

Abstract
Primary hypertension is increasingly common and is associated with significant morbidity. Here, we review the history of its discovery and rise during the last century with an emphasis on studies trying to identify its cause. Early studies identified a defect in sodium excretion by the kidney as being central to the pathogenesis. Recent studies have focused on a variety of genetic, congenital (fetal programming), and acquired mechanisms for causing the defect in natriuresis. Certain risk factors are apparent, including genetic polymorphisms that regulate sodium excretion, a congenital reduction in nephron number, obesity and hyperleptinemia, an elevated sympathetic nervous system, diet (salt and fructose), and metabolic (hyperuricemia) mechanisms. The kidney shows evidence for renal arteriolar vasoconstriction, an intrarenal inflammatory response, local oxidative stress, and intrarenal activation of the renin-angiotensin system. Recent studies suggest that intrarenal T cells have an important role in causing hypertension to be persistent, likely due to the induction of a local autoimmune response to neoantigens such as heat shock protein 70 and protein aggregates formed by isoketals resulting from lipid peroxidation. Salt retention due to impairment in pressure-diuresis leads to the release of cardiotonic steroids and central nervous system effects that cause systemic vasoconstriction and a rise in blood pressure. Some recent studies suggest that salt may increase blood pressure not simply by effects on extracellular volume but rather as a consequence of hyperosmolarity. These new insights could lead to new approaches for the prevention and treatment of this important disease.
Keywords: primary hypertension, sodium, salt, autoimmunity
the discovery of primary (“essential”) hypertension can be ascribed to Frederick Mahomed, who in the early 1870s, as a medical resident at Guy's Hospital in London, measured blood pressure (BP) in the general population. Working with a watchmaker, he made a spring-based device that could measure the tension of the radial pulse, a portable version of the sphygmograph invented by Étienne-Jules Marey a decade earlier in France. While it was known that patients with kidney disease and albuminuria could have high BP, he discovered a subset of the population that had high pressures in the absence of proteinuria (98).
Despite Mahomed's discovery, the measurement of blood pressure (BP) was not commonly performed until the 1890s, when Scipione Riva-Rocci (1863–1937) invented the BP cuff and mercury manometer (121) and Nikolai Sergeivich Korotkoff (1874–1920) used auscultation of the artery below the cuff to be sure that a complete occlusion of blood flow was achieved and permitted determination of diastolic BP (90, 132) (Fig. 1). Both Theodore Janeway and Harvey Cushing helped introduce the sphygmomanometer in the United States. Early studies by Janeway and others found systolic BP was rare >140 mmHg in adults <65 yr old (0.5–1%), whereas in those >65 the cutoff was closer to 160 mmHg (21). By 1906, insurance companies recognized that subjects with hypertension had increased mortality (21), which was confirmed by a report by the Metropolitan Life Insurance Company in 1912 (1). Soon, measurement of BP was a standard procedure by clinicians, and a cutoff of 140/90 mmHg was defined as hypertension (38). Hypertension was found not only to increase the risk for mortality, but also for stroke, congestive heart failure, and chronic kidney disease. Furthermore, a complication of congestive heart failure is that it can result in impaired perfusion to the kidney, causing kidney function to worsen, a syndrome known as cardiorenal syndrome (11).
Fig. 1.

Scipione Riva-Rocci (left) described a practical method of determining systolic blood pressure using a mercury manometer and an inflatable cuff (middle) to determine the pressure (mmHg) needed to occlude the brachial artery and thus suppress the radial pulse. The photograph of Scipione Riva-Rocci (unknown photographer, 1896) is in the public domain according to the Danish Consolidated Act Copyright of 2010 (Danish National Archive). Nicolai Korotkoff (right) described the appearance and disappearance of sounds “just below the cuff” using a stethoscope, thus allowing for the measurement of diastolic pressure. He stated that “the absence of pulsations is not indicative that the artery is completely occluded. In this respect, our hearing is a better guide” (90, 132). The photograph of Nicolai Korotkoff (unknown photographer, 1900) is in the public domain according to article 1256 of the Civil Code of the Russian Federation. The photograph of the manometer is in the public domain because its copyright has expired; the original source is Korotkoff NS, Experiments for Determining the Strength of Arterial Collaterals. St. Petersburg, Russia: Imperial Military Medical Academy, 1910. Dissertation.
The Epidemiology of Hypertension
In the past, there were differing opinions over whether hypertension, especially mild hypertension, should be treated, as some authorities viewed it as a compensatory phenomenon in response to microvascular disease (63, 105). It was not until the 1960s that the Framingham study and other definitive epidemiological investigations suggested that even mild high blood pressure could increase the risk for myocardial infarction, heart failure, stroke, and chronic renal damage when hypertension was recognized as a significant public health hazard (81).
In the early 1900s, hypertension was rare (<5% of the adult population) in most areas of the world, with the exception of western Europe and the United States, where the prevalence was between 5 and 10% (80). Over the last century, a remarkable increase in hypertension has occurred, and today the prevalence of hypertension is between 30 and 40% of adults in the United States and most areas of the world. By 2025, there will be more than 1.5 billion people with hypertension, making hypertension the most common noncommunicable disease (84). While some of the increase in hypertension, especially isolated systolic hypertension of the elderly, may be due to decreased vascular compliance due to stiffening of large arteries, hypertension is increasing in all ages, and is even becoming common among adolescents. The increasing frequency of hypertension is strongly linked with increasing obesity rates and insulin resistance. Other clinical characteristics are also predictive for the development of hypertension (Table 1).
Table 1.
Risk factors for hypertension
| Physical: Borderline and “white coat” hypertension, obesity, aging, African American, increased heart rate (>82 beats/min), increased psychosocial stress |
| Genetic: Family history, genetic polymorphisms (adducin, angiotensinogen, endothelial nitric oxide synthase, β2-adrenoceptor, others) |
| Congenital: Low birth weight and maternal malnutrition (low nephron number), maternal hypertension, preeclampsia |
| Diet/toxin: High salt intake, low potassium intake, high intake of added sugars containing fructose (high-fructose corn syrup and sucrose), low calcium intake, low dairy intake, heavy alcohol intake, low level lead or cadmium intoxication |
| Metabolic: High uric acid, insulin resistance, high hematocrit |
Adapted from Johnson RJ, Rodriguez-Iturbe B, and Bakris G. Primary hypertension. From: Comprehensive Clinical Nephrology (4th ed.), edited by Floege J, Johnson RJ, and Feehally J. St. Louis, MO: Elsevier, 2010, p. 411–420.
Renal Abnormalities in Primary Hypertension
BP is determined by the product of cardiac output and systemic vascular resistance (SVR). Most subjects with primary hypertension have a high SVR with relatively normal cardiac output. Hypertension is usually associated with disease of the small blood vessels (arterioles) with thickening and scarring (arteriolosclerosis), and is frequently accompanied by the presence of proteinaceous debris in the subendothelial space (hyalinosis) (104, 136). The kidney is especially involved in primary hypertension, with elevated renal vascular resistance, low renal blood flow, and for a relatively long time, preserved glomerular filtration rates (GFR). Renal arteriolar involvement with thickening and narrowing is present in the vast majority (98%) of subjects with primary hypertension, and the degree of narrowing of the arterioles correlates with the severity of the hypertension (136, 142). However, as many as one-third of subjects have relatively mild renal arteriolar disease (135). Nevertheless, classic functional studies done more than six decades ago have shown a reduced ratio of effective renal blood flow to functional tubular mass (53), indicating relative renal ischemia. The ischemia is due to the presence of renal arteriolar vasoconstriction and is present regardless of whether arteriolosclerosis is present (54, 146). Indeed, ischemic changes are common, including wrinkling of the basement membranes in the glomeruli and mild tubular injury and often associated with an interstitial inflammatory response, eventually resulting in the kidney appearing “contracted and granular” (136). In some subjects, glomerular and tubulointerstitial scarring develops, leading to progressive kidney failure.
Hypertension and Salt Sensitivity: A Disease of the Kidney
While a role for the kidney in driving hypertension was long suspected (75), the first supporting evidence was indirect and resulted from studies showing that salt restriction could lower BP in many hypertensive subjects (3, 4, 87). Chlorothiazide, the first modern diuretic, also lowered blood pressure, providing further evidence that salt retention played a role (147). Dahl (28) and MacGregor (96) further showed a relationship between salt intake and the prevalence of hypertension in various populations. Indeed, Dahl also developed rat models of hypertension in which BP increased markedly on exposure to high-salt diets (28).
In 1963, Borst and Borst-De Geus (10) proposed that hypertension resulted from a relative inability of the kidney to excrete salt; they hypothesized that “hypertension is part of a homeostatic reaction to deficient renal sodium output”. Guyton further developed this hypothesis, postulating that the long-term control of BP rested on the ability of the kidney to respond with an appropriate natriuresis at normal BP (57). An impairment in the pressure-natriuresis relationship required an increment in BP to maintain extracellular fluid volume within normal limits (58).
In human evolution, we have become adapted to a dramatic increment in sodium intake and a reduction in potassium intake. At the present time, salt intake ranges from 50 mg/day in the Yanomamo Indians living on their native diets (113) to 16 g in some areas of rural China (152), with an average global consumption of 9.8 g of salt daily (106). The relative inability of the kidney to meet this challenge, is more evident in the aging population; At the age of 80, as many as 30% of the total population of glomeruli may be sclerotic (82) and the kidney has a reduced ability to excrete a sodium load (36). The direct relationship between salt intake and BP has been used to identify subjects that show an exaggerated change in BP in response to salt loading or salt restriction (salt-sensitive group) vs. those who are relatively resistant to changes in salt intake (salt-resistant group). As the number of nephrons is reduced with aging, it is not surprising that these changes are associated with an impaired pressure-natriuresis response (100). While there currently are controversies related to salt intake and its mechanism for causing hypertension (which will be discussed later in Hot Topics), there is no controversy related to the fact that the frequency of salt-sensitive individuals increases with age (145) and that this condition is associated with an increased number of cardiovascular events over time (103).
Salt sensitivity may be demonstrated in hypertensive as well as in normotensive subjects and has been shown to be more frequent in African Americans, older individuals, those with the loss of the typical BP dipping at night, and those with microalbuminuria (144). Salt sensitivity and hypertension also increase in women after menopause, suggesting an important role for estrogen in pressure-natriuresis (67). Interestingly, once salt sensitivity develops the individual remains salt sensitive, as demonstrated in population studies that examined the BP response to low and high-salt intake over time (56).
