

Abstract
Wilms' tumor 1 (Wt1) is a tumor suppressor gene encoding ∼24 zinc finger transcription factors. In the mammalian testis, Wt1 is expressed mostly by Sertoli cells (SCs) involved in testis development, spermatogenesis, and adult Leydig cell (ALC) steroidogenesis. Global knockout (KO) of Wt1 is lethal in mice due to defects in embryogenesis. Herein, we showed that Wt1 is involved in regulating fetal Leydig cell (FLC) degeneration and ALC differentiation during testicular development. Using Wt1−/flox;Amh-Cre mice that specifically deleted Wt1 in the SC vs. age-matched wild-type (WT) controls, FLC-like-clusters were found in Wt1-deficient testes that remained mitotically active from postnatal day 1 (P1) to P56, and no ALC was detected at these ages. Leydig cells in mutant adult testes displayed morphological features of FLC. Also, FLC-like cells in adult mutant testes had reduced expression in ALC-associated genes Ptgds, Sult1e1, Vcam1, Hsd11b1, Hsd3b6, and Hsd17b3 but high expression of FLC-associated genes Thbs2 and Hsd3b1. Whereas serum LH and testosterone level in mutant mice were not different from controls, intratesticular testosterone level was significantly reduced. Deletion of Wt1 gene also perturbed the expression of steroidogenic enzymes Star, P450c17, Hsd3b6, Hsd3b1, Hsd17b1, and Hsd17b3. FLCs in adult mutant testes failed to convert androstenedione to testosterone due to a lack of Hsd17b3, and this defect was rescued by coculturing with fetal SCs. In summary, FLC-like cells in mutant testes are putative FLCs that remain mitotically active in adult mice, illustrating that Wt1 dictates the fate of FLC and ALC during postnatal testis development.
Keywords: Wilms' tumor 1, testis, spermatogenesis, Sertoli-Leydig cell cross-talk, germ cell development, steroidogenesis
in humans, wilms' tumor (Wt) is an embryonic malignancy of the kidney that affects around 1 in 10,000 infants, and about 5–10% of the cases are the result of an inactivation of the Wt1 (Wilms tumor 1) gene (14). Wt1 is a tumor suppressor gene and an oncogene. It is involved in cell proliferation, differentiation, apoptosis, and organ development (25). Wt1 gene encodes a transcription factor that contains four zinc finger motifs near its COOH terminus, playing an essential role in normal urogenital system development (7, 18, 24). Inactivation of Wt1 leads to gonadal dysgenesis in human males in which external genitalia do not look clearly male or female; testes are undescended, and these males are infertile. Affected females usually have normal genitalia and the conditions limited to kidney failure (49). Thus, global knockout (KO) of Wt1 in mice caused failure in kidney and gonad development and abnormal development of the mesothelium, heart, and lung, leading to embryonic lethality (33). Since Wt1 is expressed exclusively by Sertoli cells (SCs) in the seminiferous epithelium of the adult testis (16), but also endothelial cells of the microvessels and mesenchymal cells in the fetal and neonatal testicular interstitium (64), a genetic model of SC conditional KO of Wt1 in embryo was developed to examine its role in testicular development and spermatogenesis. SC-specific deletion of Wt1 using Amh-Cre that occurred at embryonic day 14.5 (E14.5) was shown to disrupt Sox9 expression, which in turn perturbed tubular architectural maintenance in the developing testis (16), which was likely mediated by a disruption of collagen α1(IV) and α2(IV) expression (11). Deletion of Wt1 in Wt1−/flox;Cre-ER adult mice using tamoxifen-induced Cre-loxP recombination approach was found to cause undifferentiated spermatogonia accumulation and meiotic progression arrest (64), disruption of SC polarity (60), and impairment of adult Leydig cell (ALC) steroidogenic function (10), leading to aspermatogenesis. However, the precise molecular mechanism(s) that leads to a failure in spermatogenesis following deletion of Wt1 in SCs, in particular its role in fetal Leydig cell (FLC) function that affects steroidogenesis, remains unresolved.
During testicular development, FLCs are gradually replaced by ALCs in the interstitium after birth (47, 61). In fetal and neonatal rat testes, FLCs are arranged in clusters and possess abundant large clustered lipid droplets (20, 21). The fate of FLC has been debated for years, and some investigators (26, 44, 57), but not others (31, 52), suggest that FLCs and ALCs arise from two distinctive precursor cell populations, with the FLCs undergoing involution and/or degeneration by postnatal day 14 (P14) and being replaced by ALCs (56). ALCs dominate the interstitial space after puberty, which appear postnatally in the sequence of stem Leydig cells (SLCs), progenitor Leydig cells (PLCs), immature Leydig cells (ILCs), and ALCs (20, 36). Another hypothesis that has gained popularity is that FLCs degenerate postnatally and are replaced by ALCs, and ALCs are derived from the same precursor cells that gave rise to FLCs, but these cells remain dormant throughout prepubertal development, and FLCs and ALCs share the same precursor cells (56). Androgens produced by FLCs are necessary for the formation of ALC precursors. Testosterone from ALC is essential to initiate, maintain, and regulate spermatogenesis (20, 61); thus, malfunctions of ALC cause reduced androgen synthesis, leading to aspermatogenesis (38, 54). FLCs synthesize only androstenedione due to the lack of 17β-hydroxysteroid dehydrogenase (17β-HSD) in these cells; fetal SCs, however, possess 17β-HSD (including Hsd17b1 and Hsd17b3), which is capable of converting androstenedione to testosterone, at least to a limited extent. As such, FLCs and SCs in fetal and neonatal testes are working in concert to produce the required level of testosterone to sustain male sexual differentiation (39, 53). In adult testes, ALCs synthesize testosterone independent of SCs via steroidogenesis (39, 53). Herein, we report findings using Wt1−/flox;Amh-Cre mice to support the concept that Wt1 is crucial to induce FLC degeneration and ALC appearance in the testis during postnatal development, in which SC Wt1 regulates spermatogenesis by switching “off” the support that maintains FLC and by turning “on” the mechanism(s) that triggers ALC development, illustrating cross-talk between Sertoli and Leydig cells.
MATERIALS AND METHODS
Mice generation and genotyping.
The use of mice for experiments reported herein was approved by the Animal Care Committee of the Institute of Zoology, The Chinese Academy of Sciences. All mice were maintained in a C57BL/6;129/SvEv mixed background. Wt1+/flox mice (16) were mated with the mice carrying the Wt1-null allele (Wt1+/−) (33) and Amh-Cre transgenic mice (34) to produce Wt1−/flox;Amh-Cre offspring. Wt1+/flox mice were mated with Wt1+/−, ROSA26LacZ/+ (55), and Amh-Cre transgenic mice to produce Wt1−/flox;ROSA26LacZ/+;Amh-Cre offspring. DNA isolated from tail biopsies was used for genotyping by PCR (16, 33, 34, 55) to confirm the desired gene KO.
Histological analysis.
For histological analysis, testes obtained following euthanization by carbon dioxide asphyxiation were fixed in 4% paraformaldehyde and embedded in paraffin. Sections 5 μm thick were cut and mounted on microscopic slides after deparaffinization and staining with hematoxylin and eosin (16).
Immunofluorescence analysis.
Testes collected from the control and the Wt1−/flox;Amh-Cre males at specified ages were fixed in 4% paraformaldehyde at 4°C overnight, stored in 70% ethanol, and embedded in paraffin. Frozen cross-sections of testes 8 μm thick were fixed in 4% paraformaldehyde for 10 min. Immunofluorescence (IF) was performed using the FITC- or tetramethylrhodamine isothiocyanate (TRITC)-conjugated secondary antibodies (Jackson ImmunoResearch). Antibodies were obtained commercially as follows: Hsd3b1 (1:400, sc-30820; Santa Cruz Biotechnology), Ki-67 (1:1,000, ab15580; Abcam), P450scc (1:500, AB1244; Chemicon/Millipore), β-galactosidase (β-gal; 1:200, MP55976; MP Biomedical), and Vcam1 (1:400, AF643; R & D Systems). IF procedures were performed as described (64). Fluorescence images were examined with a Nikon Eclipse 80i fluorescence microscope (Tokyo, Japan) and acquired by a Nikon charge-coupled device camera. IF images were merged using ImageJ software. The diameter of FLC cluster was measured by ImageJ-Analyze-Measure.
Quantitative analysis of Leydig cells.
A modified morphometric method was used to estimate the number of Leydig cells (LCs) per testis from P1 to P56 mice (57). Testes were cut into eight or 64 blocks equally in weight (8 blocks for P1, 32 blocks for P7, and 64 blocks for P14, P28, and P56), fixed in 4% paraformaldehyde, embedded in paraffin wax, sliced in 10-μm-thick serial sections, and double-stained for LC-specific marker Hsd3b1 and mitotic marker Ki-67. Cell nuclei were visualized by DAPI. FLCs were identified and distinguished from ALCs according to their histological features as described (20, 23). The round-shaped FLCs were darkly stained by Hsd3b1 and typically arranged in cell clusters with round nuclei. Adult PLCs were lightly stained by Hsd3b1 and spindle-shaped compared with round FLCs. Hsd3b1-positive ALCs were not in clusters. The Hsd3b1-positive FLCs or ALCs and Hsd3b1 plus Ki-67 double-positive LCs were counted at each section and added to the total number of tissue blocks. Three mice at each age were used for analysis, and at least three blocks were randomly selected from each testis.
Terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labeling assay.
Terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labeling (TUNEL) assays were performed using a dead-end apoptosis detection kit (Promega BioSciences, San Luis Obispo, CA) according to the manufacturer's instructions. In brief, testis sections were initially immunofluorescently stained for Hsd3b1, to be followed by an incubation with a reaction mix containing nucleotide, fluorescein-12-deoxyuridine 5-triphosphate, and terminal deoxynucleotidyl transferase for 1 h at 37°C. Cell nuclei were visualized by DAPI. The number of apoptotic cells was analyzed by random field analysis on slides by fluorescence microscopy. A minimum of 700 LCs were counted from each sample, and three testes were analyzed.
Oil red O staining.
Oil Red O (ORO) staining was performed as described (59). In brief, testes were collected from the P1 or P56 control vs. P56 Wt1−/flox;Amh-Cre males and embedded in optimal cutting temperature compound. Eight-micrometer-thick frozen sections were fixed in 4% paraformaldehyde and rinsed with 60% isopropanol. Slides were placed in freshly prepared ORO working solution, namely three parts of ORO stock [by adding 2 g (ORO; Sigma) to 500 ml of isopropanol] and two parts of double-distilled water, for 30 min, rinsed with 60% isopropanol, and counterstained with Harris hematoxylin and mounted in a water-soluble medium.
