

Abstract
Sperm freeze-drying is a revolutionary technique, which has been gaining prominence in recent years. The first related significant result was Wakayama and Yanagimachi’s demonstration in 1998 of the birth of healthy mouse offspring by Intracytoplasmic Sperm Injection (ICSI), using epididymal freeze-dried spermatozoa. Mouse, rat, and hamster models were the first small mammals born from lyophilized epididymal spermatozoa, whereas most other studies in this field used ejaculated spermatozoa. In this work, we applied this technique to ram epididymal spermatozoa, checking the correlation between DNA integrity and embryo development following ICSI. To do this, epididymal sperm from four rams was lyophilized in a trehalose, glucose, KCl, HEPES, and Trolox media. To evaluate DNA damage and fragmentation after rehydration, samples were processed for Sperm Chromatin Dispersion test (SCD), Two-Tailed Comet Assay, and were used for ICSI. Ram #2 had a higher rate of spermatozoa with intact DNA compared with rams #1, #3, and #4 (28% vs. 3.8%, 2.8%, and 5%, respectively) and the lowest rate of Single-Strand Breaks (SSBs) (70% vs. 95.9%, 92.6%, and 93% respectively). Ram #3 had a higher level of Double-Strand Breaks (DSBs) compared to Ram #1 (4.6% vs. 0.33%, respectively). Embryo development to the blastocyst stage following ICSI was only reached from rams whose sperm had higher level of intact DNA – Rams #2 and #4 (6%, 5/147 and 6.3%, 4/64, respectively). Definitively, the impact of sperm DNA damage on embryonic development depends on the balance between sperm DNA fragmentation extent, fragmentation type (SSBs or DSBs), and the oocyte’s repair capacity.
Keywords: Assisted reproduction, Comet assay, Gametes, Lyophilization, Ruminant
Sperm freeze-drying, also known as lyophilization, is a revolutionary technique that resolves many of the drawbacks of long-term storage under liquid nitrogen. Lyophilization allows storage of spermatozoa in an anhydrobiotic state, permitting ecologically friendly, non-cryogenic preservation and allowing shipping of samples in simplified and safe manner.
The first significant result of this method was provided by Wakayama and Yanagimachi in 1998, demonstrating for the first time the birth of healthy offspring from epididymal freeze-dried mouse spermatozoa [1]. Besides work on the mouse model, the birth of a healthy offspring using injected epididymal freeze-dried spermatozoa has previously only been achieved in rat and hamster models [2,3,4]. In other species, such as rabbit [5] and horse [6], where offspring were obtained, ejaculated spermatozoa were used.
To achieve fertilization, spermatozoa should maintain a certain level of DNA integrity, which would therefore be a discriminating factor in the use of lyophilized sperm. If lyophilized sperm DNA is not excessively fragmented after rehydration, embryos could be produced by Intracytoplasmic Sperm Injection (ICSI), even when using otherwise severely damaged spermatozoa that are no longer capable of physiological fertilization activity [7]. It is well established that embryonic development is strictly related to sperm quality [8]. It has also been reported that increased levels of DNA double-strand fragmentation in freeze-dried spermatozoa significantly compromises the corresponding embryonic development after ICSI in pig ejaculated spermatozoa [9, 10] and mouse epididymal spermatozoa [11]. One of the main bottlenecks to assessing the extent and type of DNA damage present in an affected cell, is the technical availability. Among all the possibilities, the Two-tail Comet Assay (2T-Comet Assay) is one of the most reliable approaches to assessing single- and double-strand breaks in the same nuclei [12, 13]. 2T-Comet Assay combines non-denaturing and denaturing conditions to the same nucleoid. Protein depleted cells are first subjected to an electrophoretic field under non-denaturing conditions to mobilize isolated free discrete DNA fragments produced from double strands breaks at the origin. This step is followed by a second electrophoresis, run perpendicular to first one but now under alkaline unwinding conditions. This second electrophoresis produce a displacement of single-strand DNA motifs contained both at the original core of the cell and at the DNA fragments that were displaced by the first electrophoresis because of the presence of double-strand breaks. Altogether, this methodology gives rise to a perfect discrimination between both types of DNA breaks at the single cell level. To the best of our knowledge, the effects of single-strand DNA fragmentation in freeze-dried spermatozoa on embryonic development following ICSI have not been studied.
Therefore, our knowledge concerning DNA fragmentation in freeze-dried epididymal spermatozoa and embryo development in large animals is lacking. This investigation was aimed to assess the impact of both single- and double-strand DNA fragmentation present in epididymal freeze-dried ram spermatozoa on embryo development after ICSI.
Material and Methods
Chemicals
Unless otherwise stated, all materials used were purchased from Sigma Aldrich (St Louis, MO, USA).
Semen collection
To assess the level of DNA fragmentation and embryonic development of epididymal freeze-dried spermatozoa, spermatozoa were collected from four different rams (Ram #1, Ram #2, Ram #3, and Ram #4) and, as a control of ICSI outcome, we used ejaculated freeze-dried spermatozoa (EJ-sperm).
Epididymal spermatozoa: Ram testicles (n = 4) were obtained at a local abattoir shortly after the animals were slaughtered, submerged in 0.9% NaCl solution and transported to the laboratory at 4ºC for processing. At the laboratory, the testicles were removed from the solution and dried. The cauda epididymis was exposed, isolated, cleaned of excess tissue and blood vessels, and was then placed in a 90 mm diameter Petri dish with 10 ml of pre-warmed ram Tris-citric acid-fructose semen extender [14] on a warm stage (37ºC). Using a sterile scalpel, cuts were made through the cauda epididymis to open it to the solution to allow the spermatozoa to swim out. The dishes with the epididymides were incubated at 37ºC for 30 min whereupon slight pressure was applied to the tissues, which were then removed and discarded. The solution with the extracted spermatozoa was aspirated, transferred into 15-ml conical-bottom tube, underlain with 2 ml of isothermal 60% iodixanol (OptiPrepTM) and centrifuged at ambient temperature (~22ºC) for 20 min at 1,000 g. The supernatant and OptiPrep were aspirated and the pellet was re-suspended in a xeroprotective medium composed of 1.0 M trehalose, 5 mM glucose, 10 mM KCl, 20 mM HEPES, and 10 µg/ml Trolox at pH 7.4 to a concentration of ~50 × 106 spermatozoa/ml and prepared for chilling, freezing, and drying.
