incubated with HIV-1 NL4.3 (8 RTU/ml) at 37°C for 1 h before infection. Analysis of cellular and HIV-1 RNAs in cells was performed 24 h after SE or vehicle treatment and HIV-1 infection. (A) Cellular viral RNA in SUPT1 cells was analyzed by qRT-PCR for Gag-Pol mRNA expression. PCR products were visualized with agarose gel ethidium bromide staining. PCR analysis was normalized to GAPDH, and vehicle was set to 1 as a reference. (B) qRT-PCR analysis of SUPT1 and TZM-bl GAPDH mRNA levels. (C) qRT-PCR analysis of SUPT1 and TZM-bl ELK1 mRNA expression. PCR analysis was normalized to GAPDH, and uninfected cells were set to 1 as a reference. Squares containing an “X” represent uninfected cells treated with SE. (D) qRT-PCR analysis of SUPT1 and TZM-bl MAZ mRNA expression. PCR analysis was normalized to GAPDH, and uninfected cells were set to 1 as a reference. (E) Agilent bioanalyzer analysis of SUPT1 cellular RNA in the presence and absence of SE. Shown is one representative profile out of three. The values are presented as averages of triplicate experiments ± standard errors of the mean. (F) SUPT1 cellular RNA concentration and RNA integrity number from Agilent bioanalyzer analysis of three independent experiments. (G) TZM-bl cellular RNA integrity was visualized with an agarose gel. Shown is one representative gel profile out of three. (H) TZM-bl cellular RNA 28S/18S ratios from three independent gel profiles quantified by ImageJ. **, P < 0.01; ns, not significant. The error bars indicate standard errors of the mean of the results of triplicate experiments.
SE reduce HIV-1-dependent, but not mitogen-dependent, LTR activity.
The results presented above suggest that SE inhibit viral RNA expression without altering production of viral RNA species (Fig. 1), reducing nucleocytoplasmic shuttling (Fig. 2), or diminishing RNA integrity (Fig. 3). Since HIV-1 LTR promoter activity is involved in viral transcription, we performed LTR-luciferase assays in cells that contain integrated HIV-1 LTR fused to β-galactosidase (β-Gal) and luciferase as reporters (TZM-bl cells) (34). The cells were exposed to HIV, SE, vehicle (phosphate-buffered saline [PBS]; negative control), or phorbol myristate acetate (PMA) (positive control) using SE posttreatment or pretreatment as previously described. Briefly, SE were added to cells 24 h before HIV or PMA (pretreatment) or SE were added to cells 24 h after HIV or PMA (posttreatment) (28). As expected, both HIV and PMA increased HIV-1 promoter activity in TZM-bl cells (increased relative light units [RLU]) (Fig. 4A and B). However, SE specifically lowered HIV-1-driven luciferase expression without decreasing PMA-induced luciferase expression (Fig. 4A and B, compare open bars in unshaded and shaded areas), regardless of the concentration and timing of PMA addition (Fig. 4C and D). The inability of SE to regulate PMA-driven promoter activity was confirmed in JLat 10.6 cells, which harbor an HIV provirus containing the green fluorescent protein (GFP) open reading frame (ORF) in place of nef and a frameshift mutation in env (35); thus, GFP expression is under the control of the HIV-1 LTR. In addition, tumor necrosis factor alpha (TNF-α) was included as a more physiologically relevant positive control. Similar to the LTR-luciferase results, SE did not decrease PMA- or TNF-α-driven GFP expression in JLat 10.6 cells, regardless of the time of SE addition (Fig. 4E to H, compare solid-line to dashed-line histograms and filled to open bars). The difference in PMA/TNF-α-driven GFP expression in JLat 10.6 cells between pre- and post-PMA/TNF-α conditions reflects the timing of addition and removal of the stimulus prior to determination of GFP expression. Cells were actively stimulated under the pre-PMA/TNF-α conditions (Fig. 4E and G), while the cells had reverted to a latent state under the post-PMA/TNF-α conditions (Fig. 4F and H). It is noteworthy that SE did not alter cell viability in the presence or absence of HIV-1, PMA, or TNF-α pre- or posttreatment (Fig. 4I to L). Together, these results suggest that SE specifically inhibit HIV-1-driven LTR promoter activation.
FIG 4.

HIV-driven LTR promoter transactivation is significantly downregulated by SE. (A) Vehicle or 100 μg/ml SE was added to TZM-bl cells for 24 h, and then the cells were washed before infection with HIV-1 NL4.3 (8 RTU/ml) or treatment with 10 ng/ml PMA (shaded) for an additional 24 h (i.e., the cells were pretreated with vehicle or SE). (B) TZM-bl cells were infected with HIV-1 NL4.3 (8 RTU/ml) or treated with 10 ng/ml PMA (shaded) for 24 h and then washed before treatment with vehicle or 100 μg/ml SE for an additional 24 h (i.e., the cells were posttreated with vehicle or SE). Naive TZM-bl indicator cells were treated with vehicle (PBS) or 100 μg/ml SE pretreatment (C) or posttreatment (D), with increasing concentrations of PMA. Each PMA concentration was normalized to vehicle treatment, where vehicle was set to 100%. RLU were assessed with SteadyGlo. (E) JLat 10.6 cells were treated with vehicle or 100 μg/ml SE and then washed at 24 h, followed by addition of 10 ng/ml PMA for 24 h. GFP expression was analyzed by flow cytometry. Shown is one representative experiment. (F) JLat 10.6 cells were treated with 10 ng/ml PMA and then washed at 24 h, followed by addition of vehicle or 100 μg/ml SE for 24 h. GFP expression was analyzed by flow cytometry. Shown is one representative experiment. (G) JLat 10.6 cells were treated as for panel E, except PMA was replaced with 10 ng/ml TNF-α. GFP expression was analyzed by flow cytometry. Shown is one representative experiment. (H) JLat 10.6 cells were treated as for panel F, except PMA was replaced with 10 ng/ml TNF-α. GFP expression was analyzed by flow cytometry. Shown is one representative experiment. (I) Cell viability was assessed by MTT assay in naive TZM-bl indicator cells that were treated with vehicle (PBS), HIV-1 NL4.3 (8 RTU/ml), 10 ng/ml PMA, or 100 μg/ml SE. (J) Latently infected GFP-expressing JLat 10.6 cells were treated with vehicle (PBS), 10 ng/ml PMA, 10 ng/ml TNF-α, or 100 μg/ml SE. (K) JLat 10.6 cells were treated with vehicle or 100 μg/ml SE before treatment with 10 ng/ml PMA or 10 ng/ml TNF-α (shaded). (L) JLat 10.6 cells were treated with 10 ng/ml PMA or 10 ng/ml TNF-α (shaded) before treatment with vehicle or 100 μg/ml SE. For all the experiments, vehicle treatment was used for normalization and set to 100%. Where indicated, cells were treated for 24 h before washing and subsequent treatment for an additional 24 h. FlowJo software was used for all flow cytometry analysis. **, P < 0.01; ns, not significant. The error bars indicate standard errors of the mean of the results of triplicate experiments.
SE do not alter basal activity of the HIV-1 promoter.
Given that SE inhibit HIV-1-driven LTR promoter activation, we examined the effect of SE on the basal activity of the HIV-1 promoter in the absence of HIV or other LTR-activating agents (PMA or TNF-α). TZM-bl cells were treated with 100 μg of SE alone or with HIV or PMA as controls and examined for luciferase expression 24 h later. While HIV and PMA increased LTR activity in TZM-bl cells, the basal activity of the HIV-1 promoter was not altered by the addition of SE (Fig. 5A). This was recapitulated using JLat 10.6 cells. As expected, PMA and TNF-α significantly activated the LTR promoter as measured by increased GFP expression (Fig. 5B, arrows). The increases in PMA- and TNF-α-mediated LTR promoter activation were ∼25% and ∼48%, respectively (Fig. 5C). In the JLat system, increased promoter activity (GFP expression) results in transcription of viral RNA, as observed in the PMA- and TNF-α-treated cells (Fig. 5D). In contrast, SE did not have an effect on GFP expression (Fig. 5B and C) and did not induce viral transcription relative to the vehicle control (Fig. 5D). To further assess the effect of SE on basal promoter activity, TZM-bl cells treated with increasing concentrations of SE for 24 h did not demonstrate altered promoter activity as measured by luciferase levels (Fig. 5E). Further, addition of SE to JLat 10.6 cells did not change basal GFP expression or cell viability (Fig. 5F to I). To ascertain that the isolation method of SE with ExoQuick reagent did not have an effect on HIV promoter activity, we repeated the promoter activation assay using ExoQuick reagent as a control. The lack of effect by residual ExoQuick reagent on the promoter activation result was verified via ExoQuick extraction of PBS (vehicle) and is shown in Fig. 5J. Thus, SE specifically repressed virus-driven LTR activation and subsequent viral transcription.