Electron microscopy.
Testes were collected from the wild-type (WT) P1 or P56 control vs. P56 Wt1−/flox;Amh-Cre males and prefixed at 4°C overnight in 2.5% glutaraldehyde, 1% paraformaldehyde in 0.1 M sucrose, and 0.1 M cacodylate, pH 7.4, at 22°C. They were then washed in phosphate buffer (pH 7.4) three times, postfixed in 1.0% osmium tetroxide in 0.1 M cacodylate, dehydrated in a graded series of ethanol, and embedded in PON 812 epoxy resin (SPI-CHEM, GS02660). Ultrathin sections were cut, stained with uranyl acetate and lead citrate, and examined with a JEM1400 transmission electron microscope (120 kV; JEOL). Diameters of lipid droplet and mitochondrion were measured by ImageJ-Analyze-Measure.
RNA extraction, reverse transcription, and quantitative PCR.
For quantitative PCR, total RNA was extracted from Percoll-purified FLCs, ALCs, and FLC-like cells or whole testes from both mutant and age-matched control mice using an RNeasy kit (Promega) according to the manufacturer's instructions. Total RNA (1–2 μg) was reverse transcribed using the M-MLV Reverse Transcriptase kit (Promega). To quantify gene expression, real-time SYBR Green assay was performed with the cDNA. Gene expression was quantified relative to the gene expression of Gapdh (glyceraldehyde-3-phosphate dehydrogenase). Primers used for the RT-PCR are listed in Table 1. Authenticity of the PCR product was confirmed by direct nucleotide sequencing.
Table 1.
Primer pairs used for qPCR to assess the steady-state mRNA level of target genes
| Gene | GenBank Accession No. | Primer Pairs (5′-3′) | Amplified Product, bp |
|---|---|---|---|
| Hsd3b1 | NM_008293.3 | Forward: AATCTGAAAGGTACCCAGAA | 499 |
| Reverse: TCATCATAGCTTTGGTGAGG | |||
| Hsd3b6 | NM_013821.3 | Forward: TTTTTTTGAGGTATTGACAAGTATTTATTG | 95 |
| Reverse: TCCCCATTCAGAGCATGTATAGC | |||
| Hsd17b1 | NM_010475.1 | Forward: ACTGTGCCAGCAAGTTTGCG | 309 |
| Reverse: AAGCGGTTCGTGGAGAAGTAG | |||
| Hsd17b3 | NM_008291.3 | Forward: ATTTTACCAGAGAAGACATCT | 366 |
| Reverse: GGGGTCAGCACCTGAATAATG | |||
| Hsd11b1 | NM_001044751.1 | Forward: TCTTCCATGACGACATCCAC | 168 |
| Reverse: GAGTAGGGAGCAATCATAGG | |||
| Thbs2 | NM_011581.3 | Forward: GTCCGGACTCTATGGCATGAC | 75 |
| Reverse: TGTGAATCAGGTGCCACCTG | |||
| Vcam1 | NM_011693.3 | Forward: TGCCGAGCTAAATTACACATTG | 122 |
| Reverse: CCTTGTGGAGGGATGTACAGA | |||
| Ptgds | NM_008963.2 | Forward: GGGAATCCCAAGAGACCCAG | 69 |
| Reverse: GCTCTGAGCAAATGGCTGC | |||
| Sult1e1 | NM_023135.2 | Forward: AAAGGGAATTATAGGAGACTGGAAGAA | 77 |
| Reverse: TGCTGCTTGTAGTGCTCATCAA | |||
| Wt1 | NM_144783.2 | Forward: CAAGGACTGCGAGAGAAGGTTT | 136 |
| Reverse: TGGTGTGGGTCTTCAGATGGT | |||
| Dhh | NM_007857.4 | Forward: CCCAACTACAACCCCGACATAA | 78 |
| Reverse: CTTTGCAACGCTCTGTCATCAG | |||
| Pdgfα | NM_008808.3 | Forward: TGAAAGAGGTCCAGGTGAGG | 215 |
| Reverse: CACGGAGGAGAACAAAGACC | |||
| Amh | NM_007445.2 | Forward: TGAAGTTCCAAGAGCCTCCACC | 168 |
| Reverse: AAGTCCACGGTTAGCACCAAATAG | |||
| Nr5a1 | NM_139051.3 | Forward: CTGTGCGTGCTGATCGAATG | 286 |
| Reverse: GCCCGAATCTGTGCTTTCTTC | |||
| Cyp17a1 | NM_007809.3 | Forward: CTTGTCGGACCAAGGAAAAGGCGT | 348 |
| Reverse: CAACCACGGGAATATGTCCACCAG | |||
| Star | NM_011485.4 | Forward: CCGGAGCAGAGTGGTGTCA | 62 |
| Reverse: CAGTGGATGAAGCACCATGC | |||
| Cyp11a1 | NM_019779.3 | Forward, CCAGTGTCCCCATGCTCAAC | 73 |
| Reverse: TGCATGGTCCTTCCAGGTCT | |||
| Gapdh | NM_001289726.1 | Forward: TTGTCTCCTGCGACTTCAACA | 98 |
| Reverse: ACCAGGAAATGAGCTTGACAAAG |
qPCR, quantitative PCR; Wt1, Wilms' tumor 1; Dhh, desert hedgehog; Pdgfα, platelet-derived growth factor-α; Amh, anti-Mullerian hormone; Star, steroid acute regulatory.
Western blot analysis.
Western blot analysis was performed as described previously (64). FLCs isolated from P1 WT control testes, ALCs from P56 WT control testes, and FLC-like cells from P56 Wt1−/flox;Amh-Cre testes were lysed in radio immunoprecipitation assay lysis buffer containing Complete Mini Protease Inhibitor Cocktail Tablets (Roche) to obtain cell lysates, and protein concentration was estimated using the Bradford assay (Bio-Rad). About 40 μg of protein was used for immunoblot analysis. Blots were incubated at 4°C overnight with primary antibody, Hsd3b1 (1:1,000, sc-30820; Santa Cruz Biotechnology), Vcam1 (1:2,000, AF643; R & D Systems), and β-tubulin (1:3,000, E7-c; DSHB, Iowa City, IA) and then incubated with an Odyssey IRDye 680CW (red) or 800CW (green) secondary antibody (1:20,000; LI-COR Bioscience). Specific signals and the corresponding band intensities were evaluated using an Odyssey Infrared Imaging system and software (version 3.0).
Serum and intratesticular hormone measurement.
Blood samples were obtained from WT control and Wt1−/flox;Amh-Cre mice, allowed to clot at 4°C overnight, and centrifuged at 1,000 g for 20 min at 4°C, and serum was frozen at −80°C until being used for radioimmunoassay (RIA). Testis samples were homogenized in ice-cold physiological saline using an IKA disperser. Homogenates were centrifuged (1,000 g, 15 min, 4°C) to remove cell debris, and the supernatant was extracted by diethyl ether, redissolved in 1 ml of PBS, and frozen at −80°C until analysis. Serum concentrations of LH, testosterone (T), progesterone (P4), and estradiol (E) as well as intratesticular T, P4, and E concentrations were quantified by corresponding specific RIAs using commercial assay kits (Beijing Chemclin Biotech, Beijing, China) according to the manufacturer's protocols. The intra- and interassay coefficients of variation (CV) were ∼10 and ∼15%, respectively. The sensitivity of the LH, T, P4, and E assay was <0.1 mIU/ml, 0.07 ng/ml, l pg/ml, and 0.1 pg/ml, respectively.
5-Bromo-4-chloro-3-indolyl β-d-galactopyranoside staining.
5-Bromo-4-chloro-3-indolyl β-d-galactopyranoside (X-gal) staining was performed as described previously (5). Briefly, testes were collected from Wt1+/flox;ROSA26LacZ/+ (negative control), ROSA26LacZ/+;Amh-Cre (positive control), and Wt1−/flox;ROSA26LacZ/+;Amh-Cre adult males. Frozen sections of 8 μm were fixed in LacZ fixative [1% formaldehyde, 0.2% glutaraldehyde, 2 mM MgCl2, 5 mM EGTA, and 0.02% Nonidet P40 (NP-40) in PBS] for 20 min on ice, washed twice in washing solution (2 mM MgCl2, 0.01% sodium deoxycholate, 0.02% NP-40, and 5 mM EGTA in PBS) for 10 min each, and stained overnight at 37°C in the dark with staining solution (5 mM potassium ferricyanide, 5 mM potassium ferrocyanide, 2 mM MgCl2, 0.01% sodium deoxycholate, 0.02% NP-40, and 1 mg/ml X-gal in PBS). Sections were counterstained with Nuclear Fast Red. Sections of negative and positive controls were processed along with the test sections in each experiment to assess putative staining.
Isolation of LCs from mouse testes.
A modified method was used to isolate primary FLCs/FLC-like cells or ALCs from P1 or P56 mouse testes (66). Briefly, testes (about 10 to 20 testes from P1 control mice and 4 to 6 testes from P56 mutant and control mice) were decapsulated and the tubules incubated in 1 mg/ml bovine serum albumin (BSA) containing 0.1% collagenase IV (wt/vol; Sigma) incubated in shaking water bath (100 rpm) at 37°C for 20 min. Tubules were precipitated and collected by centrifugation at 40 g, and the crude cell suspension that contained LCs was filtered through 200-mesh nylon membrane (note: precipitated tubules obtained in this step were used for subsequent SC isolation; see below). Cells were pelleted, washed twice, and resuspended in Dulbecco's modified Eagle's medium-Ham's F-12 nutrient mixture (DMEM-F-12). Cell suspension was separated in a discontinuous Percoll (Pharmacia) gradient of 30, 40, 50, and 60% at 800 g for 40 min. A gradient fraction containing LCs between 50 and 60% layers (1.067–1.077 g/ml) was collected and washed once in culture medium and then plated in 48-well culture dishes in DMEM-F-12 for later experiments. The purity of FLCs and ALCs isolated was >80 and 90%, respectively, based on 3β-hydroxysteroid dehydrogenase (3β-HSD) staining.
Isolation of SCs from neonatal and adult mouse testes.
