

Abstract
Idiopathic pulmonary fibrosis (IPF) is a devastating lung disease of unknown etiology. A conspicuous feature is the formation and persistence of fibroblastic/myofibroblastic foci throughout the lung parenchyma. Mechanisms remain unknown, but data indicate that fibroblasts acquire an antiapoptotic phenotype. We hypothesized that transcriptional silencing of proapoptotic genes may be implicated, and accordingly we evaluated the epigenetic regulation of p14ARF. The expression of p14ARF was analyzed by RT-PCR in IPF (n = 8) and normal derived fibroblasts (n = 4) before and after treatment with 5-aza-2′-deoxycytidine (5-aza) and trichostatin A (TSA). p14ARF gene promoter methylation was determined by methylation-specific PCR (MS-PCR) and by DNA digestion with endonuclease McrBc, which cleaves 50% of methylated CpG. Apoptosis was evaluated by Annexin-V and nuclear staining. p14ARF expression was significantly decreased in four of the eight IPF fibroblasts lines, which was restored after 5-aza treatment. No changes were found with TSA. MS-PCR of bisulfite-treated genomic DNA showed a correlation between the reduced expression of p14ARF and the presence of hypermethylated promoter. No amplification was observed in the DNA treated with the McrBc enzyme, corroborating promoter hypermethylation. p14ARF-hypermethylated IPF fibroblasts were significantly more resistant to staurosporine-and S-nitrosoglutathione-induced apoptosis compared with normal and nonmethylated IPF fibroblasts (P < 0.01) and showed reduced levels of p53. Resistance to apoptosis was provoked in fibroblasts when p14ARF expression was inhibited by siRNA (P < 0.05). These findings demonstrate that many IPF fibroblasts have reduced expression of the proapoptotic p14ARF attributable to promoter hypermethylation and indicate that epigenetic mechanisms may underlie their resistance to apoptosis.
Keywords: apoptosis, epigenetic, lung fibrosis
idiopathic pulmonary fibrosis (IPF) is a progressive and lethal disease characterized by injury and activation of lung epithelial cells, accumulation of fibroblasts/myofibroblasts, and abnormal remodeling of the lung parenchyma (9, 10, 23). Small fibroblastic foci are present in a background of collagen deposition, suggesting active fibrogenesis, whereas scarring and honeycombing indicate longstanding changes, reflecting the temporal heterogeneity of the process (9). It has been postulated that fibroblasts persist in the foci presumably because of resistance to apoptosis. Thus morphological studies usually show high rates of apoptosis and increased levels of apoptosis-related markers in alveolar epithelial cells, with relatively little evidence of apoptosis in adjacent fibroblasts/myofibroblasts (28). Moreover, myofibroblasts in fibroblastic foci of IPF lungs exhibit significantly less apoptotic activity compared with myofibroblasts in the fibromyxoid lesions of organizing pneumonia, which is usually a reversible lung disorder (14).
Epigenetic regulation of gene expression is a dynamic process that plays a key role in normal cell growth and differentiation. Alterations in DNA methylation patterns are associated with the transcriptional silencing of critical cell regulatory genes that, under normal conditions, control the cell cycle and initiate apoptotic cell death in neoplastic cells (26). Thus epigenetic inactivation of tumor suppressor genes is a common feature in human cancer.
Human p14ARF and its mouse counterpart, p19ARF, are tumor suppressors that, when upregulated, induce cell cycle arrest in both the G1 and G2 phases of the cell cycle (18, 26). p14ARF sequesters mdm2 in nucleoli, preventing mdm2-mediated p53 degradation and stabilizing a transcriptionally active p53 in the nucleoplasm (2). Thus cells injured by oncogenic insults undergo p53-dependent growth arrest or apoptosis (2, 24).
However, whereas strong evidence exists implicating epigenetic mechanisms in the pathogenesis of cancer, few such studies have been performed in lung fibrosis. Recent work has shown that hypermethylation of the Thy-1 promoter is associated with silencing of this putative fibrosis suppressor in fibroblastic foci of IPF lungs, whereas abnormalities of histone acetylation reduces the expression of cyclooxygenase-2, and consequently of PGE2, an important antifibrotic molecule (3, 22).
In this study, we evaluated the state of methylation of the promoter region of the proapoptotic gene p14ARF in fibroblasts obtained from IPF and normal lungs. Our results showed that p14ARF was hypermethylated in half of the IPF fibroblasts examined, which correlated with decreased expression of the gene and protein and increased resistance to apoptosis.
MATERIALS AND METHODS
Cell culture.
Primary human lung fibroblasts were obtained from patients with IPF (n = 8; 59 ± 7 yr) as previously described (21) after approval by the Science and BioEthics Committee of the National Institute of Respiratory Diseases. Diagnosis of IPF was made based on established criteria and confirmed by lung biopsy (19). As controls, we used three primary human lung fibroblasts obtained in our laboratory (39 ± 7 yr), derived from individuals without histological evidence of disease, and one normal human primary lung fibroblasts purchased from Clonetics, Cambrex BioScience (Walkersville, MD). Fibroblasts were cultured at 37°C in 5% CO2-95% air in Ham's F-12 medium supplemented with 10% FBS, 100 U/ml of penicillin, 100 μg/ml of streptomycin, and 2.5 mg/ml of amphotericin B. All fibroblasts were used between passages 2 and 5.
IPF and normal fibroblasts were plated 24 h before treatment with 5-aza-2′-deoxycitidine (5-aza), an inhibitor of DNA methylation, or trichostatin A (TSA), a histone deacetylase (HDAC) inhibitor (Sigma, St. Louis, MO). Cells reaching 60% of confluence were treated with 10 μM of 5-aza for 3 days or with 300 nM of TSA for 24 h. At every 24-h interval, the medium was changed to fresh medium containing the drug.
Genomic DNA isolation.
Fibroblasts were plated in 60-mm dishes, and when the cells reached ∼80% confluence, they were harvested, and genomic DNA was isolated using the Puregene Cell kit (Gentra Systems, Minneapolis, MN) according to the manufacturer's instructions. DNA concentration was determined with NanoDrop (ND-1000) spectrophotometer.
Bisulfite modification of DNA.
