

Abstract
Endotrophin is a cleavage product of collagen 6 (Col6) in adipose tissue (AT). Previously, we demonstrated that endotrophin serves as a costimulator to trigger fibrosis and inflammation within the unhealthy AT milieu. However, how endotrophin affects lipid storage and breakdown in AT and how different cell types in AT respond to endotrophin stimulation remain unknown. In the current study, by using a doxycycline-inducible mouse model, we observed significant upregulation of adipogenic genes in the white AT (WAT) of endotrophin transgenic mice. We further showed that the mice exhibited inhibited lipolysis and accelerated hypertrophy and hyperplasia in WAT. To investigate the effects of endotrophin in vitro, we incubated different cell types from AT with conditioned medium from endotrophin-overexpressing 293T cells. We found that endotrophin activated multiple pathological pathways in different cell types. Particularly in 3T3-L1 adipocytes, endotrophin triggered a fibrotic program by upregulating collagen genes and promoted abnormal lipid accumulation by downregulating hormone-sensitive lipolysis gene and decreasing HSL phosphorylation levels. In macrophages isolated from WAT, endotrophin stimulated higher expression of the collagen-linking enzyme lysyl oxidase and M1 proinflammatory marker genes. In the stromal vascular fraction isolated from WAT, endotrophin induced upregulation of both profibrotic and proinflammatory genes. In conclusion, our study provides a new perspective on the effect of endotrophin in abnormal lipid accumulation and a mechanistic insight into the roles played by adipocytes and a variety of other cell types in AT in shaping the unhealthy microenvironment upon endotrophin treatment.
Keywords: endotrophin, adipose tissue, fibrosis, inflammation, angiogenesis
overnutrition has been causally linked to the development of metabolic diseases, such as obesity, type 2 diabetes mellitus (T2DM) and cardiovascular diseases (CVD), that are associated with increased mortality (19, 23, 38, 39). Research has established that adipose tissue (AT) is the key tissue initially affected by excess caloric intake (36, 41). Multiple factors, including elevated release of free fatty acids from the fat cells, a variety of AT-secreted endocrine factors, and low-grade proinflammatory cytokines, have been suggested to influence local functions of AT, which ultimately contribute to the development of systemic metabolic dysregulations (36, 41).
Dynamic remodeling of the extracellular matrix (ECM) is essential for development, wound healing, and normal organ homeostasis (11). Life-threatening pathological conditions arise when ECM remodeling becomes excessive or uncontrolled (6, 22, 45). In this aspect, we have been focusing on how ECM remodeling contributes to AT expansion, which links tightly to obesity and obesity-related diseases. Previously, we found that Col6 is the most highly expressed fibrotic protein in differentiated adipocytes (33, 43). Further studies have demonstrated that Col6 plays an essential role in AT fibrosis, and the levels of Col6 in AT positively correlated with pathological conditions such as hyperglycemia and insulin resistance (2, 16, 22, 30). Col6 is composed of three subunits, Col6α1, -α2, and -α3. During AT expansion, these subunits undergo significant structural remodeling by folding together to form building blocks of Col6 trimers. These trimers further assemble into trimer-oligomers and are subsequently secreted into the extracellular compartment and organized into mature microfibrils to be incorporated into the ECM (5, 13). In the obese AT, local hypoxia enhances the ECM remodeling by upregulating Col6 and the collagen linking enzyme lysyl oxidase (LOX) (15, 40).
Notably, we recently reported that the globular carboxyl-terminal C5 domain of the α3 chain is proteolytically cleaved from the Col6 microfibrils during the posttranslational processing of Col6 fibrils. We refer to this small cleaved product as endotrophin (31). With specific antibodies that we generated, we detected abundant secretion of endotrophin from differentiated 3T3-L1 adipocytes (31). We further observed that endotrophin levels are upregulated in the AT of ob/ob mice compared with those of lean littermates (42). Human studies revealed a strong upregulation of endotrophin in the AT of diabetic human patients (42), supporting the concept that endotrophin has clinical relevance in the context of both tumors and obesity-associated insulin resistance (32, 42).
To investigate the direct actions of endotrophin in AT pathology, we previously generated a doxycycline (Dox)-inducible AT-specific endotrophin overexpression model (42). By using this model, we found that under high-fat diet (HFD) challenge, endotrophin served as a necessary and sufficient stimulator to trigger further enhancement of local fibrosis and inflammation, resulting in increased body weight and insulin resistance. Consistent with the overexpression results, we further demonstrated that a neutralizing antibody against endotrophin ameliorates the metabolically adverse effects induced by HFD (42). Of importance, the crucial function of endotrophin in tumor development has also been characterized in the context of adipocyte interactions with tumor and stromal cells (31).
Despite the fact that we unveiled the local profibrotic and proinflammatory effects of endotrophin in unhealthy fat pads as well as provided a fair amount of insight into the systemic pathological consequences of the excess endotrophin in an animal model, the effect of endotrophin on lipid storage and breakdown in the obese AT needs to be further elucidated. Moreover, the detailed mechanisms by which endotrophin functions directly on different cell types within the AT remain to be elucidated. In the current study, using the AT specific, Dox-inducible endotrophin transgenic (Tg) mouse model that we had previously generated (42), we observed significantly lower levels of phosphorylated HSL and more lipid contents in the adipocytes of the Tg mice, suggesting that lipolysis was impaired, which eventually leads to abnormal lipid accumulation in the AT of endotrophin Tg mice. We further found more macrophage polarization to the M1 proinflammatory phase which may explain the overall inflammatory microenvironment in the WAT of the endotrophin Tg mice. To investigate the functions of endotrophin on different cell types in AT, we used an in vitro system by culturing different cells from AT in an endotrophin-conditioned medium. We found that endotrophin triggers multiple pathological pathways in different cell types. All these different effects orchestrate to shape an unhealthy microenvironment in AT that may further lead to other local and systemic metabolic dysregulation. Our studies provide a mechanistic insight into the adverse effects of endotrophin in unhealthy AT and further establish this novel adipokine as a potential target to treat obesity and other metabolic dysfunctions.
MATERIALS AND METHODS
Chemicals and reagents.
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise indicated. The free fatty acid (FFA) assay kit was from Sigma-Aldrich (cat. no. MAK044). The glycerol assay kit was from BioAssay Systems (cat. no. EGLY-200, Harvard, CA). Cell culture medium and supplements were from Life Technologies (Carlsbad, CA). The generation of the rabbit polyclonal antibody to endotrophin has been published previously (42). The primary antibodies were purchased from Cell Signaling Technologies (Danvers, MA) unless otherwise indicated.
Animals.
The AT-specific Dox-inducible endotrophin Tg mouse model (endotrophin Tg) has been described previously (42). Briefly, to generate the double-Tg mice, TRE-endotrophin mice were crossed with adiponectin promoter-driven rtTA Tg mice (Apn-rtTA). All of these mice are of pure C57BL/6 background. All animal study protocols were reviewed and approved by the Animal Welfare Committee of the University of Texas Health Science Center at Houston as well as the Institutional Animal Care and Use Committee at University of Texas Southwestern Medical Center.
All animal experiments were conducted using littermate-controlled male mice, and Apn-rtTA Tg mice were used as control mice. All experiments were started when these mice were 7 wk old. Mice were housed in cages with 12:12-h dark-light cycles and had free access to water and a regular chow diet unless otherwise indicated.
Dox-containing HFD.
For all HFD-plus-Dox feeding experiments, mice were fed a paste diet containing 60% calories from fat and 200 mg/kg Dox (Bio-Serv, Frenchtown, NJ).
Preparation of 293T-rtTA stable cells and endotrophin-conditioned medium.
The permanent cell line 293T-rtTA, which overexpresses rtTA driven by the CMV promoter, was purchased from Clontech. Cells were cultured in DMEM full medium (high glucose DMEM supplemented with 10% FBS, 1% penicillin-streptomycin) at 37°C in a humidified 5% CO2 atmosphere; 400 μg/ml G418 was added to the culture medium for the selection of rtTA-expressing 293T cells.