FIG 5.

SE alone do not up- or downregulate basal LTR promoter activity. (A) Naive TZM-bl indicator cells were treated with vehicle (PBS), HIV-1 NL4.3 (8 RTU/ml), 10 ng/ml PMA, or 100 μg/ml SE before assessment of promoter activity. (B) Latently infected GFP-expressing JLat 10.6 cells were treated with vehicle (PBS), 10 ng/ml PMA, 10 ng/ml TNF-α, or 100 μg/ml SE. Activation was assessed by GFP flow cytometry. Shown is one representative experiment. (C) Quantification of JLat 10.6 GFP expression from three independent experiments. (D) JLat 10.6 cellular RNA was analyzed by qRT-PCR for HIV-1 Gag-Pol mRNA expression. GAPDH was used for normalization. Vehicle was set as a reference. For all experiments, cells were treated for 24 h before washing and culturing without treatment for an additional 24 h and readout. (E) TZM-bl indicator cells were treated with vehicle (PBS) or increasing concentrations of SE for 24 h before readout of promoter activity. The data are plotted as medians, and the bars are value ranges (minimum to maximum). (F) GFP expression was quantified by FlowJo analysis in latently infected GFP-expressing JLat 10.6 cells that were treated with vehicle (PBS) or 100 μg/ml SE before treatment with PBS. Shown is an average of three independent experiments. (G) Viability of JLat 10.6 cells was measured by MTT assay. (H) GFP expression was quantified in latently infected GFP-expressing JLat 10.6 cells that were treated with vehicle (PBS) before subsequent treatment with control PBS or 100 μg/ml SE. Shown is one representative experiment. (I) Viability of the JLat 10.6 cells was measured by MTT assay. For all the experiments, vehicle treatment was used for normalization and set to 100%. Where indicated, cells were treated for 24 h before washing and subsequent treatment for an additional 24 h. (J) PBS vehicle control or SE was extracted with ExoQuick reagent. Vehicle or 100 μg/ml SE was added simultaneously with HIV-1 NL4.3 to TZM-bl cells, followed by assessment of promoter activity 24 h later. For panels A, E, and J, TZM-bl promoter activity in relative light units was read by Steady-Glo (Promega). FlowJo software was used for all flow cytometry analysis. *, P < 0.05; **, P < 0.01; ns, not significant. The error bars indicate standard errors of the mean of the results of triplicate experiments.
SE inhibit binding and recruitment of host transcription factor components (NF-κB and Sp1) to the HIV-1 promoter in a cell-based assay.
The ability of SE to inhibit HIV transcription provides an experimental system in which transcriptional targets that repress HIV-1 provirus transcription can be identified. Initially, we explored whether SE affected the levels of the transcription factors Sp1 and NF-κB in infected cells. SUPT1 cells infected with HIV-1 in the presence and absence of SE were examined via RT-qPCR and Western blotting. SE had no effect on Sp1 and NF-κB p65 mRNA in HIV-1-infected cells (Fig. 6A and B). Similarly, Sp1 protein levels were unchanged in the presence and absence of SE (Fig. 6C and D). However, SE significantly decreased NF-κB p65 protein in HIV-1-infected SUPT1 cells (Fig. 6C and E). A similar trend was observed in HIV-1-infected TZM-bl cells, where SE did not reduce Sp1 and NF-κB p65 mRNA levels (Fig. 6F and G) but reduced the level of NF-κB protein (Fig. 6H and J) without altering Sp1 protein levels (Fig. 6H and I). Further, SE reduced cytoplasmic levels of NF-κB p65 in HIV-1-infected SUPT1 cells (Fig. 6K and M) and TZM-bl cells (Fig. 6L and M), although nuclear NF-κB p65 levels were more variable (Fig. 6K to M).
FIG 6.

The NF-κB/Sp1 transcription factor pathway is implicated in SE-mediated control of HIV-1 transcription. Vehicle (PBS) or semen exosomes (100 μg/ml) were incubated with HIV-1 NL4.3 (8 RTU/ml) at 37°C for 1 h before infection. (A and B) SUPT1 mRNA for Sp1 (A) and NF-κB p65 (B) by qRT-PCR. (C) Whole-cell lysates of SUPT1 cells were probed for anti-NF-κB p65, anti-Sp1, and anti-GAPDH by Western blotting. The gels shown are from one representative experiment. (D and E) Relative intensities of Sp1 (D) and NF-κB p65 (E) bands by ImageJ quantification. (F and G) TZM-bl cells were analyzed by qRT-PCR for Sp1 (F) and NF-κB p65 (G) mRNA expression. GAPDH was used for normalization. Vehicle was set as a reference. (H) TZM-bl cells were probed for anti-NF-κB p65, anti-Sp1, and anti-GAPDH by Western blotting. Shown is one representative experiment. (I and J) TZM-bl blot band intensities for Sp1 and NF-κB p65 were measured from three independent experiments by ImageJ quantification. (K and L) Cytoplasmic and nuclear extracts of SUPT1 (K) and TZM-bl (L) cells probed by Western blotting for anti-NF-κB p65. Anti-GAPDH and anti-PCNA were used as cytoplasmic and nuclear markers, respectively. (M) Relative band intensities from panels K and L by ImageJ quantification from three independent experiments. (N) ChIP assay with anti-IgG or anti-NF-κB p65 for qRT-PCR analysis of HIV-1 LTR. GAPDH was used for input control, and HIV was set to 1 as a reference. ChIP products were quantified by the fold enrichment method; shown is an average of three experiments. Products were visualized by agarose gel and ethidium bromide staining. (O) Electrophoretic mobility shift assay. TZM-bl nuclear extracts were incubated with Cy5-labeled probe in the presence of dI-dC competitor and BSA before loading on 5% nondenaturing PAGE gels. The gel shown is for one representative experiment. C1 to C5 denote NF-κB complexes. The bars are ImageJ quantifications of C5. (P) Gel mobility shift assay as in Fig. 6O, but with Sp1. The bars are ImageJ quantifications of C4. *, P < 0.05; ***, P < 0.001; ns, not significant. The error bars indicate standard errors of the mean of the results of triplicate experiments.
Direct binding of NF-κB p65 to the HIV-1 enhancer in the presence of SE was analyzed by chromatin immunoprecipitation–real-time quantitative PCR (ChIP–RT-qPCR). Chromatin was prepared from HIV-infected SUPT1 cells in the presence and absence of SE. Because we did not select cells for HIV expression, these experiments utilized a heterogeneous population of HIV-1-infected cells with a spectrum of viral gene expression. ChIP was performed using antibodies against NF-κB p65, followed by RT-qPCR for the HIV-1 LTR and host GAPDH. NF-κB p65 bound to the LTR in the absence of SE (Fig. 6N). However, in the presence of SE, the association of NF-κB p65 with the LTR was reduced by 43%, suggesting that SE may inhibit HIV-1 transcription in part through decreasing NF-κB p65 recruitment and binding to the HIV-1 LTR. We previously showed that cell-associated HIV-1 DNA is reduced in the presence of SE; thus, these data may indicate that there is less LTR available for NF-κB binding (27). Because there is less NF-κB protein in HIV-1-infected cells in the presence of SE (Fig. 6A to M), it is also plausible that there is a lower abundance of NF-κB protein available for binding.
Electrophoretic mobility shift assays (EMSA) with NF-κB p65 probe performed on nuclear extracts of TZM-bl cells infected with HIV-1 for 24 h in the presence or absence of SE provided insight into SE-dependent inhibition of NF-κB LTR association. Using an NF-κB p65 oligonucleotide probe, HIV-1 infection (Fig. 6O, lane 3) increased complex C5 and decreased complex C2 compared to uninfected cells (lane 2). HIV-1 infection enhanced C1 and C3 (lane 3) relative to uninfected cells (lane 2). These complexes (C1 and C3) were reduced when infected cells were treated with SE (compare lanes 3 and 4). In addition, SE treatment during infection opposed the HIV-1-induced increase of C5 (lanes 3 and 4), but SE alone (lane 5) did not affect C5 compared to uninfected cells (lane 2). HIV-1 infection increased the amount of complex 5, while SE decreased this complex by about 50% (Fig. 6O, bars). A similar effect was observed using an Sp1 oligonucleotide. SE significantly decreased the intensity of some complexes that were induced by HIV-1 (Fig. 6P, C4 and C5, lanes 3 and 4), and SE decreased the level of C5 by about 50% (Fig. 6P, bars).
SE directly bind to NF-κB and Sp1 sequences.