Ejaculated spermatozoa: EJ-sperm was collected as described previously [15]. Briefly, semen was collected from three adult fertile rams using an Artificial Vagina (AV) filled with warm water (40–44°C) and connected to a 15-ml tube. Immediately after collection, sperm motility was evaluated using a stereomicroscope and sperm concentration was assessed using a Burker chamber. Only ejaculates with sperm concentrations ≥ 1.8 × 109 spermatozoa/ml and motility ≥ 70% were used for the experiments.
Sperm lyophilization
Epididymal spermatozoa: Samples in 15-ml Falcon tubes were cooled to 4–5ºC at ~0.3ºC min–1 by submerging the tubes in an isothermal water bath and then placing them in a refrigerator (4ºC). Once chilled, each sample was packaged into four pre-chilled 8-ml polypropylene tubes, with 2 ml in each, and frozen in a directional freezing machine (MTG-550, IMT, Ness Ziona, Israel) as previously described [15, 16]. All frozen samples were stored under liquid nitrogen pending lyophilization and evaluation. For post-thaw evaluation, samples were thawed by holding them first at ambient temperature (~22ºC) for 90 sec, and then submerging them in a water bath at 37ºC until completely thawed (~3.5 min). After thawing, samples were kept in a water bath at 37ºC pending evaluation.
Cryopreserved samples were removed from the liquid nitrogen, stoppers removed, and the open tubes were placed in a rack inside the lyophilizer (FreeZone Plus 2.5 litre, Labconco, Kansas City, MO, USA) with the condenser pre-cooled to –84ºC. Lyophilization was performed for 24 h at 0.006 mbar. At the end of the drying process the tubes were removed from the lyophilizer, capped, and kept in the dark at room temperature pending evaluation. For rehydration, double-distilled water was added to the sample at a ratio of 2.5 µl of room temperature distilled water for every 1.0 mg of dry sample.
Ejaculated spermatozoa: EJ-sperm was lyophilized as described previously [15]. Briefly, Spermatozoa were lyophilized, using a freeze-dryer (SP Scientific-VirTis, 2.0 BenchTop, Warminster, PA, USA) following the protocol used by Wakayama and Yanagimachi [1]. For the final stage of the process, each ampule was sealed under vacuum and stored at room temperature (RT, 18–23°C) until use.
Sperm evaluation
Epididymal samples were evaluated for motility, viability, acrosome integrity, and morphology on arrival (fresh, after extraction), after thawing and after rehydration.
Motility was evaluated using a computer-aided sperm analysis (CASA) system. Samples (10 µl) were loaded onto pre-warmed (37ºC) microscope slides, covered with pre-warmed (37ºC) cover slips and analyzed using the CASA system, which consisted of an optical phase-contrast microscope (CX41; Olympus, Tokyo, Japan) with a warmed stage (37ºC), a 10 × negative phase contrast objective (PLN10XPH; Olympus), and a digital camera (avA1000-100gc, Basler, Ahrensburg, Germany). Images were captured and analyzed using SpermVision software (Minitube, Tiefenbach, Germany). Data on total and progressive sperm motility and on the rate of fast, slow, and stationary motility, and of circling spermatozoa was acquired, along with the following sperm kinetic values: curvilinear velocity (VCL, µm/sec), straight linear velocity (VSL, µm/sec), average path velocity (VAP, µm/sec), linearity (LIN = VSL/VCL), beat cross frequency (BCF, Hz), wobble (WOB = VAP/VCL), and lateral head displacement (ALH, µm). Software acquisition settings were set at a rate of 60 frames per second (60 Hz) with 30 frames acquired per field. A total of eight randomly selected fields were acquired for each sample. Sperm were considered immotile when the average orientation change (AOC) of the head was < 9.5º. Motility was considered stationary when AOC ≥ 9.5º and the distance in straight line (DSL) was under 6 µm. Motility was progressive when AOC ≥ 9.5º and DSL ≥ 6 µm.
Sperm viability was assessed by staining 10 µl of suspended sperm with eosin-nigrosin (eosin Y yellow CI 45380, nigrosin CI 50420 dissolved in 0.9% NaCl; VWR International, Darmstadt, Germany) at a 1:1 ratio, incubating at room temperature for 2 min, and then preparing smears that were air-dried before evaluation. At least 100 spermatozoa from each sample were evaluated with a light microscope (oil immersion; × 100). White (unstained) spermatozoa were classified as live and those that showed pink or red coloration were classified as dead.
Sperm aliquots were also fixed in Hancock’s fixative and assessed for acrosome integrity and sperm morphology as previously described [16, 17]. At least 100 spermatozoa were evaluated by phase-contrast microscopy (oil immersion; × 100). The acrosome was evaluated according to the following categories: normal (intact acrosome), swollen, detaching, and detached. One hundred spermatozoa were also assessed for morphology and were classified as normal, or as having a defect in the head, neck, midpiece, or endpiece. Sperm morphology included searching for a wide range of abnormalities as previously described [18].
Sperm DNA fragmentation
A portion of epididymal freeze-dried spermatozoa of each ram was used for DNA fragmentation assays.