Direct evidence that the kidney was critical to the development of hypertension was then demonstrated by the transfer of hypertension by kidney transplantation of hypertensive rats to normotensive rats (29). Other groups have also shown a key role for the kidney in models of hypertension (8, 25, 120). Similarly, Curtis et al. (27) showed that African Americans suffering from kidney failure from primary hypertension could be cured of their hypertension by receiving kidney transplants from normotensive donors. However, transplantation today is often associated with the development of hypertension in the transplant recipient, likely because of the use of calcineurin inhibitors that were not used in the original study by Curtis et al. (27). Nevertheless, these studies provide evidence that abnormalities in the kidney were sufficient to cause hypertension and that a normally functioning kidney prevents the increment in BP even when there is systemic microvascular disease present.
What are Potential Mechanisms for Renal Abnormalities in Primary Hypertension?
Genetic mechanisms.
Given that the primary defect in the kidney might involve defective excretion of sodium, genetic polymorphisms that modulate salt excretion would be excellent candidates for playing a role in hypertension (reviewed in Ref. 43). This hypothesis was supported by the discovery by Rick Lifton's group (94) that several types of hereditary hypertension are due to specific genetic defects in renal sodium handling, especially involving the epithelial sodium channel (ENaC) of the collecting duct, such as Liddle syndrome, the syndrome of apparent mineralocorticoid excess, and glucocorticoid-remediable aldosteronism. Other monogenic disorders, such as Bartter's syndrome and Gitelman's syndrome, may be associated with low BP and result in sodium wasting due to mutations in the Na-K-2Cl transporter (SLC12A1) and the Na-Cl cotransporter (SLC12A3), respectively. The strength of these observations led Lifton to postulate that hypertension is likely driven by multiple genetic polymorphisms that in aggregate result in defective sodium excretion (94). Consistent with this hypothesis, genetic polymorphisms in renal sodium handling were found to influence the frequency of hypertension in the Framingham Heart Study (74). However, some studies suggest that genetic polymorphisms in these rare disorders may account for only a small percentage of primary hypertension (22, 83).
Candidate gene approaches (2) and genome-wide association studies (GWAS) (72, 93) have also identified genetic polymorphisms associated with hypertension (reviewed in Ref. 43). Some of the candidate genes are involved in vasoconstriction or vasodilation, such as angiotensinogen (73) and endothelial nitric oxide synthase (111). However, while genetic polymorphisms involved in renal salt handling or in systemic vascular reactivity are risk factors for hypertension, nongenetic mechanisms may be more important. Indeed, a study of 635 identical twins reported that 60% of twins that were hypertensive had a twin that was normotensive (16). Thus, despite identical genetics, hypertension can be discordant. In summary, genetics play an important role in hypertension, but other mechanisms (epigenetic and acquired) may have a more critical role in the majority of patients presently grouped under the term “primary” hypertension.
Congenital reduction in the number of nephrons.
Epidemiological studies have documented an increased frequency of hypertension in adults born with low birth weight (5). The kidneys develop during the third trimester, and hence low-birth weight babies commonly have incomplete development of the kidney with a reduced number of nephrons. This led Brenner et al. (12) to suggest that a low nephron number may represent the renal abnormality that predisposes an individual to develop hypertension. Indeed, experimental studies in laboratory rats showed that maternal malnutrition can lead to small pups with reduced nephrons, and these rats show an increased propensity to develop salt-sensitive hypertension later in life (149). Furthermore, a study of subjects with hypertension who died in traffic accidents found that these subjects had a reduced number of nephrons compared with age-matched controls (86).
However, most studies suggest that the effect of birth weight on BP is modest (31) and can only explain 20% of those with hypertension (37). Other studies could not show a relationship between nephron number and BP in African Americans (71). Thus, while it is likely that stress during fetal development may lead to epigenetic changes or alterations in nephron development that increase the risk for hypertension later in life, these changes are not required for the development of primary hypertension. Thus fetal programming is more likely a risk factor than the cause of primary hypertension.
Acquired renal injury.
The observation that the kidney appears ischemic led Goldblatt (52) to propose that disease of the renal microvasculature might be responsible and that a decrease in blood flow to the kidney might result in the release of substances that raise BP. Kidney extracts were known to contain a substance (renin) that could raise BP when injected into animals (140), and hypertension due to a release of renin could be induced by mechanically constricting the renal artery of dogs (52). However, plasma renin activity tends to be normal or suppressed in most patients with hypertension, consistent with normal or increased body sodium content (91). Furthermore, while renal microvascular disease was present in most subjects, in up to one-third the severity was not enough to account for the ischemic changes (17), and since microvascular disease tends to be more severe with longer duration of hypertension (116), the vascular disease was more likely an aggravating or secondary factor rather than the primary cause.
ACQUIRED HEMODYNAMIC CHANGES IN THE KIDNEY: INTRARENAL ACTIVATION OF VASOCONSTRICTOR SYSTEMS WITH SODIUM REABSORPTION.
Despite the fact that earlier studies suggested that the histological changes in the kidney were secondary to hypertension, it has long been recognized that the sine qua non of hypertension is renal vasoconstriction, primarily of the preglomerular vasculature (55). Numerous studies over the last few decades have shown that this renal vasoconstriction is mediated by an imbalance in intrarenal vasoconstrictors and vasodilators, favoring intrarenal vasoconstriction, and that it has an important role in causing hypertension (reviewed in Refs. 126 and 146). Key participants include increased intrarenal oxidative stress, a decrease in local endothelial nitric oxide, and activation of the renal sympathetic nervous system (SNS) (33, 146). The presence of the vasoconstrictor systems is also tightly linked with sodium reabsorption via both hemodynamic and nonhemodynamic effects.
Salt-sensitive hypertension is generally associated with suppression of the systemic renin-angiotensin system (RAS) due to the tendency for sodium and volume retention, but plasma renin activity is often not fully suppressed, suggesting some continued activation of the systemic RAS (91). However, recent studies have emphasized activation of the intrarenal RAS as a primary mechanism driving renal vasoconstriction and salt-sensitive hypertension (109). Intrarenal angiotensin II levels are elevated in salt-sensitive hypertension, even in the setting of plasma volume expansion (45, 103, 110, 112). The heightened renal angiotensin activity contrasts with suppressed plasma angiotensin II levels (Fig. 2) (45). Renal angiotensin II is produced in part by angiotensinogen and ACE activity in renal tubular cells (108, 109), which then acts on AT1 receptors in the proximal tubule, collecting duct, and intercalated cells to stimulate sodium reabsorption and raise BP (reviewed in Ref. 24). Intrarenal angiotensin II levels may also be regulated by ACE2. Specifically, a decrease in ACE2 activity will lead to an increase in angiotensin II and decrease in Ang 1–7 levels, resulting in elevated renal vascular resistance and BP.
Fig. 2.

Plasma and renal angiotensin II concentrations in salt-sensitive hypertension induced with transient l-NAME administration. Studies were done after 5 wk of a high-salt diet (4% sodium diet) in rats previously treated with l-NAME orally for 3 wk. Renal angiotensin II levels were determined in renal interstitial fluid. SSHTN, salt-sensitive hypertension; MMF, rats treated similarly but receiving mycophenolate mofetil as immunosuppressive anti-inflammatory treatment. Control rats received a high (C-HSD)- or a normal (0.4%)-sodium diet (C-NSD). X-axis is shown as log 2. Values are means ± SD; n = 8–10 rats (all groups). Figure is drawn from data previously reported (44).
The intrarenal mineralocorticoid axis may also be involved in salt-sensitive hypertension. Despite suppressed plasma aldosterone levels, there is activation of the mineralocorticoid receptor, which also contributes to inappropriate sodium retention. For instance, activation of the mineralocorticoid receptor may be secondary to Rac1 (133). Rac1 is a member of the Rho-guanine triphosphate hydroxylase family and has been shown to activate the mineralocorticoid receptor via an aldosterone-independent mechanism in the Dahl salt-sensitive, but not in the salt-resistant rat. Downstream of the mineralocorticoid receptor, serum- and-glucocorticoid-inducible kinase 1 (Sgk1) is upregulated by salt loading with stimulation of the thiazide-sensitive sodium chloride cotransporter (NCC) and epithelial sodium channel (ENaC)-mediated reabsorption of sodium in the connecting and collecting duct areas of the nephron. Nevertheless, adrenalectomy suppresses Rac1 activation in salt-sensitive rats, and therefore a certain level of aldosterone is necessary for salt-induced Rac1 activation and development of salt-driven hypertension (48).
The SNS also regulates BP via both renal and extrarenal mechanisms (34). A high-salt diet results in higher plasma norepinephrine levels in patients with salt-sensitive hypertension (15, 51). In addition to causing systemic and preglomerular arteriolar vasoconstriction, activation of the SNS increases sodium reabsorption due to a reduction in the activity of the With-no-lysine kinase 4 (WNK4) (107), which is a negative (down) regulator of the NCC (150). The reduction in WNK4 activity and the resulting upregulation of the NCC is prevented by renal denervation and stimulated by a norepinephrine infusion in salt-sensitive rats. The effects of norepinephrine are reversed by propanolol and are absent in the β-adrenergic knockout mice. Therefore, the β2-adrenergic receptor plays a key function in salt-sensitive hypertension by augmenting sodium reabsorption in the distal convoluted tubule of the nephron.
Finally a role for intrarenal nitric oxide has been demonstrated in a variety of studies (6, 146). A loss of nitric oxide can lead to endothelial dysfunction, intrarenal vasoconstriction, and reduced sodium excretion (146). Reducing nitric oxide in the renal medulla of rats can result in sodium retention and an elevation in BP (101). One of the newest observations is that regulation of endothelial nitric oxide is dependent on collectrin, an ACE2 homolog (20).
While these studies show that the imbalance in intrarenal vasoconstrictors and vasodilators likely has a role in sodium handling and BP responses, they do not explain why the imbalance is occurring.
ACQUIRED INFLAMMATORY CHANGES IN THE KIDNEY: INFLAMMATION AS A MECHANISM FOR DRIVING RENAL VASOCONSTRICTION AND SODIUM RETENTION.