Consistent with the cell-based EMSA observations demonstrating that SE block binding of NF-κB p65 and Sp1 to the HIV-1 enhancer (Fig. 6O and P), a specific high-molecular-weight mobility shift was observed when SE were incubated with NF-κB p65 (Fig. 7A and B, lanes 3 and 4) and Sp1 (Fig. 7C and D, lanes 3 and 4) oligonucleotides in an in vitro EMSA. Incubation of NF-κB p65 and Sp1 with a control bovine serum albumin (BSA) protein did not demonstrate a shift (Fig. 7A to D, lanes 2). SE, both intact and with membranes disrupted, complexed with NF-κB p65 and Sp1 oligonucleotides. However, the complexes disappeared upon heating (Fig. 7A and C, lanes 5) or in the presence of 0.5% sodium dodecyl sulfate (SDS) (Fig. 7B and D, lanes 5). These results demonstrate that SE bound directly and specifically to the NF-κB and Sp1 sequences and that the complexes formed were heat-labile protein-DNA complexes. Together, these data indicate that SE-mediated downregulation of HIV-1 transcription involves inhibition of the NF-κB/Sp1 association with the viral LTR.
FIG 7.
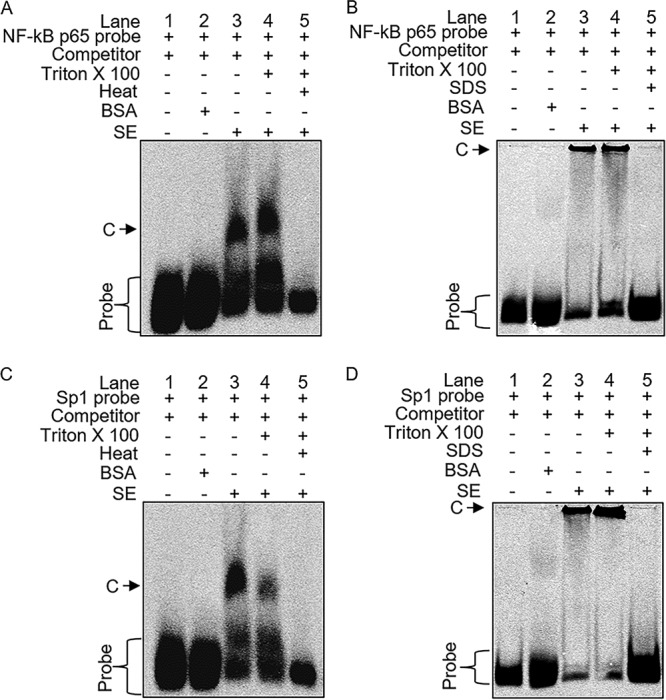
Direct binding of SE to NF-κB/Sp1. EMSA conditions were as follows: probe alone (lane 1), 100 μg BSA (lane 2), 25 μg intact SE (lane 3), SE treated with 0.25% Triton X-100 to open the membrane (lane 4), SE treated with Triton X-100 and boiled at 95°C for 30 min or treated with 0.5% SDS to denature the SE proteins (lane 5). Samples were incubated with dI-dC cold competitor (1,000×) on ice for 20 min before addition of NF-κB p65 probe (A and B) or Sp1 probe (C and D) and further incubation on ice for 20 min. Samples were separated on a 4% nondenaturing PAGE gel at 4°C protected from light. C denotes NF-κB or Sp1 complexes. The gel images show one representative experiment of three.
Binding of RNA Pol II to the HIV-1 LTR is impaired by SE.
Because SE-mediated inhibition of NF-κB recruitment to the viral LTR was incomplete, we explored additional mechanisms by which SE might repress transcription. Productive HIV-1 transcription requires recruitment of the Pol II complex to the promoter region of the HIV-1 LTR. To explore the possibility that SE decrease Pol II activity, we assessed the presence of Pol II at the HIV-1 LTR by ChIP–RT-qPCR. While Pol II is efficiently recruited to the viral LTR in HIV-1-infected SUPT1, SE decreased the amount of Pol II associated with the LTR by 58% (Fig. 8A). Reduced Pol II was associated with decreased viral mRNA (Fig. 8B). Since Pol II association with HIV-1 sequences was inhibited by SE, it is plausible that SE might inhibit HIV-1 transcription by suppressing Pol II processivity. In addition, because we previously showed less intracellular HIV-1 DNA in the presence of SE, it is also possible that there is less LTR available for binding by Pol II, leading to less HIV-1 mRNA production (27).
FIG 8.
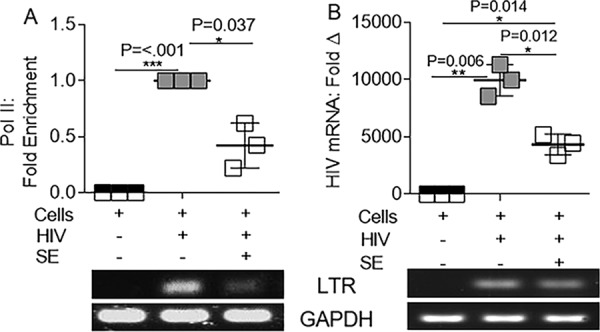
SE reduce RNA Pol II association with HIV-1 LTR. Vehicle (PBS) or semen exosomes (100 μg/ml) were incubated with HIV-1 NL4.3 (8 RTU/ml) at 37°C for 1 h before infection of SUPT1 cells. (A) ChIP assay with anti-IgG or anti-Pol II for qRT-PCR analysis of HIV-1 LTR. GAPDH was used for input control, and HIV was set to 1 as a reference. ChIP products were quantified by the fold enrichment method; shown is an average of three experiments. (B) SUPT1 mRNA was evaluated for HIV-1 LTR gene expression by qRT-PCR. Products were visualized by agarose gel ethidium bromide staining. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant. The error bars indicate standard errors of the mean of the results of triplicate experiments.
SE inhibit Tat-dependent HIV-1 LTR activity.
It is known that Tat participates in a positive-feedback mechanism that maintains high levels of proviral transcription in HIV-1-infected cells. To examine the effect of SE on Tat-dependent HIV-1 activation, TZM-bl cells were transfected with a Tat expression vector (pCMV Tat, here referred to as p-Tat) (36), followed by treatment with vehicle (PBS) or SE (100 μg/ml). SE inhibited Tat-dependent transcription by ∼54% in the luciferase assay (Fig. 9A) and by ∼55% in the β-Gal assay (Fig. 9B). SE reduced the effect of exogenous Tat protein (recombinant Tat [r-Tat]) by 66%, confirming the effect of SE on Tat-driven HIV-1 LTR activation (Fig. 9C). The kinetics of SE-mediated inhibition of HIV-1 promoter activity was evaluated by time-lapse analysis of HIV-1 LTR activity in cells infected with HIV-1 (Fig. 9D), transfected with p-Tat (Fig. 9E), or treated with r-Tat (Fig. 9F). In all three scenarios, SE inhibited HIV-1 LTR activity, and at 16 h, HIV-1-, p-Tat-, and r-Tat-driven LTR activities were inhibited by ∼46%, 71%, and 64% (Fig. 9D to F), respectively. The effect of SE on LTR activity was still observed (∼58% inhibition) 24 h after exposure to HIV-1 (Fig. 9D), p-Tat at ∼50% (Fig. 9E), and r-Tat at ∼47% (Fig. 9F). The activities of SE on HIV-1 and Tat were independent of cell viability, as measured by MTT assay (Fig. 9D to F, bottom bars). These time-lapse experiments indicate that the inhibitory effect of SE occurs early and may persist for up to 24 h. The use of p-Tat, which enables steady synthesis of Tat, revealed that SE are capable of inhibiting LTR activation of de novo Tat (Fig. 9E).
FIG 9.

SE inhibition of HIV-1 transactivation is mediated by interference with Tat protein-mediated LTR activation. TZM-bl cells were transfected with p-Tat for 3 h before removal of input plasmid, treatment with SE (100 μg/ml), and incubation for 16 h. (A and B) Promoter activity was measured by luciferase assay (A) and β-Gal assay (B). (C) TZM-bl cells were treated with r-Tat (10 μg/ml) that was preincubated with PBS (vehicle) or SE (100 μg/ml) for 1 h at 37°C prior to addition to the cells. Promoter activity was assessed by a luciferase assay at 16 h posttreatment. (D to F) Kinetics of SE inhibition of HIV-1 LTR promoter activity and MTT viability at 24 h. RLU fold changes are plotted as medians, and the bars are value ranges (minimum to maximum). (D) HIV-1 (8 RTU/ml) preincubated for 1 h at 37°C with PBS or SE (100 μg/ml). (E) Vehicle, SE alone, and p-Tat transfection 3 h before addition of SE or vehicle (PBS). (F) r-Tat (10 μg/ml) preincubated for 1 h at 37°C with PBS or SE (100 μg/ml). (G and H) TZM-bl cells were transfected with p-Tat for 3 h before treatment with SE and incubation at 37°C for 16 h. Nuclear extracts were incubated with Cy5-labeled NF-κB p65 (G) or Sp1 (H) oligonucleotides in the presence of dI-dC competitor and BSA before loading on 5% nondenaturing PAGE gels. The gel shown is for one representative experiment. C2 band intensities were quantified with ImageJ, and the error bars represent the standard errors of the mean of the results of duplicate experiments. *, P < 0.05; **, P < 0.01; ns, not significant.