Sperm Chromatin Dispersion test (SCD): DNA fragmentation was evaluated using the Halomax® kit (Halotech DNA SL, Spain, Madrid), according to the manufacturer’s instructions. Briefly, after rehydration the spermatozoa were diluted in phosphate-buffered saline (PBS) to 15 × 106 spermatozoa/ml and placed in a 0.5-ml Eppendorf tube containing agarose at 37˚C. Successively, the above solution was pipetted onto a two-well slide, covered with a cover slip, and left for 5 min at 4˚C to solidify. The cover slip was removed and the slide was incubated for 5 minutes with the lysing solution. A wash was performed for 5 min in distilled water and the slide was placed in 70% and then in 100% ethanol, 2 min in each. The sample was then stained with SYBR Green I (Molecular Probes, Leiden, the Netherlands) at 1:40 in water and mixed 1:1 with Vectashield (Vector Laboratories, Burlingame, CA, USA). The slides were analyzed using fluorescent microscopy. A minimum of 300 spermatozoa were analyzed for each ram for statistical significance.
Two-Tailed Comet Assay (2T-Comet Assay): The 2T-Comet Assay was performed as described by Enciso and colleagues [12], adjusted for ram spermatozoa. After rehydration, the spermatozoa were diluted in PBS to 1 × 106 spermatozoa/ml. An aliquot of 25 µl was mixed with 50 µl of 1% low melting point agarose (LMP; type VII) in water at 37˚C and placed on a pre-treated slide for gel adhesion (Chromacell SL, Madrid, Spain), covered with a cover slip, and transferred to an ice-cold plate to promote solidification. The cover slip was then removed and the slides were submerged sequentially in two lysing solution (LS): LS-1 [0.4 mol/l Tris-HCl, 0.8 mol/l dithiothreitol (DTT), 1% sodium dodecyl sulfate (SDS), pH 7.5] for 15 min, followed by LS-2 (0.4 mol/l Tris-HCl, 0.8 mol/l DTT, 1% SDS, 0.05 mol/l EDTA, pH 7.5) for 15 min. Then, slides were rinsed in TBE buffer (0.09 mol/l Tris-borate, 0.002 mol/l EDTA, pH 7.5) for 10 min and transferred to an electrophoresis tank where they were immersed in TBE electrophoresis buffer. Electrophoresis was performed at 20 V (1 V/cm), 12 mA for 8 min. A wash was performed in 0.9% NaCl for 2 min and slides were then incubated in unwounding solution composed of 0.03 mol/l NaOH and 1.0 mol/l NaCl for 2.5 min (keeping the solution at 4˚C till use). A second electrophoresis was performed, with the slides oriented at 90˚ to the first electrophoresis and the parameters were 20 V (1 V/cm), 12 mA for 4 min in 0.03 mol/l NaOH. Then, slides were rinsed once in a neutralization buffer (0.4 mol/l Tris-HCl, pH 7.5) for 5 min, briefly washed in TBE buffer for 2 min, dehydrated in increasing concentrations of ethanol (EtOH 70% and EtOH 100%, for 2 min in each) and air dried. Finally, DNA was stained with SYBR Green I diluted at 1:3000 in Vectashield. The type of DNA damage was classified in Single-Strand Breaks (SSBs) (up down migration) and Double-Strand Breaks (DSBs) (right left migration). For statistical significance, at least 300 spermatozoa for each ram were analyzed.
Oocyte recovery and in vitro maturation (IVM)
Sheep ovaries were obtained from the local abattoir and transferred at 37°C to the laboratory within 1–2 h after slaughter. Cumulus-Oocyte Complexes (COCs) pick-up was carried out with 21 G needles in the presence of TCM-199 medium (Gibco Life Technologies, Milan, Italy) supplemented with 0.0047% (w:v) of 4-(2- hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) and 0.005% (w:v) Heparin. Only COCs that exhibited at least 2–3 layers of compact cumulus cells were selected for IVM, which was performed in 4-well dishes containing 500 μl of IVM medium per well. The IVM medium was composed of bicarbonate-buffered TCM-199 (Gibco) supplemented with 2 mM glutamine, 0.3 mM sodium pyruvate, 100 μM cystamine, 10% fetal bovine serum (FBS) (Gibco), 5 μg/ml follicle stimulating hormone (FSH) (Ovagen, ICP, Auckland, New Zealand), 5 μg/ml luteinizing hormone (LH) and 1 μg/ml β-estradiol. Maturation was completed in a humidified atmosphere at 38.5°C and 5% CO2 in air for 24 h [19].
Intracytoplasmic Sperm Injection (ICSI)
Both freeze-dried epididymal spermatozoa and EJ-sperm were rehydrated by adding 100 μl of bi-distilled water to 40 mg of dried spermatozoa. Then 5 μl aliquot was suspended in 100 μl of H-199 + 0.4% BSA. The latter was diluted 1:1 with 12% (w:v) polyvinylpyrrolidone (PVP) and placed on the lid of a Petri dish on a warmed microscope stage and covered with mineral oil. ICSI was performed according to Anzalone and colleagues [19]. Briefly, ICSI with freeze-dried spermatozoa was carried out on an inverted microscope (Nikon Eclipse E-800) connected to a micromanipulation system (Narishige NT-88NEN, Tokyo, Japan), using a piezo micropipette driving system (PiezoXpert, Eppendorf, Milan, Italy). At 24 h from the start of IVM, the matured oocytes were injected in groups of five to avoid prolonged light exposure. PVP/sperm-containing drops were renewed every ten injected oocytes. After injection, the oocytes were chemically activated as described in the following paragraph “In vitro activation (IVA).” The whole artificial fertilization process was concluded no later than 2 h after the end of IVM.
