

Abstract
The parasitic disease onchocerciasis is the second leading cause of preventable blindness, afflicting more than 18 million people worldwide. Despite an available treatment, ivermectin, and control efforts by the World Health Organization, onchocerciasis remains a burden in many regions. With an estimated 120 million people living in areas at risk of infection, efforts are now shifting from prevention to surveillance and elimination. The lack of a robust, point-of-care diagnostic for an active Onchocerca infection has been a limiting factor in these efforts. Previously, we reported the discovery of the biomarker N-acetyl-tyramine-O-glucuronide (NATOG) in human urine samples and its ability to track treatment progression between medicated patients relative to placebo; we also established its capability to monitor disease burden in a jird model. NATOG is a human-produced metabolite of tyramine, which itself is produced as a nematode neurotransmitter. The ability of NATOG to distinguish between active and past infection overcomes the limitations of antibody biomarkers and PCR methodologies. Lateral flow immunoassay (LFIA) diagnostics offer the versatility and simplicity to be employed in the field and are inexpensive enough to be utilized in large-scale screening efforts. Herein, we report the development and assessment of a NATOG-based urine LFIA for onchocerciasis, which accurately identified 85% of analyzed patient samples (N = 27).
Keywords: point-of-care diagnostic, urine biomarker, N-acetyl-tyramine-O-glucuronide (NATOG), neglected tropical diseases, River Blindness, monoclonal antibodies
Onchocerciasis, commonly known as “River Blindness”, is a neglected tropical disease afflicting an estimated 18 million people, and 120 million live in areas at risk.1 This parasitic disease is endemic to Africa, Central and South America, and Yemen and is vector-transmitted by Simulium sp. black flies. The current treatment for onchocerciasis, ivermectin, eliminates the microfilariae of Onchocerca volvulus and suppresses their production by adult female worms for months following treatment. However, since this therapy does not readily eliminate adult worms, ivermectin has to be taken annually for years to break transmission.2 The World Health Organization has facilitated mass drug administration of ivermectin for decades. These efforts to combat onchocerciasis have been implemented in phases including: the Onchocerciasis Control Programme in West Africa (1974–2002), the African Programme for Onchocerciasis Control (1995–2015), and now the Expanded Special Project for Elimination of Neglected Tropical Diseases (2015–present).3−6
Due to the success of these prevention programs, the campaign has shifted from prevention to surveillance and elimination.7 However, these objectives depend on the availability of reliable, inexpensive, and portable diagnostic methods to monitor active infections in point-of-care areas. Currently, there are a number of tests for onchocerciasis with varied utility, sensitivity, and specificity. Most methods detect microfilariae, including the reference-standard for onchocerciasis diagnosis, a skin biopsy followed by microfilariae counting under a microscope.8 Alternatively, onchocerciasis has been diagnosed via a skin patch test based on the inflammatory response induced by diethylcarbamazine.8,9 In addition to these visual detection methods is the slit-lamp examination of the eye. While these diagnostic methods have excellent specificity, they lack sensitivity especially for populations undergoing ivermectin treatment which reduces/eliminates microfilariea.8,9 Methods that monitor adult worms include nodule palpation and biomarker analysis specific to adults. Palpation detects nodules close to the skin surface but cannot discriminate between nodules containing live or dead worms.10 Furthermore, deep tissue nodules are missed entirely by palpation. Host-produced antibodies against Ov-16 are some of the most sensitive and specific biomarkers and have even been incorporated into a portable assay.11 Unfortunately, these tests cannot distinguish between past and current infections due to the persistence of antibodies for years after infection. Recently, peptide biomarkers for adult worms have shown comparable sensitivity and specificity.12−14 Some of these peptide biomarkers have been incorporated into a lateral flow immunoassay (LFIA) with good sensitivity (63–98%) and specificity (99–100%).13 However, the healthy and cross-reactivity samples used for comparison were not from onchocerciasis endemic regions meaning further evaluation of specificity is still required.