Our group more than a decade ago (77, 79) became interested in the concept that subtle acquired injury to the kidney might be responsible for the development of hypertension. We had noted that the transient infusion of angiotensin II could induce renal microvascular lesions similar to what is observed in primary hypertension (76), raising the question of whether the renal changes might be sufficient to lead to persistent hypertension even after cessation of the angiotensin II infusion. To test this hypothesis, we administered angiotensin II to rats for 14 days, but then stopped the infusion, allowing the BP to return to the normal range. At this time, the animals had thickened renal arterioles, some evidence of tubular ischemia, mild interstitial inflammation, and reduction of the peritubular capillary network, consistent with the histological findings of primary hypertension. Indeed, when the animals were then given a high-salt diet, a marked rise in BP occurred, in association with renal vasoconstriction and the hemodynamic features of primary hypertension (46, 95). Furthermore, we were able to show that this model resulted in persistent intrarenal oxidative stress and increased intrarenal angiotensin II activity, and that translated into an impairment in pressure-natriuresis (44, 45, 123). Subsequently, we and others could induce salt-sensitive hypertension by transiently administering other vasoconstrictive agents that could induce renal microvascular and interstitial inflammatory changes (77).
The role of low-grade renal inflammation in the renal parenchyma was investigated using mycophenolate mofetil (MMF), an immunosuppressive agent. MMF was administered to the rats after the angiotensin II infusion (123). Surprisingly, this treatment blocked the subsequent renal vasoconstriction and salt-sensitive hypertension (123). Further studies showed that the infiltrating inflammatory cells consisted of both T cells and macrophages and were expressing oxidants and angiotensin II (123). The sodium retention that results from local angiotensin activity appeared to be dependent on the presence of the inflammatory cells since suppressing renal inflammation with MMF blocks the local increase in angiotensin II and subsequent salt-sensitive hypertension (45, 122). Additional experiments with a variety of immune-suppressive agents and anti-inflammatory strategies showed that blocking the interstitial inflammatory response prevented, corrected, or ameliorated hypertension in a wide variety of genetic models and acquired models of hypertension (124, 125).
More recently, evidence that the T cells were responsible for the induction of hypertension has been obtained (50, 59). While early experimental studies suggested that a normally functioning thymus was necessary for the chronic elevation of BP (138), the role of T cells in the pathogenesis of hypertension was conclusively demonstrated in the studies of Harrison and colleagues (59), who showed that adoptive transfer of T cells restored the hypertensive response to angiotensin II that was absent in mice strains devoid of lymphocytes. Subsequent studies by these and other investigators established the contribution of inflammation in the vessel walls in the development of hypertension and the protection offered by T-regulatory cells (59, 62, 129). The T cells not only produce angiotensin II, but also the inflammatory cells may respond to local angiotensin II via the angiotensin AT1 receptor to limit their inflammatory response (26). Evidence accumulated in recent years has now firmly established the role played by inflammation in the kidney, central nervous system (CNS), and arteries in hypertension and the involvement of immune reactivity in the development of these inflammatory phenomena.
In retrospect, it is evident that structural renal microvascular disease (arteriolosclerosis) is not necessary for the hypertensive response. However, it may still have an important role in altering the pressure-natriuresis response. For example, in the setting where the renal vasoconstriction is uniform, the ischemia would be expected to be evenly distributed throughout the kidney, and the rise in BP would increase renal perfusion pressure to relieve the ischemia. In these circumstances, while BP is increased, the slope of the pressure-natriuresis relationship is normal, a characteristic of salt-resistant hypertension. In contrast, if there are heterogeneous vascular lesions present, then a rise in BP and renal perfusion pressure might result in overperfusion of some regions of the kidney and underperfusion of others. The presence of persistent ischemia in some regions of the kidney might favor greater BP variability in response to high and low salt intakes than that observed in normal subjects (77, 131). Furthermore, as arterioles become fibrotic, the ability to autoregulate may also be impaired, leading to greater transmission of pressure to the glomerulus, favoring progression of kidney disease (127).
Current Hot Topics
Could primary hypertension be an autoimmune disease?
Our studies showed that the transient administration of agents that caused renal vasoconstriction (such as angiotensin II or blockade of the nitric oxide system) resulted in an infiltration of T cells and macrophages that cause persistent vasoconstriction, an impairment in pressure-natriuresis, and the development of salt-sensitive hypertension. Initially, our thought was that ischemia was stimulating the nonspecific release of chemokines that was eliciting the immune response and renal vasoconstriction. However, what was the stimulus for maintaining the renal vasoconstriction?
One possibility was that the T cells were reacting to neoantigens expressed in the ischemic tissue, resulting in an autoimmune response (117). Experimental studies have suggested that the accumulation of γ-ketoaldehydes (isoketals) induced by lipid peroxidation resulting from oxidative stress cause aggregation of proteins in the dendritic cells that are then capable of activating T cells and stimulating the production of cytokines (IL-6, IL-1β, and IL-23) and an increase in co-stimulatory proteins (CD80 and CD86) that predispose to autoimmunity (88). One of the autoantigens that has been identified in animal models of salt-sensitive hypertension is heat shock protein-70 (117). Tolerizing rats with a HSP-70 peptide can protect rats from salt-sensitive hypertension that occurs following inhibition of nitric oxide synthesis (117).
Studies of the role of the immune system in human hypertension are limited, but there is some evidence that patients with primary hypertension have both T cell reactivity and antibodies to HSP-70 (117) as well as isoketal-modified proteins in circulating monocytes and dendritic cells (88). There is also one study that suggests that MMF can reduce BP in hypertensive subjects suffering from psoriasis and rheumatoid arthritis (68). Furthermore, subjects with autoimmune deficiency syndrome who have a low CD4 count have a lower prevalence of hypertension compared with those on retroviral therapy with improved CD4 counts (130).
Continued controversies related to salt.
The average salt intake in most countries in the world is in the range of 9–12 g/day (14). A reduction of 2–5 g of salt/day has been estimated to reduce cardiovascular events and stroke by ∼20% in meta-analyses (64–66). The benefit of a reduction in salt intake has led to recommendations by the World Health Organization to restrict sodium intake to 1,500–2,000 mg daily, equivalent to 4–6 g of salt each day. Recent studies estimate that 1 of every 10 deaths from cardiovascular causes may be attributed to salt ingestion >2 g daily, and 5 of these deaths occurred in individuals younger than 70 yr of age (106). In contrast, a large population study concluded that a higher daily intake of salt (between 3 and 6 g of sodium and >1.5 g of potassium) was associated with a lower risk of death and cardiovascular events (112).
While controversy continues to exist related to the degree of salt restriction, there is also evidence that it may not be the amount of salt that is ingested, but rather the balance of salt and water intake. The original concept was that salt might act to increase BP by increasing blood volume, but more recent studies suggest that salt may also raise BP through its effects on osmolarity (32, 78). For example, a study reported that the administration of 6 g of salt in soup to normotensive volunteers resulted in an acute increase in serum sodium levels of 3 mmol/l associated with a 5.7 mmHg rise in systolic BP (137). It has been proposed that the rise in serum and cerebrospinal sodium activates the SNS in the CNS, resulting in the release of cardiotonic steroids (ouabain and marinobufagenin) from the adrenal gland. In turn, these substances block Na+-K+-ATPases in vascular smooth muscle cells, leading to intracellular sodium that activates the Na/Ca exchanger, resulting in intracellular calcium accumulation and contraction of the vascular smooth muscle (9). The rise in serum sodium also activates macrophages, resulting in a stimulation of vascular endothelial growth factor that increases tissue permeability that results in the sequestration of sodium in the interstitial space (141).
Thus, if correct, these studies suggest that the hyperosmolar mechanism by which salt intake raises BP might be mitigated provided sufficient water was ingested to prevent the development of hypertonicity. Studies investigating this potential mechanism are encouraged as it would help separate the effects of volume expansion from hyperosmolarity in driving the hypertensive response. In addition to the direct effects of salt on BP, recent evidence also links salt with the development of autoimmunity since a high-salt diet results in the induction of pathogenic IL-17-producing CD4 (helper) T cells (89).
Role of obesity and metabolic syndrome in hypertension.
The marked rise in obesity over the last century parallels the rise in hypertension, and indeed ∼65–75% of primary hypertension today may be accounted for by obesity (148). Obesity is associated with impaired natriuresis and salt retention (60), but the specific reason this occurs remains unclear. Several factors might be involved (Table 2) (30).
Table 2.
Potential pathogenic factors associated with hypertension in obesity
| Endothelial dysfunction (impaired endothelial nitric oxide bioavailability) |
| Hyperinsulinemia |
| Hyperleptinemia |
| Hyperuricemia |
| Hypoadiponectinemia |
| Impaired baroreflex sensitivity |
| Increased CNS SNS activation |
| Intrarenal SNS activation |
| Physical compression of the kidney by fat |
| Renin-angiotensin system activation (intrarenal) |
| Sleep apnea (obstructive) |
| Sugar intake (fructose) |
CNS, central nervous system; SNS, sympathetic nervous system.
First, there is evidence for increased SNS activity both in the brain and in the kidney. Activation of the SNS might be due to a variety of factors, including coexistent obstructive sleep apnea, impaired baroreflex sensitivity, hyperinsulinemia, or alterations in adiponectin or leptin. Recent studies suggest leptin signaling may play an important role (30). Leptin is an adipokine (a hormone released from adipose tissue) that has a primary function to stimulate satiety following ingestion of a meal. Leptin is released after food ingestion and then binds to leptin receptors in the hypothalamus that lead to a reduction in food intake and increase energy expenditure, but in many individuals with obesity a state of leptin resistance occurs. As a consequence, serum leptin levels rise. However, while the satiety centers are resistant to leptin, other parts of the brain remain sensitive to the increasing leptin levels (119). Specifically, leptin binds to leptin receptors in pro-opiomelanocortin-expressing cells in the arcuate nucleus of the hypothalamus, stimulating the release of α-melanocyte-stimulating hormone (α-MSH) that binds melanocortin-4 receptors in the paraventricular nucleus and lateral hypothalamic area to stimulate SNS activity, that stimulates renal SNS activity, resulting in sodium reabsorption and an impairment in pressure-natriuresis. Indeed, hypertension in obese animals can be prevented if various components of this pathway are blocked (35, 61, 139). Leptin may also activate the intracerebral RAS to activate CNS and SNS activity (69).