In vitro activation (IVA): As a control for oocyte quality, 210 oocytes were in vitro activated (IVA) for the production of parthenogenic embryos as previously described by Loi and colleagues [20]. Briefly, at the end of IVM the oocytes intended for IVA embryo production and the oocytes fertilized by ICSI were activated in 5 μM of Ionomycin dissolved in H-199 + 0.4% BSA for 5 min, washed in H-199 + 0.4% BSA, and incubated for 3 h in bicarbonate-buffered synthetic oviductal fluid (SOF) enriched with 2% (v:v) basal medium Eagle essential amino acids (EAA), 1% (v:v) minimum essential medium (MEM)-nonessential amino acids (NEAA) (Gibco), 1 mM glutamine, and 8 mg/ml fatty acid-free BSA plus 2 mM of 6-dimethylaminopurine (6-DMAP). Finally, oocytes were washed and cultured as described below.
Embryo culture
All presumptive zygotes were cultured in 20 μl drops of SOF enriched with 2% (v:v) basal medium Eagle EAA, 1% (v:v) MEM-NEAA, 1 mM glutamine, and 8 mg/ml fatty acid-free BSA, covered by mineral oil. The medium was renewed on day 3 [SOF supplemented with 0.27 mg/ml glucose (SOF+), 2% EAA, 1% NEAA], on day 5 [SOF+ with 10% charcoal stripped FBS (CS-FBS), 2% EAA, 1% NEAA], and on day 6 (1:1 MEM/M199 enriched with 10% CS-FBS, 2.5 μg/ml gentamicin and 1% sodium pyruvate). In vitro development was evaluated on day 1 (fragmented, not-divided or 2-Cells stage) and day 6/7 (blastocyst stage) of embryo culture.
Statistical analysis
Data were analyzed using GraphPad Prism for Windows (Version 6.01, GraphPad software, CA, USA). Statistical analyses for embryo development, done using a non-parametric Mann-Whitney test, were based on five replicates per experiment. Data are reported as means ± standard error mean (SEM). To compare between sperm evaluations and levels of intact sperm in the fresh and lyophilized samples, pair-wise comparisons were performed using the Wilcoxon signed-rank test and the chi-square test, respectively. P values for the Wilcoxon signed-rank test were computed using an exact methodology, which is why degrees of freedom are not reported. The level of significance was set at P < 0.05.
Results
Sperm evaluation
Seminal parameters evaluated in the fresh samples are reported in Table 1. After thawing, 28.0 ± 1.22% of the ram spermatozoa still showed intact membranes, as evaluated by eosin-nigrosin staining, but were immotile. After rehydration, no motility was observed. Viability (intact plasma membrane) after rehydration ranged between zero and 1%. The majority (84.5 ± 3.0%) of dried spermatozoa showed normal morphology. The main morphological defects in ram spermatozoa, noted also in the fresh samples, were proximal and distal cytoplasmic droplets. After desiccation, defects also included loss of the entire tail or only the end-piece. Intact acrosome was found in 74.5 ± 4.3% and 64.5 ± 7.5% of the ram spermatozoa after thawing and after rehydration of freeze-dried samples, respectively.
Table 1. Fresh ram spermatozoa after extraction from the cauda epididymides.
| Concentration (× 106/ml) |
CASA total motility (%) |
CASA progressive motility (%) |
Viability (%) |
Morphology (%) |
Acrosome (%) |
|
|---|---|---|---|---|---|---|
| Ram #1 | 3915 | 89.9 | 81.6 | 93 | 91 | 98 |
| Ram #2 | 1400 | 95.8 | 89.4 | 95 | 76 | 96 |
| Ram #3 | 935 | 91.2 | 86.4 | 94 | 33 | 84 |
| Ram #4 | 1905 | 88.4 | 79.4 | 93 | 61 | 96 |
Sperm DNA fragmentation
Sperm samples processed for the chromatin dispersion test to assess DNA fragmentation gave rise to two different nucleoid phenotypes. Nucleoids displaying a small and compact halo of chromatin dispersion (baseline halos) corresponded to spermatozoa with unfragmented DNA (white arrows in Fig. 1A and 1B). In contrast, nuclei exhibiting a moderate or large halo of DNA diffusion (red arrows in Fig. 1A and 1B) contained fragmented DNA molecules.
Fig. 1.

Visualization of ram sperm DNA damage using the sperm chromatin dispersion (SCD) test (a, b) and a 2T-Comet Assay (c). Low (a) and high (b) levels of sperm DNA fragmentation in two different rams using the sperm chromatin dispersion test. Red arrows: nucleoids with fragmented DNA. White arrows: nucleoids containing non-fragmented DNA. (c) 2-Tails comet assay showing electrophoretic fields mapping the different SSBs and DSBs positioning. Scale bars represent 10 µm.
Using the 2T-Comet Assay, a continuous rate of DNA damage was observed when different spermatozoa were compared (Fig. 2). Spermatozoa without DNA damage displayed a compact chromatin displacement in the alkaline migration (Y-axis in Fig. 1C, labelled as structural). This Comet was due to the presence of alkali-labile sites in all spermatozoa and is represented in the comet image showing an absence of detectable DNA damage under our experimental conditions. This comet would be equivalent to the baseline halo observed after using the sperm chromatin dispersion test.
Fig. 2.

Structural comet. (a–b) Using black and white filter. (a´–b´) Using electronic filtering to show density of SSBs (highlighted in blue) as the comet is larger. (a–a´) Structural comet in fresh-frozen sperm. (b–b´) Different degrees of SSBs in lyophilized sperm comet. Scale bars represent 10 µm.
Migration on the X-axis of DNA fragments corresponds to the presence DSBs at the origin. SSBs were represented as extra DNA migration beyond the structural comet with displacement on the Y-axis. Fragments of DNA mobilized on the X-axis during the first electrophoresis, denature and migrate also on the Y-axis (Fig. 1C).
The DNA fragmentation analyses brought to light the following comparison of the different types of DNA breaks (SSBs and DSBs) in the four rams shown in Table 2. Ram #2 had a considerably higher proportion of sperm with intact DNA compared to the other three rams, while Ram #3 had the highest rate of DSBs and the lowest rate of spermatozoa with intact DNA. Moreover, data detected in fresh spermatozoa of all rams showed higher rates of intact DNA compered to freeze-dried spermatozoa (P < 0.0001) (Table 2).