One microgram of DNA was treated with sodium bisulfite to convert unmethylated cytosine (but not methylated cytosines) to uracil. The sodium bisulfite modification of DNA was performed by using the DNA modification kit EpiTect (Qiagen, Valencia, CA) according to the manufacturer's instructions. Bisulfite-modified DNA was used immediately or stored at −20°C.
Methylation-specific PCR.
The methylation status of the p14ARF gene promoter in fibroblasts was determined by methylation-specific PCR (MS-PCR). The primers used to distinguish between methylated and unmethylated DNA sequences were: methylated primers 5′-GTGTTAAAGGGCGGCGTAGC-3′ (sense), 5′-AAAACCCTCACTCGCGACGA-3′ (antisense); unmethylated primers 5′-TTTTTGGTGTTAAAGGGTGGTGTAGT-3′ (sense), 5′-CACAAAAACCCTCACTC ACAACAA-3′ (antisense). The MS-PCR was carried out with ∼50 ng of bisulfite-modified DNA and Taq Platinum (Invitrogen, Carlsbad, CA). The amplification reactions for both methylated and unmethylated sequences were carried out for 31 cycles at 95°C for 30 s, 58°C for 30 s, and 72°C for 30 s, followed by extension at 72°C for 7 min. Unmethylated and methylated DNA controls (Chemicon, La Jolla, CA) were included in each reaction.
Genomic DNA digestion and promoter amplification.
Genomic DNA (250 ng) was digested 1 h at 37°C with three units of the endonuclease McrBC (New England Biolaboratories, Beverly, MA) supplemented with 100 μg/ml BSA and 1 mM guanosine triphosphate. After incubation, the reaction was heat inactivated 20 min at 65°C. We amplified by PCR four regions (R1-R4) of the p14ARF promoter as previously described (1). PCR amplification was performed with 20 ng of undigested or digested DNA with Accuprime Taq DNA polymerase (Invitrogen). The primers used for amplification after digestion were: R1: 5′-GAAGAATGGAAGACTTTCGACG-3′ (sense), 5′-TAACTGCAGACTGGGACCCA-3′ (antisense); R2: 5′-GCGAC TCCACCTACCTAGT-3′ (sense), 5′-CCACTTTCCCGCCCTGTG-3′ (antisense); R3: 5′-GGCACACACCCACCCAC-3′(sense), 5′-ACTAGGTAGGTGGAGTCGCA-3′(antisense); R4: 5′-CTGAAGGTGGTGGTAGGAG-3′ (sense), 5′-TGGGTGGGTG TGTGCC-3′ (antisense) (13). The amplification reaction for R1 was carried out for 35 cycles at 95°C for 30 s, 52°C for 30 s, and 72°C for 45 s, followed by extension at 72°C for 5 min. For R2–4, the conditions were 35 cycles at 95°C for 20 s, 55°C for 30 s, and 72°C for 1 min, followed by extension at 68°C for 5 min. All reactions were visualized on 1% agarose gels stained with etidium bromide.
Quantitative analysis was performed by real-time PCR using i-Cycler iQ Detection System with 50 ng of digested or undigested DNA in SYBR master mix (Applied Biosystems, Foster City, CA) containing the primers previously described for R2. The reaction was carried out for 40 cycles of 95°C for 15 s, and 60°C for 1 min. As an endogenous control, 18S was amplified with an eukaryotic 18S probe (Applied Biosystems). The results were analyzed by relative quantitation using the Δ cycle threshold method (ΔCt). Experiments were done twice in duplicate.
Quantitative real-time PCR.
p14ARF gene expression was analyzed in IPF and control fibroblasts before and after treatment with 5-aza or TSA by quantitative real-time PCR. Total RNA from fibroblasts was isolated using the RNeasy mini-kit (Qiagen). One microgram of total RNA was reverse transcribed using Advantage RT-for-PCR Kit (Clontech, Palo Alto, CA). Quantitative real-time PCR amplification was performed in a mixture containing 3 μl of cDNA, Taq Gold polymerase (Roche, Branchburg, NJ), and TaqMan probe-FAM dye-labeled assay on Demand Hs00233365_m1 (Applied Biosystems), using i-Cycler iQ Detection System (Bio-Rad, Hercules, CA). A dynamic range was built with each product of PCR on copy number serial dilutions of 1 × 1010, 1 × 108, 1 × 106, 1 × 104, and 1 × 102; all PCRs were performed in triplicate. Results were expressed as the number of copies of the target gene normalized to 18S rRNA (Human Euk 18s rRNA).
Western blot.
Proteins from cells were extracted with lysis buffer, and aliquots containing 40 μg of protein were separated in 10% SDS-polyacrylamide gels. Proteins were electroblotted onto nitrocellulose membranes (Hybond ECL; Amersham Biosciences, Cambridge, UK), and the membranes were blocked with 5% (wt/vol) nonfat dried milk. After incubation overnight at 4°C with antibodies against p53 (1:1,000; Cell Signaling, Beverly, MA), the membranes were incubated 1 h at room temperature with anti-mouse-horseradish peroxidase (1:5,000; Amersham). The signal was detected with the enhanced chemiluminescence (ECL) detection system (Amersham) using ChemiDoc-XRS+ imagin system (Bio-Rad). Detection of human GAPDH (1 μg/ml; Santa Cruz Biotechnology, Santa Cruz, CA) was used to test equal loading.
siRNA silencing of p14ARF.
p14ARF was silenced in fibroblasts by siRNA transfection procedure. Human p14ARF was purchased as a prevalidated Silencer Selected siRNA (Ambion, Austin, TX). siRNA was transfected into fibroblasts using the siPORT NeoFX transfection reagent (Ambion) according to the manufacturer's protocol. After 24 h of transfection in 12-well dishes, the medium was replaced by fresh culture medium, and 24 h later cells were used for apoptosis assay as described below. Previously a dose-response experiment was carried out to test the impact of transfection with 0, 2, 5, or 10 nM of siRNA on the mRNA level of p14ARF. Subsequently, 5 nM of siRNA was used in transfection experiments to test the effect of this siRNA on the fibroblast apoptosis. The positive and negative controls for transfection were analyzed in parallel. Transfection efficiency was determined by RT-PCR analysis. Two independent experiments were performed by duplicate.