While activation of the CNS and renal SNS activity appears to be important in obesity-associated hypertension in animal models, the initial enthusiasm for performing denervation of the renal SNS has waned following the results of the Simplicity HTN-3 trial (7). In this trial, subjects with resistant hypertension were randomized to sham operation or catheter-based renal artery denervation. Despite exciting results from earlier trials, this well-done study could not show a benefit on BP reduction at the 6-mo end point (7). It remains possible that the renal denervation procedure was not fully successful, as no specific measures were performed to address whether the renal SNS activity was blocked (7). Nevertheless, it raises the question of whether other mechanisms may contribute to the hypertensive response in subjects with difficult-to-control hypertension (7).
One emerging candidate is hyperuricemia, which is common in subjects with obesity, particularly those with the metabolic syndrome, and has been found to consistently predict the development of hypertension (40). Relatively higher uric acid levels are also seen in low-birth-weight children and have been correlated with endothelial dysfunction and higher BP (47, 114). Raising uric acid in rats causes renal vasoconstriction and hypertension in association with the development of microvascular disease and interstitial inflammation (102, 128). Initially, the hypertension responds to the lowering of uric acid, but once the microvascular disease and interstitial inflammation develop the animals will develop salt-sensitive hypertension even if the serum uric acid returns to the normal range (143). These studies suggest that an elevation in uric acid might be more important in the initiation of hypertension, but that once the microvascular and interstitial inflammation take hold, the kidney will drive the hypertensive response. Indeed, elevated uric acid is present in almost 90% of adolescents with primary hypertension (39), and pilot clinical studies also suggest lowering uric acid might be of benefit in improving BP in prehypertensive and hypertensive adolescents (42, 134). While uric acid is a good candidate for driving hypertension, some studies suggest that the hypertension may be mediated more by xanthine oxidase, which produces both oxidants and uric acid (49). Furthermore, while some genetic studies have linked genetic polymorphisms that increase uric acid levels with hypertension (99, 115), others have not (18, 151). Hence, more clinical evidence is needed.
There have also been a number of studies linking intake of added sugars, particularly soft drinks, with the development of hypertension (70, 85, 110). One potential mechanism may relate to the fructose component in high-fructose corn syrup (HFCS) and sucrose, as fructose is known to raise uric acid in humans (23, 92) and also to increase BP (13). While studies linking fructose intake with hypertension have been mixed (97), there is evidence that reducing sugar intake can result in lowering of BP (19). Further studies are necessary to better evaluate this relationship.
Summary
We have made great progress since Mahomed discovered primary hypertension over 140 years ago. A figure summarizing the current proposed mechanism for hypertension is shown (Fig. 3). We propose that hypertension is initiated by a variety of mechanisms that result in renal vasoconstriction. The vasoconstriction induces renal ischemia that brings in inflammatory (T cells and macrophages) cells that drive continuing intrarenal vasoconstriction and oxidative stress that lead to impaired pressure-natriuresis. The ischemia results in the expression of neoantigens such as HSP-70 that result in an autoimmune reaction that maintains the local inflammatory reaction, leading to persistent renal vasoconstriction and salt sensitivity.
Fig. 3.
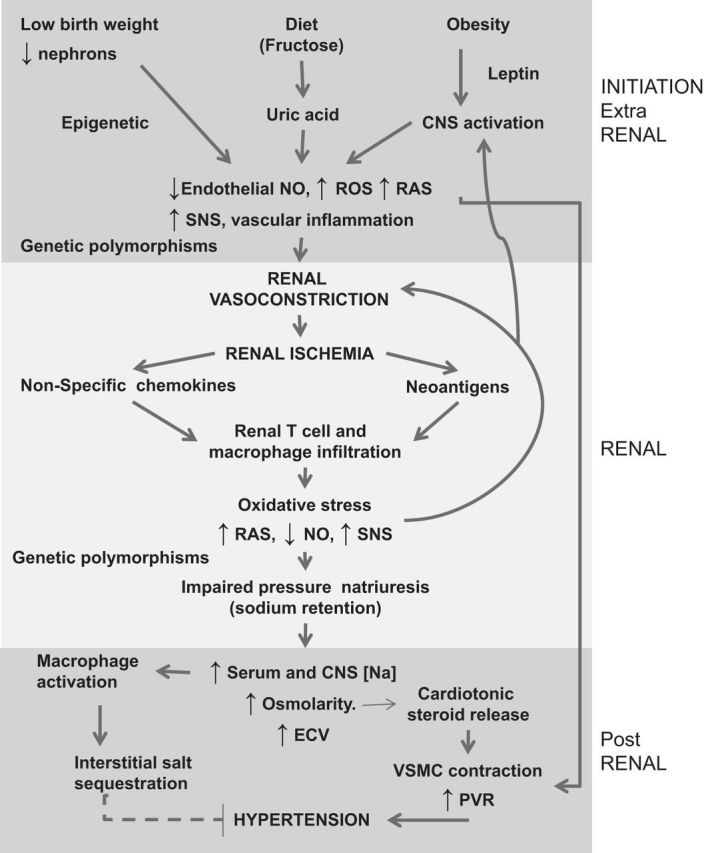
Proposed mechanism for the development of hypertension. Certain risk factors, such as low birth weight, diets high in fructose or salt, and obesity may increase the risk for hypertension. Low birth weight and dietary intake of added sugars, for example, can both result in mild hyperuricemia that may result in generation of reactive oxygen species (ROS), activation of the renin-angiotensin system (RAS), and endothelial dysfunction with low nitric oxide (NO) levels (41). Elevated leptin levels present in subjects with obesity or metabolic syndrome can also stimulate sympathetic outflow from the central nervous system (CNS). The generation of these vasoconstrictors results in renal vasoconstriction that causes mild ischemia, leading to nonspecific chemokine release as well as the induction of neoantigens [heat shock protein (HSP) 70, isoketal-induced protein aggregates] that activates dendritic cells and an autoimmune response. The local inflammation results in intrarenal generation of vasoconstrictors that perpetuate the ischemia and block pressure-natriuresis. Sodium is retained, resulting in an increase in serum osmolarity that leads to CNS sympathetic nervous system (SNS) activation and the release of cardiotonic steroids such as ouabain and marinobufagenin that cause vascular smooth muscle cell (VSMC) constriction and a rise in systemic vascular resistance (SVR). As blood pressure increases, the increase in renal perfusion pressure relieves tubular ischemia, allowing sodium handling to reset but with a parallel shift in the pressure-natriuresis curve (salt-resistant state). However, with the development of microvascular disease the relief of the ischemia is not uniform, leading to persistent ischemic areas that result in a rightward shift and change in slope of the pressure-natriuresis curve, resulting in salt-sensitive hypertension. ECV, extracellular volume.
The factors initiating the hypertensive response likely involve a variety of genetic, epigenetic, and acquired mechanisms. The potential role of fructose and uric acid also deserves further study. Once the cause is delineated, we should expect major advances in both prevention and treatment of this very important disease.
DISCLOSURES
R. J. Johnson and M. A. Lanaspa are inventors with several patent applications related to blocking fructose metabolism in metabolic and renal diseases (University of Colorado). R. J. Johnson, M. A. Lanaspa, and L. G. Sánchez-Lozada are also founders of Colorado Research Partners. R. J. Johnson is also on the Scientific Board of Amway and XORT Therapeutics.
AUTHOR CONTRIBUTIONS
Author contributions: R.J.J and B.R.-I. prepared figures; R.J.J and B.R.-I. drafted manuscript; R.J.J, M.A.L., L.G.S.-L., and B.R.-I. edited and revised manuscript; R.J.J, M.A.L., L.G.S.-L., and B.R.-I. approved final version of manuscript.