Table 2. Sperm DNA fragmentation evaluation outcome.
| Spermatozoa with Intact DNA (%) |
Freeze-dried spermatozoa with fragmented DNA (%) |
|||
|---|---|---|---|---|
| Fresh | Freeze-dried | SSBs | DSBs | |
| Ram #1 | 95.7 | 3.8 * | 95.9 | 0.3 |
| Ram #2 | 95.7 | 28.0 * | 70.0 | 2.0 |
| Ram #3 | 95.4 | 2.8 * | 92.6 | 4.6 |
| Ram #4 | 93.7 | 5.0 * | 93.0 | 2.0 |
DNA fragmentation levels in four rams in fresh and freeze-dried sperm samples. Freeze-dried spermatozoa were evaluated for SSBs and DSBs by comet assay based on indication of the existence of SSBs during the Sperm Chromatin Dispersion (SCD) test. No such SSBs indication was noted in the fresh samples when evaluated only by SCD test, suggesting a level of < 10% of SSBs that would not be visible in comet assay. Comet assay values for the fresh samples are thus based on SSBs and DSBs combined. Values of spermatozoa with intact DNA differed significantly between fresh and freeze-dried samples within each ram. All values are based on the evaluation of 300 spermatozoa. Asterisks (*) indicate a significant difference between mean intact DNA values within each row at P < 0.0001.
Embryo development
Embryo development results are reported in Table 3. At 24 h post activation, Ram #1 had lower numbers of fragmented embryos compared to Ram #2, Ram #4, EJ-sperm, and IVA (8.3% vs. 26.5%, 25%, 23.6%, and 24.8%, respectively, P < 0.05). IVA embryos showed lower numbers of not-divided oocytes compared to Ram #1, Ram #2, Ram #3, and Ram #4 (21% vs. 79.2%, 45.8%, 50%, and 56.3%, respectively, P < 0.05) and higher numbers of 2-Cell stage embryos compared to all groups (54.3% vs. 12.5%, 27.7%, 29.3%, 18.8%, and 32.2% in Ram #1, Ram #2, Ram #3, Ram #4, and EJ-sperm, respectively, P < 0.05). On day seven of embryo culture, only spermatozoa from Ram #2, Ram #4, and EJ-sperm were able to direct embryonic development to the expanded blastocyst stage (6%, 6.3%, and 9.5%, respectively) (Fig. 3). The IVA group had a higher percentage of expanded blastocyst compared to all groups (20% vs. 0%, 6%, 0%, 6.3%, and 9.5% respectively, P < 0.05) (Fig. 3).
Table 3. Embryo development outcome.
| N. oocytes | Fragmented (%) | Not-divided (%) | 2-Cells (%) | Expanded blastocyst (%) | |
|---|---|---|---|---|---|
| Ram #1 | 72 | 6 (8.3) a | 57 (79.2) a | 9 (12.5) a | 0 (0) a |
| Ram #2 | 83 | 22 (26.5) b | 38 (45.8) bcd | 23 (27.7) bc | 5 (6.0) abc |
| Ram #3 | 58 | 12 (20.7) ab | 29 (50.0) b | 17 (29.3) bc | 0 (0) ab |
| Ram #4 | 64 | 16 (25.0) b | 36 (56.3) bc | 12 (18.8) ab | 4 (6.3) bc |
| EJ-sperm | 242 | 57 (23.6) b | 107 (44.2) bc | 78 (32.2) c | 23 (9.5) c |
| IVA | 210 | 52 (24.8) b | 44 (21.0) d | 114 (54.3) d | 42 (20.0) d |
Comparisons were performed within each column (all evaluations are based on five replicates). Values not sharing a common letter differ significantly (P < 0.05).
Fig. 3.
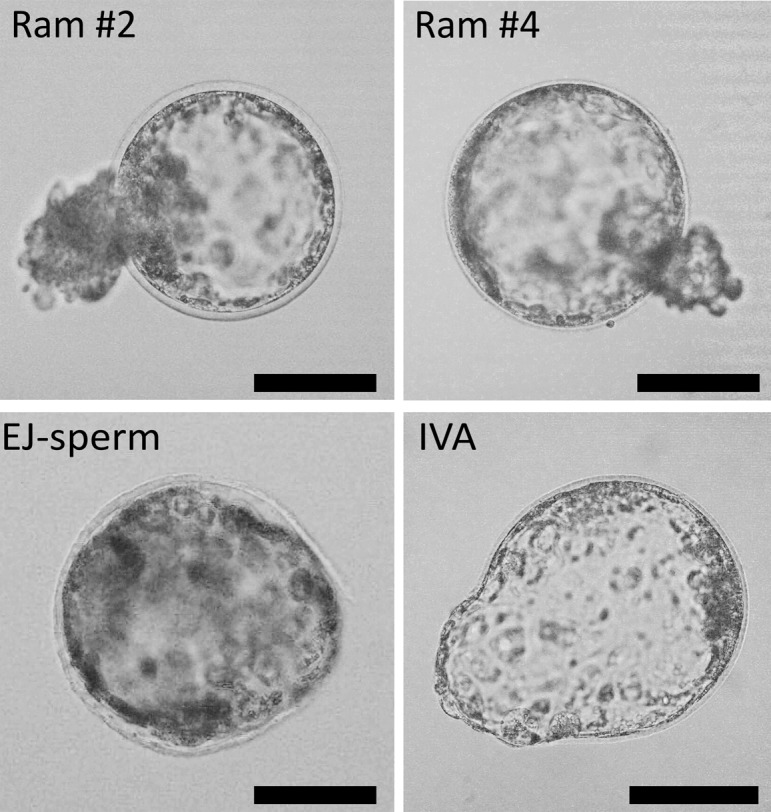
Embryo development of Ram #2, Ram #4, EJ-sperm, and IVA group. Images of expanded blastocyst at the 8th day of culture obtained from ICSI (Ram #2, Ram #4, and EJ-sperm) and IVA. Scale bars represent 100 µm.