Precipitated tubules used for isolating LCs as described above were used for SC isolation, as described previously (35). Tubules were washed twice and further digested in 0.1% collagenase IV and 0.1% hyaluronidase (Sigma) in shaking water bath (at 100 rpm) at 37°C for 10 min. Tubules were precipitated, washed twice, and further incubated in 0.1% collagenase IV, 0.1% hyaluronidase, 0.1% trypsin, and 0.1% DNase I in shaking water bath (at 100 rpm) at 37°C for 20–30 min. Cells containing primarily SCs and type A spermatogonia were suspended, washed twice, and placed in a 48-well dish containing DMEM-F-12. Residual germ cells were removed by hypotonic treatment in which SC cultures were exposed to 20 mM Tris (pH 7.4) that lyzed contaminating germ cells for 2 min, as described (15). SCs were allowed to recover for 24 h after the hypotonic treatment before they were used for analyses of steroidogenic enzyme activities. The purity of SCs was >90% for P1 and >80% for P56 SC cultures when assessed by Amh, Sox9, and Wt1 staining.
Analyses of steroidogenic enzyme activities.
Steroidogenic enzyme activities of the cells were determined as described (53), with minor modifications. In brief, purified FLCs/FLC-like cells, fetal SCs, ALCs, and adult SCs were either cultured separately or cocultured in DMEM-F-12 containing 10% fetal bovine serum (Excell, Genetimes Technology), 100 U/ml penicillin, and 100 μg/ml streptomycin (Gibco) in 5% CO2-95% air (vol/vol) at 37 (fetal cells) or 33°C (adult cells), respectively. After cells were attached to culture dishes, culture medium was replaced by serum-free DMEM-F-12 containing 1 mg/ml BSA, 10 μg/ml bovine insulin, and 5 μg/ml human transferrin with or without 100 ng/ml LH. Pregnenolone, P4, or androstenedione (Sigma) dissolved in DMSO was added to the culture media to a desired concentration of 1 µM. Six hours after progesterone or 4 h after androstenedione or P4 was added, culture media were collected and frozen at −80°C until they were used for RIA.
Statistical analysis.
Experiments were repeated at least three times using different mice or cultures. Data were evaluated for statistical differences using Student's t-test or one-way ANOVA. Differences were considered significant with a P value of <0.05.
RESULTS
Sertoli cell-specific Wt1 deletion leads to a disruption of testis architecture, persistence of FLC-like clusters, failure of ALC appearance, and loss of expression of SC genes pertinent to LC development.
We used SC-expressed Amh-Cre transgene, the Wt1-null allele (Wt1+/−), and the Wt1flox strains to obtain SC-specific Wt1 ablation in testes of Wt1−/flox;Amh-Cre males at E14.5. SC-specific KO of Wt1 was confirmed by genotyping. Studies have shown that SC-specific deletion of Wt1 leads to pseudohermaphroditism, with defects in the differentiation of Müllerian duct and Wolffian duct system, reduced testis size, testicular cord disruption, and gradual loss of Sertoli and germ cells until LCs are the only cells found in adult testes (16). Since LCs synthesize testosterone, which is essential to maintain spermatogenesis, we sought to investigate the precise changes in LCs in these mutant testes vs. the age-matched WT control during development. Histological analysis from P1 to adult testes in control WT mice illustrates normal testicular architecture containing testicular cords in which SCs support spermatogenesis and LCs support development in the interstitium (Fig. 1A, top). More importantly, FLCs were gradually replaced by ALCs from P14 to P28 (Fig. 1A, top). In contrast, mutant testes lacked the normal tubular architecture (Fig. 1A, bottom), consistent with an earlier report (16). The eosinophilic cells displayed hyperplasia and formed FLC-like cell clusters from P28 to P56 (Fig. 1A, bottom). Based on the KO efficiency of Wt1 under the combination of Wt1flox and Amh-Cre transgene in this mouse model, Wt1 mRNA level was not affected at E14.5, but it was downregulated from P1 mutant testes following its specific deletion in SCs (Fig. 1B). The residual Wt1 detected in the testis following SC-specific Wt1 KO as shown in Fig. 1B was from the endothelial cells of the microvessels, as well as some resident mesenchymal cells (64), and perhaps resident macrophages and fibroblasts. It is known that Desert hedgehog (Dhh) (12, 62), platelet-derived growth factor-α (Pdgfα) (6, 19), and anti-Mullerian hormone (Amh) (4, 45, 48) are SC-derived paracrine factors that coordinate FLC and ALC differentiation and functions, and deletion of Wt1 also led to downregulation on the mRNA levels of Dhh, Pdgfα, and Amh in the mutant testes vs. the age-matched WT controls (Fig. 1B). It is noted that testis weight was reduced considerably in mutant mice vs. the age-matched WT controls. For instance, P56 mutant mice were just ∼2% of the aged-matched WT control testes, such that the per pair testes weight was 3.30 ± 0.99 in P56 mutant mouse vs. 217.86 ± 8.5 in the age-matched WT control mouse (n = 8 mice), and the cellular contents, such as relative ratios of SC vs. different germ cell types, are different between mutant and age-matched control testes, as reflected in data shown in Fig. 1A and consistent with findings from an earlier report (16).
Fig. 1.

Sertoli cell-specific deletion of Wilms' tumor 1 (Wt1) in Wt1−/flox;Amh-Cre mice affects Leydig cell (LC) development and the expression of Desert hedgehog (Dhh), platelet-derived growth factor-α (Pdgfα), and anti-Mullerian hormone (Amh). A: hematoxylin and eosin staining of cross-sections of testes from mutant (Wt1−/flox;Amh-Cre) vs. age-matched wild-type (WT) control mice from postnatal day (P)1 to P56. In WT control testes, normal microvessels (annotated by “V”) and interstitial LCs were found (arrowheads). Fetal Leydig cells (FLCs) are encircled in yellow box and adult Leydig cells (ALCs) in red box. In mutant testes, Sertoli cell-specific deletion of Wt1 led to the appearance of multiple FLC-like cell clusters (encircled in yellow dashed line); hyperplastic/eosinophilic cells are annotated by arrowheads. Spermatocytes and round spermatids were not detected in mutant testes, illustrating meiosis arrest in these mice, and the mutant testis weight (per pair testes) was only ∼2% of the age-matched WT controls in P56 mice, consistent with an earlier report (16). Insets are the corresponding magnified views of the colored boxed areas. B: quantitative real-time PCR analysis of Wt1 and Sertoli cell-derived paracrine factors coordinating LC differentiation genes Dhh, Amh, and Pdgfα in Wt1−/flox;Amh-Cre mice testes vs. WT controls. Scale bar, 50 μm (10 μm in insets), which applies to all micrographs. *P < 0.05; **P < 0.01. E14.5, embryonic day 14.5.
SC-specific Wt1 KO disrupts replacement of FLCs by ALCs in the testis during postnatal development.
By dual-labeled IF using LC marker Hsd3b1 (3β-HSD type 1) and mitotic marker Ki67, cells in LC clusters were found to remain mitotically active from P1 to P56 in mutant testes (Fig. 2A, yellow boxes in mutants vs. control) in contrast to the WT testis. In control mice, the oval-shaped, mitotically inactive FLCs formed clusters in P1, P3, P7, and P10 testes (Fig. 2A, yellow boxes), and these FLCs gradually disintegrated and degenerated from P14 through P21 (Fig. 2A; see yellow boxes in P14 and P21). The adult type spindle-shaped PLCs differentiate in the interstitium near seminiferous tubules from P7 onward (Fig. 2A, red boxes in P7–P21 control); among them, some were Ki-67 positive (Fig. 2A, red boxes in P21 control). PLCs gradually enlarged and became round-shaped, with reduced proliferative capacity, and transformed to ILCs in P28 WT testes (Fig. 2A, red box in P28 control). ALCs differentiated from ILCs dominated the interstitial space in the adult WT testes (Fig. 2A, red box in P56 control). FLC-like cells arranged in cell clusters in P1 to P10 in mutant testes, and some were Ki-67 positive (Fig. 2A, yellow boxes in P1–P10 mutants). In contrast, round-shaped FLC-like cells persisted and remained proliferative from P14 to P56 in mutant testes (Fig. 2A, yellow boxes in P14–P56 mutants). The apparent LC hyperplasia in mutant testes could be the result of an increase in LC number or due to tubule disruption associated with germ cell loss without considerable changes in LC number. To determine whether the LC number was altered in mutant testes, morphometric analysis was performed. In WT control testes, the FLC population stayed relatively stable from P1 to P7 but declined from P14 to P28 and was nondetectable by P56; however, FLCs and FLC-like cells were maintained from P1 to P14 and increased significantly in P28 and P56 in mutant testes (Fig. 2B). The ratio of Ki-67-positive FLC/FLC-like cells to FLC/FLC-like cells that estimated FLC/FLC-like cell mitotic activity (mitotic index) was found to be induced from P1 to P14 and reduced by P56 in mutant testes vs. the age-matched WT controls (Fig. 2C). Furthermore, FLC-like cell clusters were enlarged in P28 and P56 in mutant testes vs. the age-matched WT controls when their diameters were quantified (Fig. 2D). Total LCs (i.e., ALC and FLC combined) also increased significantly in WT control testes beginning on P14 (Fig. 2E), and the increased ALC number accounted for the overall increase in total LC population in control mouse testes (Fig. 2, F and G). However, no spindle-shaped Hsd3b1-positive PLCs were detected in any mutant testes vs. the age-matched WT controls (Fig. 2, A and G). We next used TUNEL assay to investigate any changes in cell apoptosis in the relative population of ALC and FLC/FLC-like cells in the mutant vs. control testes. No difference in apoptosis was detected in these mice (Fig. 3 and Table 2), illustrating that changes in the relative population of FLC/FLC-like cells or ALCs in the mutant vs. control testes are the result of mitosis.
Fig. 2.