SE directly counteract association of Tat with NF-κB p65 and Sp1.
Tat directly associates with the p65 subunit of NF-κB, thereby increasing p65 DNA binding and transcriptional activities (19). Thus, we hypothesized that SE would repress Tat association with NF-κB p65 and Sp1. Since SE had a significant impact on Tat-mediated activation by 16 h, we conducted our studies at that time. TZM-bl cells were transfected with p-Tat, and cells were treated with either vehicle or SE. Nuclear extracts were analyzed by EMSA following incubation with labeled NF-κB p65 or Sp1 oligonucleotides. Two complexes (C1 and C2) were observed. C2, but not C1, intensity was reduced by SE, and therefore, C2 was quantified. Although the affinity of Tat for NF-κB p65 was not increased beyond basal levels, SE significantly decreased Tat binding to NF-κB p65 oligonucleotides (Fig. 9G, C2, and compare lanes 3 and 4, gray and white bars). In contrast to Tat–NF-κB p65 binding, Tat significantly increased binding of Sp1 to DNA, but SE potently suppressed Tat-Sp1 association (Fig. 9H, C2, and compare lanes 3 and 4, gray and white bars). Together, these results suggest that SE repress HIV-1 transcriptional activation by reducing Tat association with host transcription factors NF-κB p65 and Sp1.
DISCUSSION
The HIV-1 provirus transcribes its genes in actively infected cells, but proviral transcription is halted and suppressed in latent infection. It is known that HIV-1 transcription is regulated by disparate positive and negative regulators whose activities control viral replication in host cells (3). Since HIV-1 transcription is regulated by the relative levels of positive and negative regulators, any factor capable of inhibiting positive regulators of transcription may lead to the suppression of proviral transcription.
Possible links between SE and HIV-1 transcription have previously been inferred. In 2014 and 2015, Madison et al. (26, 27) observed that, among other functions, SE induced a decrease in HIV-1 RNA expression, though it was not determined if SE per se repressed viral transcription. Here, we provide evidence that SE repress viral transcription by blocking human and viral transcription factor access to the HIV-1 promoter. We hypothesize that this is accomplished through either steric hindrance or epigenetic modifications. Transcription of the HIV-1 provirus is regulated at different levels, including recruitment of cellular transcription factors, chromatin organization, transcription initiation, and transcription elongation. Our data strongly suggest that SE potently induce a transcription-repressed state in the HIV-1 promoter leading to inefficient viral RNA expression. The exact effect of SE on the viral promoter is still unknown, but given that SE bind to NF-κB and Sp1; impair NF-κB, Sp1, and Pol II recruitment to the HIV-1 promoter; and block Tat binding to NF-κB and Sp1, it is evident that SE negatively regulate HIV-1 transcription.
SE reduced interactions between cellular and viral transcription factors and the HIV-1 promoter. These observations suggest that SE may be involved in HIV-1 transcription at the level of initiation. Previous studies linked NF-κB to the initiation of HIV-1 transcription (37). NF-κB is required for HIV-1 transcription initiation and chromatin remodeling and plays both positive and negative roles in HIV-1 transcription and latency. Induction of NF-κB p65 and recruitment of other host factors, such as histone acetyltransferases, are central to reversing proviral transcription in latency (38). We observed that SE modestly reduced NF-κB p65 protein levels but significantly inhibited NF-κB p65 and Sp1 binding to the HIV-1 LTR. Further, biochemical analysis by gel shift assays (EMSA) suggested that SE may directly bind NF-κB p65 and Sp1 through protein-protein interaction, since binding was obliterated by increasing the temperature or addition of SDS. However, this hypothesis remains to be validated once the SE-specific component(s) mediating these processes has been identified.
Using SE to repress HIV-1 transcription allowed us to identify transcription elongation as another target of SE in provirus transcription. To our knowledge, this is the first report to suggest that Pol II recruitment/binding is targeted by exosomes. In the presence of SE, ChIP analysis demonstrated reduced Pol II enrichment at the HIV-1 LTR. In the same cells, we observed decreased HIV-1 RNA, suggesting an association between inhibition of Pol II recruitment and reduced RNA expression.
Following transcription initiation, the maintenance of highly efficient viral gene expression and viral replication is directed by Tat (4). Although a viral protein, Tat associates with Sp1, NF-κB, P-TEFb, and the trans-activator-responsive element TAR at the HIV-1 LTR (19, 39). Interaction of Tat with NF-κB occurs via the p65 subunit of NF-κB (19). SE blocked Tat–NF-κB interactions in the absence of other cellular cofactors. Thus, in addition to reducing transcription initiation, SE also serve as negative regulators of HIV-1 transcription elongation via inhibition of Tat. Indeed, in our system, SE reduced levels of both Tat binding to NF-κB p65 and Sp1 sequences and Tat-induced Luc and β-Gal gene expression under HIV-1 LTR control. In addition to the role of Tat as the master regulator of HIV-1 gene expression and latency, Tat regulates Pol II-mediated activation of HIV-1 transcription and controls alternative mechanisms of viral replication by regulating chromatin remodeling (40), mRNA splicing (41), Rev function (42), microRNA biogenesis (43), and reverse transcriptase function (5, 6, 44–46). Interestingly, we previously found that SE regulate the ratio of HIV-1 reverse transcriptase subunits (27). Whether the effects of SE on reverse transcriptase and Tat utilize distinct mechanisms of inhibition has yet to be determined. However, it is clear that two important HIV-1 proteins, reverse transcriptase and Tat, are susceptible to the antiviral effects of SE, and there may be multiple SE-driven mechanisms that negatively regulate HIV-1 infection and replication.
Because we have yet to identify the factor(s) contributing to our observations, we are hesitant to conclusively link these effects solely to exosomes and recognize that the effects may be due to a host factor that is associated with exosomes during purification. However, we believe this is unlikely, given that purification of semen exosomes by different methods has resulted in the same phenotype (27, 47). Characterization of the protein and nucleic acid content of SE is needed to identify the component(s) mediating the inhibitory activity on the HIV-1 LTR.
Our study extends our understanding of the role of exosomes in microbial pathogenesis. In the context of HIV-1 infection, the source of exosomes dictates their role in HIV-1 infection (48). While exosomes produced from some HIV-1-infected cell lines may enhance viral infection, exosomes from body fluids, including breast milk, vaginal lavage fluid, and semen, inhibit HIV-1 infection (26, 27, 49, 50). These diverse functions highlight the complexity associated with exosomes and their functions. Because of the significant role of exosomes in intercellular communication, the therapeutic potential of exosomes continues to be increasingly investigated and developed. Novel therapies against HIV-1 infection are needed to combat drug resistance adaptations by the virus and interaction with illicit substances. Exosomes with antiviral function offer the unique opportunity to harness a naturally evolved resistance to HIV-1 infection that has thus far not been overcome during HIV-1 evolution.
On the basis of our findings, we suggest that SE may fit this critical need. SE contain known antiviral factors, such as the APOBEC family; preferentially inhibit HIV-1 infection; and alter the balance of HIV-1 reverse transcriptase subunits (27). Here, we show that SE interfere with the critical quadripartite complex Tat–NF-κB–Sp1–Pol II circuitry, resulting in impaired proviral transcription and replication. Thus, the translational implication of our findings is the potential use of SE or its cargo to inhibit Tat function. The HIV-1 Tat protein is expressed early; therefore, its inhibition would result in early termination of viral replication. Although secreted, Tat inhibition may result in little or no cytotoxicity, since it has no cellular homologs. Blockade of Tat is expected to prevent Tat-mediated feedback loops that enhance viral transcription and the production of viral progeny and blocking of viral reactivation to promote prolonged silencing of latent viruses. Indeed, the ability of SE to inhibit the activities of two different classes of transcription factors (host and viral) involved in HIV-1 gene expression explains why SE is such a powerful negative regulator of HIV-1 replication.
MATERIALS AND METHODS
Ethical approvals.
The University of Iowa Institutional Review Board (IRB) approved the use of human specimens. HIV-1-negative and -positive subjects consented to participate in this study via written informed consent. All samples were received unlinked to any identifiers. All experiments were performed in accordance with the approved university guidelines and regulations.
Purification of exosomes from human semen.