Flow cytometry.
Fibroblasts were grown to ∼75% confluence in 12-well culture plates. Apoptosis was induced by 1 μM staurosporine (sts, Sigma) at times indicated in the figures. In parallel experiments, IPF fibroblasts with and without methylated p14ARF promoter were exposed to S-nitrosoglutathione (GSNO, 500 μM) for 24 h (31). Apoptosis was determined by Annexin-V staining assessed by flow cytometry as follows: harvested cells were incubated with Annexin V-PE and 7AAD (BD Bioscience, San Diego, CA) following manufacturer's instructions. Apoptotic cell death (Annexin-V+, 7AAD-) was measured by flow cytometry using a FACSAria cytometer (BD Bioscience). The results were analyzed with Flow Jo software. Two independent experiments were performed in triplicate.
Nuclei staining.
Fibroblasts (1 × 104/cm2) from a normal (N1) and IPF cell line (IPF-5) were plated on coverslips. After 24 h, the cells were incubated with DMSO (control) or 1 μM staurosporine for 6 h to induce apoptosis and then fixed 1 h with 2% paraformaldehyde. Cells were stained with 1 mg/ml of Hoechst 33342 (Invitrogen) for 20 min at room temperature. The coverslips were washed with PBS, mounted on glass slides and analyzed by fluorescence microscopy. The number of apoptotic cells was determined in two slides of each cell line counting the cells in seven random fields. Photographs were taken with a ×60 objective on Olympus IX81 interfaced to a DP71 digital camera system.
Statistical analysis.
The results were expressed as means ± SD. Comparisons were analyzed using ANOVA and Dunnett's multiple-comparison test. Values of P < 0.05 were considered statistically significant.
RESULTS
Methylation of p14ARF in IPF fibroblasts.
The DNA methylation status of the p14ARF gene promoter was analyzed in eight IPF and four normal fibroblast cell lines by MS-PCR. Four of the eight IPF fibroblasts exhibited hypermethylation of the p14ARF promoter (M band), whereas the four normal and the other four IPF fibroblasts remained unmethylated (U band). Figure 1A illustrates a representative example of MS-PCR assay showing the unmethylated PCR (U band ∼132 pb) and methylated PCR (M band ∼122 pb) products. Treatment with 5-aza for 3 days induced the loss of methylated PCR products found in the four IPF fibroblasts cell lines (Fig. 1B). No effect was observed with TSA treatment (data not shown).
Fig. 1.

Methylation of p14ARF in idiopathic pulmonary fibrosis (IPF) fibroblasts by methylation-specific PCR (MS-PCR) analysis. DNA extracted from normal and IPF fibroblasts before (A) and after (B) treatment with 5-aza was amplified by PCR with primers specific to the unmethylated (U) or the methylated (M) CpG islands of the p14ARF gene after modification with sodium bisulfite. The expected sizes of PCR products of p14ARF were 132 bp with U primers and 122 bp with M primers and were identified with commercially available unmethylated and methylated DNA. Methylation products were amplified in four IPF fibroblasts (IPF-5 to 8).
The methylation status of p14ARF was also evaluated by amplification of the promoter in two of the IPF cell lines that showed hypermethylation by MS-PCR analysis (IPF-5 and IPF-6) and in one of the IPF fibroblasts that did not (IPF-2). The amplification of the promoter was carried out in DNA digested with the endonuclease McrBc, which cleaves 50% of all mCpG (1). Thus the methylated regions are cut by the enzyme and therefore do not amplify, whereas nonmethylated regions remain uncut, and they are amplified. The promoter was screened and amplified by PCR using four different amplicons (Fig. 2A). Virtually none of the amplicons were amplified in the DNA treated with the McrBc enzyme in IPF-5 and IPF-6 fibroblasts, further corroborating that their promoters are methylated, whereas, in IPF-2, DNA was not cleaved, and the products of amplification were visible (Fig. 2A). To quantify methylation levels, we analyzed the amplification of R2 of the promoter of digested or undigested DNA by SYBR Green-based real-time PCR. For this assay, methylation levels were calculated using the ratio of McrBc digested and undigested DNA in each sample that was taken as 100%. This approach corroborated that, after incubation with McrBc, significantly less amplification is observed in the IPF-5 and IPF-6 fibroblasts (Fig. 2B).
Fig. 2.
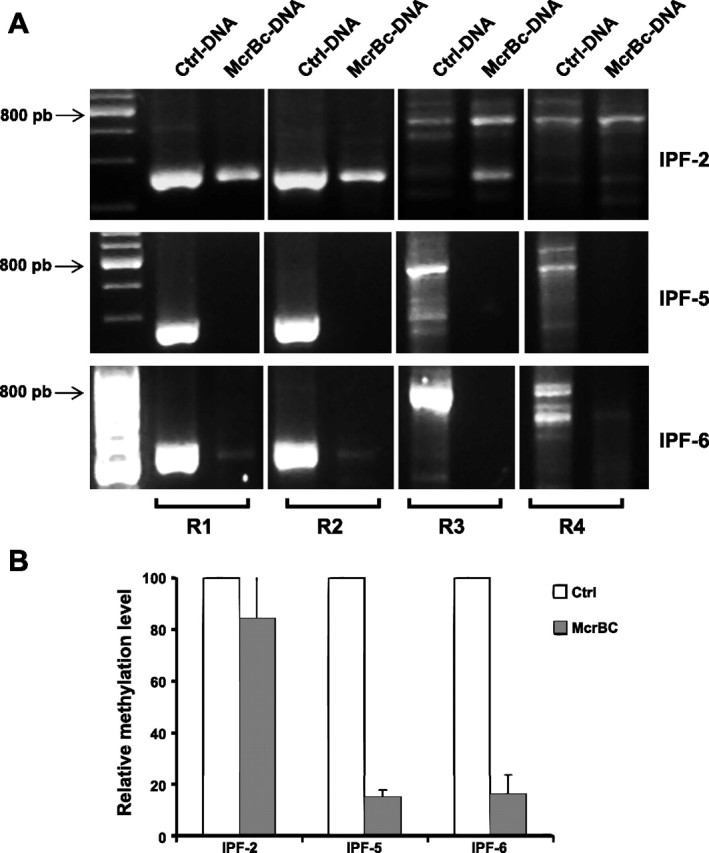
Fibroblast p14ARF promoter amplification. Genomic DNA extracted from IPF fibroblasts was digested with the enzyme McrBc as described in materials and methods. A: p14ARF promoter was amplified by PCR in undigested DNA (Ctrl-DNA) and DNA cleaved by McrBc (McrBc-DNA) using 4 different amplicons (R1-R4). Absence of amplification product in McrBc-DNA from IPF-5 and IPF-6 indicates methylation of corresponding region. B: methylation status of p14ARF promoter by relative quantitation of R2 amplicon using real-time-PCR after digestion with McrBc against its undigested cells. White spaces represent a reassembly of noncontiguous gel.