REFERENCES
- 1.Anonymous. The medico-actuarial mortality investigation. The Association of Life Insurance Medical Directors Actuarial Society of America. 1912. Obes Res : 1995. [PubMed] [Google Scholar]
- 2.Agarwal A, Williams GH, Fisher ND. Genetics of human hypertension. Trends Endocrinol Metab : 127–133, 2005. [DOI] [PubMed] [Google Scholar]
- 3.Allen FM, Sherrill JW. The treatment of arterial hypertension. J Metab Res : 429–545, 1922. [Google Scholar]
- 4.Ambard L, Beaujard L. Causes de l'hypertension arterielle. Arch Gen Med : 786–792, 1904. [Google Scholar]
- 5.Barker DJ, Osmond C, Golding J, Kuh D, Wadsworth ME. Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. BMJ : 564–567, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baylis C. Nitric oxide deficiency in chronic kidney disease. Am J Physiol Renal Physiol : F1–F9, 2008. [DOI] [PubMed] [Google Scholar]
- 7.Bhatt DL, Kandzari DE, O'Neill WW, D'Agostino R, Flack JM, Katzen BT, Leon MB, Liu M, Mauri L, Negoita M, Cohen SA, Oparil S, Rocha-Singh K, Townsend RR, Bakris GL; SIMPLICITY HTN-3 Investigators. A controlled trial of renal denervation for resistant hypertension. N Engl J Med : 1393–1401, 2014. [DOI] [PubMed] [Google Scholar]
- 8.Bianchi G, Fox U, Di Francesco GF, Giovanetti AM, Pagetti D. Blood pressure changes produced by kidney cross-transplantation between spontaneously hypertensive rats and normotensive rats. Clin Sci Mol Med : 435–448, 1974. [DOI] [PubMed] [Google Scholar]
- 9.Blaustein MP, Leenen FH, Chen L, Golovina VA, Hamlyn JM, Pallone TL, Van Huysse JW, Zhang J, Wier WG. How NaCl raises blood pressure: a new paradigm for the pathogenesis of salt-dependent hypertension. Am J Physiol Heart Circ Physiol : H1031–H1049, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Borst JG, Borst-De Geus A. Hypertension explained by Starling's theory of circulatory homoeostasis. Lancet : 677–682, 1963. [DOI] [PubMed] [Google Scholar]
- 11.Braam B, Joles JA, Danishwar AH, Gaillard CA. Cardiorenal syndrome—current understanding and future perspectives. Nat Rev Nephrol : 48–55, 2014. [DOI] [PubMed] [Google Scholar]
- 12.Brenner BM, Garcia DL, Anderson S. Glomeruli and blood pressure. Less of one, more the other? Am J Hypertens : 335–347, 1988. [DOI] [PubMed] [Google Scholar]
- 13.Brown CM, Dulloo AG, Yepuri G, Montani JP. Fructose ingestion acutely elevates blood pressure in healthy young humans. Am J Physiol Regul Integr Comp Physiol : R730–R737, 2008. [DOI] [PubMed] [Google Scholar]
- 14.Brown IJ, Tzoulaki I, Candeias V, Elliott P. Salt intakes around the world: implications for public health. Int J Epidemiol : 791–813, 2009. [DOI] [PubMed] [Google Scholar]
- 15.Campese VM, Romoff MS, Levitan D, Saglikes Y, Friedler RM, Massry SG. Abnormal relationship between sodium intake and sympathetic nervous system activity in salt-sensitive patients with essential hypertension. Kidney Int : 371–378, 1982. [DOI] [PubMed] [Google Scholar]
- 16.Carmelli D, Cardon LR, Fabsitz R. Clustering of hypertension, diabetes, and obesity in adult male twins: same genes or same environments? Am J Hum Genet : 566–573, 1994. [PMC free article] [PubMed] [Google Scholar]
- 17.Castleman B, Smithwick RH. The relation of vascular disease to the hypertensive state; the adequacy of the renal biopsy as determined from a study of 500 patients. N Engl J Med : 729–732, 1948. [DOI] [PubMed] [Google Scholar]
- 18.Caulfield MJ, Munroe PB, O'Neill D, Witkowska K, Charchar FJ, Doblado M, Evans S, Eyheramendy S, Onipinla A, Howard P, Shaw-Hawkins S, Dobson RJ, Wallace C, Newhouse SJ, Brown M, Connell JM, Dominiczak A, Farrall M, Lathrop GM, Samani NJ, Kumari M, Marmot M, Brunner E, Chambers J, Elliott P, Kooner J, Laan M, Org E, Veldre G, Viigimaa M, Cappuccio FP, Ji C, Iacone R, Strazzullo P, Moley KH, Cheeseman C. SLC2A9 is a high-capacity urate transporter in humans. PLoS Med : e197, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen L, Caballero B, Mitchell DC, Loria C, Lin PH, Champagne CM, Elmer PJ, Ard JD, Batch BC, Anderson CA, Appel LJ. Reducing consumption of sugar-sweetened beverages is associated with reduced blood pressure: a prospective study among United States adults. Circulation : 2398–2406, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chu PL, Le TH. Role of collectrin, an ACE2 homologue, in blood pressure homeostasis. Curr Hypertens Rep : 490, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cook HW. Blood Pressure in Prognosis. New York: Wood, 1911. [Google Scholar]
- 22.Cowley AW., Jr The genetic dissection of essential hypertension. Nat Rev Genet : 829–840, 2006. [DOI] [PubMed] [Google Scholar]
- 23.Cox CL, Stanhope KL, Schwarz JM, Graham JL, Hatcher B, Griffen SC, Bremer AA, Berglund L, McGahan JP, Keim NL, Havel PJ. Consumption of fructose- but not glucose-sweetened beverages for 10 weeks increases circulating concentrations of uric acid, retinol binding protein-4, and gamma-glutamyl transferase activity in overweight/obese humans. Nutr Metab (Lond) : 68, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crowley SD, Coffman TM. The inextricable role of the kidney in hypertension. J Clin Invest : 2341–2347, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crowley SD, Gurley SB, Herrera MJ, Ruiz P, Griffiths R, Kumar AP, Kim HS, Smithies O, Le TH, Coffman TM. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci USA : 17985–17990, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crowley SD, Song YS, Sprung G, Griffiths R, Sparks M, Yan M, Burchette JL, Howell DN, Lin EE, Okeiyi B, Stegbauer J, Yang Y, Tharaux PL, Ruiz P. A role for angiotensin II type 1 receptors on bone marrow-derived cells in the pathogenesis of angiotensin II-dependent hypertension. Hypertension : 99–108, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Curtis JJ, Luke RG, Dustan HP, Kashgarian M, Whelchel JD, Jones P, Diethelm AG. Remission of essential hypertension after renal transplantation. N Engl J Med : 1009–1015, 1983. [DOI] [PubMed] [Google Scholar]
- 28.Dahl LK. Possible role of salt intake in the development of essential hypertension. In: Essential Hypertension—An International Symposium, edited by Cottier P and Bock K. Berlin: Springer, 1960, p. 53–65. [Google Scholar]
- 29.Dahl LK, Heine M. Primary role of renal homografts in setting chronic blood pressure levels in rats. Circ Res : 692–696, 1975. [DOI] [PubMed] [Google Scholar]
- 30.da Silva AA, do Carmo JM, Hall JE. Role of leptin and central nervous system melanocortins in obesity hypertension. Curr Opin Nephrol Hypertens : 135–140, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Jong F, Monuteaux MC, van Elburg RM, Gillman MW, Belfort MB. Systematic review and meta-analysis of preterm birth and later systolic blood pressure. Hypertension : 226–234, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Wardener HE, He FJ, MacGregor GA. Plasma sodium and hypertension. Kidney Int : 2454–2466, 2004. [DOI] [PubMed] [Google Scholar]
- 33.DiBona GF. Renal neural mechanisms in salt-sensitive hypertension. Blood Press Suppl : 81–87, 1995. [PubMed] [Google Scholar]
- 34.DiBona GF, Kopp UC. Neural control of renal function. Physiol Rev : 75–197, 1997. [DOI] [PubMed] [Google Scholar]
- 35.do Carmo JM, da Silva AA, Cai Z, Lin S, Dubinion JH, Hall JE. Control of blood pressure, appetite, and glucose by leptin in mice lacking leptin receptors in proopiomelanocortin neurons. Hypertension : 918–926, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Epstein M, Hollenberg NK. Age as a determinant of renal sodium conservation in normal man. J Lab Clin Med : 411–417, 1976. [PubMed] [Google Scholar]
- 37.Eriksson J, Forsen T, Tuomilehto J, Osmond C, Barker D. Fetal and childhood growth and hypertension in adult life. Hypertension : 790–794, 2000. [DOI] [PubMed] [Google Scholar]
- 38.Faught FA. Blood Pressure Primer. Philadelphia, PA: Pilling, 1914. [Google Scholar]
- 39.Feig DI, Johnson RJ. Hyperuricemia in childhood primary hypertension. Hypertension : 247–252, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feig DI, Kang DH, Johnson RJ. Uric acid and cardiovascular risk. N Engl J Med : 1811–1821, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Feig DI, Madero M, Jalal DI, Sanchez-Lozada LG, Johnson RJ. Uric acid and the origins of hypertension. J Pediatr : 896–902, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Feig DI, Soletsky B, Johnson RJ. Effect of allopurinol on blood pressure of adolescents with newly diagnosed essential hypertension: a randomized trial. JAMA : 924–932, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Franceschini N, Le TH. Genetics of hypertension: discoveries from the bench to human populations. Am J Physiol Renal Physiol : F1–F11, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Franco M, Martinez F, Quiroz Y, Galicia O, Bautista R, Johnson RJ, Rodriguez-Iturbe B. Renal angiotensin II concentration and interstitial infiltration of immune cells are correlated with blood pressure levels in salt-sensitive hypertension. Am J Physiol Regul Integr Comp Physiol : R251–R256, 2007. [DOI] [PubMed] [Google Scholar]