Semen samples received from the University of Iowa In Vitro Fertilization and Reproductive Testing Laboratories were collected and stored at −80°C until isolation. All the donors had no history of HIV, hepatitis B virus (HBV), or hepatitis C virus (HCV) infection. Semen exosomes were isolated using a previously described protocol (26–28, 47) in which, prior to isolation, 12 individual semen samples were thawed at room temperature and centrifuged (10,000 × g; 30 min; 4°C) to pellet cellular debris, after which the supernatants, or seminal plasma, from individual donors were pooled, placed in a new tube, mixed by inversion with ExoQuick reagent (SBI) at a ratio of 4:1 (seminal plasma to ExoQuick), and incubated at 4°C overnight according to the manufacturer's instructions. To pellet the exosomes, the mixture was centrifuged at 1,500 × g for 30 min at 4°C, and the ExoQuick/exosome-free supernatant was removed. Residual ExoQuick was removed by centrifuging the exosome pellet at 1,500 × g for 5 min and discarding the supernatant. The exosome pellet was resuspended in PBS in 1/10 of the original volume of semen and is here referred to as SE; 100 μg/ml SE was used for all cell treatments.
Cells and plasmids.
TZM-bl, SUPT1, JLat full-length (JLat 10.6), and HIV-1 lymphadenopathy-associated virus (LAV)-infected Jurkat E6 (J1.1) cells were obtained through the NIH AIDS Reagent Program. 293T cells were purchased from the ATCC. TZM-bl and 293T cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (Gibco-BRL/Life Technologies) and SUPT1, JLat 10.6, and Jurkat E6 (J1.1) cells in RPMI (Gibco-BRL/Life Technologies) containing 5% exosome-depleted fetal bovine serum (FBS) (Gibco), 100 U/ml penicillin, 100 μg/ml streptomycin, sodium pyruvate, and 0.3 mg/ml l-glutamine (Invitrogen, Molecular Probes), as previously described (27). The pNL4.3 plasmid was obtained from the NIH AIDS Reagent Program, and the pCMV Tat plasmid (p-Tat) was a kind gift from Francesca Di Nunzio (Institut Pasteur, Paris, France).
HIV-1 production.
HIV-1 was produced either by purification of supernatants from HIV-1 latently infected Jurkat cells or by 293T cell transfection with pNL4.3. Briefly, cell culture supernatant of J1.1 cells was clarified by centrifugation at 2,000 × g for 10 min, and virus was concentrated by ultracentrifugation at 100,000 × g for 2 h at 4°C and resuspended in RPMI medium. 293T cells were seeded in a 6-well tissue culture-treated plate with 2 ml antibiotic-free medium 24 h before transfection. Ten microliters of Lipofectamine 2000 (Invitrogen) was combined with 150 μl Opti-MEM for 5 min at room temperature, and 4 μg/well pNL4.3 plasmid was diluted in 150 μl Opti-MEM. DNA and Lipofectamine were combined for 20 min at room temperature, and 250 μl of DNA-Lipofectamine complex was added to the cells; 24 h posttransfection, the input plasmid was removed by replacing the transfection medium with antibiotic-free DMEM. Forty-eight to 72 h posttransfection, supernatants were collected and clarified of cell debris by centrifugation at 2,000 × g for 10 min before determination of the viral titer. The viral titer was determined by EnzChek reverse transcriptase assay (Life Technologies) (27).
LTR promoter activation assay.
Vehicle (PBS), HIV-1 NL4.3 (8 reverse transcriptase units [RTU]/ml), 100 μg/ml SE, or 10 ng/ml PMA (Sigma-Aldrich) was added to TZM-bl indicator cells. Where indicated, treatments were added to cells for 16 or 24 h before removal, washing, and treatment or culturing for an additional 24 h, after which the cells were lysed and measured for luciferase reporter activity with Steady-Glo (Promega). Vehicle (PBS), 100 μg/ml SE, 10 ng/ml PMA (Sigma-Aldrich), or 10 ng/ml TNF-α (Sigma-Aldrich) was added to JLat 10.6 indicator cells. Where indicated, the treatments were added to the cells for 24 h before removal, washing, and treatment or culturing for an additional 24 h, after which the cells were measured for GFP activity by flow cytometry analysis and lysed for RNA evaluation.
Western blot analysis.
Whole-cell lysates or cells fractionated into cytoplasmic and nuclear compartments via NE-PER nuclear and cytoplasmic extraction reagents (Thermo Fisher, Life Technologies) according to the manufacturer's protocol were probed with the following primary antibodies: anti-GAPDH, anti-NF-κB p65, anti-Sp1, and anti-PCNA. The appropriate secondary IRDye antibodies were used for Odyssey infrared imaging (Li-Cor Biosciences) (51–53). Image J was used to the quantify blots.
ChIP assay.
SUPT1 cells were treated with vehicle or infected with HIV in the presence and absence of SE for 24 h. For NF-κB p65, SUPT1 cells were treated with vehicle or infected with HIV-1 in the presence and absence of SE (100 μg/ml) or 10 ng/ml PMA (Sigma-Aldrich) for 24 h. The ChIP assay was performed using a Pierce ChIP assay kit (Thermo Fisher, Life Technologies). Immunoprecipitations (IP) were performed with lysates from 1 × 106 cells per reaction with 2 μg mock anti-IgG antibody, 5 μg anti-Pol II antibody (Thermo Fisher, Life Technologies), or 5 μg anti-NF-κB p65 (Thermo Fisher, Life Technologies). The extracted DNA was subjected to PCR with SYBR green master mix using LTR and GAPDH primers (Table 1). The products were resolved on 3% agarose gels. The data were normalized via fold enrichment quantification [step 1, (IP CT) − (mock CT); step 2, 2−ΔΔCT] (Thermo Fisher, Life Technologies), where IgG served as the mock threshold cycle (CT) and was subtracted from each IP CT value.
p24 quantification.
The progeny virion p24 released from infected cells was quantified by p24 enzyme-linked immunosorbent assay (ELISA) (XpressBio) according to the manufacturer's instructions. Intracellular p24 was evaluated by flow cytometry analysis via cell permeabilization and addition of primary antibody p24 (4121; NIH AIDS Reagent Repository) before incubation with secondary antibody, Alexa Fluor-488 (Jackson ImmunoResearch).
RNA evaluation.
Cells were fractionated into cytoplasmic and nuclear compartments with NE-PER nuclear and cytoplasmic extraction reagents (Thermo Fisher, Life Technologies) according to the manufacturer's protocol. RNA was extracted from cells or nuclear and cytoplasmic fractions using an RNeasy kit (Qiagen) according to the manufacturer's instructions. The RNA was subjected to treatment with DNase (Qiagen) as previously described (27). Viral RNA was extracted from virions using a viral RNA isolation kit (Zymogen). RNA integrity was determined with an Agilent bioanalyzer. The RNA concentration was determined by NanoDrop RNA absorbance, and an equivalent concentration of RNA (1 ng/μl) was used for cDNA synthesis (ABI). Gene-specific primers were used to amplify GAPDH, as previously described (27), and pre-GAPDH, as well as HIV-1 LTR, Gag-Pol, U5-Gag, Vif, Vpr, Tat-Rev, NF-κB p65, and Sp1 (Table 1). Gag-Pol-specific primers were used for gRNA quantification (Table 1). qRT-PCR products were analyzed by relative quantification for all transcripts; each transcript was first normalized to the corresponding GAPDH value and, second, where indicated, to either HIV-infected vehicle-treated cells or uninfected vehicle-treated cells set to 1 as a reference value. PCR amplicons were visualized where indicated using a 2% agarose gel and ethidium bromide staining. PCR analysis was performed using 7500 Fast qRT-PCR (ABI) and droplet digital PCR (ddPCR) (Bio-Rad). For ddPCR, RNA extraction and cDNA synthesis were performed as described above. The ddPCR reaction mixture was prepared using 100 ng cDNA, QX200 EvaGreen Supermix (Bio-Rad), and the primers described above according to the manufacturer's protocol. A Bio-Rad QX200 droplet generator formed each PCR mixture into droplets. The PCR was subsequently carried out with a C1000 Touch thermal cycler (Bio-Rad) with cycling conditions as suggested by the manufacturer. The PCR mixture was quantified using a Bio-Rad QX200 droplet reader. The data were analyzed with QuantaSoft software (Bio-Rad), and reactions with less than 10,000 droplets were excluded. For qRT-PCR and ddPCR, fold change values were used for statistical analysis.
SE inhibition of HIV-1.
SE (100 μg/ml) or an equivalent volume of vehicle (PBS) was preincubated with 8 RTU/ml HIV-1 at 37°C for 1 h prior to addition to target cells. The effects of SE on HIV-1 were determined 24 h later. Progeny virion assays were performed by infection in the presence and absence of SE for 4 h before removal, washing, and subsequent culture in medium without treatment until 24 h postinfection. Progeny virion released from infected cells was determined by incubation with naive TZM-bl cells for 24 h before measurement of infectivity by Steady-Glo (Promega) luminescence.
EMSA.