Discussion
We have demonstrated for the first time the ability of epidydimal freeze-dried ram spermatozoa to reach the blastocyst stage through ICSI. Moreover, we have expanded our understanding of the association between sperm DNA fragmentation and embryonic development.
Sperm DNA fragmentation is a remarkable barrier to proper embryonic development, especially when using lyophilized spermatozoa [7, 21]. Nevertheless, if an intact or relatively intact freeze-dried spermatozoon nucleus is injected into an oocyte by ICSI, the latter is potentially able to develop to produce a live offspring, as can be assumed to have happened in mice [1], rats [2, 3], hamsters [4], rabbits [5], and horses [6]. With the exception of a single offspring in a horse, all studies using freeze-dried spermatozoa from large animal models (cattle [22], pig [9, 10, 23, 24], and sheep [15]) were confined to in vitro embryonic development up to the blastocyst stage. Dried epididymal spermatozoa have never been used in large animal models. Furthermore, in those studies that evaluated DNA fragmentation in lyophilized spermatozoa, only DSBs were evaluated [9, 10, 22, 25,26,27].
The blastocyst stage reached in our experiment when using lyophilized spermatozoa, precisely reflects the normal embryonic development time in sheep (Ram #2 6%, Ram #4 6.3%, and EJ-sperm 9.5% of expanded blastocyst at day seven after fertilization). Good embryonic development is strictly timing-dependent [28, 29]. It therefore follows that epididymal freeze-dried ram spermatozoa are capable of directing fertilization and normal embryo development. With the exception of the murine model, where ICSI with lyophilized spermatozoa resulted in high developmental rates [1, 30], blastocyst rates in large animals (horse, cattle, and pig) was reported to be around 10–12% [6, 22, 23]. Our embryo developmental rate is comparable to the results achieved previously in these domestic species. Several human pathologies cause a requirement for the exclusive use of epidydimal spermatozoa [31]. This work can give a likely opportunity to expand the application of the lyophilization field in that direction.
Hypothetically, high integrity of sperm DNA may be required for normal embryonic development, thus achieving a successful pregnancy. The use of epididymal sperm samples provides several advantages with respect to ejaculated samples in terms of the observed levels of sperm DNA fragmentation. Most comparative studies between these two stages of sperm maturation have been performed in humans and have recurrently demonstrated that spermatozoa retrieved from the testis have better DNA quality than those obtained from ejaculated samples in the same subject [32, 33]. Moreover, in males with severe oligospermia, the use of testicular-retrieved spermatozoa not only shows a tendency for an improved clinical pregnancy, but also produces decreased rates of miscarriage [34].
The consequences of sperm DNA damage in embryonic development is connected to the balance between the amount, type, and complexity of the DNA damage present in the fertilizing spermatozoon, and the oocyte’s repair capacity. After penetration of the spermatozoon into the oocyte, extensive DSBs associated with apoptotic-like processes or breaks of other etiology, may surpass the repair capacity of the oocyte. These situations lead to delayed paternal DNA replication and probably to paternal DNA degradation, with the subsequent arrest of embryonic development [35]. It is interesting to note that DNA repair mechanisms are operative even in cases of severe sperm DNA damage, as may have been the case with the dried spermatozoa used in our study, where high rates of SSBs have been detected. When the fertilizing spermatozoon contains a low level of structural basic sites, moderate DSBs, SSBs, and/or alternative base analogues to the orthodox ATGC, the oocyte may respond with specific DNA repair pathways to reconstitute a stable DNA molecule. If this is the case, the whole DNA of the male pronucleus should function normally during syngamy and early embryonic development. Within this scenario, it is possible that some mis-repaired or unrepaired DNA lesions could still potentially lead to mutations or chromosome aberrations that may take place during embryonic development. Unrepaired SSBs as well as other lesions types may also result in DSBs at the time of DNA replication. These types of situations, as well as the maintenance of unrepaired DSBs, are prone to yielding chromosome structural abnormalities in early embryonic development [36]. Most of these chromosome alterations are highly unstable, affecting mitotic chromosome segregation, resulting in chromosome imbalances in the newly produced cells, with subsequent cell death and poor or inhibited embryonic development. Moreover, the induction of DSBs caused by the lyophilization process was also evaluated in sheep lymphocytes [37]. Iuso et al. [37] proved that the formation of DSBs in lymphocytes is a consequence of the lyophilization. In our work, where spermatozoa were used, the levels of DSBs were relatively low (a range between 0.3% and 4.6%), suggesting that sperm DNA structure responds better to freeze-drying (in terms of DSBs).
The most interesting result in this study is the fact that the only two rams capable of directing embryonic development to the blastocyst stage (Rams #2 and #4) showed higher proportions of normal DNA (28% and 5%, respectively), contrarily to Rams #1 and #3, which had lower scores of DNA integrity (3.8% and 2.8%, respectively) and produced no blastocysts. In terms of DNA fragmentation, Rams #1 and #3 had a considerably higher rate of SSBs (95.9% and 92.6%, respectively) compared to Ram #2 (70%). The oocyte is endowed with the ability to repair the DNA of the dried sperm nucleus [37,38,39]. Previous studies in mice have shown that paternal chromosome aberrations caused by the presence of DSBs can be repaired completely or partially by maternal DSB repair mechanisms in the early zygotes [36, 40]. Definitively, the implication of sperm DNA damage in fertilization and embryonic development should depend on the balance between the extent of sperm DNA damage and the oocyte’s repair capacity. Therefore, oocyte’s restoration of DSBs and SSBs might explain why Rams #2 and #4 produced blastocysts while Rams #1 and #3 did not.
The conclusive demonstration, i.e. the birth of the first lamb from freeze-dried spermatozoa, is still pending. In light of recent results, this is highly desirable.