Expression of p14ARF in IPF and normal fibroblasts.
To determine differences in the pattern of expression of the p14ARF gene between IPF and normal fibroblasts, we analyzed the mRNA levels by quantitative real-time PCR in both IPF and normal cell lines. As shown in Fig. 3 (solid bars), the basal level of p14ARF gene expression was significantly reduced in the four IPF fibroblasts that had the gene hypermethylated (IPF-5, 6, 7, and 8) compared with the remaining cell lines (P < 0.01).
Fig. 3.

Levels of expression of the p14ARF in normal and IPF fibroblasts. Fibroblasts were cultured alone or in presence of 10 μM of 5-aza-2′-deoxycitidine (5-aza) for 3 days, and mRNA expression was analyzed by real-time RT-PCR. IPF fibroblasts with methylated promoter (IPF-5 to IPF-8) showed a significant reduced expression of p14ARF at basal level compared with the others fibroblasts (solid bars), and the expression was restored after 5-aza treatment (open bars). Bars represent the means ± SD of copy number of p14ARF mRNA normalized to 18S rRNA expression of 2 independent experiments made by triplicate. *P < 0.05; **P < 0.01.
To evaluate whether the decreased expression of p14ARF was provoked by epigenetic changes, fibroblasts were treated with 5-aza, an inhibitor of DNA methylation, or TSA, a histone deacetylase inhibitor. Treatment with 5-aza restored the expression of p14ARF in the four cell lines (Fig. 3). Compared with the basal levels, the fold change induced by 5-aza in IPF-5, IPF-6, IPF-7, and IPF-8 was ∼2.9 (P < 0.01), ∼0.7 (P < 0.05), ∼1.2 (P < 0.01), and ∼1.9 (P < 0.01), respectively. The expression of p14ARF did not show changes with TSA treatment (data not shown).
Apoptosis in normal and IPF fibroblasts.
P14ARF is considered to be a proapoptotic gene. The reduced expression in the IPF fibroblasts with hypermethylated promoter indicates the possibility that this epigenetic change could confer resistance to apoptosis. To test this possibility, apoptosis was induced with 1 μM of sts in fibroblasts with nonmethylated (N1, IPF-4) and methylated p14ARF (IPF-5). Apoptosis was determined by Annexin-V staining 6 h later by flow cytometry. Compared with the fibroblasts that did not have P14ARF hypermethylation, IPF-5 fibroblasts were significantly more resistant to apoptosis induced by sts (11.3 ± 4.4% vs. 29.0 ± 3.1% in normal fibroblasts; and 37.8 ± 9.0 in IPF-4; P < 0.01; Fig. 4A). Additionally, nuclear fragmentation and condensation, characteristic changes of cells undergoing apoptosis, were evaluated by Hoechst staining. We found that fibroblasts without p14ARF promoter hypermethylation stimulated with sts showed an apoptotic level of 12.5 ± 5.1%, whereas IPF-5 fibroblasts that have the promoter p14ARF gene methylated showed 7.5 ± 2.1% (P = 0.05) (Fig. 4B).
Fig. 4.
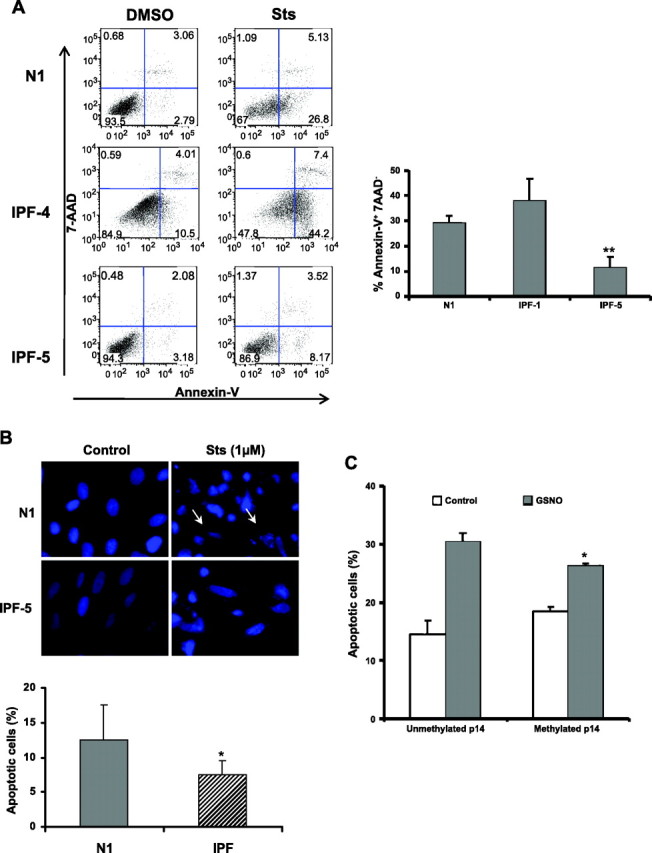
Increased resistance to apoptosis in p14ARF hypermethylated IPF fibroblasts. A: fibroblast with negative methylation (N1 and IPF-4) and 1 IPF with positive methylation (IPF-5) were cultured in presence of DMSO (control) or staurosporine (sts) for 6 h, and apoptosis was analyzed by flow cytometry. Representative charts of flow cytometry experiments. Cells negative for 7AAD staining and positive for Annexin-V binding represent apoptotic cells. Right: analysis of apoptotic cells showing the percent of apoptosis obtained in 2 independent experiments. B: fibroblasts grown on coverslips were incubated with DMSO (control) or 1 μM of sts for 6 h and fixed with 2% paraformaldehyde. Cells were stained with Hoechst 33342 (1 μg/ml) for 20 min at room temperature and analyzed by fluorescence microscopy. Left: representative photomicrograph of apoptotic changes in normal (N1) and 1 of IPF fibroblast cell lines with p14ARF promoter hypermethylation (IPF-5). Arrows indicate cells with fragmented nuclei. Bottom: percentage of cells with apoptotic changes (nuclear fragmentation) of 7 random fields. C: fibroblasts with methylated and nonmethylated p14ARF promoter were cultured alone or in presence of S-nitrosoglutathione (GSNO, 500 μM) for 24 h, and apoptosis was analyzed by flow cytometry. *P < 0.05; **P < 0.01.