Nuclear extracts from TZM-bl (1 × 106 to 2 × 106) or SUPT1 (4 × 106 to 5 × 106) cells were prepared using nuclear and cytoplasmic extraction reagents (NE-PER; Thermo Fisher), following the manufacturer's instructions, and the proteins were quantified using Bradford reagent (Bio-Rad). 5′-Cy5-labeled NF-κB p65 and Sp1 forward oligonucleotides (probes) (Table 2), as well as their complementary reverse primers, were synthesized and high-performance liquid chromatography (HPLC) purified by Integrated DNA Technologies (Coralville, IA). Sequences were as follows: NF-κB p65 consensus oligonucleotide, forward, 5′-Cy5-AGTTGAGGGGACTTTCCCAGGC-3′, and reverse, 3′-TCAACTCCCCTGAAAGGGTCCG-5′; Sp1 consensus oligonucleotide, forward, 5′-Cy5-ATTCGATCGGGGCGGGGCGAGC-3′, and reverse, 3′-TAAGCTAGCCCCGCCCCGCTCG-5′; the underlined regions are the binding sites for NF-κB and Sp1 transcription factors, respectively (Table 2). Equal amounts (1 pmol) of forward and reverse primers were annealed in 100 μl annealing buffer (50 mM HEPES, pH 7.9, 100 mM NaCl, 1 mM EDTA) by heating at 95°C for 1 min and slowly decreasing the temperature to 25°C (2.5% decrease rate in a Veriti thermal cycler [Applied Biosystems]). In a 19-μl total volume, 40 μg nuclear extracts was mixed with 5 μl 4× binding buffer (20 mM HEPES, pH 7.9, 1 mM dithiothreitol [DTT], 0.1 mM EDTA, 50 mM KCl, 5% [vol/vol] glycerol, 0.2 mg/ml BSA) and 1 μl dI-dC competitor (1 μg/μl) and incubated on ice for 20 min. One microliter of annealed probe (20 fmol) was then added, and the mixture was further incubated on ice for 20 min; 2 μl of 10× orange loading dye (Li-Cor) was finally added before loading on a 6% nondenaturing PAGE gel. The gel was separated for 90 min (400 V; 25 mA; 5 W) in a cold room and protected from light. Gel visualization was performed using the Odyssey infrared imaging system (Li-Cor), and gel bands were quantified using ImageJ software.
TABLE 2.
Probe sequences
| Target | Probe sequence |
|
|---|---|---|
| Forward | Reverse | |
| NF-κB p65 consensus oligonucleotide | 5′–Cy5-AGTTGAGGGGACTTTCCCAGGC–3′ | 5′-GCCTGGGAAAGTCCCCTCAACT-3′ |
| Sp1 consensus oligonucleotide | 5′–Cy5-ATTCGATCGGGGCGGGGCGAGC–3′ | 5′-GCTCGCCCCGCCCCGATCGAAT-3′ |
The underlined regions are the binding sites for NF-κB and Sp1.
SE inhibition of Tat.
TZM-bl cells were transfected with p-Tat plasmid using EndoFectin Max transfection reagent (GeneCopoeia) at a ratio of 2 μl EndoFectin to 1 μg plasmid in reduced-serum medium (Opti-MEM). Three hours posttransfection, the medium was replaced with complete DMEM containing 100 μg/ml SE or an equivalent volume of vehicle (PBS). For recombinant Tat, the protein (10 μg/ml) was preincubated with 100 μg/ml SE or an equivalent volume of vehicle (PBS) at 37°C for 1 h prior to addition to target cells. The effects of SE on Tat were determined 16 h later or at the indicated times by measuring promoter activity using the Steady-Glo (Promega) luminescence assay or the β-galactosidase assay reagent (Pierce, Thermo Fisher).
Cell viability.
Cell viability was determined by MTT assay as previously described (54).
Statistics.
Two-tailed paired Student t test P value (GraphPad Prism) calculations determined statistical significance. The error bars represent standard errors of the mean (SEM) across independent experiments unless otherwise indicated. Exact P values for nonsignificant values are reported in Table 3.
TABLE 3.
Exact P values for nonsignificant values
| Figure |
P value |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HIV vs. HIV + SE | Cells vs. HIV | Cells vs. HIV + SE | Cells vs. SE | PMA vs. SE | TNF-α vs. SE | Cells vs. PMA | Cells vs. TNF-α | HIV vs. SE | p-Tat vs. SE | r-Tat vs. SE | Cells vs. p-Tat | |
| 1O | 0.065 | |||||||||||
| 1P | 0.53 | |||||||||||
| 2D | 0.67 | |||||||||||
| 2E | 0.28 | |||||||||||
| 3B (SUPT1) | 0.60 | 0.064 | ||||||||||
| 3B (TZM-bl) | 0.063 | 0.11 | ||||||||||
| 3C (SUPT1) | 0.12 | 0.69 | 0.95 | |||||||||
| 3C (TZM-bl) | 0.52 | 0.16 | 0.10 | |||||||||
| 3D (SUPT1) | 0.27 | 0.16 | 0.22 | |||||||||
| 3D (TZM-bl) | 0.11 | 0.98 | 0.59 | |||||||||
| 3F (RNA concn) | 0.44 | 0.10 | 0.25 | |||||||||
| 3F (RNA integrity no.) | 0.67 | 0.46 | 0.094 | |||||||||
| 3H | 0.22 | 0.72 | 0.54 | |||||||||
| 4A | 0.72 | |||||||||||
| 4B | 0.64 | |||||||||||
| 4C | 0.22–0.99a | |||||||||||
| 4D | 0.10–0.26a | |||||||||||
| 4E | 0.86 | |||||||||||
| 4F | 0.24 | |||||||||||
| 4G | 0.76 | |||||||||||
| 4H | 0.83 | |||||||||||
| 4I | 0.94 | 0.57 | 0.53 | |||||||||
| 4J | 0.31 | 0.88 | 0.080 | |||||||||
| 4K | 0.72 | 0.19 | ||||||||||
| 4L | 0.67 | 0.93 | ||||||||||
| 5A | 0.25 | |||||||||||
| 5C | 0.26 | |||||||||||
| 5D | 0.50 | |||||||||||
| 5E | 0.066–0.87a | |||||||||||
| 5F | 0.069 | |||||||||||
| 5G | 0.45 | |||||||||||
| 5H | 0.96 | |||||||||||
| 5I | 0.76 | |||||||||||
| 6A | 0.58 | |||||||||||
| 6B | 0.43 | |||||||||||
| 6D | 0.85 | 0.19 | 0.48 | |||||||||
| 6E | 0.46 | 0.38 | ||||||||||
| 6F | 0.23 | |||||||||||
| 6G | 0.43 | |||||||||||
| 6I | 0.60 | 0.29 | 0.28 | |||||||||
| 6J | 0.31 | 0.62 | ||||||||||
| 6M Nucleus TZM-bl | 0.087 | |||||||||||
| 6O | 0.69 | |||||||||||
| 6P | 0.27 | |||||||||||
| 9D (0.5 h) | 0.29 | |||||||||||
| 9D (8 h) | 0.77 | |||||||||||
| 9D (16 h) | 0.059 | |||||||||||
| 9D (Viability) | 0.28 | |||||||||||
| 9E (3 h) | 0.19 | |||||||||||
| 9E (8 h) | 0.10 | |||||||||||
| 9E (Viability) | 0.80 | |||||||||||
| 9F (0.5 h) | 0.38 | |||||||||||
| 9F (8 h) | 0.24 | |||||||||||
| 9F (Viability) | 0.12 | |||||||||||
| 9G | 0.45 | 0.61 | ||||||||||
| 9H | 0.29 | 0.11 | ||||||||||
Contact the corresponding author for individual values.
ACKNOWLEDGMENTS
This work was supported by National Institute on Drug Abuse (NIDA) grant 1R01DA042348-01 (to C.M.O.), VA Merit Review BX000207 (to J.T.S.), and National Institutes of Health (NIH) grant 5T32AI007533-18 (to J.L.W.).
We thank core services of the University of Iowa, including the DNA core. We also thank the semen and blood donors and the University of Iowa HIV clinic and the In Vitro Fertilization and Reproductive Testing Laboratories for providing samples.
C.M.O. conceived the studies. C.M.O., J.T.S., P.M.S., H.K., and J.L.W. designed the studies. J.L.W. and H.K. executed the experiments. C.M.O., J.T.S., H.K., and J.L.W. analyzed data. C.M.O., J.T.S., P.M.S., H.K., and J.L.W. wrote and revised the manuscript.
We declare no competing interests.