The generation of the Dox-inducible endotrophin overexpression plasmid (pTRE-endotrophin) has been previously described (42). To allow endotrophin to secrete from the cells, the prolactin signal (PLS) sequence was fused with the NH2 terminus of the endotrophin coding sequence (42).
To prepare the endotrophin-enriched conditioned medium, 293T-rtTA cells were plated onto 100-mm dishes 1 day before transfection. For the transfection, 15 µg of pTRE-endotrophin plasmid was added to the lipofectamine transfection reagent (ThermoFisher Scientific, Carlsbad, CA). The same amount of pTRE empty plasmid was used for control dishes. To wash off the untransfected plasmids in the medium, 5 h posttransfection, the medium was changed with 6 ml of new medium plus 1 μg/ml Dox. Twenty four hours after the replacement medium was added, the medium was carefully collected and centrifuged to remove the dead cells. The medium was then assayed for secreted endotrophin levels by Western blot analysis. The medium was then kept at 4°C for future treatment of different cells. At the same time, the 293T-rtTA cells were collected to check the cellular overexpression levels of endotrophin.
Culture and differentiation of 3T3-L1 cells.
The mouse fibroblast 3T3-L1 cells were purchased from ATCC. To differentiate the cell into mature fat cells, 3T3-L1 preadipocytes were maintained and grown to 70% confluence in DMEM full medium at 37°C in a humidified 5% CO2 atmosphere. After two days of post-confluence, the media were changed to induction medium (DMEM medium with 1 μM dexamethasone, 5 μg/mL insulin, and 0.5 mM 3-isobutyl-1-methylxanthine). Two days later, the media were replaced with differentiation medium (DMEM full medium with 5 μg/mL insulin). After three days in differentiation medium, cells were dissociated from the plate by 0.25% trypsin and were seeded onto a new 12-well plate. Twenty-four hours later, cells were treated with control- or endotrophin-conditioned medium for the indicated periods of time.
Isolation of SVF from the WAT of wild-type C57BL/6 mice.
Six-week old wild-type C57BL/6 mice were euthanized, and the subcutaneous WAT (sWAT) was collected and washed immediately three times in sterile PBS and minced into small pieces in collagenase type I solution (1 mg/ml; Worthington Biochemical, Lakewood, NJ). The samples were then transferred into a 50-ml conical centrifuge tube and incubated at 37°C on an orbital shaker for 30–60 min for tissue digestion. After the digestion, samples were passed through a sterile 100-μm cell strainer (BD Biosciences, Durham, NC), and 4–5 times volume of full medium was added to the samples. The suspension was then centrifuged at 1,000 rpm for 10 min at 4°C, and the pelleted cells were suspended and seeded onto 12-well plates with DMEM full medium and cultured at 37°C in a humidified 5% CO2 atmosphere; 48 h later, the cells were treated with control or endotrophin-conditioned medium.
Isolation of primary macrophages.
The “Immorto/GFP” strain was created by crossing GFP mice with H-2K(b)-tsA58 “Immorto mice” (17). Monocytes were isolated as CD45+ cells by fluorescence-activated cell sorting (FACS) with anti-CD45 antibody from WAT of Immorto-GFP mice by adherence to plastic. Macrophages were transiently expanded in RPMI containing 10% FCS and 10% LTK cell-conditioned medium at 33°C. For experiments, macrophages were cultured at 33°C with DMEM full medium in a humidified 5% CO2 atmosphere. After one passage, the cells were treated with control or endotrophin-conditioned medium.
Conditioned medium treatment.
Fresh control or endotrophin-conditioned media were used to treat the differentiated 3T3-L1 cells and SVF cells, as well as primary macrophages. The endotrophin levels in the conditioned media were determined by SDS-PAGE followed by Western blotting with anti-endotrophin antibody. All cells were maintained at 70% confluence before the treatment with conditioned medium. Twenty-four hours after the treatment, the cells were collected for further analysis.
FFA and glycerol analysis.
For secreted FFA measurement, 50 µl of the cultured medium was collected and measured by FFA assay kit (Sigma-Aldrich) following the instructions from the company. For secreted glycerol measurement, 10 µl of the cultured medium was collected and measured by glycerol assay kit (BioAssay Systems) following the instructions from the company.
Oil red O staining.
Differentiating 3T3-L1 cells were treated with fresh control or endotrophin-conditioned media. Seventy-two hours later, the cells were washed twice with PBS and fixed for 10 min with 10% PBS-buffered formalin (pH 7.4). Cells were then stained with 0.5% Oil red O (Cambridge, MA) in isopropanol for 30–60 min and washed three times with PBS. After pictures were taken, the Oil red O dye retained in the cells was extracted using an equal volume of 100% isopropanol. The optical density at 500 nm (OD500) was determined and normalized with the levels of total DNA in the cells.
Histology.
The sWAT (collected from the peri-inguinal depot) and the epididymal WAT (eWAT) were immediately excised after the mice were euthanized and fixed in 10% PBS-buffered formalin for 24 h. For immunohistochemistry (IHC), following paraffin embedding, the tissue sections were stained with primary antibodies against total perilipin, total HSL, or phosphorylated HSL (Ser563 or 660 sites) followed by biotinylated secondary antibodies. Binding of second antibodies was visualized using DAB chromogen A by following the methodology described in the company’s instructions (ThermoFisher Scientific) (40). Counterstaining was performed with hematoxylin. All the images were acquired with Axio Scope A1 microscope and ZEISS microscope software ZEN2 (Carl Zeiss, Oberkochen, Germany). For immunofluorescence staining, formalin-fixed paraffin-embedded tissues were sectioned and analyzed by immunofluorescence as described (1). The antibodies were as follows: rabbit anti-CD11C (cat. no. 553802, BD Pharmigen, 1:80); mouse anti-CD206 (cat. no. ab8918, ABCAM, 1:50); Alexa fluor 488 AffiniPure F(ab')2 Fragment Donkey Anti-Rabbit IgG (H+L) (cat. no. 711546152, Jackson Immuno Research Laboratories, 1:200) and Alexa fluor 594 AffiniPure Donkey Anti-Mouse IgG (H+L; cat. no. 715585150, Jackson Immuno Research Laboratories, 1:200). Nuclei were visualized with 4,6-diamidino-2-phenylindole (DAPI) staining. Immunofluorescence images were captured with a Zeiss LSM 710 confocal microscope and analyzed using ImageJ software.
Lipid extraction from adipocytes.
A total of 300 mg of WAT excised from mice was placed into a 1.5-ml Eppendorf tube with 500 μl of PBS at room temperature and homogenized by MagNA Lyser (Roche Diagnostics, Switzerland). The homogenized samples were centrifuged at 6,000 rpm for 15 min at room temperature, and the lipid layer was removed and weighed. The lipid levels were normalized to the total DNA levels in adipocytes.
Total RNA extraction and quantitative real-time quantitative PCR.
Total RNA was extracted from tissues or the cultured cells by TRIzol following the company’s instructions (Invitrogen, La Jolla, CA). The contaminating genomic DNAs were digested by DNase I (5 PRIME, Gaithersburg, MD). Total mRNAs were then purified using the RNeasy RNA extraction kit (Qiagen, Hilden, Germany). The quality and quantity of the RNA were determined by measuring the 260/280 ratios using the Nanodrop 2000 (ThermoFisher Scientific). cDNA was prepared by reverse transcribing 1 μg of RNA with the iScript Select cDNA Synthesis Kit (Bio-Rad Laboratories, Hercules, CA). qPCR reactions were carried out on the Bio-Rad CFX96 system (Bio-Rad Laboratories, La Jolla, CA). The primer sequences are listed in Table 1. Results were calculated using the 2−ΔΔCt method, normalized to glyceraldehyde-3-phosphate dehydrogenase (Gapdh) levels.
Table 1.