An adult worm biomarker assay would overcome many of the limitations of microfilariae monitoring methods while also providing a rapid and noninvasive means to track and diagnose active onchocerciasis. For decades, the efforts to identify biomarkers with the necessary selectivity and sensitivity have been largely unsuccessful due to population inconsistencies and cross-reactivity.8 We have reported upon a biomarker we have termed, N-acetyltyramine-O-glucuronide (NATOG), which was uncovered through a metabolomics-mining approach (Figure 1).15,16 We have posited this urine biomarker to arise from a combination of metabolic steps in both the nematode and the host. Thus, tyramine, a nematode neurotransmitter, is acetylated by the nematode to form the metabolite N-acetyltyramine, which is glucuronidated by the host following excretion.16 Not only have we demonstrated that NATOG values are significantly increased in onchocerciasis patients relative to controls, but also we have established its disease monitoring potential by tracking doxycycline and ivermectin treatments relative to placebo.16 Furthermore, the progression of infection has been observed in a rodent model monitoring NATOG urine concentrations in jirds infected with the closely related nematode Litomosoides sigmodontis.17 This capability to track active infection is unique among most biomarkers for onchocerciasis and could have a significant impact on surveillance programs.8,12 Moreover, NATOG has even been shown to be species-specific when compared to Mansonella perstans and Loa loa infections, two human filarial nematodes known to be coendemic with O. volvulus. Although average NATOG concentrations are higher in both L. loa (14.7 ± 2.5 μM) and M. perstans (13.6 ± 2.5 μM) than the negative control (6.4 ± 0.7 μM), they are still significantly reduced in comparison to O. volvulus positive patients (42.8 ± 3.7 μM).16 Investigations into the stability characteristics of NATOG have shown it withstands sun exposure, temperature changes, and pH ranges (4–8), making it ideally suited for use in tropical settings.18
Figure 1.
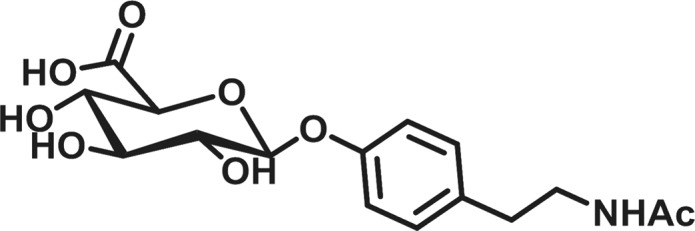
N-acetyltyramine-O-glucuronide (NATOG).
In order to translate these findings into an operational field test, we sought to employ NATOG in a semiquantifiable competitive LFIA. LFIA are inexpensive, portable, and simple to use making them an ideal screening platform for field work in neglected regions. These immunoassays utilize three main materials: a nitrocellulose membrane (platform), colored nanoparticles (reporter), and antibodies (biomarker selector) (Figure 2). While success in the LFIA arena requires all three components to be optimized, it is the biomarker selector that provides the greatest challenge as obtaining a high affinity/selective antibody to a carbohydrate conjugate as the antigen is fraught with uncertainty. Accordingly, monoclonal antibodies (mAbs) were produced by immunization of mice with a unique keyhole limpet hemocyanin (KLH) hapten conjugate. Remarkably, even with the high concentration of glucuronidated metabolites in human urine, mAbs selective for NATOG were identified by competition enzyme-linked immunosorbent assay (ELISA) against structurally related analogs. With the biomarker selector difficulty overcome, antibodies were conjugated with collidal gold nanoparticles and used in the development of the LFIA. Under optimized conditions, urine samples spiked with NATOG were tested in triplicate, analyzed, and found to have a linear NATOG dependency (R2 = 0.91). Analyses of O. volvulus positive (N = 293) and negative (N = 255) patient urine samples were used to generate a distribution of NATOG concentrations for the two populations. On the basis of this distribution, a cutoff (25 μM) was selected to allow for 95% confidence in a positive diagnosis. Patient samples (N = 27) were accessed employing this cutoff, and 85% was identified accurately.
Figure 2.
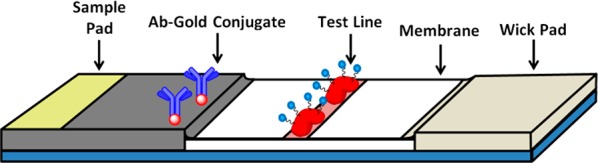
Lateral flow immunoassay diagram displaying the urine application site (sample pad), gold antibody conjugates (biomarker selector), the biomarker protein conjugates (test line), the nitrocellulose membrane, and the absorbent wick pad.