REFERENCES
- 1.Griffin GE, Leung K, Folks TM, Kunkel S, Nabel GJ. 1989. Activation of HIV gene expression during monocyte differentiation by induction of NF-kappa B Nature 339:70–73. [DOI] [PubMed] [Google Scholar]
- 2.Henderson AJ, Connor RI, Calame KL. 1996. C/EBP activators are required for HIV-1 replication and proviral induction in monocytic cell lines. Immunity 5:91–101. doi: 10.1016/S1074-7613(00)80313-1. [DOI] [PubMed] [Google Scholar]
- 3.Schiralli Lester GM, Henderson AJ. 2012. Mechanisms of HIV transcriptional regulation and their contribution to latency. Mol Biol Int 2012:614120. doi: 10.1155/2012/614120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karn J, Stoltzfus CM. 2012. Transcriptional and posttranscriptional regulation of HIV-1 gene expression. Cold Spring Harb Perspect Med 2:a006916. doi: 10.1101/cshperspect.a006916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Apolloni A, Meredith LW, Suhrbier A, Kiernan R, Harrich D. 2007. The HIV-1 Tat protein stimulates reverse transcription in vitro. Curr HIV Res 5:473–483. doi: 10.2174/157016207781662443. [DOI] [PubMed] [Google Scholar]
- 6.Harrich D, Ulich C, Garcia-Martinez LF, Gaynor RB. 1997. Tat is required for efficient HIV-1 reverse transcription. EMBO J 16:1224–1235. doi: 10.1093/emboj/16.6.1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin MH, Sivakumaran H, Apolloni A, Wei T, Jans DA, Harrich D. 2012. Nullbasic, a potent anti-HIV tat mutant, induces CRM1-dependent disruption of HIV rev trafficking. PLoS One 7:e51466. doi: 10.1371/journal.pone.0051466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arya SK, Guo C, Josephs SF, Wong-Staal F. 1985. Trans-activator gene of human T-lymphotropic virus type III (HTLV-III). Science 229:69–73. doi: 10.1126/science.2990040. [DOI] [PubMed] [Google Scholar]
- 9.Berkhout B, Silverman RH, Jeang KT. 1989. Tat trans-activates the human immunodeficiency virus through a nascent RNA target. Cell 59:273–282. doi: 10.1016/0092-8674(89)90289-4. [DOI] [PubMed] [Google Scholar]
- 10.Kashanchi F, Piras G, Radonovich MF, Duvall JF, Fattaey A, Chiang CM, Roeder RG, Brady JN. 1994. Direct interaction of human TFIID with the HIV-1 transactivator tat. Nature 367:295–299. doi: 10.1038/367295a0. [DOI] [PubMed] [Google Scholar]
- 11.Veschambre P, Roisin A, Jalinot P. 1997. Biochemical and functional interaction of the human immunodeficiency virus type 1 Tat transactivator with the general transcription factor TFIIB. J Gen Virol 78:2235–2245. doi: 10.1099/0022-1317-78-9-2235. [DOI] [PubMed] [Google Scholar]
- 12.Abraham S, Sweet T, Sawaya BE, Rappaport J, Khalili K, Amini S. 2005. Cooperative interaction of C/EBP beta and Tat modulates MCP-1 gene transcription in astrocytes. J Neuroimmunol 160:219–227. doi: 10.1016/j.jneuroim.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 13.Ambrosino C, Palmieri C, Puca A, Trimboli F, Schiavone M, Olimpico F, Ruocco MR, di Leva F, Toriello M, Quinto I, Venuta S, Scala G. 2002. Physical and functional interaction of HIV-1 Tat with E2F-4, a transcriptional regulator of mammalian cell cycle. J Biol Chem 277:31448–31458. doi: 10.1074/jbc.M112398200. [DOI] [PubMed] [Google Scholar]
- 14.Wei P, Garber ME, Fang SM, Fischer WH, Jones KA. 1998. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell 92:451–462. doi: 10.1016/S0092-8674(00)80939-3. [DOI] [PubMed] [Google Scholar]
- 15.Kashanchi F, Khleif SN, Duvall JF, Sadaie MR, Radonovich MF, Cho M, Martin MA, Chen SY, Weinmann R, Brady JN. 1996. Interaction of human immunodeficiency virus type 1 Tat with a unique site of TFIID inhibits negative cofactor Dr1 and stabilizes the TFIID-TFIIA complex. J Virol 70:5503–5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hottiger MO, Nabel GJ. 1998. Interaction of human immunodeficiency virus type 1 Tat with the transcriptional coactivators p300 and CREB binding protein. J Virol 72:8252–8256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jeang KT, Chun R, Lin NH, Gatignol A, Glabe CG, Fan H. 1993. In vitro and in vivo binding of human immunodeficiency virus type 1 Tat protein and Sp1 transcription factor. J Virol 67:6224–6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Loregian A, Bortolozzo K, Boso S, Caputo A, Palu G. 2003. Interaction of Sp1 transcription factor with HIV-1 Tat protein: looking for cellular partners. FEBS Lett 543:61–65. doi: 10.1016/S0014-5793(03)00399-5. [DOI] [PubMed] [Google Scholar]
- 19.Fiume G, Vecchio E, De Laurentiis A, Trimboli F, Palmieri C, Pisano A, Falcone C, Pontoriero M, Rossi A, Scialdone A, Fasanella Masci F, Scala G, Quinto I. 2012. Human immunodeficiency virus-1 Tat activates NF-kappaB via physical interaction with IkappaB-alpha and p65. Nucleic Acids Res 40:3548–3562. doi: 10.1093/nar/gkr1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kwon HS, Brent MM, Getachew R, Jayakumar P, Chen LF, Schnolzer M, McBurney MW, Marmorstein R, Greene WC, Ott M. 2008. Human immunodeficiency virus type 1 Tat protein inhibits the SIRT1 deacetylase and induces T cell hyperactivation. Cell Host Microbe 3:158–167. doi: 10.1016/j.chom.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kilareski EM, Shah S, Nonnemacher MR, Wigdahl B. 2009. Regulation of HIV-1 transcription in cells of the monocyte-macrophage lineage. Retrovirology 6:118. doi: 10.1186/1742-4690-6-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Demarchi F, d'Adda di Fagagna F, Falaschi A, Giacca M. 1996. Activation of transcription factor NF-kappaB by the Tat protein of human immunodeficiency virus type 1. J Virol 70:4427–4437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perkins ND, Edwards NL, Duckett CS, Agranoff AB, Schmid RM, Nabel GJ. 1993. A cooperative interaction between NF-kappa B and Sp1 is required for HIV-1 enhancer activation. EMBO J 12:3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hiscott J, Kwon H, Genin P. 2001. Hostile takeovers: viral appropriation of the NF-kappaB pathway. J Clin Invest 107:143–151. doi: 10.1172/JCI11918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pazin MJ, Sheridan PL, Cannon K, Cao Z, Keck JG, Kadonaga JT, Jones KA. 1996. NF-kappa B-mediated chromatin reconfiguration and transcriptional activation of the HIV-1 enhancer in vitro. Genes Dev 10:37–49. doi: 10.1101/gad.10.1.37. [DOI] [PubMed] [Google Scholar]
- 26.Madison MN, Jones PH, Okeoma CM. 2015. Exosomes in human semen restrict HIV-1 transmission by vaginal cells and block intravaginal replication of LP-BM5 murine AIDS virus complex. Virology 482:189–201. doi: 10.1016/j.virol.2015.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Madison MN, Roller RJ, Okeoma CM. 2014. Human semen contains exosomes with potent anti-HIV-1 activity. Retrovirology 11:102. doi: 10.1186/s12977-014-0102-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Welch JL, Madison MN, Margolick JB, Galvin S, Gupta P, Martinez-Maza O, Dash C, Okeoma CM. 2017. Effect of prolonged freezing of semen on exosome recovery and biologic activity. Sci Rep 7:45034. doi: 10.1038/srep45034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boily MC, Baggaley RF, Wang L, Masse B, White RG, Hayes RJ, Alary M. 2009. Heterosexual risk of HIV-1 infection per sexual act: systematic review and meta-analysis of observational studies. Lancet Infect Dis 9:118–129. doi: 10.1016/S1473-3099(09)70021-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ajamian L, Abrahamyan L, Milev M, Ivanov PV, Kulozik AE, Gehring NH, Mouland AJ. 2008. Unexpected roles for UPF1 in HIV-1 RNA metabolism and translation. RNA 14:914–927. doi: 10.1261/rna.829208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Butsch M, Boris-Lawrie K. 2000. Translation is not required to generate virion precursor RNA in human immunodeficiency virus type 1-infected T cells. J Virol 74:11531–11537. doi: 10.1128/JVI.74.24.11531-11537.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grewe B, Ehrhardt K, Hoffmann B, Blissenbach M, Brandt S, Uberla K. 2012. The HIV-1 Rev protein enhances encapsidation of unspliced and spliced, RRE-containing lentiviral vector RNA. PLoS One 7:e48688. doi: 10.1371/journal.pone.0048688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mohammadi P, Desfarges S, Bartha I, Joos B, Zangger N, Munoz M, Gunthard HF, Beerenwinkel N, Telenti A, Ciuffi A. 2013. 