Sequences of the primers used for qPCR
| Gene Name | Forward Primer (5′–3′) | Reverse Primer (5′–3′) |
|---|---|---|
| Acc | GATGAACCATCTCCGTTGGC | GACCCAATTATGAATCGGGAGTG |
| Adipoq | TGTTCCTCTTAATCCTGCCCA | CCAACCTGCACAAGTTCCCTT |
| Atgl | ACCACCCTTTCCAACATGCTA | GGCAGAGTATAGGGCACCA |
| Cd163 | TCCACACGTCCAGAACAGTC | CCTTGGAAACAGAGACAGGC |
| Cd206 | CTCTGTTCAGCTATTGGACGC | CGGAATTTCTGGGATTCAGCTTC |
| Cd36 | AGATGACGTGGCAAAGAACAG | CCTTGGCTAGATAACGAACTCTG |
| Cd40 | TGTCATCTGTGAAAAGGTGGTC | ACTGGAGCAGCGGTGTTATG |
| Cd86 | TGTTTCCGTGGAGACGCAAG | TTGAGCCTTTGTAAATGGGCA |
| Col1α1 | GTGCTCCTGGTATTGCTGGT | GGCTCCTCGTTTTCCTTCTT |
| Col3α1 | GGGTTTCCCTGGTCCTAAAG | CCTGGTTTCCCATTTTCTCC |
| Col6α1 | GATGAGGGTGAAGTGGGAGA | CAGCACGAAGAGGATGTCAA |
| F4/80 | CTTTGGCTATGGGCTTCCAGTC | GCAAGGAGGACAGAGTTTATCGTG |
| Fabp4 | AAGGTGAAGAGCATCATAACCCT | TCACGCCTTTCATAACACATTCC |
| Fasn | GGAGGTGGTGATAGCCGGTAT | TGGGTAATCCATAGAGCCCAG |
| Gapdh | AGGTCGGTGTGAACGGATTTG | TGTAGACCATGTAGTTGAGGTCA |
| Hsl | TGGCACACCATTTTGACCTG | TTGCGGTTAGAAGCCACATAG |
| Il-1β | CAACCAACAAGTGATATTCTCCATG | GATCCACACTCTCCAGCTGCA |
| Il-6 | ACAACGATGATGCACTT | CTTGGTCCTTAGCCACT |
| Lox | CCACAGCATGGACGAATTCA | AGCTTGCTTTGTGGCCTTCA |
| Lpl | GGGAGTTTGGCTCCAGAGTTT | TGTGTCTTCAGGGGTCCTTAG |
| Mgl | ACCATGCTGTGATGCTCTCTG | CAAACGCCTCGGGGATAACC |
| Mmp12 | ACATTTCGCCTCTCTGCTGATGAC | CAGAAACCTTCAGCCAGAAGAACC |
| Nlrp3 | ATTACCCGCCCGAGAAAGG | TCGCAGCAAAGATCCACACAG |
| Plin1 | CAAGCACCTCTGACAAGGTTC | GTTGGCGGCATATTCTGCTG |
| Pparg | TCGCTGATGCACTGCCTATG | GAGAGGTCCACAGAGCTGATT |
| Pref-1 | CAGTGCATCTGCAAGGATGGCTG | CTTGTGCTGGCAGTCCTTTCCAG |
| Srebp1 | GCAGCCACCATCTAGCCTG | CAGCAGTGAGTCTGCCTTGAT |
| Tgfβ1 | CGCCATCTATGAGAAAACC | GTAACGCCAGGAATTGT |
| Tgfβ R2 | CATTTGGTTCCAAGGTGC | TGGTAGTGTTCAGCGAG |
| Tlr4 | ATGGCATGGCTTACACCACC | GAGGCCAATTTTGTCTCCACA |
| Tnfα | CACGTCGTAGCAAACCACCAAGTGGA | TGGGAGTAGACAAGGTACAACCC |
| Zfp423 | GAGCCAGCACGCACAGTGAG | GCACACTAGCTGGAGCAGGAC |
Western blotting.
After 24-h conditioned-medium treatment, total proteins were extracted with a cell lysis buffer (25 mM Tris·HCl, 150 mM NaCl, 1% Triton X-100, 1% Tween 20, and 1% SDS, pH 7.4). The protein concentration was measured with a BCA assay kit (ThermoFisher Scientific). Thirty micrograms of total cell lysates or 20 µl of cell supernants were then separated with 4–20% Bis-Tris gels (Invitrogen) and transferred to a PVDF membrane (Millipore). For immunoblotting, the primary antibodies for endotrophin (42), peroxisome proliferator-activated receptor-γ (PPARγ, cat. no. sc-7196; Santa Cruz Biotechnology, 1:500), β-actin (cat. no. sc-47778, Santa Cruz Biotechnology, 1:2,000), and adiponectin (1:1,000) were used, followed by incubation with secondary antibodies labeled with infrared dye emitting at 700 nm or 800 nm (Li-Cor Bioscience). The blots were analyzed with the Odyssey software (Li-Cor Bioscience). The blot bands were quantified with the ImageJ software.
To detect the overexpression levels of endotrophin, 24 h after performing the transfection, 35 μl of conditioned medium and 30 µg of cell lysates were analyzed. The rabbit polyclonal anti-endotrophin has been described previously (31, 42).
Statistical analysis.
All data are represented as means ± SE. Statistical analysis was performed with GraphPad Prism (GraphPad Software). Student’s t-test was applied for statistical analysis. A P value of <0.05 was considered statistically significant.
RESULTS
Locally derived endotrophin in WAT stimulates enhanced lipid accumulation.
We have previously reported local fibrosis and inflammation in the endotrophin Tg mice (42). Here, we sought to determine the abnormal lipid storage and breakdown in the Tg mice. To achieve this aim, the adipocyte-specific endotrophin Tg mice and their littermate controls were challenged with HFD plus Dox induction for 8 wk (Fig. 1A). The qPCR results showed that the levels of adipogenic genes, i.e., Pparγ, fatty acid-binding protein-4 (Fabp4), sterol regulatory element-binding protein-1 (Srebp1), as well as preadipocyte factor 1 (Pref-1) were significantly upregulated in the sWAT of endotrophin Tg mice (Fig. 1B). These results were subsequently confirmed by the protein levels of the adipogenic marker PPARγ in the sWAT of endotrophin Tg mice by Western blotting with anti-PPARγ (Fig. 1C). Since the balance between lipogenesis (lipid synthesis) and lipolysis (lipid breakdown) determines the total lipid levels in adipocytes (21), we further sought to examine the levels of key lipogenesis- and lipolysis-regulating genes. To our surprise, the qPCR results indicated that neither the expression of the lipogenic genes, such as acetyl-CoA carboxylase-α (Acc) and fatty acid synthase (Fasn) nor the lipolytic genes, namely adipose triglyceride lipase (Atgl), hormone-sensitive lipase (Hsl), and monoacylglycerol lipase (Mgl) changed significantly (Fig. 1, D and E). Nevertheless, the level of phosphorylated HSL on Ser660 (pSer660), which is the critical modification for enhanced lipolysis (25), was dramatically decreased in the endotrophin Tg mice, as shown by IHC (Fig. 1F, quantitative measurement in Fig. 1G), and Western blot (Fig. 1H), whereas the total protein levels of HSL did not change. Of note, the levels of CD36, the key fatty acid transporter in AT (10), were also significantly decreased in the sWAT of endotrophin Tg mice (Fig. 1I). Of note, we also analyzed gene and protein expression in eWAT of endotrophin Tg mice and observed similar results (data not shown). In line with all the changes in adipogenesis and lipolysis, the lipid amounts in adipocytes isolated from the eWAT of endotrophin Tg mice were significantly higher (Fig. 1J).
Fig. 1.