- 45.Franco M, Tapia E, Bautista R, Pacheco U, Santamaria J, Quiroz Y, Johnson RJ, Rodriguez-Iturbe B. Impaired pressure natriuresis resulting in salt-sensitive hypertension is caused by tubulointerstitial immune cell infiltration in the kidney. Am J Physiol Renal Physiol : F982–F990, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Franco M, Tapia E, Santamaria J, Zafra I, Garcia-Torres R, Gordon KL, Pons H, Rodriguez-Iturbe B, Johnson RJ, Herrera-Acosta J. Renal cortical vasoconstriction contributes to development of salt-sensitive hypertension after angiotensin II exposure. J Am Soc Nephrol : 2263–2271, 2001. [DOI] [PubMed] [Google Scholar]
- 47.Franco MC, Christofalo DM, Sawaya AL, Ajzen SA, Sesso R. Effects of low birth weight in 8- to 13-year-old children: implications in endothelial function and uric acid levels. Hypertension : 45–50, 2006. [DOI] [PubMed] [Google Scholar]
- 48.Fujita T. Mechanism of salt-sensitive hypertension: focus on adrenal and sympathetic nervous systems. J Am Soc Nephrol : 1148–1155, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.George J, Carr E, Davies J, Belch JJ, Struthers A. High-dose allopurinol improves endothelial function by profoundly reducing vascular oxidative stress and not by lowering uric acid. Circulation : 2508–2516, 2006. [DOI] [PubMed] [Google Scholar]
- 50.Gersch M, Heinig M, Clapp W, Rodriguez-Iturbe B, Johnson RJ. Immunodeficient mice are protected from salt sensitive hypertension. J Am Soc Nephrol : abstract SU-FC019, 2007. [Google Scholar]
- 51.Gill JR Jr, Gullner G, Lake CR, Lakatua DJ, Lan G. Plasma and urinary catecholamines in salt-sensitive idiopathic hypertension. Hypertension : 312–319, 1988. [DOI] [PubMed] [Google Scholar]
- 52.Goldblatt H. The renal origin of hypertension. Physiol Rev : 120–165, 1947. [DOI] [PubMed] [Google Scholar]
- 53.Goldring W, Chasis H, Ranges HA, Smith HW. Effective renal blood flow in subjects with essential hypertension. J Clin Invest : 637–653, 1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gomez-Alamillo C, Sanchez-Casajus A, Sierra M, Huarte E, Diez J. Vasoconstriction of the afferent arteriole and defective renal synthesis of nitric oxide in essential hypertension. Kidney Int Suppl : S129–S131, 1996. [PubMed] [Google Scholar]
- 55.Gomez DM. Evaluation of renal resistances, with special reference to changes in essential hypertension. J Clin Invest : 1143–1155, 1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gu D, Zhao Q, Chen J, Chen JC, Huang J, Bazzano LA, Lu F, Mu J, Li J, Cao J, Mills K, Chen CS, Rice T, Hamm LL, He J. Reproducibility of blood pressure responses to dietary sodium and potassium interventions: the GenSalt study. Hypertension : 499–505, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guyton AC, Coleman TG, Cowley AV Jr, Scheel KW, Manning RD Jr, Norman RA Jr.. Arterial pressure regulation. Overriding dominance of the kidneys in long-term regulation and in hypertension. Am J Med : 584–594, 1972. [DOI] [PubMed] [Google Scholar]
- 58.Guyton AC, Coleman TG, Fourcade JC, Navar LG. Physiologic control of arterial pressure. Bull NY Acad Med : 811–830, 1969. [PMC free article] [PubMed] [Google Scholar]
- 59.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med : 2449–2460, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hall JE, Crook ED, Jones DW, Wofford MR, Dubbert PM. Mechanisms of obesity-associated cardiovascular and renal disease. Am J Med Sci : 127–137, 2002. [DOI] [PubMed] [Google Scholar]
- 61.Harlan SM, Morgan DA, Agassandian K, Guo DF, Cassell MD, Sigmund CD, Mark AL, Rahmouni K. Ablation of the leptin receptor in the hypothalamic arcuate nucleus abrogates leptin-induced sympathetic activation. Circ Res : 808–812, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Harrison DG, Vinh A, Lob H, Madhur MS. Role of the adaptive immune system in hypertension. Curr Opin Pharmacol : 203–207, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hay J. The significance of a raised blood pressure. Br Med J : 43–47, 1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.He FJ, MacGregor GA. A comprehensive review on salt and health and current experience of worldwide salt reduction programmes. J Hum Hypertens : 363–384, 2009. [DOI] [PubMed] [Google Scholar]
- 65.He FJ, MacGregor GA. How far should salt intake be reduced? Hypertension : 1093–1099, 2003. [DOI] [PubMed] [Google Scholar]
- 66.He FJ, MacGregor GA. Salt reduction lowers cardiovascular risk: meta-analysis of outcome trials. Lancet : 380–382, 2011. [DOI] [PubMed] [Google Scholar]
- 67.Hernandez Schulman I, Raij L. Salt sensitivity and hypertension after menopause: role of nitric oxide and angiotensin II. Am J Nephrol : 170–180, 2006. [DOI] [PubMed] [Google Scholar]
- 68.Herrera J, Ferrebuz A, Macgregor EG, Rodriguez-Iturbe B. Mycophenolate mofetil treatment improves hypertension in patients with psoriasis and rheumatoid arthritis. J Am Soc Nephrol : S218–S225, 2006. [DOI] [PubMed] [Google Scholar]
- 69.Hilzendeger AM, Morgan DA, Brooks L, Dellsperger D, Liu X, Grobe JL, Rahmouni K, Sigmund CD, Mark AL. A brain leptin-renin angiotensin system interaction in the regulation of sympathetic nerve activity. Am J Physiol Heart Circ Physiol : H197–H206, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hostmark AT. The Oslo health study: soft drink intake is associated with the metabolic syndrome. Appl Physiol Nutr Metab : 635–642, 2010. [DOI] [PubMed] [Google Scholar]
- 71.Hughson MD, Douglas-Denton R, Bertram JF, Hoy WE. Hypertension, glomerular number, and birth weight in African Americans and white subjects in the southeastern United States. Kidney Int : 671–678, 2006. [DOI] [PubMed] [Google Scholar]
- 72.International Consortium for Blood Pressure Genome-Wide Association Studies; Ehret GB, Munroe PB, Rice KM, Bochud M, Johnson AD, Chasman DI, Smith AV, Tobin MD, Verwoert GC, Hwang SJ, Pihur V, Vollenweider P, O'Reilly PF, Amin N, Bragg-Gresham JL, Teumer A, Glazer NL, Launer L, Zhao JH, Aulchenko Y, Heath S, Sober S, Parsa A, Luan J, Arora P, Dehghan A, Zhang F, Lucas G, Hicks AA, Jackson AU, Peden JF, Tanaka T, Wild SH, Rudan I, Igl W, Milaneschi Y, Parker AN, Fava C, Chambers JC, Fox ER, Kumari M, Go MJ, van der Harst P, Kao WH, Sjogren M, Vinay DG, Alexander M, Tabara Y, Shaw-Hawkins S, Whincup PH, Liu Y, Shi G, Kuusisto J, Tayo B, Seielstad M, Sim X, Nguyen KD, Lehtimaki T, Matullo G, Wu Y, Gaunt TR, Onland-Moret NC, Cooper MN, Platou CG, Org E, Hardy R, Dahgam S, Palmen J, Vitart V, Braund PS, Kuznetsova T, Uiterwaal CS, Adeyemo A, Palmas W, Campbell H, Ludwig B, Tomaszewski M, Tzoulaki I, Palmer ND; CARDiogram Consortium; CKDGen Consortium; KidneyGen Consortium; EchoGen consortium; consortium CHARGE-HF consortium; Aspelund T, Garcia M, Chang YP, O'Connell JR, Steinle NI, Grobbee DE, Arking DE, Kardia SL, Morrison AC, Hernandez D, Najjar S, McArdle WL, Hadley D, Brown MJ, Connell JM, Hingorani AD, Day IN, Lawlor DA, Beilby JP, Lawrence RW, Clarke R, Hopewell JC, Ongen H, Dreisbach AW, Li Y, Young JH, Bis JC, Kahonen M, Viikari J, Adair LS, Lee NR, Chen MH, Olden M, Pattaro C, Bolton JA, Kottgen A, Bergmann S, Mooser V, Chaturvedi N, Frayling TM, Islam M, Jafar TH, Erdmann J, Kulkarni SR, Bornstein SR, Grassler J, Groop L, Voight BF, Kettunen J, Howard P, Taylor A, Guarrera S, Ricceri F, Emilsson V, Plump A, Barroso I, Khaw KT, Weder AB, Hunt SC, Sun YV, Bergman RN, Collins FS, Bonnycastle LL, Scott LJ, Stringham HM, Peltonen L, Perola M, Vartiainen E, Brand SM, Staessen JA, Wang TJ, Burton PR, Soler Artigas M, Dong Y, Snieder H, Wang X, Zhu H, Lohman KK, Rudock ME, Heckbert SR, Smith NL, Wiggins KL, Doumatey A, Shriner D, Veldre G, Viigimaa M, Kinra S, Prabhakaran D, Tripathy V, Langefeld CD, Rosengren A, Thelle DS, Corsi AM, Singleton A, Forrester T, Hilton G, McKenzie CA, Salako T, Iwai N, Kita Y, Ogihara T, Ohkubo T, Okamura T, Ueshima H, Umemura S, Eyheramendy S, Meitinger T, Wichmann HE, Cho YS, Kim HL, Lee JY, Scott J, Sehmi JS, Zhang W, Hedblad B, Nilsson P, Smith GD, Wong A, Narisu N, Stancakova A, Raffel LJ, Yao J, Kathiresan S, O'Donnell CJ, Schwartz SM, Ikram MA, Longstreth WT Jr, Mosley TH, Seshadri S, Shrine NR, Wain LV, Morken MA, Swift AJ, Laitinen J, Prokopenko I, Zitting P, Cooper JA, Humphries SE, Danesh J, Rasheed A, Goel A, Hamsten A, Watkins H, Bakker SJ, van Gilst WH, Janipalli CS, Mani KR, Yajnik CS, Hofman A, Mattace-Raso FU, Oostra BA, Demirkan A, Isaacs A, Rivadeneira F, Lakatta EG, Orru M, Scuteri A, Ala-Korpela M, Kangas AJ, Lyytikainen LP, Soininen P, Tukiainen T, Wurtz P, Ong RT, Dorr M, Kroemer HK, Volker U, Volzke H, Galan P, Hercberg S, Lathrop M, Zelenika D, Deloukas P, Mangino M, Spector TD, Zhai G, Meschia JF, Nalls MA, Sharma P, Terzic J, Kumar MV, Denniff M, Zukowska-Szczechowska E, Wagenknecht LE, Fowkes FG, Charchar FJ, Schwarz PE, Hayward C, Guo X, Rotimi C, Bots ML, Brand E, Samani NJ, Polasek O, Talmud PJ, Nyberg F, Kuh D, Laan M, Hveem K, Palmer LJ, van der Schouw YT, Casas JP, Mohlke KL, Vineis P, Raitakari O, Ganesh SK, Wong TY, Tai ES, Cooper RS, Laakso M, Rao DC, Harris TB, Morris RW, Dominiczak AF, Kivimaki M, Marmot MG, Miki T, Saleheen D, Chandak GR, Coresh J, Navis G, Salomaa V, Han BG, Zhu X, Kooner JS, Melander O, Ridker PM, Bandinelli S, Gyllensten UB, Wright AF, Wilson JF, Ferrucci L, Farrall M, Tuomilehto J, Pramstaller PP, Elosua R, Soranzo N, Sijbrands EJ, Altshuler D, Loos RJ, Shuldiner AR, Gieger C, Meneton P, Uitterlinden AG, Wareham NJ, Gudnason V, Rotter JI, Rettig R, Uda M, Strachan DP, Witteman JC, Hartikainen AL, Beckmann JS, Boerwinkle E, Vasan RS, Boehnke M, Larson MG, Jarvelin MR, Psaty BM, Abecasis GR, Chakravarti A, Elliott P, van Duijn CM, Newton-Cheh C, Levy D, Caulfield MJ, Johnson T. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature : 103–109, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jeunemaitre X, Soubrier F, Kotelevtsev YV, Lifton RP, Williams CS, Charru A, Hunt SC, Hopkins PN, Williams RR, Lalouel JM, Courvel P. Molecular basis of human hypertension: role of angiotensinogen. Cell : 169–180, 1992. [DOI] [PubMed] [Google Scholar]