It has been suggested that p14ARF is involved in apoptosis induced by nitric oxide (NO) (31). To investigate whether the absence of p14ARF prevents the cell death induced by NO, fibroblasts with the unmethylated and methylated promoter were exposed to GSNO (donor of NO) during 24 h, and the percentage of apoptotic cells was determined by flow cytometry. As shown in Fig. 4C, the treatment with GSNO induced apoptosis in both fibroblasts lines. However, in fibroblasts that have hypermethylation of the p14ARF promoter, the percentage of NO-induced apoptosis was significantly lower. In p14ARF unmethylated fibroblasts, the percentage of apoptotic cells increased from 14.6 ± 2.3% (control) to 30.4 ± 1.6% after GSNO treatment. By contrast, in fibroblasts with p14ARF methylation, this percentage increased from 18.5 ± 0.7% control to 26.3 ± 0.4%; P < 0.05.
To determine whether the in vitro silencing of p14 has an effect on apoptosis resistance, we turned off the expression of this gene by siRNA transfection, after which apoptosis was induced by sts and measured as described before. By this procedure, the expression of p14 in fibroblasts that normally express this gene was reduced about 70% (Fig. 5A). The analysis by Annexin-V staining showed that the apoptosis in the fibroblasts transfected with siRNA was reduced about 35% compared with fibroblasts transfected with control siRNA, from 24 ± 2.6% to 15.6 ± 3.2% (P < 0.05) (Fig. 5B).
Fig. 5.
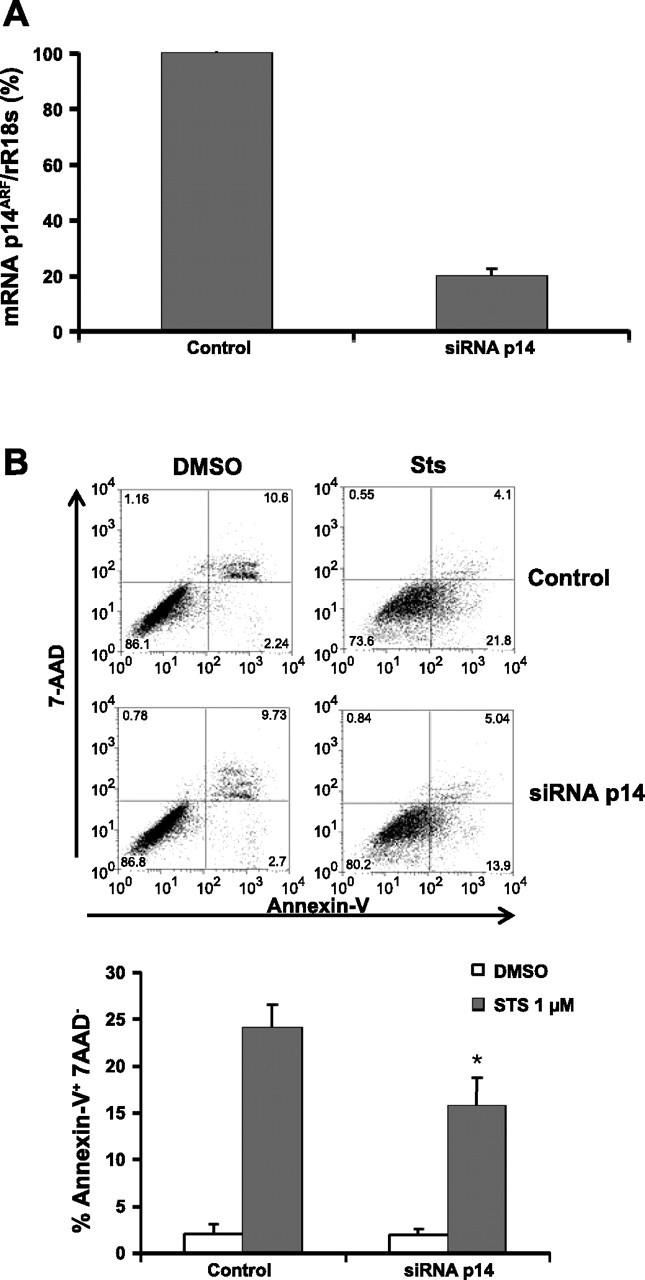
Silencing of p14ARF increases resistance to apoptosis in fibroblasts. Normal fibroblasts were transfected with 5 nM of siRNA for p14ARF. After 48 h, fibroblasts were exposed to DMSO or 1 μM of sts for 6 h, and apoptosis was analyzed by flow cytometry. A: relative quantitation of gene expression in control and transfected fibroblasts by real time-PCR. B: representative charts of cells negative for 7AAD staining and positive for Annexin-V binding represent apoptotic cells obtained in 1 of 2 independent experiments. The graph represents the percentage of apoptosis obtained in 2 independent experiments. *P < 0.05.
p53 levels in methylated and unmethylated IPF fibroblasts.
It has been demonstrated that p14ARF promotes the stabilization and accumulation of p53 (25). To determine whether this process is affected with the p14ARF methylation, cell lysates of two p14ARF unmethylated (IPF-1 and IPF-3) and two methylated (IPF-5 and IPF-7) fibroblasts were analyzed by Western blot. As shown in Fig. 6A, p53 protein is decreased in the fibroblasts that have hypermethylation of the p14ARF promoter compared with unmethylated fibroblasts. However, the decrease of p14ARF expression by using siRNA induced a small and nonsignificant decrease of p53 (Fig. 6B).