Locally derived endotrophin in white adipose tissue (WAT) stimulates abnormal lipid accumulation under a high-fat dfiet (HF challenge. A: schematic representation of the protocol for the experiments. B: qPCR analysis of mRNA levels of adipogenic genes Pparγ, Fabp4, Srebp1, Pref-1, and Zfp423 in sWAT of endotrophin transgenic (Tg) mice and their littermate controls (n = 4 per group, Student’s t-test, * P < 0.05; N.S., no significant difference). C: Western blot analysis for PPARγ in sWAT of endotrophin Tg and their littermate controls (n = 2 per group). Results were normalized with β-actin. D: qPCR analysis of mRNA levels of lipogenic genes Acc and Fasn in sWAT of endotrophin Tg mice and their littermate controls (n = 4 per group, Student’s t-test). E: qPCR analysis of mRNA levels of lipolytic genes Atgl and Hsl in sWAT of endotrophin Tg mice and their littermate controls (n = 4 per group, Student’s t-test). F–H: immunohistochemical staining (IHC; F) and quantification of IHC staining by ImageJ software (G), and Western blot analysis (H) of phosphorylated HSL at Ser660 (Pser660) and total HSL levels in sWAT of endotrophin Tg mice and their littermate controls (representative of 4 samples per group, Student's t-test, * P < 0.05). I: qPCR analysis of levels of fatty acid transporter gene Cd36 in sWAT of endotrophin Tg mice and their littermate controls (n = 4 per group, Student’s t-test, * P < 0.05). J: total lipid contents in sWAT and eWAT of endotrophin Tg mice and their littermate controls. (data are means ± SE; n = 4–5. Student’s t-test, *P < 0.05; scale bar, 50 μm).
Taken together, our data suggest that overexpression of endotrophin may cause abnormal lipid accumulation by promoting adipogenesis as well as by inhibiting lipolysis in adipose tissue.
Locally derived endotrophin in WAT induces M1 polarization of macrophages.
Previously, we showed that endotrophin overexpression in WAT exacerbates local inflammation by inducing macrophage accumulation (42). Here, we sought to determine whether endotrophin also triggers macrophage polarization. We compared the numbers of M1 proinflammatory and M2 anti-inflammatory macrophages by coimmunofluorescent staining with CD11c (M1 macrophage marker, green) and CD206 (M2 macrophage marker, red). The results indicated that the presence of CD11c-positive cells was significantly higher than of CD206-positive cells in the sWAT of endotrophin Tg mice but not of control mice (Fig. 2A). Quantification further confirmed that the ratio of M1 to M2 macrophages was significantly higher in the sWAT of endotrohpin Tg mice (Fig. 2B). We also performed the coimmunofluorescence staining in eWAT of endotrophin Tg mice and obtained similar results as in the sWAT (data not shown).
Fig. 2.

Locally derived endotrophin in sWAT induces M1 macrophage polarization under a HFD challenge. A: coimmunofluorescent staining of CD11c (M1 macrophage marker) and CD206 (M2 macrophage marker) on sWAT of endotrophin Tg mice and their littermate controls, showing increased M1 macrophages (green) and decreased M2 macrophages (red) in the endotrophin Tg group vs. controls (cell nuclei were visualized by staining with DAPI; scale bar, 50 μm). B: quantification of M1/M2 ratio for the area of positive stains in sWAT by ImageJ software (3 different fields from n = 2 per group, Student’s t-test, ** P < 0.01).
These results, combined with the previous observations (42), clearly demonstrated that locally overexpressed endotrophin triggers inflammation by inducing macrophage accumulation and polarization to M1 proinflammatory state.
Generation of Dox-inducible endotrophin overexpression and secretion model in vitro.
To further investigate the detailed molecular mechanisms by which endotrophin exerts its functions on different cell types in AT, we developed an in vitro endotrophin-overexpressing model by employing a Dox-inducible system at the base of 293T-rtTA cells. In the model system, the TRE-endotrophin plasmids were transiently transfected into HEK 293T cells, which constitutively overexpress the transcriptional factor reverse tetracycline-dependent transcriptional activator (rtTA) (3). Upon Dox treatment, rtTA binds to the TRE and activates the overexpression of endotrophin. To allow the endotrophin to secrete into the culture medium, a stretch encoding the prolactin signal peptide (PRL, a secretory signal peptide) was fused into the encoding sequences of endotrophin at its NH2 terminus (Fig. 3A) (42).
Fig. 3.

Endotrophin upregulates proinflammatory and fibrotic genes in stromal vascular fraction (SVF). A: schematic representation of doxycyclene (Dox)-inducible endotrophin overexpression cell model. Permanent rtTA-overexpressing stable 293T cells were used for generating endotrophin overexpression model. Tetracycline response element (TRE)-endotrophin construct was transiently transfected in the cells. Upon transfection, the TRE promoter is regulated by the reverse tetracycline-dependent transcriptional activator (rtTA), which is activated by binding with Dox. In the TRE-endotrophin construct, the endotrophin encoding sequence was fused to a secretory signal (PRL) at the NH2 terminus. Thus, the expressed endotrophin was released into the medium. B: Western blot analysis of endotrophin overexpression levels in medium and cell lysates 24 h after transfection and Dox induction. Results were normalized with β-actin. C: protocol for in vitro experiments with collected endotrophin conditioned medium. D: qPCR analysis of mRNA levels of fibrotic genes Col1α1, Col3α1, Col6α1, Lox, Tgfβ1, and TgfβR2 in SVF treated with control or endotrophin-conditioned medium (data are means ± SE; n = 3. Student’s t-test, * P < 0.05.). E: qPCR analysis of mRNA levels of inflammatory genes Il-1β, IL-6, Nlrp3, and Tlr4 in SVF treated with control or endotrophin-conditioned medium (data are means ± SE; n = 3. Student t-test, *P < 0.05).
Endotrophin is overexpressed and secreted into the culture media upon Dox treatment.
To test whether endotrophin is overexpressed by Dox induction in the in vitro system, we transiently transfected TRE-endotrophin or the empty TRE vector into 293T-rtTA cells and then induced overexpression by Dox. We titrated the amount of Dox to find a balance between cell toxicity and the efficiency of gene expression, and we chose 1 µg/ml Dox as the proper dose for the treatment. Twenty-four hours after Dox treatment, high levels of endotrophin were detected in both cell lysates and the medium in the TRE-endotrophin-transfected group but not in the control group (Fig. 3B), confirming that the in vitro endotrophin overexpression system worked. We then collected the medium from both endotrophin-overexpressing cells and the controls for further experiments (Fig. 3C). To standardize the condition among different samples, one single large batch of the conditional medium was produced for all the treatments.
Endotrophin upregulates both fibrotic and proinflammatory genes in the SVF.
Since the SVF is an essential component in AT (36), we first used the endotrophin-conditioned medium to treat the SVF isolated from WAT. Twenty-four hours after the treatment, we found that the mRNA levels of Col1α1 and the collagen cross-linking enzyme Lox were upregulated (Fig. 3D). Interestingly, we also found that the mRNA levels of transforming growth factor (TGF)β1 and its receptor TGFβR2 were both upregulated in endotrophin-conditioned medium-treated cells (Fig. 3D), in agreement with our previous report that endotrophin simulates the TGFβ signaling in the WAT of Tg mice (42).
Since endotrophin triggers local inflammation in vivo (42), we next determined the levels of inflammatory genes in the endotrophin-treated SVF. As anticipated, we found that endotrophin stimulated upregulation of proinflammatory genes such as Toll-like receptor 4 (Tlr4) and NLR family pyrin domain containing 3 (Nlrp3) in SVF (Fig. 3E).
Taken together, our results suggest that endotrophin functions on the SVF by triggering both fibrotic and proinflammatory genes.
Endotrophin stimulates fibrosis in differentiating 3T3-L1 adipocytes.
To investigate the function of endotrophin in adipocytes, we used the endotrophin-conditioned medium to treat the differentiating 3T3-L1 cells. Twenty-four hours after the treatment, we found that the mRNA levels of the major ECM components in AT (Col1α1, Col3α1, and Col6α1) were all upregulated, whereas Lox showed no change (Fig. 4A). Moreover, the mRNA levels of TGFβ1 and TGFβR2 were also upregulated (Fig. 4A).