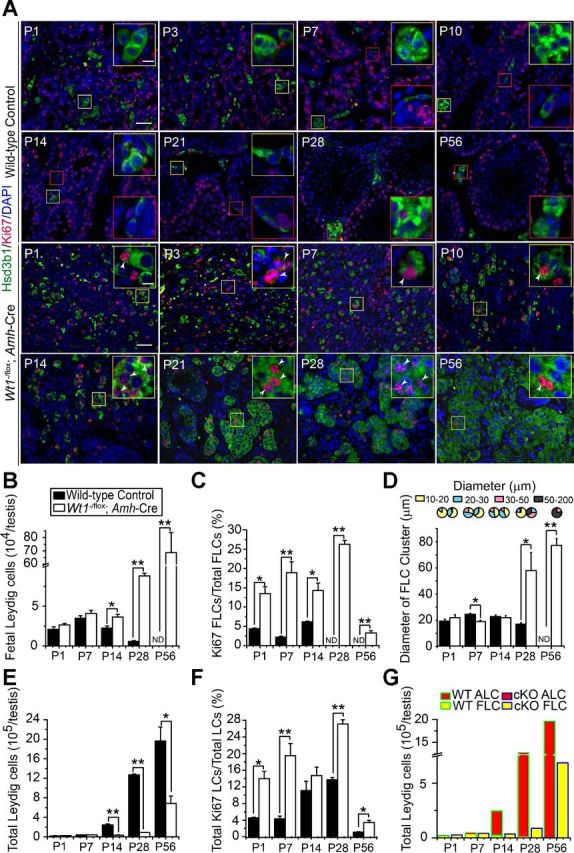
Persistence of FLC-like cell clusters and FLC proliferation in Wt1−/flox;Amh-Cre mutant vs. WT control testes from newborn to adult mice. A: immunofluorescence analysis of LC marker Hsd3b1 (FITC; green fluorescence) and proliferation marker Ki-67 [tetramethylrhodamine isothiocyanate (TRITC); red fluorescence] in testes of WT control vs. mutant mice. Insets are the corresponding magnified views of the colored boxed areas. LCs displayed a typical development pattern in control testes from P1 to P56 (yellow boxes, FLCs; red boxes, ALCs). FLCs formed clusters in the P1, P3, P7, and P10 testes, which were mitotically inactive, and they gradually disintegrated and decreased from P14 to P21. ALC progenitors [progenitor Leydig cells (PLCs)] were seen in the interstitium near seminiferous tubules from P7 onward (red boxes in P7–P21), which are mitotically active (P21; red box). Immature Leydig cells (ILCs) appeared in P28 testes (red box in P28), and the distinct ALCs dominated the interstitial space in adult testes (red box in P56). FLCs/FLC-like cells in the mutant testes persisted (yellow boxes in P1–P56) and remained mitotically active (arrowheads in P1–P56) from P1 to adulthood vs. their age-matched controls. No typical PLC or ALC was found in the mutant testis from P7 to P56 mice. Scale bar, 50 μm (10 μm in inset), which applies to all other micrographs and insets. B: composite data that estimate FLC population in the testis of mutant vs. the age-matched WT control mice. In control testes, FLC population remained relatively stable from P1 to P7 but declined from P14. In mutant testes, FLC/FLC-like cell population was stable from P1 to P14 but significantly induced in P28–P56, consistent with data shown in A. C: mitotic index that estimates mitotic activity of FLC/FLC-like cells and is expressed as the ratio of Ki-67-positive FLC/FLC-like cells to total FLC/FLC-like cells. FLCs in control testes were mitotically inactive; however, the mitotic index of FLC/FLC-like cells in mutant testes was significantly higher and declined on P56. D: pie chart and histogram summarize the cluster size and average diameter of FLC/FLC-like cell clusters in control and mutant testes. The diameter of FLC clusters was significantly increased from P28 in the mutant testes vs. the age-matched WT controls. E: total LC (i.e., ALC and FLC combined) population in the mouse testis in both groups was assessed. Total LCs were found to increase significantly in control testes beginning on P14. In mutant mice, total LCs also increased from P28, but this was not as significant as that of control testes. F: mitotic index that estimates mitotic activity of total LCs and is expressed as the ratio of total Ki-67-positive LCs to total LCs. Total LCs in control testes remained mitotically active from P14 to P28, which was likely due to differentiation and proliferation of PLCs and ILCs. However, the mitotic index of total LCs in mutant testes was still higher, including P14, except that at this age it was not statistically significant. G: total LC population was further analyzed by assessing changes in the relative composition of ALC vs. FLC in mutant testes vs. controls. The increasing ALC number accounted for the overall increase in total LC population in the control mouse testis, and no differentiated ALC was found in the mutant testis. Each bar is the mean ± SE of n = 3 mice. *P < 0.05; **P < 0.01.
Fig. 3.

Changes in the relative population of ALC and FLC in the Wt1−/flox;Amh-Cre mutant vs. the age-matched WT control testes are not the result of differential apoptosis. A: Hsd3b1 (red fluorescence), a marker of LCs, and apoptotic LCs [green fluorescence; based on terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labeling (TUNEL) assay] in P1 to P28 control and mutant testes were assessed. Insets are the corresponding magnified views of the colored boxed areas. TUNEL-positive cells did not overlay with Hsd3b1-positive LCs in all ages examined. TUNEL signals were located in the interstitial space but associated with non-LCs in P1, P7, and P28 control testes and in the testicular cords in P14 control testes. Scale bar, 100 μm in micrograph and 10 μm in inset, which applies to other micrographs and insets. These findings are consistent with data shown in Table 2.
Table 2.
Effects of Sertoli cell-specific deletion of Wt1 on Leydig cell apoptosis
| Postnatal Day | Control | Wt1−/flox;Amh-Cre |
|---|---|---|
| 1 | 1/890 | 2/946 |
| 7 | 0/810 | 0/734 |
| 14 | 0/948 | 0/834 |
| 28 | 0/956 | 0/784 |
No. of terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labeling- and Hsd3b1-positive Leydig cells/no. of Hsd3b1-positive Leydig cells counted.
LC clusters in Wt1−/flox;Amh-Cre testes exhibit biochemical, ultrastructural, and molecular features of FLCs.
To investigate whether LC clusters that persisted in mutant testes were composed of FLCs when Wt1 was specifically inactivated in SCs, biochemical, ultrastructural, and molecular analyses were performed in mutant testes vs. the age-matched WT controls.
Biochemical features.
Lipid droplets (LDs) in Leydig cell cytosols are known to serve as a lipid pool for storage of cholesterol esters for steroidogenesis, and a disruption of LD homeostasis is known to affect steroid hormone production by LCs (37). ORO was used to specifically stain LDs in P1 FLCs and P56 ALCs in WT control testes vs. P56 FLC-like cells in mutant testes. In control testes, large LDs were found in FLCs at P1, and small LDs were detected in ALCs at P56. In contrast, more extensive LDs persisted in FLC-like cells in mutant mouse testes on P56 (Fig. 4A).
Fig. 4.

FLC-like cell clusters in Wt1−/flox;Amh-Cre adult mutant testes contain large nos. of clustered lipid droplets (LDs), analogous to FLCs in neonatal WT control testes. A: Oil Red O (ORO) is known to stain LDs (59); large LDs were found in FLCs in WT control testis on P1, and small LDs were detected in the ALCs on P56. In contrast, extensive large LDs persisted and were found in FLC-like cells in Wt1−/flox;Amh-Cre mutant mouse testes on P56. Insets are the corresponding magnified views of the boxed areas. Scale bar, 50 μm in micrograph and 10 μm in inset, which applies to other micrographs and insets. Transmission electron micrograph (TEM) of FLCs and ALCs in WT control mouse testes on P1 and P56, respectively, vs. Wt1−/flox;Amh-Cre adult mouse testes on P56. FLCs possessed distinctive ultrastructure with large clustered LDs (average diameter: 0.97 μm). However, ALCs were found to have significantly fewer small LDs (average diameter: 0.52 μm). In contrast, FLC-like cells in mutant testes were characterized by the presence of large number of LDs (average diameter: 1.1 μm) in the cytosol. N, nucleus. Scale bar, 2 μm, which applies to other micrographs. B: ORO signals in P1 FLCs and P56 ALCs in WT control mouse testes and P56 FLC-like cells in mutant testes were quantified and compared. Image analysis of ORO signals illustrate an increase in ORO intensity in mutant testes. The pie chart summarizes the relative sizes of LDs in diameter (μm) in mutant vs. WT control testes at specified time points. Each bar represents the average diameter of LDs. Diameter of LDs in FLC-like cells in the mutant testes was significantly larger vs. control testes. Data are presented as means ± SE of n = 3 mice. Different letters indicate significant differences, 1-way ANOVA, P < 0.05.
Ultrastructural features.
Findings based on transmission electron microscope shown in Fig. 4A, bottom, are consistent with ORO staining data, illustrating numerous LDs in FLC-like cells in P56 mutant testes. Furthermore, results based on analysis of the ORO staining intensity and the relative diameter of LD in LC clusters in mutant testes on P56 are consistent with the notion that these are FLC-like cells, which are analogous to FLCs found in P1 control testes (Fig. 4B).
Molecular features.
To further confirm the notion that FLC-like cells in mutant testes are indeed originated from FLCs, we examined the steady-state levels of markers associated with FLC and ALC (41) using Percoll-purified LCs isolated from P56 mutant testes vs. P1 and P56 WT control testes. Results depicted in Fig. 5A illustrate that the mRNA levels of the ALC-enriching genes Ptgds, Sult1e1, Vcam1, Hsd11b1, Hsd3b6, and Hsd17b3 were significantly reduced, whereas the FLC-associated genes Thbs2 and Hsd3b1 were increased in LCs isolated from mutant testes on P56 vs. WT control testes at P56. In short, the expression pattern of these genes in FLC-like cells from mutant testes on P56 was similar to FLCs isolated from control testes at P1 (Fig. 5A). We next examined the ALC-associated marker Vcam1 and FLC and ALC marker P450scc by dual-labeled IF to further analyze the molecular feature of LCs in the mutant testes. We found that the Vcam1 was not expressed in FLCs but located in the interstitial undifferentiated cells in P1 WT control testes (Fig. 5B, left). Vcam1 was detected in ALCs and was colocalized with P450scc in adult testes on P56 WT control testis (Fig. 5B, middle). In contrast, the P450scc-positive FLC-like cells expressed almost no Vcam1 in P56 mutant testes (Fig. 5B, right). We also detected a few Vcam1-positive cells that may be the interstitial cells that are similar to the undifferentiated cells in P1 testes (Fig. 5B, right). Consistent with the results showed in Fig. 5, A and B, the protein level of ALC-associated marker Vcam1 was downregulated, but the FLC marker Hsd3b1 was upregulated in FLC-like cells isolated from P56 mutant testes vs. ALCs from WT control testes on P56 (Fig. 5C). The Vcam1 signals in FLCs from P1 testes might be derived from interstitial undifferentiated cells that also expressed Vcam1 in these testes. These findings thus further support the notion that cells in the FLC-like cell clusters in mutant testes on P56 were putative FLCs.
Fig. 5.