- 74.Ji W, Foo JN, O'Roak BJ, Zhao H, Larson MG, Simon DB, Newton-Cheh C, State MW, Levy D, Lifton RP. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat Genet : 592–599, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Johnson G. The Anatomy of Bright's Disease: The “Arterio-Capillary Fibrosis” of Sir Wm. Gull and Dr. Sutton. Br Med J I: 604–605, 1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Johnson RJ, Alpers CE, Yoshimura A, Lombardi D, Pritzl P, Floege J, Schwartz SM. Renal injury from angiotensin II-mediated hypertension. Hypertension : 464–474, 1992. [DOI] [PubMed] [Google Scholar]
- 77.Johnson RJ, Herrera-Acosta J, Schreiner GF, Rodriguez-Iturbe B. Subtle acquired renal injury as a mechanism of salt-sensitive hypertension. N Engl J Med : 913–923, 2002. [DOI] [PubMed] [Google Scholar]
- 78.Johnson RJ, Rodriguez-Iturbe B, Roncal-Jimenez C, Lanaspa MA, Ishimoto T, Nakagawa T, Correa-Rotter R, Wesseling C, Bankir L, Sanchez-Lozada LG. Hyperosmolarity drives hypertension and CKD—water and salt revisited. Nat Rev Nephrol : 415–420, 2014. [DOI] [PubMed] [Google Scholar]
- 79.Johnson RJ, Schreiner GF. Hypothesis: the role of acquired tubulointerstitial disease in the pathogenesis of salt-dependent hypertension. Kidney Int : 1169–1179, 1997. [DOI] [PubMed] [Google Scholar]
- 80.Johnson RJ, Titte S, Cade JR, Rideout BA, Oliver WJ. Uric acid, evolution and primitive cultures. Semin Nephrol : 3–8, 2005. [DOI] [PubMed] [Google Scholar]
- 81.Kannel WB, Schwartz MJ, McNamara PM. Blood pressure and risk of coronary heart disease: the Framingham study. Dis Chest : 43–52, 1969. [DOI] [PubMed] [Google Scholar]
- 82.Kaplan C, Pasternack B, Shah H, Gallo G. Age-related incidence of sclerotic glomeruli in human kidneys. Am J Pathol : 227–234, 1975. [PMC free article] [PubMed] [Google Scholar]
- 83.Kato N, Julier C. Linkage mapping for hypertension susceptibility genes. Curr Hypertens Rep : 15–24, 1999. [DOI] [PubMed] [Google Scholar]
- 84.Kearney PM, Whelton M, Reynolds K, Muntner P, Whelton PK, He J. Global burden of hypertension: analysis of worldwide data. Lancet : 217–223, 2005. [DOI] [PubMed] [Google Scholar]
- 85.Kell KP, Cardel MI, Bohan Brown MM, Fernandez JR. Added sugars in the diet are positively associated with diastolic blood pressure and triglycerides in children. Am J Clin Nutr : 46–52, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Keller G, Zimmer G, Mall G, Ritz E, Amann K. Nephron number in patients with primary hypertension. N Engl J Med : 101–108, 2003. [DOI] [PubMed] [Google Scholar]
- 87.Kempner W. Radical dietary treatment of hypertensive and arteriosclerotic vascular disease, heart and kidney disease, and vascular retinopathy. GP : 71–92, 1954. [PubMed] [Google Scholar]
- 88.Kirabo A, Fontana V, de Faria AP, Loperena R, Galindo CL, Wu J, Bikineyeva AT, Dikalov S, Xiao L, Chen W, Saleh MA, Trott DW, Itani HA, Vinh A, Amarnath V, Amarnath K, Guzik TJ, Bernstein KE, Shen XZ, Shyr Y, Chen SC, Mernaugh RL, Laffer CL, Elijovich F, Davies SS, Moreno H, Madhur MS, Roberts J 2nd, Harrison DG. DC isoketal-modified proteins activate T cells and promote hypertension. J Clin Invest : 4642–4656, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, Muller DN, Hafler DA. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature : 518–522, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Korotkoff NC. On methods of studying blood pressure. Bull Imperial Acad Med (St Petersburg) : 1905. [Google Scholar]
- 91.Laragh JH, Sealey JE. The plasma renin test reveals the contribution of body sodium-volume content (V) and renin-angiotensin (R) vasoconstriction to long-term blood pressure. Am J Hypertens : 1164–1180, 2011. [DOI] [PubMed] [Google Scholar]
- 92.Le MT, Frye RF, Rivard CJ, Cheng J, McFann KK, Segal MS, Johnson RJ, Johnson JA. Effects of high-fructose corn syrup and sucrose on the pharmacokinetics of fructose and acute metabolic and hemodynamic responses in healthy subjects. Metabolism : 641–651, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Levy D, Ehret GB, Rice K, Verwoert GC, Launer LJ, Dehghan A, Glazer NL, Morrison AC, Johnson AD, Aspelund T, Aulchenko Y, Lumley T, Kottgen A, Vasan RS, Rivadeneira F, Eiriksdottir G, Guo X, Arking DE, Mitchell GF, Mattace-Raso FU, Smith AV, Taylor K, Scharpf RB, Hwang SJ, Sijbrands EJ, Bis J, Harris TB, Ganesh SK, O'Donnell CJ, Hofman A, Rotter JI, Coresh J, Benjamin EJ, Uitterlinden AG, Heiss G, Fox CS, Witteman JC, Boerwinkle E, Wang TJ, Gudnason V, Larson MG, Chakravarti A, Psaty BM, van Duijn CM. Genome-wide association study of blood pressure and hypertension. Nat Genet : 677–687, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lifton RP. Genetic dissection of human blood pressure variation: common pathways from rare phenotypes. Harvey Lect : 71–101, 2004. [PubMed] [Google Scholar]
- 95.Lombardi D, Gordon KL, Polinsky P, Suga S, Schwartz SM, Johnson RJ. Salt-sensitive hypertension develops after short-term exposure to angiotensin II. Hypertension : 1013–1019, 1999. [DOI] [PubMed] [Google Scholar]
- 96.MacGregor GA. Sodium is more important than calcium in essential hypertension. Hypertension : 628–640, 1985. [DOI] [PubMed] [Google Scholar]
- 97.Madero M, Perez-Pozo SE, Jalal D, Johnson RJ, Sanchez-Lozada LG. Dietary fructose and hypertension. Curr Hypertens Rep : 29–35, 2011. [DOI] [PubMed] [Google Scholar]
- 98.Mahomed FA. The etiology of Bright's disease and the prealbuminuric state. Med Chir Trans : 197–228, 1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mallamaci F, Testa A, Leonardis D, Tripepi R, Pisano A, Spoto B, Sanguedolce MC, Parlongo RM, Tripepi G, Zoccali C. A polymorphism in the major gene regulating serum uric acid associates with clinic SBP and the white-coat effect in a family-based study. J Hypertens 2014. [DOI] [PubMed] [Google Scholar]
- 100.Masilamani S, Zhang XZ, Baylis C. Blunted pressure natriuretic response in the old rat: participation of the renal nerves. Am J Kidney Dis : 605–610, 1998. [DOI] [PubMed] [Google Scholar]
- 101.Mattson DL, Lu S, Nakanishi K, Papanek PE, Cowley AW Jr. Effect of chronic renal medullary nitric oxide inhibition on blood pressure. Am J Physiol Heart Circ Physiol : H1918–H1926, 1994. [DOI] [PubMed] [Google Scholar]
- 102.Mazzali M, Hughes J, Kim YG, Jefferson JA, Kang DH, Gordon KL, Lan HY, Kivlighn S, Johnson RJ. Elevated uric acid increases blood pressure in the rat by a novel crystal-independent mechanism. Hypertension : 1101–1106, 2001. [DOI] [PubMed] [Google Scholar]
- 103.Morimoto A, Uzu T, Fujii T, Nishimura M, Kuroda S, Nakamura S, Inenaga T, Kimura G. Sodium sensitivity and cardiovascular events in patients with essential hypertension. Lancet : 1734–1737, 1997. [DOI] [PubMed] [Google Scholar]
- 104.Moritz A, Oldt M. Arteriolar sclerosis in hypertensive and nonhypertensive individuals. Am J Pathol : 679–728, 1937. [PMC free article] [PubMed] [Google Scholar]
- 105.Moser M. Historical perspectives on the management of hypertension. J Clin Hypertens : 15–20, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mozaffarian D, Fahimi S, Singh GM, Micha R, Khatibzadeh S, Engell RE, Lim S, Danaei G, Ezzati M, Powles J; Global Burden of Diseases Nutrition and Chronic Diseases Expert Group. Global sodium consumption and death from cardiovascular causes. N Engl J Med : 624–634, 2014. [DOI] [PubMed] [Google Scholar]
- 107.Mu S, Shimosawa T, Ogura S, Wang H, Uetake Y, Kawakami-Mori F, Marumo T, Yatomi Y, Geller DS, Tanaka H, Fujita T. Epigenetic modulation of the renal beta-adrenergic-WNK4 pathway in salt-sensitive hypertension. Nat Med : 573–580, 2011. [DOI] [PubMed] [Google Scholar]
- 108.Navar LG, Harrison-Bernard LM, Nishiyama A, Kobori H. Regulation of intrarenal angiotensin II in hypertension. Hypertension : 316–322, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Navar LG, Kobori H, Prieto-Carrasquero M. Intrarenal angiotensin II and hypertension. Curr Hypertens Rep : 135–143, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nguyen S, Choi HK, Lustig RH, Hsu CY. Sugar-sweetened beverages, serum uric acid, and blood pressure in adolescents. J Pediatr : 807–813, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Niu W, Qi Y. An updated meta-analysis of endothelial nitric oxide synthase gene: three well-characterized polymorphisms with hypertension. PloS One : e24266, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.O'Donnell M, Mente A, Rangarajan S, McQueen MJ, Wang X, Liu L, Yan H, Lee SF, Mony P, Devanath A, Rosengren A, Lopez-Jaramillo P, Diaz R, Avezum A, Lanas F, Yusoff K, Iqbal R, Ilow R, Mohammadifard N, Gulec S, Yusufali AH, Kruger L, Yusuf R, Chifamba J, Kabali C, Dagenais G, Lear SA, Teo K, Yusuf S; PURE Investigators. Urinary sodium and potassium excretion, mortality, and cardiovascular events. N Engl J Med : 612–623, 2014. [DOI] [PubMed] [Google Scholar]