Fig. 4.

Endotrophin stimulates fibrosis and lipid accumulation in differentiating 3T3-L1 adipocytes. A: qPCR analysis of mRNA levels of fibrotic genes Col1α1, Col3α1, Col6α1, Lox, Tgfβ1, and TgfβR2 in differentiating 3T3-L1 cells treated with control or endotrophin-conditioned medium (data are means ± SE; n = 3. Student’s t-test, *P < 0.05). B: qPCR analysis of mRNA levels of inflammatory genes IL1β, IL-6, and Tlr4 in differentiating 3T3-L1 cells treated with control or endotrophin-conditioned medium (data are means ± SE; n = 3. Student’s t-test). C: qPCR analysis of mRNA levels of adiponectin in differentiating 3T3-L1 adipocytes treated with control or endotrophin-conditioned medium (data are means; ± SE n = 3. Student’s t-test, **P < 0.01). D and E: Western blot analysis (D) and quantification by density of the bands on the Western blot by ImageJ software (E) for adiponectin in differentiating 3T3-L1 adipocytes treated with control or endotrophin-conditioned medium. Results were normalized with β-actin (data are means ± SE; n = 3. Student’s t-test, *P < 0.05). F: qPCR analysis of mRNA levels of adipogenic factors Fabp4, Pparγ, Srebp1, and Zfp423 in differentiating 3T3-L1 cells treated with control or endotrophin-conditioned medium (data are means ± SE; n = 3. Student’s t-test, *P < 0.05, **P < 0.01). G: qPCR analysis of mRNA levels of Cd36 in differentiating 3T3-L1 cells treated with control or endotrophin-conditioned medium (data are means ± SE; n = 3. Student’s t-test). H: qPCR analysis of mRNA levels of lipogenic genes Acc and Fasn in differentiating 3T3-L1 cells treated with control or endotrophin-conditioned medium (data are means ± SE; n = 3. Student’s t-test). I: qPCR analysis of mRNA levels of lipolytic genes Atgl, Hsl, and Mgl in differentiating 3T3-L1 cells treated with control or endotrophin-conditioned medium (data are means ± SE; n = 3. Student’s t-test, *P < 0.05). J and K: visualization (J) and quantification by ImageJ software (K) of lipid droplets by Oil red O staining. (data are means ± SE; n = 3. Student’s t-test, **P < 0.01, scale bar, 100 μm). L: analysis of FFA levels in cultured medium for 3T3-L1 cells treated with control or endotrophin-conditioned medium (data are means ± SE; n = 3. Student’s t-test, *P < 0.05). M: analysis of glycerol levels in cultured medium for 3T3-L1 cells treated with control or endotrophin-conditioned medium (data are means ± SE; n = 3. Student’s t-test, *P < 0.05).
We next determined the levels of proinflammatory genes in the endotrophin treated 3T3-L1 cells. Unexpectedly, we found that endotrophin treatment had no significant effect on the mRNA levels of these genes (Fig. 4B). Instead, adiponectin, an essential hormone secreted exclusively by adipocytes (7), was significantly downregulated at both mRNA and protein levels (Fig. 4, C and D, quantitative measurement in Fig. 4E), suggesting that endotrophin insult may lead to adipocytes losing their proper endocrine functions.
Collectively, our results suggest that endotrophin may trigger fibrosis and cell dysfunction, but not inflammation, in adipocytes.
Endotrophin stimulates adipocyte differentiation and lipid accumulation.
To further explore the direct effects of endotrophin on adipogenesis and lipid storage capacity in adipocytes, we sought to determine the expression levels of adipogenic, as well as lipogenic and lipolytic genes in endotrophin treated 3T3-L1 cells. To achieve this goal, we used early-stage differentiating 3T3-L1 cells that had been induced in adipogenesis-induction medium for only 3 days. We found that the mRNA levels of adipogenic genes, i.e., Pparγ, Zfp423, Srebp1, and Fabp4, were significantly upregulated (Fig. 4F). On the other hand, Cd36 showed a trend of decrease (Fig. 4G). The key lipogenic genes, such as ACC and Fasn, had no significant changes (Fig. 4H), whereas the mRNA levels of the lipases, namely Atgl, Hsl, and Mgl, were all dramatically downregulated in the endotrophin-treated cells (Fig. 4I).
In line with the gene expression results, the Oil red O staining showed that endotrophin stimulates abnormal lipid accumulation in the endotrophin treated 3T3-L1 cells (Fig. 4, J and K). Moreover, the levels of secreted FFA and glycerol in the cultured medium were significantly lower in the endotrophin-treated group (Fig. 4, L and M), which confirms impairment of lipolysis in the treated 3T3-L1 cells.
In summary, our results demonstrate that endotrophin treatment causes lipid accumulation in 3T3-L1 cells largely due to increased adipogenesis and impaired lipolysis. These results are in agreement with the lipid profiles in the WAT of endotrophin Tg mice that we showed above.
Endotrophin stimulates fibrosis and inflammation in macrophages.
Previously, we reported that locally overexpressed endotrophin attracts and activates macrophages in WAT in vivo (42). To investigate how endotrophin triggers the activation of the macrophages, we treated the isolated macrophages from WAT with endotrophin-conditioned medium. We found that the mRNA levels of a range of collagens had no changes in response to endotrophin treatment. Instead, the collagen linking enzyme Lox levels were significantly upregulated (Fig. 5A). As anticipated, we found that endotrophin treatment significantly upregulated the mRNA levels of the macrophage marker, F4/80, as well as that of proinflammatory cytokines, such as IL-1β and TNFα (Fig. 5B). Importantly, we found that the levels of classically activated macrophage (M1) markers, including Cd40 and Cd86 were dramatically upregulated, whereas the levels of alternatively activated macrophage (M2) markers, such as Cd163 and Cd206, had no change or had the trend to decrease (Fig. 5, C and D), suggesting that endotrophin may trigger a major shift in the M1/M2 ratio, favoring a proinflammatory state. The result is in agreement with the M1 macrophage polarization effect in the endotrophin Tg mice (Fig. 2). Taken together, our results demonstrate that endotrophin may trigger fibrosis in macrophages by upregulating LOX. More importantly, endotrophin activates M1 macrophages to induce local inflammation in WAT.
Fig. 5.

Endotrophin upregulated LOX and proinflammatory and classically activated marker (M1) genes in macrophages. A: qPCR analysis of mRNA levels of fibrotic genes Col1α1, Col6α1, Lox, Tgfβ1, and TgfβR2 in macrophages treated with control or endotrophin-conditioned medium (data are means ± SE; n = 3. Student’s t-test, *P < 0.05). B: qPCR analysis of mRNA levels of inflammatory genes IL-1β, IL-6, TNFα, and F4/80 in macrophages treated with control or endotrophin-conditioned medium (data are means ± SE; n = 3. Student’s t-test, *P < 0.05, **P < 0.01). C: qPCR analysis of mRNA levels of M1 macrophage markers Cd40 and Cd86 in macrophages treated with control or endotrophin-conditioned medium (data are means ± SE; n = 3. Student’s t-test, *P < 0.05). D: qPCR analysis of mRNA levels of M2 macrophage markers Cd163 and Cd206 in macrophages treated with control or endotrophin-conditioned medium (data are means ± SE; n = 3. Student’s t-test).