Differential expression of ALC- and FLC-associated mRNAs and proteins in mutant testes on P56 vs. control testes on P1 and P56. A: real-time PCR analysis to compare the steady-state mRNA levels of ALC (Ptgds, Sult1e1, Vcam1, Hsd11b1, Hsd3b6, and Hsd17b3) and FLC (Thbs2 and Hsd3b1) marker genes using LCs isolated from WT control mice testes on P1 and P56 vs. mutant testes on P56. FLC-like cells isolated from mutant testes on P56 shared more similarities with FLCs in control testes on P1. Data are presented as means ± SE of n = 3 mice. Different letters indicate significant differences; P < 0.05. B: immunofluorescence analysis of LC marker P450scc (FITC; green fluorescence) and ALC-enriched marker Vcam1 (TRITC; red fluorescence) in cross-sections of P1 and P56 WT control vs. P56 mutant mouse testes. Insets are the corresponding magnified views of the colored boxed areas. Consistent with the findings shown in A, most of the P450scc-positive FLCs (green box) did not overlay with the Vcam1-positive cells (red box): Vcam1-positive undifferentiated interstitial cells in P1 WT control testes (but the P450scc-positive ALCs were also strong). Orange box: Vcam1-positive cells overlaid with P450scc positive ALCs in P56 WT control testes. In the P56 Wt1−/flox;Amh-Cre mutant testes, most of the FLC-like cells were only P450scc positive (green box); interestingly, a few red Vcam1 signals (orange box) were detected in putative undifferentiated interstitial cells. Scale bar, 50 μm in micrograph and 10 μm in inset, which applies to other micrographs and insets. C: Western blot analysis of the ALC-associated protein Vcam1 and FLC-associated protein Hsd3b1 using LCs isolated from P1 and P56 WT control testes vs. P56 mutant testes. Consistent with the findings shown in A and B, the steady-state levels of proteins in FLC-like cells isolated from P56 mutant testes share more similarities with FLCs from P1 WT control testes.
In short, steroidogenic function of FLC-like cells in adult mutant testes resemble FLCs, instead of ALCs, in WT control testes.
Serum and intratesticular hormones.
Serum and intratesticular steroid levels were compared between Wt1−/flox;Amh-Cre mutant and age-matched control mice. Levels of serum LH and testosterone were not significantly different between mutant and WT control mice, whereas serum P4 and E levels were significantly upregulated in mutant mice (Fig. 6A). However, intratesticular T was significantly reduced from P1 to P56 mice, whereas the intratesticular P4 and E levels were significantly upregulated at P56 in mutant mice vs. controls (Fig. 6A), illustrating a disruption of T synthesis and an upregulation of T metabolism in adult mutant testes.
Fig. 6.

Comparison of serum and intratesticular steroid levels, steady-state mRNA levels of steroidogenic-related genes in mutant vs. age-matched control mice, and tracing of Wt1-knockout Sertoli cells in Wt1−/flox;ROSA26LacZ/+;Amh-Cre testes. A: levels of serum LH and testosterone were not significantly altered in mutant vs. WT control mice, whereas the progesterone and estradiol levels were significantly upregulated. However, intratesticular testosterone level was reduced significantly in the mutant mice, whereas the progesterone and estradiol levels were significantly upregulated in mutant mice. B: real-time PCR analysis of the steady-state mRNA levels of selected steroidogenic related genes. The mRNA levels of steroidogenic acute regulatory protein (Star) and Cyp17a1 were downregulated, Hsd17b1 was upregulated, and there was no change in Nr5a1 and Cyp11a1 in FLC-like cells in P56 Wt1−/flox;Amh-Cre mouse testes vs. the age-matched controls. Data are means ± SE of n = 3 mice, using RNA extracted from LCs for quantitative PCR. *P < 0.05; **P < 0.01. C: Sertoli cells were traced by 5-bromo-4-chloro-3-indolyl β-d-galactopyranoside staining, which marks the β-galactosidase (β-gal; LacZ) sites activated by the Amh-Cre. Insets are the corresponding magnified views of the boxed areas. The P56 negative control testes (Wt1−/flox;ROSA26LacZ/+) revealed no β-gal signal. In the P56 positive control testes (ROSA26LacZ/+;Amh-Cre), the β-gal-positive Sertoli cells displayed cyan perinuclear dots. In the adult Wt1 mutant testes (Wt1−/flox;ROSA26+/LacZ;Amh-Cre), the β-gal-positive Sertoli cells formed clusters that displayed cyan dots perinuclear and in cytosol (top). Immunofluorescence analysis of LC marker Hsd3b1 (TRITC; red) and Sertoli cell tracing signal β-gal (FIITC; green) in cross-sections of negative control and positive control vs. mutant mouse testes. Insets are the corresponding magnified views of the boxed areas. Consistent with A, there was no β-gal-positive SCs in the negative control testes. The β-gal-positive SCs did not overlay with the Hsd3b1-positive ALCs in the positive control testes. The β-gal-positive SCs overlay with the Hsd3b1 signals in the Wt1−/flox;ROSA26+/LacZ;Amh-Cre testes. Scale bar, 50 μm (25 μm in inset), which applies to all other micrographs and insets (bottom).
Steroidogenic metabolizing enzymes.
We next examined the mRNA levels of several key metabolizing enzymes that regulate testosterone biosynthesis, namely Nr5a1, Star, Cyp11a1, Cyp17a1, Hsd3b1, Hsd3b6, Hsd17b1, and Hsd17b3, in FLC-like cells isolated from mutant testes on P56 vs. FLCs and ALCs isolated from P1 and P56 control testes. It is noted that the orphan nuclear receptor steroidogenic factor 1, encoded by the Nr5a1 gene and associated with P450scc (encoded by Cyp11a1) and the steroidogenic acute regulatory (Star) protein, is crucial for LC steroidogenesis (28). Star is involved in the initiate step of steroidogenesis that is localized to the mitochondria (1). P450scc, P450c17 (encoded by Cyp17a1), Hsd3b1/Hsd3b6, and Hsd17b1/Hsd17b3 are key enzymes in the biosynthesis of testosterone (43). The mRNA levels of Star and Cyp17a1 were downregulated, but there was no change in the expression of Nr5a1 and Cyp11a1 in FLC-like cells vs. control FLCs and ALCs (Fig. 6B). Hsd3b1 (Fig. 5A), which is expressed mainly by FLCs, and Hsd17b1 (Fig. 6B), which is expressed by fetal SCs, were upregulated. In contrast, Hsd3b6 and Hsd17b3, which are expressed predominantly by ALCs, were downregulated in the mutant mouse testes (Fig. 5A). To further investigate the source of 17β-HSD, we used Wt1−/flox;ROSA26+/LacZ;Amh-Cre mutant mouse testes to trace SCs by X-gal staining, which mark the β-gal (LacZ) sites activated by the Amh-Cre. SCs were arranged around the inner side of the tubules in positive control testes (ROSA26+/LacZ;Amh-Cre), whereas SCs resided in the tubular remnants in the mutant testes (Fig. 6C, top). Also, remnant SCs expressed Hsd3b1, indicating partial steroidogenesis (Fig. 6C, bottom). FLCs, which expressed all of the steroidogenic-related metabolizing enzymes, except for 17β-HSD, require fetal SCs, which expressed 17β-HSD, for T synthesis in the fetal mouse testis (Fig. 7A). To confirm the origin of T in Wt1−/flox;Amh-Cre mutant testes, FLCs, FSCs, and ALCs isolated in WT control testes and FLC-like cells in mutant testes were incubated with pregnenolone (P5) and androstenedione (adione), respectively. Consistent with the location of 17β-HSD in fetal and adult testes, ALCs and FSCs were capable of converting adione to T. In contrast, FLCs and FLC-like cells produced lower levels of T (Fig. 7B). ALCs, FLCs, and FLC-like cells synthesized similar levels of progesterone (P4) from P5, indicating no changes in 3β-HSD activity (Fig. 7B). We further examined whether the capacity of synthesizing T could be restored when FLC-like cells were cocultured with FSCs using a FLC-like cell/FSC ratio of 1:10. As expected, FLC-like cells/FSCs produced significant amount of T, almost fivefold more than FLC-like cells alone (Fig. 7B).
Fig. 7.
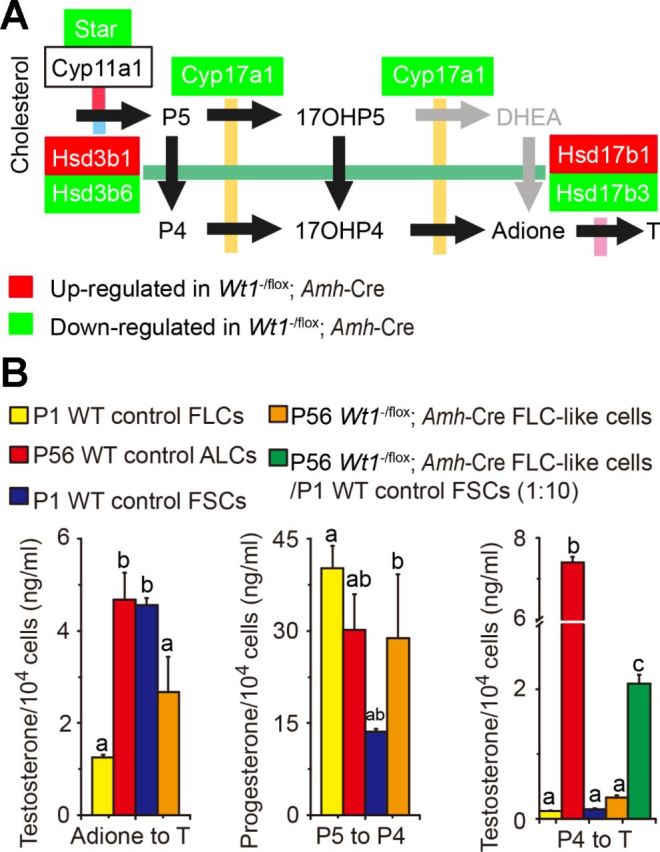
Steroidogenic activities of FLC-like cells isolated from testes of P56 mutant mice are similar to FLCs from P1 WT control mice. A: a schematic drawing that illustrates the synthetic pathway from cholesterol to testosterone in murine fetal testes. Star protein transfers cholesterol from the outer to the inner mitochondrial membrane, where the enzyme P450 side-chain cleavage (P450scc) resides. Pregnenolone (P5) is ultimately transferred to smooth endoplasmic reticulum, where testosterone is synthesized. Reaction 1, P450scc; reaction 2, 3β-hydroxysteroid dehydrogenase (Hsd3b1/Hsd3b6); reaction 3, cytochrome P450 17β-hydroxylase (P450c17); reaction 4, 17β-hydroxysteroid dehydrogenase (Hsd17b1/Hsd17b3). Since 17β-HSD is not expressed in mouse FLCs but FSCs (fetal Sertoli cells), reaction 4 can take place only in FSCs to produce T in immature mice. Black arrows illustrate the preferred pathways of steroidogenesis in murine fetal testis. Gray arrows are for human fetal testes, indicating a degree of flexibility in the pathways (50). Red-shaded boxes indicate that the transcript was upregulated in mutant mouse testis FLCs, whereas green-shaded boxes indicate that the transcript was downregulated. B: equivalent nos. of 104 cells of FLCs, ALCs, and FSCs in WT control testes and FLC-like cells in adult mutant testes were incubated in the presence of P5, androstenedione (adione), or progesterone (P4). FLCs were cocultured with FSCs at a 1:10 cell ratio. Steroids in the medium were quantified by corresponding specific radioimmunoassay. Data are means ± SE of n = 3 independent experiments using different preparations of cell cultures. Different letters indicate significant differences, 1-way ANOVA, P < 0.05. 17OHP4, 17β-hydroxyprogesterone; 17OHP5, 17β-hydroxypregnenolone; DHEA, dihydroepiandrosterone.