- 113.Oliver WJ, Cohen EL, Neel JV. Blood pressure, sodium intake, and sodium related hormones in the Yanomamo Indians, a “no-salt” culture. Circulation : 146–151, 1975. [DOI] [PubMed] [Google Scholar]
- 114.Park B, Park E, Cho SJ, Kim Y, Lee H, Min J, Ha E, Kang D, Park H. The association between fetal and postnatal growth status and serum levels of uric acid in children at 3 years of age. Am J Hypertens : 403–408, 2009. [DOI] [PubMed] [Google Scholar]
- 115.Parsa A, Brown E, Weir MR, Fink JC, Shuldiner AR, Mitchell BD, McArdle PF. Genotype-based changes in serum uric acid affect blood pressure. Kidney Int : 502–507, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Perera GA. Hypertensive vascular disease; description and natural history. J Chronic Dis : 33–42, 1955. [DOI] [PubMed] [Google Scholar]
- 117.Pons H, Ferrebuz A, Quiroz Y, Romero-Vasquez F, Parra G, Johnson RJ, Rodriguez-Iturbe B. Immune reactivity to heat shock protein 70 expressed in the kidney is cause of salt-sensitive hypertension. Am J Physiol Renal Physiol : F289–F299, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Prior LJ, Eikelis N, Armitage JA, Davern PJ, Burke SL, Montani JP, Barzel B, Head GA. Exposure to a high-fat diet alters leptin sensitivity and elevates renal sympathetic nerve activity and arterial pressure in rabbits. Hypertension : 862–868, 2010. [DOI] [PubMed] [Google Scholar]
- 120.Rey FE, Cifuentes ME, Kiarash A, Quinn MT, Pagano PJ. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O2− and systolic blood pressure in mice. Circ Res : 408–414, 2001. [DOI] [PubMed] [Google Scholar]
- 121.Riva-Rocci S. Un nuovo sfingomanometro. Gazz Med Torino 50–51: 1001–1007, 1896. [Google Scholar]
- 122.Rodriguez-Iturbe B, Franco M, Johnson RJ. Impaired pressure natriuresis is associated with interstitial inflammation in salt-sensitive hypertension. Curr Opin Nephrol Hypertens : 37–44, 2013. [DOI] [PubMed] [Google Scholar]
- 123.Rodriguez-Iturbe B, Pons H, Quiroz Y, Gordon K, Rincon J, Chavez M, Parra G, Herrera-Acosta J, Gomez-Garre D, Largo R, Egido J, Johnson RJ. Mycophenolate mofetil prevents salt-sensitive hypertension resulting from angiotensin II exposure. Kidney Int : 2222–2232, 2001. [DOI] [PubMed] [Google Scholar]
- 124.Rodriguez-Iturbe B, Pons H, Quiroz Y, Johnson RJ. The immunological basis of hypertension. Am J Hypertens : 1327–1337, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rodriguez-Iturbe B, Pons H, Quiroz Y, Lanaspa MA, Johnson RJ. Autoimmunity in the pathogenesis of hypertension. Nat Rev Nephrol : 56–62, 2014. [DOI] [PubMed] [Google Scholar]
- 126.Rodriguez-Iturbe B, Romero F, Johnson RJ. Pathophysiological mechanisms of salt-dependent hypertension. Am J Kidney Dis : 655–672, 2007. [DOI] [PubMed] [Google Scholar]
- 127.Sanchez-Lozada LG, Tapia E, Johnson RJ, Rodriguez-Iturbe B, Herrera-Acosta J. Glomerular hemodynamic changes associated with arteriolar lesions and tubulointerstitial inflammation. Kidney Int Suppl: S9–S14, 2003. [DOI] [PubMed] [Google Scholar]
- 128.Sanchez-Lozada LG, Tapia E, Santamaria J, Avila-Casado C, Soto V, Nepomuceno T, Rodriguez-Iturbe B, Johnson RJ, Herrera-Acosta J. Mild hyperuricemia induces vasoconstriction and maintains glomerular hypertension in normal and remnant kidney rats. Kidney Int : 237–247, 2005. [DOI] [PubMed] [Google Scholar]
- 129.Schiffrin EL. T lymphocytes: a role in hypertension? Curr Opin Nephrol Hypertens : 181–186, 2010. [DOI] [PubMed] [Google Scholar]
- 130.Seaberg EC, Munoz A, Lu M, Detels R, Margolick JB, Riddler SA, Williams CM, Phair JP, Multicenter AIDS Cohort Study. Association between highly active antiretroviral therapy and hypertension in a large cohort of men followed from 1984 to 2003. AIDS : 953–960, 2005. [DOI] [PubMed] [Google Scholar]
- 131.Sealey JE, Blumenfeld JD, Bell GM, Pecker MS, Sommers SC, Laragh JH. On the renal basis for essential hypertension: nephron heterogeneity with discordant renin secretion and sodium excretion causing a hypertensive vasoconstriction-volume relationship. J Hypertens : 763–777, 1988. [DOI] [PubMed] [Google Scholar]
- 132.Segall HN. How Korotkoff, the surgeon, discovered the auscultatory method of measuring arterial pressure. Ann Intern Med : 561–562, 1975. [DOI] [PubMed] [Google Scholar]
- 133.Shibata S, Mu S, Kawarazaki H, Muraoka K, Ishizawa K, Yoshida S, Kawarazaki W, Takeuchi M, Ayuzawa N, Miyoshi J, Takai Y, Ishikawa A, Shimosawa T, Ando K, Nagase M, Fujita T. Rac1 GTPase in rodent kidneys is essential for salt-sensitive hypertension via a mineralocorticoid receptor-dependent pathway. J Clin Invest : 3233–3243, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Soletsky B, Feig DI. Uric acid reduction rectifies prehypertension in obese adolescents. Hypertension : 1148–1156, 2012. [DOI] [PubMed] [Google Scholar]
- 135.Sommers SC, Melamed J. Renal pathology of essential hypertension. Am J Hypertens : 583–587, 1990. [DOI] [PubMed] [Google Scholar]
- 136.Sommers SC, Relman AS, Smithwick RH. Histologic studies of kidney biopsy specimens from patients with hypertension. Am J Pathol : 685–715, 1958. [PMC free article] [PubMed] [Google Scholar]
- 137.Suckling RJ, He FJ, Markandu ND, MacGregor GA. Dietary salt influences postprandial plasma sodium concentration and systolic blood pressure. Kidney Int : 407–411, 2012. [DOI] [PubMed] [Google Scholar]
- 138.Svendsen UG. Evidence for an initial, thymus independent and a chronic, thymus dependent phase of DOCA and salt hypertension in mice. Acta Pathol Microbiol Scand A : 523–528, 1976. [DOI] [PubMed] [Google Scholar]
- 139.Tallam LS, Stec DE, Willis MA, da Silva AA, Hall JE. Melanocortin-4 receptor-deficient mice are not hypertensive or salt-sensitive despite obesity, hyperinsulinemia, and hyperleptinemia. Hypertension : 326–332, 2005. [DOI] [PubMed] [Google Scholar]
- 140.Tigerstedt R, Bergman PG. Niere und kreislauft. Scand Arch Physiol : 223–270, 1898. [Google Scholar]
- 141.Titze J, Machnik A. Sodium sensing in the interstitium and relationship to hypertension. Curr Opin Nephrol Hypertens : 385–392, 2010. [DOI] [PubMed] [Google Scholar]
- 142.Tracy RE, Overll EO. Arterioles of perfusion-fixed hypertensive and aged kidneys. Arch Pathol : 526–534, 1966. [PubMed] [Google Scholar]
- 143.Watanabe S, Kang DH, Feng L, Nakagawa T, Kanellis J, Lan H, Mazzali M, Johnson RJ. Uric acid, hominoid evolution, and the pathogenesis of salt-sensitivity. Hypertension : 355–360, 2002. [DOI] [PubMed] [Google Scholar]
- 144.Weinberger MH. Salt sensitivity of blood pressure in humans. Hypertension : 481–490, 1996. [DOI] [PubMed] [Google Scholar]
- 145.Weinberger MH, Fineberg NS. Sodium and volume sensitivity of blood pressure. Age and pressure change over time. Hypertension : 67–71, 1991. [DOI] [PubMed] [Google Scholar]
- 146.Wilcox CS. Oxidative stress and nitric oxide deficiency in the kidney: a critical link to hypertension? Am J Physiol Regul Integr Comp Physiol : R913–R935, 2005. [DOI] [PubMed] [Google Scholar]
- 147.Wilkins RW. New drugs for hypertension, with special reference to chlorothiazide. N Engl J Med : 1026–1030, 1957. [DOI] [PubMed] [Google Scholar]
- 148.Wilson PW, D'Agostino RB, Sullivan L, Parise H, Kannel WB. Overweight and obesity as determinants of cardiovascular risk: the Framingham experience. Arch Intern Med : 1867–1872, 2002. [DOI] [PubMed] [Google Scholar]
- 149.Woods LL, Weeks DA, Rasch R. Programming of adult blood pressure by maternal protein restriction: role of nephrogenesis. Kidney Int : 1339–1348, 2004. [DOI] [PubMed] [Google Scholar]
- 150.Yang CL, Angell J, Mitchell R, Ellison DH. WNK kinases regulate thiazide-sensitive Na-Cl cotransport. J Clin Invest : 1039–1045, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Yang Q, Kottgen A, Dehghan A, Smith AV, Glazer NL, Chen MH, Chasman DI, Aspelund T, Eiriksdottir G, Harris TB, Launer L, Nalls M, Hernandez D, Arking DE, Boerwinkle E, Grove ML, Li M, Linda Kao WH, Chonchol M, Haritunians T, Li G, Lumley T, Psaty BM, Shlipak M, Hwang SJ, Larson MG, O'Donnell CJ, Upadhyay A, van Duijn CM, Hofman A, Rivadeneira F, Stricker B, Uitterlinden AG, Pare G, Parker AN, Ridker PM, Siscovick DS, Gudnason V, Witteman JC, Fox CS, Coresh J. Multiple genetic loci influence serum urate levels and their relationship with gout and cardiovascular disease risk factors. Circ Cardiovasc Genet : 523–530, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zhou BF, Stamler J, Dennis B, Moag-Stahlberg A, Okuda N, Robertson C, Zhao L, Chan Q, Elliott P, INTERMAP Research Group. Nutrient intakes of middle-aged men and women in China, Japan, United Kingdom, and United States in the late 1990s: the INTERMAP study. J Hum Hypertens : 623–630, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]