DISCUSSION
In the current study, we applied an in vitro system by culturing different cells from AT in an endotrophin-containing medium to investigate the effects of endotrophin on the different cell populations. We found that endotrophin activates distinct pathological pathways in different cell types. In 3T3-L1 adipocytes, endotrophin triggers a fibrotic program by upregulating several collagen proteins, including Col1, Col3, and Col6. Moreover, it downregulates the expression of critical lipolytic genes, such as Atgl, Hsl, Mgl, and adiponectin. As a result, the endotrophin-treated 3T3-L1 cells accumulate larger lipid droplets. In macrophages isolated from WAT, endotrophin treatment stimulates the expression of the proinflammatory genes IL-1β and TNFα and genes of collagen-linking enzymes, such as LOX, whereas it does not increase the expression of collagen genes. Importantly, endotrophin treatment also induced the macrophages switch to M1 proinflammatory state. This might be a mechanism by which endotrophin exerts its proinflammatory effect. We further confirmed the endotrophin effects in an AT-specific, Dox-inducible endotrophin Tg mouse model. Consistent with the in vitro results, in addition to previously reported local fibrosis and inflammation in WAT of endotrophin Tg mice (42), we found significantly reduced phosphorylated HSL levels and increased lipid content in adipocytes of endotrophin Tg mice compared with control mice, suggesting that the lipolysis was inhibited in the endotrophin Tg AT. Moreover, we report that the accumulated macrophages were polarized to M1 proinflammatory state in WAT of the endotrophin Tg mice, suggesting a possible mechanism by which endotrophin shapes an inflammatory microenvironment in the Tg mice. Combined, our results provide a mechanistic insight into the adverse effects of endotrophin in unhealthy AT and further establish this novel adipokine as a potential target to treat obesity and other metabolic dysfunctions.
The link between fibrosis and inflammation has been well established (8, 15, 40, 41). However, the causal relationship between them in the context of obesity and lipodystrophy remains unclear. Moreover, the quantitative contributions of different cell types toward fibrotic deposits and the stepwise progression from a nonfibrotic to a profibrotic and proinflammatory state are not well established (9, 43). Here, using both in vitro and in vivo endotrophin-induced fibrotic and inflammatory models, we clearly show that endotrophin may directly stimulate collagen proteins in adipocytes and the SVF to provide fibrotic materials. Intriguingly, LOX, the critical enzyme that links these collagens together to form mature fibrils (6), is upregulated in endotrophin-treated macrophages and the SVF but not in the 3T3-L1 adipocytes. This suggests that it is the macrophages in the SVF that provide the critical enzyme (LOX) to link the collagens to form the mature ECM. Our results support the notion that both adipocytes and macrophages contribute to local fibrosis in AT in a stepwise manner, but their roles could be distinct: adipocytes are the major cellular origin of the fibrotic collagens, while macrophages serve as the master “regulator” of the fibrosis through upregulated LOX function.
WAT expansion is positively associated with chronic inflammation (46). Our results suggest that macrophages are predominantly responsible for the chronic inflammation associated with increased endotrophin. In obesity, macrophages populate WAT and play a central role in adipose pathology (47, 48). In agreement, we previously reported that the total number of macrophages in the WAT of endotrophin Tg mice were significantly increased (42). Recent literature highlights the importance of not only the numbers of infiltrating macrophages, but also of their activation in the maintenance of the inflammation (4). A switch in polarization of the macrophages from anti-inflammatory M2 to proinflammatory M1 state is a key feature of WAT dysfunction in obesity (26, 27, 35). In this study, we found that endotrophin stimulates this switch that is associated with chronic inflammation locally in WAT and ultimately leads to systemic insulin resistance. Interestingly, in our study, M1 macrophage polarization was found to be positively associated with lipid accumulation in adipocytes both in vivo and in vitro. This is likely explained by impaired lipolysis in endotrophin-treated adipocytes, as reduced lipid breakdown leads to its accumulation and adipocyte hypertrophy. It should be noted that the strict subdivision into M1 and M2 macrophages has recently been challenged (28). The proinflammatory phenotypic changes caused by endotrophin are likely to be complex and key molecular pathways, such as those mediated by NF-κB and microRNA-155 (14, 20, 44), remain to be investigated.
WAT is an active endocrine organ that secretes numerous protein hormones, including leptin, adiponectin, and resistin (12, 29, 34). To determine whether endotrophin adversely affects the endocrine function of adipocytes, we measured the adiponectin levels in the endotrophin-treated 3T3-L1 cells. We found that endotrophin promoted downregulation of adiponectin at both mRNA and protein levels. This could have resulted from endotrophin causing abnormal ECM deposition, which leads to local fibrosis and triggers the necrosis of adipocytes (43). Since adiponectin fulfills a critical role as an important messenger to communicate between AT and other organs, the downregulation of adiponectin by endotrophin suggests a possible mechanism by which endotrophin impairs metabolic homeostasis in obese individuals.
Adipocytes are the major lipid-storing cells. The stored lipid comes from two sources: one is the diet and the other is de novo lipogenesis (36). We examined the activity of critical lipogenic enzymes, including FAS and ACC in endotrophin-treated 3T3-L1 cells and endotrophin-overexpressed AT. Somewhat surprisingly, all these lipogenic factors were unaffected by endotrophin both in vitro and in vivo. To determine whether it is the impaired breakdown of lipids stored in the fat cells that causes the abnormal lipid metabolism in adipocytes by endotrophin, we further investigated the effects of endotrophin on the lipolysis process. Classically, the lipolytic machinery consists of three major enzymes: ATGL, HSL, and MGL (24). Since these three lipases account for more than 90% of the total lipase activity in AT (49), we measured the levels of the lipases in fat cells after endotrophin treatment. Of note, all three lipases were dramatically downregulated in the endotrophin-treated fat cells. In accord with the in vitro data, the endotrophin-overexpressed fat pads also expressed lower levels of these lipases. Of note, in sWAT of mice, endotrophin overexpression did not alter the total expression levels of HSL but only the phosphorylation status. In contrast, in differentiating 3T3-L1 cells there was also a dramatic downregulation of Hsl mRNA. This apparent discrepancy could be because in addition to adipocytes WAT contains other cell types expressing HSL constitutively. Indeed, we did not observe the alteration of mRNA levels of HSL in SVF in response to endotrophin treatment (data not shown). The impaired lipolytic pathway might lead to abnormal lipid droplets in the cells. In agreement with this premise, we detected higher levels of lipid accumulation in endotrophin-treated differentiating 3T3-L1 cells. Moreover, we also detected higher lipid content in the WAT of endotrophi-overexpressing mice when fed a HFD.
Endotrophin-expressing mice gain more body weight upon HFD feeding (42). This is largely due to more adipose tissue mass in the endotrophin Tg mice. In obesity, WAT expands via hyperplasia (cell number increase) and hypertrophy (cell size increase) (18, 37). Our data show that endotrophin causes more lipid accumulation and larger adipocytes. Moreover, it stimulates upregulation of adipogenic genes, such as PPARγ, SREBP1, and ZFP423, in the differentiating 3T3-L1 cells and in WAT of endotrophin Tg mice. Furthermore, we observed increased in small to medium size adipocytes near areas of endotrophin in the sWAT of the endotrophin Tg mice (data not shown), suggesting that endotrophin stimulates recruitment of new adipocyte progenitors. These results suggest that endotrophin may cause WAT expansion via both hyperplasia and hypotrophy. In addition to the direct effects of endotrophin that we identified, adipocyte dysfunction in WAT of endotrophin-overexpressing mice could indirectly contribute to inflammation and insulin resistance.
The endothelium plays a critical role in adipose tissue remodeling during obesity development. It is important to investigate the effects of endotrophin on endothelial cells in WAT. In this regard, Park et, al. (31) incubated MS-1 cells (a mouse microvascular endothelial cell line) with conditioned medium from endotrophin-overexpressing 293T cells, and they found that the endotrophin-treated MS-1 cells migrated and organized vascular structures much more actively, suggesting that endotrophin may stimulate angiogenesis through the activation of endothelial cells in obese AT. This finding further supports our hypothesis that endotrophin regulates AT pathological remodeling at multiple levels in different cell types.
In conclusion, our study demonstrates that endotrophin functions as a stimulator and exerts profound pathological functions by triggering multiple distinct signaling pathways in different cell populations in obese AT. Combining all the data, we propose a model in which the endotrophin-insulted cells cross-talk and exert profound adverse effects locally in obese AT, which ultimately leads to systemic inflammation and whole body insulin resistance (Fig. 6). Of note, all the results were obtained from different cell types isolated from mouse AT; thus, these observations would pertain only to murine WAT. Understanding the complex events occurring in the rapidly remodeled AT caused by endotrophin will help us to design novel strategies to deal with obesity and obesity-related metabolic dysfunctions by targeting this new adipokine.