DISCUSSION
Unlike SCs, which provide direct structural and nourishment support to germ cells during spermatogenesis in the seminiferous epithelium, LCs residing in the interstitium support spermatogenesis by producing testosterone, which defines the males by masculinization in fetus and adulthood. Malfunctioning of the LC, however, is associated with aspermatogenesis in adult males since intratesticular testosterone must be maintained at a level ∼100-fold higher than the systemic circulation to sustain spermatogenesis (27, 65). In the present study, FLC-like cells were found to persist in the testis of adult mutant mice following specific KO of Wt1 in SCs, whereas ALCs failed to differentiate after birth in age-matched WT mouse testes, and FLC-like cells were the steroid-producing cells in adult mutant testes.
FLCs were first identified in pig embryos in 1904, and it was hypothesized that there were two distinct LC populations in the testis during development (47, 61). However, the fate of FLC remains controversial. Based on histomorphological, ultrastructural, and quantitative analysis of embryonic and postnatal mouse, rat, and human testes, it is believed that FLCs undergo gradual atrophy after birth and vanish in adult testes, and this process does not involve apoptosis (13, 26, 44, 57). A decline in androgen production during the neonatal and prepubertal period also supports atrophy of FLCs (40). However, this dogma has been challenged in other studies in which cells with typical FLC ultrastructure are detected in adult testes, and the absolute volume of these FLCs per testis is almost similar across all ages in the rat (31). Nonetheless, testosterone from adult testes is produced mainly by ALCs. In the present study, we found that FLCs in WT testes maintained their population until P7 and gradually declined in numbers, consistent with a recent report (23) illustrating that total FLC numbers reached the maximum in neonatal mice but then declined in pubertal and adult testes. Data from TUNEL assay reported herein also support the observation that the disappearance of FLC is not the result of apoptosis, and this observation is also consistent with an earlier report suggesting that FLCs in neonatal mouse testes are associated with an involution or degeneration process (13). In the present study, ALCs were absent in neonatal testes in the WT mice but then progressively increased from P7 to adulthood, with a marked rise at P28, which resembles the ALC developmental process reported in normal mouse testes (23). ALCs, however, undergo differentiation from P7, most likely from the same precursor cells that gave rise to the FLCs after lying dormant throughout prepubertal development, not the preexisting or remaining FLCs, and gradually replace the FLC population.
In the SC-specific Wt1 inactivation mouse testes, FLCs/FLC-like cells were mitotically active from P1 to P28, with their population reaching their peak on P56, and remained relatively stable thereafter. Studies have reported that FLCs in normal mice are out of the active cell cycle (42), and their population is supplied by the interstitial undifferentiated cells from the testis progenitor pool (9). Thus, FLCs/FLC-like cells in Wt1−/flox;Amh-Cre mouse testes that displayed high mitotic activity from P1 to P28 are likely due to a malfunctioning of SCs since SCs are known to regulate FLC development via secreted ligands. As such, mRNAs encoding SC-derived ligands that regulate FLC differentiation were examined, which included Dhh, Pdgfα, and Amh. Dhh is a SC product (62), and Pdgfα is associated mostly with SCs in mouse testes (3). Both Dhh (62) and Pdgfα (6) are involved in FLC lineage specification and differentiation but have limited effects on the maintenance or expansion of FLC population (6, 62). On the other hand, Amh by SCs serves as a negative modulator in FLC steroidogenic activity without affecting the number of FLC (48). In the fetal testis of Wt1 mutant mice, when FLC was found to be mitotically active, a significant downregulation of Dhh, Pdgfα, and Amh from E14.5 to P28 vs. the age-matched control testes was detected. These findings thus support the notion that an inactivation of Wt1 in the SC impedes the regulatory function of SCs on the FLCs, causing defects in FLC degeneration. Collectively, these findings suggest that SCs likely act as the organizing center during fetal testis differentiation, affecting the differentiation and function of FLCs.
We failed to detect any ALC or signs of ALC differentiation at any age in the mutant testis. ALC is known to originate from the spindle-shaped stem/progenitor cell in the interstitium, which may share the same precursor cell that gives rise to FLCs (32). The onset of an ALC establishment program also requires the SC factors Dhh (12) and Pdgfα (19). In Wt1−/flox;Amh-Cre mutant testes the expression of Dhh and Pdgfα remained downregulated after birth, which may impede ALC differentiation. In addition, androgens are required for the differentiation of ALC precursors (22). A significantly reduced intratesticular testosterone in the mutant mouse testis during the neonatal stage apparently restrains the onset of ALC precursor differentiation.
FLCs are distinguished from ALCs mainly by their morphology (20, 21), specific markers (41), and reduced capacity of steroidogenesis due to the lack of 17β-HSD in FLCs (39, 53). Interestingly, FLC-like cells in adult Wt1 mutant testes exhibit histological, ultrastructural, and functional features of FLC in neonatal WT testes. We also noted that the expression of ALC markers was significantly downregulated, whereas FLC markers were highly expressed in FLC-like cells of adult mutant mouse testes. However, the marker expression pattern of FLC-like cells in Wt1 mutant testes was not identical to FLCs, and thus we referred these cells to FLC-like cells. These differences may be the result of a disrupted testicular microenvironment that occurred after the loss of SC Wt1, which thus contributed to changes in gene expression and function of FLC-like cells in the Wt1 mutant testes vs. putative FLCs. It is known that LDs serve as the lipid pool in the cytosol of these steroid-producing cells to supply the substrate (e.g., cholesterol) for androgen synthesis. FLCs contain large clustered LDs, whereas ALCs have small scattered LDs. Larger LDs were indeed found in Wt1 mutant mouse testes, thus supporting the notion that LCs in these mutant testes are putative FLCs, and further indicated the impaired steroid-producing function. In this context, it is of interest to note that ethane dimethane sulphonate (EDS) treatment in adult rats was capable of inducing regeneration of ALCs from FLCs (29, 30), illustrating the possibility that new FLCs can arise in response to changes in environment, such as a toxicant assault induced by EDS or an architectural/structural damage to the tubule following SC Wt1 KO, in which FLCs can differentiate to ALCs. As such, it is plausible that in Wt1 mutant testes, a few ALCs might have been differentiated from FLCs, which could assume the FLC morphology despite the downregulation of Dhh and Pdgfα. Thus the FLC-like cells in the mutant adult testis may be a combination of FLCs that are present in majority plus a small subset of ALCs. Thus, the overall LC population in the mutant testes displays a few ALC features, such as the expression of Vcam1 and Hsd3b6.
The mRNAs of Star, Cyp11a1, and Cyp17a1, which are fundamental to ALC function (41), were all shown to be downregulated, supporting the notion that the majority of the FLC-like cells are indeed FLCs in essence in adult mutant testes. However, the trend of changes in some steriodogenic-related genes was different between Wt1−/flox;Cre-ER (deletion of Wt1 induced by tamoxifen injection in 2-mo-old mice) (10) and Wt1−/flox;Amh-Cre testes. In the adult testes of tamoxifen-induced Wt1 deletion, ALCs in these mice were found to become differentiated, and eventually they replaced FLCs, whereas, in Wt1−/flox;Amh-Cre testes, the FLC-like cell clusters that likely originated from FLCs were different from ALCs based on the expression profile of the ALC- and FLC-associated genes. The equivalent transformation rate of pregnenolone to progesterone among ALCs, FLCs, and FLC-like cells indicated that the Hsd3b1, which was expressed mainly by FLCs, may compensate for the Hsd3b6, which is found predominantly in ALCs. However, FLC-like cells were not as effective as ALCs in synthesizing testosterone from androstenedione, illustrating the lack of Hsd17b3 in these FLC-like cells. The progesterone level was upregulated in the Wt1-ablated testes, which also indicated disrupted testosterone synthesis in the adult Wt1 mutant mouse. Hsd17b3 that converts androstenedione to testosterone during steriodogenesis (17) is highly expressed by ALCs, but not FLCs; however, it is also expressed by fetal SCs, which have ∼10% of the capacity vs. the ALCs (53). Indeed, the mRNA level of Hsd17b3, which encodes Hsd17b3, is downregulated in mutant testes, thereby impeding the ability to generate testosterone from androstenedione. However, it remains to be investigated whether tamoxifen, an estrogen receptor antagonist, is responsible for the appearance of ALCs and/or conversion of FLC-like cells to ALCs.