Fig. 6.
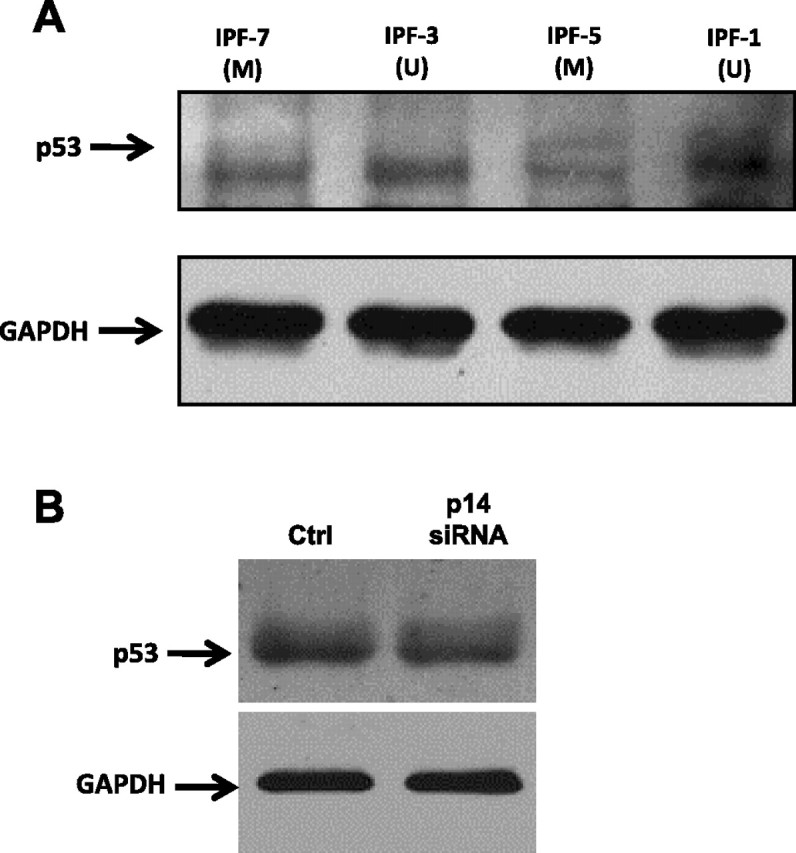
Western blot analysis of p53 in IPF fibroblasts. Fibroblasts with methylated (M) and unmethylated (U) p14ARF promoter (A) and fibroblasts treated with p14ARF siRNA or nontreated (Ctrl) (B) were plated in 60-mm dishes and cultured in Ham's F-12 medium supplemented with 10% FBS until ∼80% confluence was reached. Cell lysates were analyzed by immunoblotting using anti-human p53 antibody. Detection of GAPDH was used to test equal loading.
DISCUSSION
Resolution of alveolar damage is associated with successful reepithelialization and the eventual removal of myofibroblasts by a mechanism that likely involves apoptosis. In this context, it has been postulated that persistence of myofibroblasts in fibroblastic foci is a critical process in the pathogenesis and irreversibility of IPF (11). However, the mechanisms involved remain unclear. TGF-β1 has been shown to promote myofibroblast resistance to apoptosis, and recent findings indicate that endothelin-1 may have a similar role because it activates the prosurvival PI3K/AKT signaling pathway in normal and fibrotic human lung fibroblasts (7, 12). Some evidence also supports a role of the tumor suppressor phosphatase and tensin homologue (PTEN), a nonredundant, evolutionarily conserved phosphatase. PTEN decreases cellular proliferation, which is associated with an induction of apoptosis. Importantly, fibroblasts from fibroblastic foci exhibit a distinct loss of PTEN, which has been related to the induction of a migratory phenotype, increased proliferation, resistance to apoptosis, and myofibroblast differentiation (29, 30). However, the putative resistance to apoptosis by IPF fibroblasts is likely associated with multiple mechanisms.
Growing evidence indicates that some aging-associated phenotypes/diseases have an epigenetic basis. Thus epigenetic dysregulation by abnormal DNA methylation, as well as aberrant histone modifications, appears to contribute to different human diseases including autoimmunity and cancer (15). Studies in IPF, an aging-associated disease, are scanty, but recent work indicates that epigenetic downregulation of Thy-1 in IPF fibroblasts provoked by hypermethylation of the gene promoter correlates with a more profibrotic myofibroblast phenotype (22). Likewise, it has been demonstrated that defective histone acetylation prevents activated transcription factors from binding to the cyclooxygenase (COX)-2 promoter, resulting in diminished COX-2 gene transcription in IPF (3).
In this study, we found that hypermethylation of CpG islands in the promoter region of the proapoptotic gene P14ARF occurs often in fibroblasts obtained from IPF lungs. This process was demonstrated by the restored expression of the P14ARF gene after treatment with the DNA methyltransferase inhibitor 5-aza and further corroborated by using restriction digestion with McrBc, which showed a high level of methylation of the p14ARF promoter in the IPF fibroblast primary lines. The latter method allows examination the methylation status in a more global sense compared with that of a few CpG sites and also avoids the process of treating the genomic DNA with sodium bisulfite (26). Deacetylation of histones by histone deacetylase produces compact chromatin, which is also able to inhibit gene transcription. However, treatment of IPF fibroblasts with the pan-HDAC inhibitor trichostatin A had no effect on P14ARF expression.
P14ARF was selected for a number of reasons. It has been shown that it is able to activate intrinsic mitochondrial apoptosis, and in IPF lungs mitochondria-mediated apoptotic pathways are activated in alveolar epithelial cells but not in fibroblasts (6, 13). Also, some evidence indicates that NO induces the upregulation of P14ARF, which in turn activates p53, leading to apoptosis (31). In IPF, nitrative stress induced by NO participates in the pathogenesis of epithelial cell damage and aberrant regeneration but does not affect fibroblasts (27).
In addition, it is well known that p14ARF is regulated mainly at the transcriptional level, and its methylation-induced silencing has been reported in several types of cancer (4, 8, 16, 17). In these cases, alterations in p14ARF seem to occur somewhat later during the development of cancer, affecting tumor progression rather than carcinogenesis (8). p14ARF induces cell cycle arrest in G1 and G2 phases and enhances p53-dependent transactivation and apoptosis. Moreover, it also induces apoptosis in a p53-independent manner interacting with the transcription factor E2F1 and inhibiting its transcriptional activity (5).