Fig. 6.
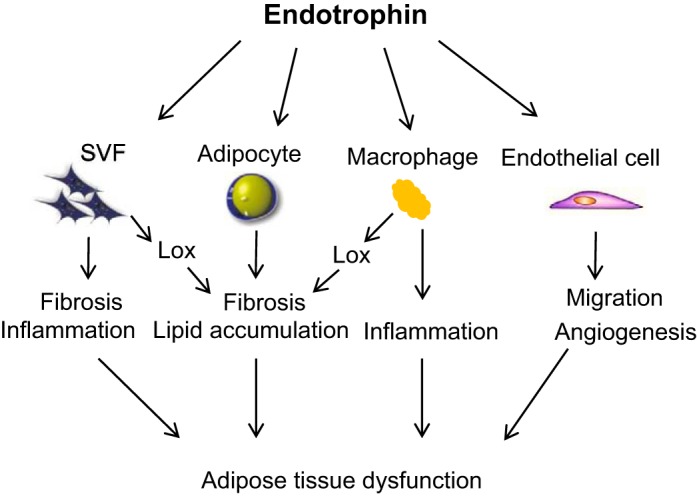
Working model for the functions of endotrophin in different cell populations of WAT. The complex events that occur in different cell types in endotrophin-insulted AT, including abnormal lipid accumulation, fibrosis, angiogenesis, and inflammation. An orchestrated set of interactions between these critical cell types takes place during the rapidly remodeling process in response to overnutrition, which eventually leads to systemic metabolic dysfunction.
GRANTS
This work was funded by a pilot award from Center of Clinical and Translational Studies (CCTS) at University of Texas Health Science Center at Houston (CTSA UL1 TR000371 for K. Sun).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Y.Z., X.G., and K.S. performed experiments; Y.Z., N.Z., M.G.K., Z.A., and K.S. analyzed data; Y.Z. and K.S. interpreted results of experiments; Y.Z. and K.S. prepared figures; N.Z., M.G.K., Z.A., and K.S. edited and revised manuscript; K.S. conception and design of research; K.S. drafted manuscript; K.S. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Philipp E. Scherer’s laboratory at University of Texas Southwestern Medical Center at Dallas for offering reagents and technical assistance. We also thank Dr. Eva M. Zsigmond for assistance with manuscript preparation.
REFERENCES
- 1.Azhdarinia A, Daquinag AC, Tseng C, Ghosh SC, Ghosh P, Amaya-Manzanares F, Sevick-Muraca E, Kolonin MG. A peptide probe for targeted brown adipose tissue imaging. Nat Commun : 2472, 2013. doi: 10.1038/ncomms3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berria R, Wang L, Richardson DK, Finlayson J, Belfort R, Pratipanawatr T, De Filippis EA, Kashyap S, Mandarino LJ. Increased collagen content in insulin-resistant skeletal muscle. Am J Physiol Endocrinol Metab : E560–E565, 2005. doi: 10.1152/ajpendo.00202.2005. [DOI] [PubMed] [Google Scholar]
- 3.Bueno AC, Sun K, Martins CS, Elias Junior J, Miranda W, Tao C, Foss-Freitas MC, Barbieri MA, Bettiol H, de Castro M, Scherer PE, Antonini SR. A novel ADIPOQ mutation (p.M40K) impairs assembly of high-molecular-weight adiponectin and is associated with early-onset obesity and metabolic syndrome. J Clin Endocrinol Metab : E683–E693, 2014. doi: 10.1210/jc.2013-3009. [DOI] [PubMed] [Google Scholar]
- 4.Chinetti-Gbaguidi G, Staels B. Macrophage polarization in metabolic disorders. Curr Opin Lipidol : 365–372, 2011. doi: 10.1097/MOL.0b013e32834a77b4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chu ML, Conway D, Pan TC, Baldwin C, Mann K, Deutzmann R, Timpl R. Amino acid sequence of the triple-helical domain of human collagen type VI. J Biol Chem : 18601–18606, 1988. [PubMed] [Google Scholar]
- 6.Cox TR, Erler JT. Remodeling and homeostasis of the extracellular matrix: implications for fibrotic diseases and cancer. Dis Model Mech : 165–178, 2011. doi: 10.1242/dmm.004077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deng Y, Scherer PE. Adipokines as novel biomarkers and regulators of the metabolic syndrome. Ann N Y Acad Sci : E1–E19, 2010. doi: 10.1111/j.1749-6632.2010.05875.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Divoux A, Moutel S, Poitou C, Lacasa D, Veyrie N, Aissat A, Arock M, Guerre-Millo M, Clément K. Mast cells in human adipose tissue: link with morbid obesity, inflammatory status, and diabetes. J Clin Endocrinol Metab : E1677–E1685, 2012. doi: 10.1210/jc.2012-1532. [DOI] [PubMed] [Google Scholar]
- 9.Divoux A, Tordjman J, Lacasa D, Veyrie N, Hugol D, Aissat A, Basdevant A, Guerre-Millo M, Poitou C, Zucker JD, Bedossa P, Clément K. Fibrosis in human adipose tissue: composition, distribution, and link with lipid metabolism and fat mass loss. Diabetes : 2817–2825, 2010. doi: 10.2337/db10-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Febbraio M, Hajjar DP, Silverstein RL. CD36: a class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J Clin Invest : 785–791, 2001. doi: 10.1172/JCI14006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. J Cell Sci : 4195–4200, 2010. doi: 10.1242/jcs.023820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Funahashi T, Nakamura T, Shimomura I, Maeda K, Kuriyama H, Takahashi M, Arita Y, Kihara S, Matsuzawa Y. Role of adipocytokines on the pathogenesis of atherosclerosis in visceral obesity. Intern Med : 202–206, 1999. doi: 10.2169/internalmedicine.38.202. [DOI] [PubMed] [Google Scholar]
- 13.Furthmayr H, Wiedemann H, Timpl R, Odermatt E, Engel J. Electron-microscopical approach to a structural model of intima collagen. Biochem J : 303–311, 1983. doi: 10.1042/bj2110303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao Z, Zhang J, Henagan TM, Lee JH, Ye X, Wang H, Ye J. P65 inactivation in adipocytes and macrophages attenuates adipose inflammatory response in lean but not in obese mice. Am J Physiol Endocrinol Metab : E496–E505, 2015. doi: 10.1152/ajpendo.00532.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Halberg N, Khan T, Trujillo ME, Wernstedt-Asterholm I, Attie AD, Sherwani S, Wang ZV, Landskroner-Eiger S, Dineen S, Magalang UJ, Brekken RA, Scherer PE. Hypoxia-inducible factor 1alpha induces fibrosis and insulin resistance in white adipose tissue. Mol Cell Biol : 4467–4483, 2009. doi: 10.1128/MCB.00192-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iyengar P, Espina V, Williams TW, Lin Y, Berry D, Jelicks LA, Lee H, Temple K, Graves R, Pollard J, Chopra N, Russell RG, Sasisekharan R, Trock BJ, Lippman M, Calvert VS, Petricoin EF III, Liotta L, Dadachova E, Pestell RG, Lisanti MP, Bonaldo P, Scherer PE. Adipocyte-derived collagen VI affects early mammary tumor progression in vivo, demonstrating a critical interaction in the tumor/stroma microenvironment. J Clin Invest : 1163–1176, 2005. doi: 10.1172/JCI23424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jat PS, Noble MD, Ataliotis P, Tanaka Y, Yannoutsos N, Larsen L, Kioussis D. Direct derivation of conditionally immortal cell lines from an H-2Kb-tsA58 transgenic mouse. Proc Natl Acad Sci USA : 5096–5100, 1991. doi: 10.1073/pnas.88.12.5096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jo J, Gavrilova O, Pack S, Jou W, Mullen S, Sumner AE, Cushman SW, Periwal V. Hypertrophy and/or hyperplasia: dynamics of adipose tissue growth. PLOS Comput Biol : e1000324, 2009. doi: 10.1371/journal.pcbi.1000324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature : 840–846, 2006. doi: 10.1038/nature05482. [DOI] [PubMed] [Google Scholar]