Acknowledgments
This project has received funding from the European Union’s Horizon 2020 Research and Innovation Programme under the Marie Skłodowska-Curie grant agreements No. 734434 and No. 749537, and under Twinning grant agreement No. 698165. Moreover, this investigation was part of experimental protocols conducted under the research projects BFU-2013-44290-R and RTC-2016-4733-1. The authors also participate in the COST Action (CA16119).
The authors wish to thank Francisca Arroyo for technical support.
References
- 1.Wakayama T, Yanagimachi R. Development of normal mice from oocytes injected with freeze-dried spermatozoa. Nat Biotechnol 1998; 16: 639–641. [DOI] [PubMed] [Google Scholar]
- 2.Hirabayashi M, Kato M, Ito J, Hochi S. Viable rat offspring derived from oocytes intracytoplasmically injected with freeze-dried sperm heads. Zygote 2005; 13: 79–85. [DOI] [PubMed] [Google Scholar]
- 3.Hochi S, Watanabe K, Kato M, Hirabayashi M. Live rats resulting from injection of oocytes with spermatozoa freeze-dried and stored for one year. Mol Reprod Dev 2008; 75: 890–894. [DOI] [PubMed] [Google Scholar]
- 4.Muneto N, Horiuchi T. Full-term development of hamster embryos produced by injecting freeze-dried spermatozoa into oocytes. J Mamm Ova Res 2011; 28: 32–39. [Google Scholar]
- 5.Liu JL, Kusakabe H, Chang CC, Suzuki H, Schmidt DW, Julian M, Pfeffer R, Bormann CL, Tian XC, Yanagimachi R, Yang X. Freeze-dried sperm fertilization leads to full-term development in rabbits. Biol Reprod 2004; 70: 1776–1781. [DOI] [PubMed] [Google Scholar]
- 6.Choi YH, Varner DD, Love CC, Hartman DL, Hinrichs K. Production of live foals via intracytoplasmic injection of lyophilized sperm and sperm extract in the horse. Reproduction 2011; 142: 529–538. [DOI] [PubMed] [Google Scholar]
- 7.Kusakabe H, Szczygiel MA, Whittingham DG, Yanagimachi R. Maintenance of genetic integrity in frozen and freeze-dried mouse spermatozoa. Proc Natl Acad Sci USA 2001; 98: 13501–13506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Janny L, Menezo YJ. Evidence for a strong paternal effect on human preimplantation embryo development and blastocyst formation. Mol Reprod Dev 1994; 38: 36–42. [DOI] [PubMed] [Google Scholar]
- 9.Men NT, Kikuchi K, Nakai M, Fukuda A, Tanihara F, Noguchi J, Kaneko H, Linh NV, Nguyen BX, Nagai T, Tajima A. Effect of trehalose on DNA integrity of freeze-dried boar sperm, fertilization, and embryo development after intracytoplasmic sperm injection. Theriogenology 2013; 80: 1033–1044. [DOI] [PubMed] [Google Scholar]
- 10.Olaciregui M, Luño V, González N, Domingo P, de Blas I, Gil L. Chelating agents in combination with rosmarinic acid for boar sperm freeze-drying. Reprod Biol 2017; 17: 193–198. [DOI] [PubMed] [Google Scholar]
- 11.Kawase Y, Wada NA, Jishage K. Evaluation of DNA fragmentation of freeze-dried mouse sperm using a modified sperm chromatin structure assay. Theriogenology 2009; 72: 1047–1053. [DOI] [PubMed] [Google Scholar]
- 12.Enciso M, Sarasa J, Agarwal A, Fernández JL, Gosálvez J. A two-tailed Comet assay for assessing DNA damage in spermatozoa. Reprod Biomed Online 2009; 18: 609–616. [DOI] [PubMed] [Google Scholar]
- 13.Cortés-Gutiérrez EI, López-Fernández C, Fernández JL, Dávila-Rodríguez MI, Johnston SD, Gosálvez J. Interpreting sperm DNA damage in a diverse range of mammalian sperm by means of the two-tailed comet assay. Front Genet 2014; 5: 404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Salamon S, Maxwell WMC. Storage of ram semen. Anim Reprod Sci 2000; 62: 77–111. [DOI] [PubMed] [Google Scholar]
- 15.Anzalone DA, Palazzese L, Iuso D, Martino G, Loi P. Freeze-dried spermatozoa: An alternative biobanking option for endangered species. Anim Reprod Sci 2018; 190: 85–93. [DOI] [PubMed] [Google Scholar]
- 16.Saragusty J, Hildebrandt TB, Behr B, Knieriem A, Kruse J, Hermes R. Successful cryopreservation of Asian elephant (Elephas maximus) spermatozoa. Anim Reprod Sci 2009; 115: 255–266. [DOI] [PubMed] [Google Scholar]
- 17.Saragusty J. Directional freezing for large volume cryopreservation. In: Wolkers WF, Oldenhof H (eds.), Methods in Cryopreservation and Freeze-Drying. New York: Springer Verlarg; 2015: 381−397. [DOI] [PubMed] [Google Scholar]
- 18.Saragusty J, Walzer C, Petit T, Stalder G, Horowitz I, Hermes R. Cooling and freezing of epididymal sperm in the common hippopotamus (Hippopotamus amphibius). Theriogenology 2010; 74: 1256–1263. [DOI] [PubMed] [Google Scholar]
- 19.Anzalone DA, Iuso D, Czernik M, Ptak G, Loi P. Plasma membrane and acrosome loss before ICSI is required for sheep embryonic development. J Assist Reprod Genet 2016; 33: 757–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loi P, Ledda S, Fulka J, Jr, Cappai P, Moor RM. Development of parthenogenetic and cloned ovine embryos: effect of activation protocols. Biol Reprod 1998; 58: 1177–1187. [DOI] [PubMed] [Google Scholar]