24 hours in the life of HIV-1 in a T cell line. PLoS Pathog 9:e1003161. doi: 10.1371/journal.ppat.1003161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wei X, Decker JM, Liu H, Zhang Z, Arani RB, Kilby JM, Saag MS, Wu X, Shaw GM, Kappes JC. 2002. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob Agents Chemother 46:1896–1905. doi: 10.1128/AAC.46.6.1896-1905.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jordan A, Bisgrove D, Verdin E. 2003. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J 22:1868–1877. doi: 10.1093/emboj/cdg188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lelek M, Casartelli N, Pellin D, Rizzi E, Souque P, Severgnini M, Di Serio C, Fricke T, Diaz-Griffero F, Zimmer C, Charneau P, Di Nunzio F. 2015. Chromatin organization at the nuclear pore favours HIV replication. Nat Commun 6:6483. doi: 10.1038/ncomms7483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mousseau G, Valente S. 2012. Strategies to block HIV transcription: focus on small molecule Tat inhibitors. Biology 1:668–697. doi: 10.3390/biology1030668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brooks DG, Arlen PA, Gao L, Kitchen CM, Zack JA. 2003. Identification of T cell-signaling pathways that stimulate latent HIV in primary cells. Proc Natl Acad Sci U S A 100:12955–12960. doi: 10.1073/pnas.2233345100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim YS, Kim JM, Jung DL, Kang JE, Lee S, Kim JS, Seol W, Shin HC, Kwon HS, Van Lint C, Hernandez N, Hur MW. 2005. Artificial zinc finger fusions targeting Sp1-binding sites and the trans-activator-responsive element potently repress transcription and replication of HIV-1. J Biol Chem 280:21545–21552. doi: 10.1074/jbc.M414136200. [DOI] [PubMed] [Google Scholar]
- 40.Treand C, du Chene I, Bres V, Kiernan R, Benarous R, Benkirane M, Emiliani S. 2006. Requirement for SWI/SNF chromatin-remodeling complex in Tat-mediated activation of the HIV-1 promoter. EMBO J 25:1690–1699. doi: 10.1038/sj.emboj.7601074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jablonski JA, Amelio AL, Giacca M, Caputi M. 2010. The transcriptional transactivator Tat selectively regulates viral splicing. Nucleic Acids Res 38:1249–1260. doi: 10.1093/nar/gkp1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin MH, Sivakumaran H, Jones A, Li D, Harper C, Wei T, Jin H, Rustanti L, Meunier FA, Spann K, Harrich D. 2014. A HIV-1 Tat mutant protein disrupts HIV-1 Rev function by targeting the DEAD-box RNA helicase DDX1. Retrovirology 11:121. doi: 10.1186/s12977-014-0121-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hayes AM, Qian S, Yu L, Boris-Lawrie K. 2011. Tat RNA silencing suppressor activity contributes to perturbation of lymphocyte miRNA by HIV-1. Retrovirology 8:36. doi: 10.1186/1742-4690-8-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Apolloni A, Hooker CW, Mak J, Harrich D. 2003. Human immunodeficiency virus type 1 protease regulation of tat activity is essential for efficient reverse transcription and replication. J Virol 77:9912–9921. doi: 10.1128/JVI.77.18.9912-9921.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hooker CW, Scott J, Apolloni A, Parry E, Harrich D. 2002. Human immunodeficiency virus type 1 reverse transcription is stimulated by tat from other lentiviruses. Virology 300:226–235. doi: 10.1006/viro.2002.1554. [DOI] [PubMed] [Google Scholar]
- 46.Ulich C, Dunne A, Parry E, Hooker CW, Gaynor RB, Harrich D. 1999. Functional domains of Tat required for efficient human immunodeficiency virus type 1 reverse transcription. J Virol 73:2499–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Madison MN, Welch JL, Okeoma CM. 2017. Isolation of exosomes from semen for in vitro uptake and HIV-1 infection assays. Bio Protoc 7:e2216. doi: 10.21769/BioProtoc.2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Madison MN, Okeoma CM. 2015. Exosomes: implications in HIV-1 pathogenesis. Viruses 7:4093–4118. doi: 10.3390/v7072810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Naslund TI, Paquin-Proulx D, Paredes PT, Vallhov H, Sandberg JK, Gabrielsson S. 2014. Exosomes from breast milk inhibit HIV-1 infection of dendritic cells and subsequent viral transfer to CD4+ T cells. AIDS 28:171–180. doi: 10.1097/QAD.0000000000000159. [DOI] [PubMed] [Google Scholar]
- 50.Smith JA, Daniel R. 2016. Human vaginal fluid contains exosomes that have an inhibitory effect on an early step of the HIV-1 life cycle. AIDS 30:2611–2616. doi: 10.1097/QAD.0000000000001236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jones PH, Mahauad-Fernandez WD, Madison MN, Okeoma CM. 2013. BST-2/tetherin is overexpressed in mammary gland and tumor tissues in MMTV-induced mammary cancer. Virology 444:124–139. doi: 10.1016/j.virol.2013.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jones PH, Maric M, Madison MN, Maury W, Roller RJ, Okeoma CM. 2013. BST-2/tetherin-mediated restriction of chikungunya (CHIKV) VLP budding is counteracted by CHIKV non-structural protein 1 (nsP1). Virology 438:37–49. doi: 10.1016/j.virol.2013.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jones PH, Mehta HV, Maric M, Roller RJ, Okeoma CM. 2012. Bone marrow stromal cell antigen 2 (BST-2) restricts mouse mammary tumor virus (MMTV) replication in vivo. Retrovirology 9:10. doi: 10.1186/1742-4690-9-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mahauad-Fernandez WD, DeMali KA, Olivier AK, Okeoma CM. 2014. Bone marrow stromal antigen 2 expressed in cancer cells promotes mammary tumor growth and metastasis. Breast Cancer Res 16:493. doi: 10.1186/s13058-014-0493-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Okeoma CM, Huegel AL, Lingappa J, Feldman MD, Ross SR. 2010. APOBEC3 proteins expressed in mammary epithelial cells are packaged into retroviruses and can restrict transmission of milk-borne virions. Cell Host Microbe 8:534–543. doi: 10.1016/j.chom.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mousseau G, Kessing CF, Fromentin R, Trautmann L, Chomont N, Valente ST. 2015. The Tat inhibitor didehydro-cortistatin A prevents HIV-1 reactivation from latency. mBio 6:e00465. doi: 10.1128/mBio.00465-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Asin SN, Fanger MW, Wildt-Perinic D, Ware PL, Wira CR, Howell AL. 2004. Transmission of HIV-1 by primary human uterine epithelial cells and stromal fibroblasts. J Infect Dis 190:236–245. doi: 10.1086/421910. [DOI] [PubMed] [Google Scholar]
- 58.Li XY, Guo F, Zhang L, Kleiman L, Cen S. 2007. APOBEC3G inhibits DNA strand transfer during HIV-1 reverse transcription. J Biol Chem 282:32065–32074. doi: 10.1074/jbc.M703423200. [DOI] [PubMed] [Google Scholar]
- 59.Badran YR, Dedeoglu F, Leyva Castillo JM, Bainter W, Ohsumi TK, Bousvaros A, Goldsmith JD, Geha RS, Chou J. 2017. Human RELA haploinsufficiency results in autosomal-dominant chronic mucocutaneous ulceration. J Exp Med 214:1937–1947. doi: 10.1084/jem.20160724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zelko IN, Mueller MR, Folz RJ. 2008. Transcription factors sp1 and sp3 regulate expression of human extracellular superoxide dismutase in lung fibroblasts. Am J Respir Cell Mol Biol 39:243–251. doi: 10.1165/rcmb.2007-0378OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Blissenbach M, Grewe B, Hoffmann B, Brandt S, Uberla K. 2010. Nuclear RNA export and packaging functions of HIV-1 Rev revisited. J Virol 84:6598–6604. doi: 10.1128/JVI.02264-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Turrini F, Marelli S, Kajaste-Rudnitski A, Lusic M, Van Lint C, Das AT, Harwig A, Berkhout B, Vicenzi E. 2015. HIV-1 transcriptional silencing caused by TRIM22 inhibition of Sp1 binding to the viral promoter. Retrovirology 12:104. doi: 10.1186/s12977-015-0230-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shan J, Donelan W, Hayner JN, Zhang F, Dudenhausen EE, Kilberg MS. 2015. MAPK signaling triggers transcriptional induction of cFOS during amino acid limitation of HepG2 cells. Biochim Biophys Acta 1853:539–548. doi: 10.1016/j.bbamcr.2014.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kenny PJ, Zhou H, Kim M, Skariah G, Khetani RS, Drnevich J, Arcila ML, Kosik KS, Ceman S. 2014. MOV10 and FMRP regulate AGO2 association with microRNA recognition elements. Cell Rep 9:1729–1741. doi: 10.1016/j.celrep.2014.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]