- 20.Karkeni E, Astier J, Tourniaire F, El Abed M, Romier B, Gouranton E, Wan L, Borel P, Salles J, Walrand S, Ye J, Landrier JF. Obesity-associated inflammation Induces microRNA-155 expression in adipocytes and adipose tissue: outcome on adipocyte function. J Clin Endocrinol Metab : 1615–1626, 2016. doi: 10.1210/jc.2015-3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kersten S. Mechanisms of nutritional and hormonal regulation of lipogenesis. EMBO Rep : 282–286, 2001. doi: 10.1093/embo-reports/kve071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khan T, Muise ES, Iyengar P, Wang ZV, Chandalia M, Abate N, Zhang BB, Bonaldo P, Chua S, Scherer PE. Metabolic dysregulation and adipose tissue fibrosis: role of collagen VI. Mol Cell Biol : 1575–1591, 2009. doi: 10.1128/MCB.01300-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Knowler WC, Barrett-Connor E, Fowler SE, Hamman RF, Lachin JM, Walker EA, Nathan DM; Diabetes Prevention Program Research Group . Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med : 393–403, 2002. doi: 10.1056/NEJMoa012512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Langin D. Adipose tissue lipolysis as a metabolic pathway to define pharmacological strategies against obesity and the metabolic syndrome. Pharmacol Res : 482–491, 2006. doi: 10.1016/j.phrs.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 25.Lass A, Zimmermann R, Oberer M, Zechner R. Lipolysis—a highly regulated multi-enzyme complex mediates the catabolism of cellular fat stores. Prog Lipid Res : 14–27, 2011. doi: 10.1016/j.plipres.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest : 175–184, 2007. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lumeng CN, Deyoung SM, Bodzin JL, Saltiel AR. Increased inflammatory properties of adipose tissue macrophages recruited during diet-induced obesity. Diabetes : 16–23, 2007. doi: 10.2337/db06-1076. [DOI] [PubMed] [Google Scholar]
- 28.Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep : 13, 2014. doi: 10.12703/P6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsuzawa Y, Funahashi T, Nakamura T. Molecular mechanism of metabolic syndrome X: contribution of adipocytokines adipocyte-derived bioactive substances. Ann N Y Acad Sci : 146–154, 1999. doi: 10.1111/j.1749-6632.1999.tb07793.x. [DOI] [PubMed] [Google Scholar]
- 30.Muona P, Jaakkola S, Zhang RZ, Pan TC, Pelliniemi L, Risteli L, Chu ML, Uitto J, Peltonen J. Hyperglycemic glucose concentrations up-regulate the expression of type VI collagen in vitro. Relevance to alterations of peripheral nerves in diabetes mellitus. Am J Pathol : 1586–1597, 1993. [PMC free article] [PubMed] [Google Scholar]
- 31.Park J, Scherer PE. Adipocyte-derived endotrophin promotes malignant tumor progression. J Clin Invest : 4243–4256, 2012. doi: 10.1172/JCI63930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park J, Scherer PE. Endotrophin - a novel factor linking obesity with aggressive tumor growth. Oncotarget : 1487–1488, 2012. doi: 10.18632/oncotarget.796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pasarica M, Gowronska-Kozak B, Burk D, Remedios I, Hymel D, Gimble J, Ravussin E, Bray GA, Smith SR. Adipose tissue collagen VI in obesity. J Clin Endocrinol Metab : 5155–5162, 2009. doi: 10.1210/jc.2009-0947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pelleymounter MA, Cullen MJ, Baker MB, Hecht R, Winters D, Boone T, Collins F. Effects of the obese gene product on body weight regulation in ob/ob mice. Science : 540–543, 1995. doi: 10.1126/science.7624776. [DOI] [PubMed] [Google Scholar]
- 35.Prieur X, Mok CY, Velagapudi VR, Núñez V, Fuentes L, Montaner D, Ishikawa K, Camacho A, Barbarroja N, O’Rahilly S, Sethi JK, Dopazo J, Orešič M, Ricote M, Vidal-Puig A. Differential lipid partitioning between adipocytes and tissue macrophages modulates macrophage lipotoxicity and M2/M1 polarization in obese mice. Diabetes : 797–809, 2011. doi: 10.2337/db10-0705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rosen ED, Spiegelman BM. What we talk about when we talk about fat. Cell : 20–44, 2014. doi: 10.1016/j.cell.2013.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rutkowski JM, Stern JH, Scherer PE. The cell biology of fat expansion. J Cell Biol : 501–512, 2015. doi: 10.1083/jcb.201409063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Song X, Jousilahti P, Stehouwer CD, Söderberg S, Onat A, Laatikainen T, Yudkin JS, Dankner R, Morris R, Tuomilehto J, Qiao Q; DECODE Study Group . Cardiovascular and all-cause mortality in relation to various anthropometric measures of obesity in Europeans. Nutrition, metabolism, and cardiovascular diseases. Nutr Metab Cardiovasc Dis : 295–304, 2015. doi: 10.1016/j.numecd.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 39.Song X, Tabák AG, Zethelius B, Yudkin JS, Söderberg S, Laatikainen T, Stehouwer CD, Dankner R, Jousilahti P, Onat A, Nilsson PM, Satman I, Vaccaro O, Tuomilehto J, Qiao Q; DECODE Study Group . Obesity attenuates gender differences in cardiovascular mortality. Cardiovasc Diabetol : 144, 2014. doi: 10.1186/s12933-014-0144-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun K, Halberg N, Khan M, Magalang UJ, Scherer PE. Selective inhibition of hypoxia-inducible factor 1α ameliorates adipose tissue dysfunction. Mol Cell Biol : 904–917, 2013. doi: 10.1128/MCB.00951-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun K, Kusminski CM, Scherer PE. Adipose tissue remodeling and obesity. J Clin Invest : 2094–2101, 2011. doi: 10.1172/JCI45887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun K, Park J, Gupta OT, Holland WL, Auerbach P, Zhang N, Goncalves Marangoni R, Nicoloro SM, Czech MP, Varga J, Ploug T, An Z, Scherer PE. Endotrophin triggers adipose tissue fibrosis and metabolic dysfunction. Nat Commun : 3485, 2014. doi: 10.1038/ncomms4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sun K, Tordjman J, Clément K, Scherer PE. Fibrosis and adipose tissue dysfunction. Cell Metab : 470–477, 2013. doi: 10.1016/j.cmet.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tourniaire F, Romier-Crouzet B, Lee JH, Marcotorchino J, Gouranton E, Salles J, Malezet C, Astier J, Darmon P, Blouin E, Walrand S, Ye J, Landrier JF. Chemokine Expression in Inflamed Adipose Tissue Is Mainly Mediated by NF-κB. PLoS One : e66515, 2013. doi: 10.1371/journal.pone.0066515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tyagi SC. Physiology and homeostasis of extracellular matrix: cardiovascular adaptation and remodeling. Pathophysiology : 177–182, 2000. doi: 10.1016/S0928-4680(00)00046-8. [DOI] [PubMed] [Google Scholar]
- 46.Wang H, Ye J. Regulation of energy balance by inflammation: common theme in physiology and pathology. Rev Endocr Metab Disord : 47–54, 2015. doi: 10.1007/s11154-014-9306-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest : 1796–1808, 2003. doi: 10.1172/JCI200319246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest : 1821–1830, 2003. doi: 10.1172/JCI200319451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Young SG, Zechner R. Biochemistry and pathophysiology of intravascular and intracellular lipolysis. Genes Dev : 459–484, 2013. doi: 10.1101/gad.209296.112. [DOI] [PMC free article] [PubMed] [Google Scholar]