Elimination of myofibroblasts by apoptosis is essential during normal wound healing, a process that seems to be hindered in IPF. In this context, understanding the mechanisms implicated in the resistance to cell death is critical to design new therapeutic strategies for this and other fibrotic lung disorders. In our study, we found that the P14ARF gene promoter region of IPF fibroblasts appeared to frequently undergo CpG island hypermethylation, which in turn contributed to a significant downregulation of gene and protein expression. Importantly, this gene silencing conferred high resistance to sts-induced apoptosis and also to the cell death induced by NO, demonstrating that P14ARF is involved in regulating lung fibroblast survival.
The fact that this mechanism of apoptosis resistance was only seen in four out of eight IPF fibroblast cultures tested could have several possible explanations. First, IPF is a spatially and temporally heterogeneous disease, and it is not known whether particular cultures represent fibroblasts specifically derived from fibroblastic foci. Secondly, it is possible that there are a variety of epigenetic and nonepigenetic pathways that can predispose to fibrosis and that different patients may represent distinct pathogenic alterations.
It has been established that p14ARF works like a positive regulator of p53, and the propensity of oncogenes to trigger the p53-pathway of apoptosis is at least partially mediated through this gene (25). Thus expression of p14ARF stabilizes p53, favoring its accumulation, which leads to cell cycle arrest or apoptosis. In this context, we found that p53 protein is decreased in hypermethylated p14ARF fibroblasts, indicating that in these cells the loss of p14ARF may increase p53 degradation with the subsequent reduction of protein, as we observed by Western blot. Thus decreased expression of p14ARF by its promoter hypermethylation affects p53-mediated apoptosis. However, this effect was not observed when p14ARF gene expression was attenuated by the specific siRNA.
In a previous report, we found that fibroblasts from patients with IPF showed increased apoptosis compared with normal fibroblasts (20). However, the study design differed, in that the previous study measured spontaneous apoptosis vs. induced apoptosis in this study. Furthermore, in the previous study, we used fibroblasts in more advanced passages, and we have observed that promoter hypermethylation might be lost in advanced passages. Also, the controls in the prior study were derived from uninvolved tissue in tumor resections, whereas in this study controls were from normal lung.
In summary, we report for the first time that the P14ARF gene promoter is frequently epigenetically inactivated in fibroblasts derived from IPF lungs. Several pathways may contribute to the resistance to apoptosis and subsequent persistence of fibroblasts/myofibroblasts within the fibroblast focus, and our findings indicate that hypermethylation-induced silencing of p14ARF gene may be one of them. The reversibility of this profibrotic phenotypic feature with a drug that has been used clinically suggests that epigenetic silencing of apoptosis suppressors or fibrosis suppressors may be a primary or secondary target for novel antifibrotic therapeutics.
GRANTS
This work was partially supported by Universidad Nacional Autónoma de México: SDI.PTID.05.6.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: J.C. and M.S. conception and design of research; J.C., M.C., B.O.-Q., M.N., I.H., and C.R. performed experiments; J.C., J.H., M.C., B.O.-Q., M.N., I.H., C.R., and A.P. analyzed data; J.C., M.N., C.R., and A.P. interpreted results of experiments; J.C. prepared figures; J.C., J.H., M.C., B.O.-Q., M.N., I.H., C.R., A.P., and M.S. approved final version of manuscript; J.H., A.P., and M.S. edited and revised manuscript; M.S. drafted manuscript.
REFERENCES
- 1.Badal V , Menendez S , Coomber D , Lane DP. Regulation of the p14ARF promoter by DNA methylation. Cell Cycle : 112–119, 2008. [DOI] [PubMed] [Google Scholar]
- 2.Charles Sherr J. The Pezcoller lecture: cancer cell cycles revisited. Cancer Res : 3689–3695, 2000. [PubMed] [Google Scholar]
- 3.Coward WR , Watts K , Feghali-Bostwick CA , Knox A , Pang L. Defective histone acetylation is responsible for the diminished expression of cyclooxygenase 2 in idiopathic pulmonary fibrosis. Mol Cell Biol : 4325–4339, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Esteller M , Tortola S , Toyota M , Capella G , Peinado MA , Baylin SB , Herman JG. Hypermethylation-associated inactivation of p14ARF is independent of p16INK4a methylation and p53 mutational status. Cancer Res : 129–133, 2000. [PubMed] [Google Scholar]
- 5.Eymin B , Karayan L , Séité P , Brambilla C , Brambilla E , Larsen CJ , Gazzéri S. Human ARF binds E2F1 and inhibits its transcriptional activity. Oncogene : 1033–1041, 2001. [DOI] [PubMed] [Google Scholar]
- 6.Hemmati PG , Normand G , Gillissen B , Wendt J , Dörken B , Daniel PT. Cooperative effect of p21Cip1/WAF-1 and 14–3-3sigma on cell cycle arrest and apoptosis induction by p14ARF. Oncogene : 6707–6719, 2008. [DOI] [PubMed] [Google Scholar]
- 7.Horowitz JC , Rogers DS , Sharma V , Vittal R , White ES , Cui Z , Thannickal VJ. Combinatorial activation of Fak and Akt by transforming growth factor-beta1 confers an anoikis-resistant phenotype to myofibroblasts. Cell Signal : 761–771, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ishida E , Nakamura M , Ikuta M , Shimada K , Matsuyoshi S , Kirita T , Konishi N. Promotor hypermethylation of p14ARF is a key alteration for progression of oral squamous cell carcinoma. Oral Oncol : 614–622, 2005. [DOI] [PubMed] [Google Scholar]