- 21.Martins CF, Dode MN, Báo SN, Rumpf R. The use of the acridine orange test and the TUNEL assay to assess the integrity of freeze-dried bovine spermatozoa DNA. Genet Mol Res 2007; 6: 94–104. [PubMed] [Google Scholar]
- 22.Keskintepe L, Pacholczyk G, Machnicka A, Norris K, Curuk MA, Khan I, Brackett BG. Bovine blastocyst development from oocytes injected with freeze-dried spermatozoa. Biol Reprod 2002; 67: 409–415. [DOI] [PubMed] [Google Scholar]
- 23.Kwon IK, Park KE, Niwa K. Activation, pronuclear formation, and development in vitro of pig oocytes following intracytoplasmic injection of freeze-dried spermatozoa. Biol Reprod 2004; 71: 1430–1436. [DOI] [PubMed] [Google Scholar]
- 24.Nakai M, Kashiwazaki N, Takizawa A, Maedomari N, Ozawa M, Noguchi J, Kaneko H, Shino M, Kikuchi K. Effects of chelating agents during freeze-drying of boar spermatozoa on DNA fragmentation and on developmental ability in vitro and in vivo after intracytoplasmic sperm head injection. Zygote 2007; 15: 15–24. [DOI] [PubMed] [Google Scholar]
- 25.Olaciregui M, Luño V, Martí JI, Aramayona J, Gil L. Freeze-dried stallion spermatozoa: evaluation of two chelating agents and comparative analysis of three sperm DNA damage assays. Andrologia 2016; 48: 900–906. [DOI] [PubMed] [Google Scholar]
- 26.Oldenhof H, Zhang M, Narten K, Bigalk J, Sydykov B, Wolkers WF, Sieme H. Freezing-induced uptake of disaccharides for preservation of chromatin in freeze-dried stallion sperm during accelerated aging. Biol Reprod 2017; 97: 892–901. [DOI] [PubMed] [Google Scholar]
- 27.Arav A, Saragusty J. Directional freezing of sperm and associated derived technologies. Anim Reprod Sci 2016; 169: 6–13. [DOI] [PubMed] [Google Scholar]
- 28.Mio Y, Maeda K. Time-lapse cinematography of dynamic changes occurring during in vitro development of human embryos. Am J Obstet Gynecol 2008; 199: 660.e1–660.e5. [DOI] [PubMed] [Google Scholar]
- 29.Park MJ, Kim EY, Kang MJ, Lee JB, Jeong CJ, Park SP. Investigation of the developmental potential and developmental kinetics of bovine parthenogenetic and somatic cell nuclear transfer embryos using a time-lapse monitoring system. Cell Reprogram 2017; 19: 245–254. [DOI] [PubMed] [Google Scholar]
- 30.Kusakabe H, Yanagimachi R, Kamiguchi Y. Mouse and human spermatozoa can be freeze-dried without damaging their chromosomes. Hum Reprod 2008; 23: 233–239. [DOI] [PubMed] [Google Scholar]
- 31.Hayden RP, Wright DL, Toth TL, Tanrikut C. Selective use of percutaneous testis biopsy to optimize IVF-ICSI outcomes: a case series. Fertil Res Pract 2016; 2: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Greco E, Scarselli F, Iacobelli M, Rienzi L, Ubaldi F, Ferrero S, Franco G, Anniballo N, Mendoza C, Tesarik J. Efficient treatment of infertility due to sperm DNA damage by ICSI with testicular spermatozoa. Hum Reprod 2005; 20: 226–230. [DOI] [PubMed] [Google Scholar]
- 33.Moskovtsev SI, Jarvi K, Mullen JB, Cadesky KI, Hannam T, Lo KC. Testicular spermatozoa have statistically significantly lower DNA damage compared with ejaculated spermatozoa in patients with unsuccessful oral antioxidant treatment. Fertil Steril 2010; 93: 1142–1146. [DOI] [PubMed] [Google Scholar]
- 34.Esteves SC, Sánchez-Martín F, Sánchez-Martín P, Schneider DT, Gosálvez J. Comparison of reproductive outcome in oligozoospermic men with high sperm DNA fragmentation undergoing intracytoplasmic sperm injection with ejaculated and testicular sperm. Fertil Steril 2015; 104: 1398–1405. [DOI] [PubMed] [Google Scholar]
- 35.Gawecka JE, Marh J, Ortega M, Yamauchi Y, Ward MA, Ward WS. Mouse zygotes respond to severe sperm DNA damage by delaying paternal DNA replication and embryonic development. PLoS One 2013; 8: e56385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marchetti F, Essers J, Kanaar R, Wyrobek AJ. Disruption of maternal DNA repair increases sperm-derived chromosomal aberrations. Proc Natl Acad Sci USA 2007; 104: 17725–17729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Iuso D, Czernik M, Di Egidio F, Sampino S, Zacchini F, Bochenek M, Smorag Z, Modlinski JA, Ptak G, Loi P. Genomic stability of lyophilized sheep somatic cells before and after nuclear transfer. PLoS One 2013; 8: e51317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Loi P, Matzukawa K, Ptak G, Natan Y, Fulka J, Jr, Arav A. Nuclear transfer of freeze-dried somatic cells into enucleated sheep oocytes. Reprod Domest Anim 2008; 43(Suppl 2): 417–422. [DOI] [PubMed] [Google Scholar]
- 39.Loi P, Matsukawa K, Ptak G, Clinton M, Fulka J, Jr, Nathan Y, Arav A. Freeze-dried somatic cells direct embryonic development after nuclear transfer. PLoS One 2008; 3: e2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Derijck A, van der Heijden G, Giele M, Philippens M, de Boer P. DNA double-strand break repair in parental chromatin of mouse zygotes, the first cell cycle as an origin of de novo mutation. Hum Mol Genet 2008; 17: 1922–1937. [DOI] [PubMed] [Google Scholar]