It is noted that the serum testosterone level in mutant mice was not significantly different from WT control mice, which is likely due to the remnant SCs that express Hsd17b1 and Hsd17b3 to compensate for the lost testosterone and the fact that Hsd17b1 is capable of converting androstenedione to testosterone, but at a lower efficiency vs. Hsd17b3 in ALCs (51). Our lineage tracing system using induced ROSA26+/LacZ has indeed demonstrated the presence of SCs in the mutant testes, which may express Hsd17b1 and/or Hsd17b3 to cooperate with the FLCs to synthesize testosterone. Besides, in the Wt1 mutant mouse, other organs such as adrenal glands and kidneys may also synthesize testosterone from androstenedione. A small number of ALCs that morphologically resemble FLC may also be present in the mutant adult testis. Thus, SC Hsd17b1 and Hsd17b3 in mutant adult testes, nongondal steroidogenetic pathways, and FLC-like ALCs could produce sufficient testosterone to maintain the serum testosterone level. However, the intratesticular testosterone level, which usually maintains at ∼100-fold over the serum level in rodents and humans (27, 65), failed to be maintained, and it was reduced almost eightfold vs. control testes. It is noted that the main function of Hsd17b1 is to catalyze the conversion of estrone to estradiol in ovary, placenta, and mammary gland, which would also reduce testosterone level (43). The lack of Hsd17b3 and overexpression of Hsd17b1 in Wt1 mutant testes would also reduce the intratesticular testosterone level. However, in Wt1 mutant testes, Hsd17b1 could be potent in testosterone synthesis rather than metabolism. Furthermore, the upregulation of Hsd17b1 activity and the downregulation of estrogen sulfotransferase (encoded by Sult1e1), which catalyzes the sulfoconjugation and inactivation of estrogens (58), may also account for the increase in estradiol level in the Wt1 mutant mouse, which is known to induce LC hyperplasia (63). In short, our studies demonstrate that the FLC-like cells and remnant SCs in Wt1 mutant testes are inclined to keep up the steroidogenic function of Hsd3b1 and Hsd17b1, instead of Hsd3b6 and Hsd17b3 by ALCs, to maintain the serum testosterone level. However, the low testosterone synthesis efficiency of FLC-like cells in Wt1 mutant testes fails to maintain the normal intratesticular testosterone level, thereby impeding spermatogenesis.
In this study, we investigated the role of Wt1 in the fate determination of FLC and ALC in the testis during postnatal development. Because the testicular architecture failed to form in the mutant adult testis (16) and the SC paracrine factors Pdgfα and Dhh, which are known to play key roles in LC development, were downregulated, the persistence of FLC-like cells may relate to a disruption of testicular microenvironment coupled with a loss of SC functions. It is noted that conditional knockout of Sox9 (Sox9flox/flox;Amh-Cre) (2), Sox9, and Sox8 double-knockout (Sox9flox/flox;Sox8−/−;Amh-Cre) (2) and β-catenin [β-catenin+/flox(e3);Amh-Cre] (8) in SCs also display similar phenotypes as Wt1 mutant testes, in which the testicular architecture was deformed in Sox9/Sox8 or β-catenin mutant but not Sox9 mutants. Since Sox9, Sox8, and β-catenin are downstream targets of Wt1 (8, 16), it is likely that the transcript factor Wt1, which was expressed as early as E10.5 in testicular stromal cells (46), may act as an upstream regulator to maintain testicular architecture and LC function. Needless to say, this possibility requires additional investigation in future studies.
In summary, we provide new insights regarding the role of Wt1 in regulating survival and proliferation of FLC-like cells in mouse testes. Although these FLC-like cells maintain normal serum testosterone levels, their inability to differentiate into ALCs fails to maintain the intratesticular testosterone level to support spermatogenesis. These findings also illustrate a functional cross-talk between SCs and LCs to maintain spermatogenesis via Wt1.
DISCLOSURES
The authors have nothing to disclose.
AUTHOR CONTRIBUTIONS
Q.W., F.G., C.Y.C., and Y.-X.L. conception and design of research; Q.W. performed experiments; Q.W., Q.-S.Z., X.-X.L., Z.-Y.H., and C.Y.C. analyzed data; Q.W., Q.-S.Z., X.-X.L., and C.Y.C. interpreted results of experiments; Q.W. and C.Y.C. prepared figures; Q.W. and C.Y.C. drafted manuscript; Q.W., C.Y.C., and Y.-X.L. edited and revised manuscript; Q.W., Q.-S.Z., X.-X.L., Z.-Y.H., F.G., C.Y.C., and Y.-X.L. approved final version of manuscript.
REFERENCES
- 1.Arakane F, Kallen CB, Watari H, Foster JA, Sepuri NB, Pain D, Stayrook SE, Lewis M, Gerton GL, Strauss JF. The mechanism of action of steroidogenic acute regulatory protein (StAR). StAR acts on the outside of mitochondria to stimulate steroidogenesis. J Biol Chem : 16339–16345, 1998. [DOI] [PubMed] [Google Scholar]
- 2.Barrionuevo F, Georg I, Scherthan H, Lecureuil C, Guillou F, Wegner M, Scherer G. Testis cord differentiation after the sex determination stage is independent of Sox9 but fails in the combined absence of Sox9 and Sox8. Dev Biol : 301–312, 2009. [DOI] [PubMed] [Google Scholar]
- 3.Basciani S, Mariani S, Spera G, Gnessi L. Role of platelet-derived growth factors in the testis. Endocr Rev : 916–939, 2010. [DOI] [PubMed] [Google Scholar]
- 4.Behringer RR, Finegold MJ, Cate RL. Mullerian-inhibiting substance function during mammalian sexual development. Cell : 415–425, 1994. [DOI] [PubMed] [Google Scholar]
- 5.Bell P, Limberis M, Gao G, Wu D, Bove MS, Sanmiguel JC, Wilson JM. An optimized protocol for detection of E. coli beta-galactosidase in lung tissue following gene transfer. Histochem Cell Biol : 77–85, 2005. [DOI] [PubMed] [Google Scholar]
- 6.Brennan J, Tilmann C, Capel B. Pdgfr-alpha mediates testis cord organization and fetal Leydig cell development in the XY gonad. Genes Dev : 800–810, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Call KM, Glaser T, Ito CY, Buckler AJ, Pelletier J, Haber DA, Rose EA, Kral A, Yeger H, Lewis WH. Isolation and chracterziation of a zinc finger polypeptdie gene at the human chromosome 11 Wilms' tumor locus. Cell : 509–520, 1990. [DOI] [PubMed] [Google Scholar]
- 8.Chang H, Gao F, Guillou F, Taketo MM, Huff V, Behringer RR. Wt1 negatively regulates beta-catenin signaling during testis development. Development : 1875–1885, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen H, Stanley E, Jin S, Zirkin BR. Stem Leydig cells: from fetal to aged animals. Birth Defects Res C Embryo Today : 272–283, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen M, Wang X, Wang Y, Zhang L, Xu B, Lv L, Cui X, Li W, Gao F. Wt1 is involved in leydig cell steroid hormone biosynthesis by regulating paracrine factor expression in mice. Biol Reprod : 71, 2014. [DOI] [PubMed] [Google Scholar]
- 11.Chen SR, Chen M, Wang XN, Zhang J, Wen Q, Ji SY, Zheng QS, Gao F, Liu YX. The Wilms tumor gene, Wt1, maintains testicular cord integrity by regulating the expression of Col4a1 and Col4a2. Biol Reprod : 56, 2013. [DOI] [PubMed] [Google Scholar]
- 12.Clark AM, Garland KK, Russell LD. Desert hedgehog (Dhh) gene is required in the mouse testis for formation of adult-type Leydig cells and normal development of peritubular cells and seminiferous tubules. Biol Reprod : 1825–1838, 2000. [DOI] [PubMed] [Google Scholar]
- 13.Faria MJ, Simões ZL, Lunardi LO, Hartfelder K. Apoptosis process in mouse Leydig cells during postnatal development. Microsc Microanal : 68–73, 2003. [DOI] [PubMed] [Google Scholar]
- 14.Friedman AD. Wilms tumor. Pediatr Rev : 328–330; discussion 330, 2013. [DOI] [PubMed] [Google Scholar]
- 15.Galdieri M, Zani B. Hormonal induced changes in sertoli cell glycoproteins. Cell Biol Int Rep : 111, 1981. [DOI] [PubMed] [Google Scholar]
- 16.Gao F, Maiti S, Alam N, Zhang Z, Deng JM, Behringer RR, Lecureuil C, Guillou F, Huff V. The Wilms tumor gene, Wt1, is required for Sox9 expression and maintenance of tubular architecture in the developing testis. Proc Natl Acad Sci USA : 11987–11992, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Geissler WM, Davis DL, Wu L, Bradshaw KD, Patel S, Mendonca BB, Elliston KO, Wilson JD, Russell DW, Andersson S. Male pseudohermaphroditism caused by mutations of testicular 17 beta-hydroxysteroid dehydrogenase 3. Nat Genet : 34–39, 1994. [DOI] [PubMed] [Google Scholar]
- 18.Gessler M, Poustka A, Cavenee W, Neve RL, Orkin SH, Bruns GA. Homozygous deletion in Wilms tumours of a zinc-finger gene identified by chromosome jumping. Nature : 774–778, 1990. [DOI] [PubMed] [Google Scholar]
- 19.Gnessi L, Basciani S, Mariani S, Arizzi M, Spera G, Wang C, Bondjers C, Karlsson L, Betsholtz C. Leydig cell loss and spermatogenic arrest in platelet-derived growth factor (PDGF)-A-deficient mice. J Cell Biol : 1019–1026, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haider SG. Cell biology of Leydig cells in the testis. Int Rev Cytol : 181–241, 2004. [DOI] [PubMed] [Google Scholar]
- 21.Haider SG, Servos G, Tran N. Structural and histological analysis of leydig cell steroidogenic function. In: The Leydig Cell in Health and Disease, edited by , Payne AH, Hardy MP. Totowa, NJ: Humana, 2007, p. 33–45. [Google Scholar]
- 22.Hardy MP, Kelce WR, Klinefelter GR, Ewing LL. Differentiation of Leydig cell precursors in vitro: a role for androgen. Endocrinology : 488–490, 1990. [DOI] [PubMed] [Google Scholar]
- 23.Hazra R, Jimenez M, Desai R, Handelsman DJ, Allan CM. Sertoli cell androgen receptor expression regulates temporal fetal and adult Leydig cell differentiation, function, and population size. Endocrinology : 3410–3422, 2013. [DOI] [PubMed] [Google Scholar]