- 9.Katzenstein AL , Mukhopadhyay S , Myers JL. Diagnosis of usual interstitial pneumonia and distinction from other fibrosing interstitial lung diseases. Hum Pathol : 1275–1294, 2008. [DOI] [PubMed] [Google Scholar]
- 10.King TE , Pardo A , Selman M. Idiopathic pulmonary fibrosis. Lancet : 1949–1961, 2011. [DOI] [PubMed] [Google Scholar]
- 11.Kis K , Liu X , Hagood JS. Myofibroblast differentiation and survival in fibrotic disease. Expert Rev Mol Med : e27, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kulasekaren P , Scavone CA , Rogers DS , Arenberg DA , Thannickal VJ , Horowitz JC. Endothelin-1 and TGF-beta independently induce fibroblast resistance to apoptosis via AKT activation. Am J Respir Cell Mol Biol : 484–493, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuwano K , Hagimoto N , Maeyama T , Fujita M , Yoshimi M , Inoshima I , Nakashima N , Hamada N , Watanabe K , Hara N. Mitochondria-mediated apoptosis of lung epithelial cells in idiopathic interstitial pneumonias. Lab Invest : 1695–1706, 2002. [DOI] [PubMed] [Google Scholar]
- 14.Lappi-Blanco E , Soini Y , Paakko P. Apoptotic activity is increased in the newly formed fibromyxoid connective tissue in bronchiolitis obliterans organizing pneumonia. Lung : 367–376, 1999. [DOI] [PubMed] [Google Scholar]
- 15.Lu Q , Qiu X , Hu N , Wen H , Su Y , Richardson BC. Epigenetics, disease, and therapeutic interventions. Ageing Res Rev : 449–467, 2006. [DOI] [PubMed] [Google Scholar]
- 16.Nakamura M , Watanabe T , Klangby U , Asker C , Wiman K , Yonekawa Y , Kleihues P , Ohgaki H. p14ARF deletion and methylation in genetic pathways to glioblastomas. Brain Pathol : 159–168, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Negrini S , Gorgoulis VG , Halazonetis TD. Genomic instability–an evolving hallmark of cancer. Nat Rev Mol Cell Biol : 220–228, 2010. [DOI] [PubMed] [Google Scholar]
- 18.Perrone F , Tabano S , Colombo F , Dagrada G , Birindelli S , Gronchi A , Colecchia M , Pierotti MA , Pilotti S. p15INK4b, p14ARF, and p16INK4a inactivation in sporadic and neurofibromatosis type 1-related malignant peripheral nerve sheath tumors. Clin Cancer Res : 4132–4138, 2003. [PubMed] [Google Scholar]
- 19.Raghu G , Collard HR , Egan JJ , Martinez FJ , Behr J , Brown KK , Colby TV , Cordier JF , Flaherty KR , Lasky JA , Lynch DA , Ryu JH , Swigris JJ , Wells AU , Ancochea J , Bouros D , Carvalho C , Costabel U , Ebina M , Hansell DM , Johkoh T , Kim DS , King TE , Kondoh Y , Myers J , Müller NL , Nicholson AG , Richeldi L , Selman M , Dudden RF , Griss BS , Protzko SL , Schünemann HJ; ATS/ERS/JRS/ALAT. Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med : 788–824, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ramos C , Montaño M , García-Alvarez J , Ruiz V , Uhal BD , Selman M , Pardo A. Fibroblasts from idiopathic pulmonary fibrosis and normal lungs differ in growth rate, apoptosis, and tissue inhibitor of metalloproteinases expression. Am J Respir Cell Mol Biol : 591–598, 2001. [DOI] [PubMed] [Google Scholar]
- 21.Ramos C , Montaño M , Becerril C , Cisneros-Lira J , Barrera L , Ruíz V , Pardo A , Selman M. Acidic fibroblast growth factor decreases α-smooth muscle actin expression and induces apoptosis in human normal lung fibroblasts. Am J Physiol Lung Cell Mol Physiol : L871–L879, 2006. [DOI] [PubMed] [Google Scholar]
- 22.Sanders YY , Pardo A , Selman M , Nuovo GJ , Tollefsbol TO , Siegal GP , Hagood JS. Thy-1 promoter hypermethylation: a novel epigenetic pathogenic mechanism in pulmonary fibrosis. Am J Respir Cell Mol Biol : 610–618, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Selman M , King TE , Pardo A. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med : 136–151, 2001. [DOI] [PubMed] [Google Scholar]
- 24.Sherr CJ. Tumor surveillance via the ARF-p53 pathway. Genes Dev : 2984–2991, 1998. [DOI] [PubMed] [Google Scholar]
- 25.Sherr CJ , Bertwistle D , Besten W DEN , Kuo ML , Sugimoto M , Tago K , Williams RT , Zindy F , Roussel MF. p53-Dependent and -independent functions of the Arf tumor suppressor. Cold Spring Harb Symp Quant Biol : 129–37, 2005. [DOI] [PubMed] [Google Scholar]
- 26.Sherr CJ. Divorcing ARF and p53: an unsettled case. Nat Rev Cancer : 663–673, 2006. [DOI] [PubMed] [Google Scholar]
- 27.Terasaki Y , Akuta T , Terasaki M , Sawa T , Mori T , Okamoto T , Ozaki M , Takeya M , Akaike T. Guanine nitration in idiopathic pulmonary fibrosis and its implication for carcinogenesis. Am J Respir Crit Care Med : 665–673, 2006. [DOI] [PubMed] [Google Scholar]
- 28.Thannickal VJ , Horowitz JC. Evolving concepts of apoptosis in idiopathic pulmonary fibrosis. Proc Am Thorac Soc : 350–356, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.White ES , Atrasz RG , Hu B , Phan SH , Stambolic V , Mak TW , Hogaboam CM , Flaherty KR , Martinez FJ , Kontos CD , Toews GB. Negative regulation of myofibroblast differentiation by PTEN (Phosphatase and Tensin Homolog Deleted on chromosome 10). Am J Respir Crit Care Med : 112–121, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xia H , Diebold D , Nho R , Perlman D , Kleidon J , Kahm J , Avdulov S , Peterson M , Nerva J , Bitterman P , Henke C. Pathological integrin signaling enhances proliferation of primary lung fibroblasts from patients with idiopathic pulmonary fibrosis. J Exp Med : 1659–1672, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zeini M , Través PG , López-Fontal R , Pantoja C , Matheu A , Serrano M , Boscá L , Hortelano S. Specific contribution of p19(ARF) to nitric oxide-dependent apoptosis. J Immunol : 3327–3336, 2006. [DOI] [PubMed] [Google Scholar]