- 24.Huang A, Campbell CE, Bonetta L, McAndrews-Hill MS, Chilton-MacNeill S, Coppes MJ, Law DJ, Feinberg AP, Yeger H, Williams BR. Tissue, developmental, and timor-specific expression of divergent transcripts in Wilms tumor. Science : 991–994, 1990. [DOI] [PubMed] [Google Scholar]
- 25.Huff V. Wilms' tumours: about tumour suppressor genes, an oncogene and a chameleon gene. Nat Rev Cancer : 111–121, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huhtaniemi I, Pelliniemi LJ. Fetal Leydig cells: cellular origin, morphology, life span, and special functional features. Proc Soc Exp Biol Med : 125–140, 1992. [DOI] [PubMed] [Google Scholar]
- 27.Jarow JP, Zirkin BR. The androgen microenvironment of the human testis and hormonal control of spermatogenesis. Ann NY Acad Sci : 208–220, 2005. [DOI] [PubMed] [Google Scholar]
- 28.Jeyasuria P, Ikeda Y, Jamin SP, Zhao L, De Rooij DG, Themmen AP, Behringer RR, Parker KL. Cell-specific knockout of steroidogenic factor 1 reveals its essential roles in gonadal function. Mol Endocrinol : 1610–1619, 2004. [DOI] [PubMed] [Google Scholar]
- 29.Kerr JB, Bartlett JM, Donachie K, Sharpe RM. Origin of regenerating Leydig cells in the testis of the adult rat. An ultrastructural, morphometric and hormonal assay study. Cell Tissue Res : 367–377, 1987. [DOI] [PubMed] [Google Scholar]
- 30.Kerr JB, Donachie K, Rommerts FF. Selective destruction and regeneration of rat Leydig cells in vivo. A new method for the study of seminiferous tubular-interstitial tissue interaction. Cell Tissue Res : 145–156, 1985. [DOI] [PubMed] [Google Scholar]
- 31.Kerr JB, Knell CM. The fate of fetal Leydig cells during the development of the fetal and postnatal rat testis. Development : 535–544, 1988. [DOI] [PubMed] [Google Scholar]
- 32.Kilcoyne KR, Smith LB, Atanassova N, Macpherson S, McKinnell C, van den Driesche S, Jobling MS, Chambers TJ, De Gendt K, Verhoeven G, O'Hara L, Platts S, Renato de Franca L, Lara NL, Anderson RA, Sharpe RM. Fetal programming of adult Leydig cell function by androgenic effects on stem/progenitor cells. Proc Natl Acad Sci USA : E1924–E1932, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kreidberg JA, Sariola H, Loring JM, Maeda M, Pelletier J, Housman D, Jaenisch R. WT-1 is required for early kidney development. Cell : 679–691, 1993. [DOI] [PubMed] [Google Scholar]
- 34.Lecureuil C, Fontaine I, Crepieux P, Guillou F. Sertoli and granulosa cell-specific Cre recombinase activity in transgenic mice. Genesis : 114–118, 2002. [DOI] [PubMed] [Google Scholar]
- 35.Li XX, Chen SR, Shen B, Yang JL, Ji SY, Wen Q, Zheng QS, Li L, Zhang J, Hu ZY, Huang XX, Liu YX. The Heat-Induced Reversible Change in the Blood-Testis Barrier (BTB) Is Regulated by the Androgen Receptor (AR) via the Partitioning-Defective Protein (Par) Polarity Complex in the Mouse. Biol Reprod. In press. [DOI] [PubMed] [Google Scholar]
- 36.Mendis-Handagama SM, Ariyaratne HB. Differentiation of the adult Leydig cell population in the postnatal testis. Biol Reprod : 660–671, 2001. [DOI] [PubMed] [Google Scholar]
- 37.Miller WL, Bose HS. Early steps in steroidogenesis: intracellular cholesterol trafficking. J Lipid Res : 2111–2135, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O'Shaughnessy PJ. Hormonal control of germ cell development and spermatogenesis. Semin Cell Dev Biol : 55–65, 2014. [DOI] [PubMed] [Google Scholar]
- 39.O'Shaughnessy PJ, Baker PJ, Heikkila M, Vainio S, McMahon AP. Localization of 17beta-hydroxysteroid dehydrogenase/17-ketosteroid reductase isoform expression in the developing mouse testis—androstenedione is the major androgen secreted by fetal/neonatal leydig cells. Endocrinology : 2631–2637, 2000. [DOI] [PubMed] [Google Scholar]
- 40.O'Shaughnessy PJ, Baker PJ, Johnston H. The foetal Leydig cell— differentiation, function and regulation. Int J Androl : 90–95; discussion 105–108, 2006. [DOI] [PubMed] [Google Scholar]
- 41.O'Shaughnessy PJ, Willerton L, Baker PJ. Changes in Leydig cell gene expression during development in the mouse. Biol Reprod : 966–975, 2002. [DOI] [PubMed] [Google Scholar]
- 42.Orth JM. Proliferation of Sertoli cells in fetal and postnatal rats: a quantitative autoradiographic study. Anat Rec : 485–492, 1982. [DOI] [PubMed] [Google Scholar]
- 43.Payne AH, Hales DB. Overview of steroidogenic enzymes in the pathway from cholesterol to active steroid hormones. Endocr Rev : 947–970, 2004. [DOI] [PubMed] [Google Scholar]
- 44.Prince FP. Ultrastructural evidence of mature Leydig cells and Leydig cell regression in the neonatal human testis. Anat Rec : 405–417, 1990. [DOI] [PubMed] [Google Scholar]
- 45.Racine C, Rey R, Forest MG, Louis F, Ferré A, Huhtaniemi I, Josso N, di Clemente N. Receptors for anti-müllerian hormone on Leydig cells are responsible for its effects on steroidogenesis and cell differentiation. Proc Natl Acad Sci USA : 594–599, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rackley RR, Flenniken AM, Kuriyan NP, Kessler PM, Stoler MH, Williams BR. Expression of the Wilms' tumor suppressor gene WT1 during mouse embryogenesis. Cell Growth Differ : 1023–1031, 1993. [PubMed] [Google Scholar]
- 47.Roosen-Runge EC, Anderson D. The development of the interstitial cells in the testis of the albino rat. Acta Anat (Basel) : 125–137, 1959. [DOI] [PubMed] [Google Scholar]
- 48.Rouiller-Fabre V, Carmona S, Merhi RA, Cate R, Habert R, Vigier B. Effect of anti-Mullerian hormone on Sertoli and Leydig cell functions in fetal and immature rats. Endocrinology : 1213–1220, 1998. [DOI] [PubMed] [Google Scholar]
- 49.Scharnhorst V, van der Eb AJ, Jochemsen AG. WT1 proteins: functions in growth and differentiation. Gene : 141–161, 2001. [DOI] [PubMed] [Google Scholar]
- 50.Scott HM, Mason JI, Sharpe RM. Steroidogenesis in the fetal testis and its susceptibility to disruption by exogenous compounds. Endocr Rev : 883–925, 2009. [DOI] [PubMed] [Google Scholar]
- 51.Sha J, Baker P, O'Shaughnessy PJ. Both reductive forms of 17 beta-hydroxysteroid dehydrogenase (types 1 and 3) are expressed during development in the mouse testis. Biochem Biophys Res Commun : 90–94, 1996. [DOI] [PubMed] [Google Scholar]
- 52.Shima Y, Miyabayashi K, Baba T, Otake H, Katsura Y, Oka S, Zubair M, Morohashi K. Identification of an enhancer in the Ad4BP/SF-1 gene specific for fetal Leydig cells. Endocrinology : 417–425, 2012. [DOI] [PubMed] [Google Scholar]
- 53.Shima Y, Miyabayashi K, Haraguchi S, Arakawa T, Otake H, Baba T, Matsuzaki S, Shishido Y, Akiyama H, Tachibana T, Tsutsui K, Morohashi K. Contribution of Leydig and Sertoli cells to testosterone production in mouse fetal testes. Mol Endocrinol : 63–73, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smith LB, Walker WH. The regulation of spermatogenesis by androgens. Semin Cell Dev Biol : 2–13, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet : 70–71, 1999. [DOI] [PubMed] [Google Scholar]
- 56.Svingen T, Koopman P. Building the mammalian testis: origins, differentiation, and assembly of the component cell populations. Genes Dev : 2409–2426, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tapanainen J, Kuopio T, Pelliniemi LJ, Huhtaniemi I. Rat testicular endogenous steroids and number of Leydig cells between the fetal period and sexual maturity. Biol Reprod : 1027–1035, 1984. [DOI] [PubMed] [Google Scholar]
- 58.Tong MH, Jiang H, Liu P, Lawson JA, Brass LF, Song WC. Spontaneous fetal loss caused by placental thrombosis in estrogen sulfotransferase-deficient mice. Nat Med : 153–159, 2005. [DOI] [PubMed] [Google Scholar]
- 59.Wang H, Wang H, Xiong W, Chen Y, Ma Q, Ma J, Ge Y, Han D. Evaluation on the phagocytosis of apoptotic spermatogenic cells by Sertoli cells in vitro through detecting lipid droplet formation by Oil Red O staining. Reproduction : 485–492, 2006. [DOI] [PubMed] [Google Scholar]
- 60.Wang XN, Li ZS, Ren Y, Jiang T, Wang YQ, Chen M, Zhang J, Hao JX, Wang YB, Sha RN, Huang Y, Liu X, Hu JC, Sun GQ, Li HG, Xiong CL, Xie J, Jiang ZM, Cai ZM, Wang J, Wang J, Huff V, Gui YT, Gao F. The Wilms tumor gene, Wt1, is critical for mouse spermatogenesis via regulation of sertoli cell polarity and is associated with non-obstructive azoospermia in humans. PLoS Genet : e1003645, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yao HH, Barsoum I. Fetal leydig cells. In: The Leydig Cell in Health and Disease, edited by , Payne AH, Hardy MP. Totowa, NJ: Humana, 2007, p. 47–54. [Google Scholar]
- 62.Yao HH, Whoriskey W, Capel B. Desert Hedgehog/Patched 1 signaling specifies fetal Leydig cell fate in testis organogenesis. Genes Dev : 1433–1440, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yu W, Zheng H, Lin W, Tajima A, Zhang Y, Zhang X, Zhang H, Wu J, Han D, Rahman NA, Korach KS, Gao GF, Inoue I, Li X. Estrogen promotes Leydig cell engulfment by macrophages in male infertility. J Clin Invest : 2709–2721, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zheng QS, Wang XN, Wen Q, Zhang Y, Chen SR, Zhang J, Li XX, Sha RN, Hu ZY, Gao F, Liu YX. Wt1 deficiency causes undifferentiated spermatogonia accumulation and meiotic progression disruption in neonatal mice. Reproduction : 45–52, 2014. [DOI] [PubMed] [Google Scholar]
- 65.Zirkin BR. Spermatogenesis: its regulation by testosterone and FSH. Semin Cell Dev Biol : 417–421, 1998. [DOI] [PubMed] [Google Scholar]
- 66.Zwain IH, Morris PL, Cheng CY. Identification of an inhibitory factor from a Sertoli clonal cell line (TM4) that modulates adult rat Leydig cell steroidogenesis. Mol Cell Endocrinol : 115–126, 1991. [DOI] [PubMed] [Google Scholar]