Results and Discussion
Hapten and Test Line Conjugate Synthesis
Production of mAbs for NATOG detection requires the synthesis of a small molecule hapten. Due to the number and variety of glucuronidated compounds present in urine, it was vital to obtain mAbs with high selectivity for NATOG.19 However, on the basis of literature precedence, mAbs raised against small molecule carbohydrates often have low affinity.20 In addition, the selection of an “immunologically silent” linker becomes increasingly more difficult to achieve as the size of the carbohydrate epitope becomes smaller, resulting in immunodominant responses directed toward the tether and not the compound of interest.20−22 An immunodominant response for the linker would contribute to preferential binding of the test line, limiting the sensitivity of the LFIA. To maximize recognition, our hapten was designed with a simple modification to the amide to append a thiol linkage site while maintaining the core NATOG structure (Figure 3). Furthermore, from this site, an asymmetric maleimide linker could be conjugated to a carrier protein in order to enhance the immune response toward the small carbohydrate hapten.22,23 Therefore, this hapten referred to as DG1 was synthesized from commercially available tyramine and acetobromo-α-d-glucuronic acid methyl ester (Figure 3). Tyramine was Boc protected in the presence of Boc anhydride to yield tert-butyl (4-hydroxyphenethyl)carbamate which was then coupled to the glucuronic acid donor via activation by silver trifluoromethanesulfonate. Trifluoroacetic acid deprotection of the Boc amine followed by EDC coupling with 2-(triphenylmethylthio)ethanoic acid and base catalyzed deprotection of the acyl and methoxy protecting groups afforded the hapten DG1. Conjugation of DG1 to the carrier protein KLH was achieved via a maleimide linker, and the hapten–carrier conjugate was used for mAb procurement. Following the same procedure, a bovine serum album (BSA)–DG1 hapten–protein conjugate was also constructed for mAb characterization and to serve as the test line portion of our LFIA system. To explore the possibility that linker effects could alter the LFIA, a second hapten was synthesized through coupling of the free amine with diethyl squarate followed by conjugation to BSA to yield the BSA–SQ1. Due to the amino-selectivity and its wide use in carbohydrate chemistry, the squaric acid linker was selected to provide a structurally distinct comparison to the maleimide linker utilized in DG1 (Figure S1).24,25 With this additional BSA conjugate, tether specificity could be evaluated and the test line could be further varied during optimization of the LFIA.
Figure 3.

Synthesis of N-acetyl-tyramine-O-glucuronide (NATOG) hapten derivatives DG1 and SQ1 with a triphenylmethyl protected thiol linkage site for conjugation with maleimide primed proteins and a square amide linker site for lysine conjugation, respectively.
To ensure consistency between the ELISA, surface plasmon resonance (SPR), and LFIA optimization experiments, a single batch of NATOG needed to be synthesized for all experimentation. However, the previous synthetic route for NATOG was not amenable to larger scales.15 To achieve a robust synthetic strategy for NATOG (see Figure S2), the more sensitive glycosyl bromide was replaced with a glycosyl trichloroacetimidate and the amide in the donor was converted to an inactive azide. With these adjustments, NATOG was synthesized consistently and could be readily scaled. Starting from commercially available d-glucurono-6,3-lactone and 4-hydroxyphenethyl bromide, NATOG was synthesized. d-Glucurono-6,3-lactone was converted to the glycosylation acceptor methyl 2,3,4-tri-O-acetyl-1-O-(trichloroacetimidoyl)-α-d-glucopyranuronate, and 4-hydroxyphenethyl bromide was converted to the donor 4-(2-azidoethyl)phenol, on the basis of literature procedures.26,27 This donor/acceptor pair was coupled in the presence of the activator boron trifluoride etherate at −78 °C and warmed to room temperature to yield 2-(4-(2-azidoethyl)phenoxy)-6-(methoxycarbonyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate. NATOG was isolated as a white powder following azide conversion to the amide in the presence of thioacetic acid and base catalyzed deprotection of the acyl and methoxy protecting groups. NATOG produced through this synthetic route was utilized in competitive ELISA and SPR experiments as well as in the optimization of our LFIA.
Monoclonal Antibody Selection
The first phase of mAb selection focused on the affinity for NATOG. A large panel of mAbs supernatants was screened using competition ELISA with BSA–DG1 and free NATOG. On the basis of these initial findings, a set of 8 antibodies were advanced to the next phase of selection where NATOG specificity was investigated using structural analogs of NATOG and compounds containing various NATOG moieties. Consistent with the literature, the production of mAbs for a small glycosylated antigen generated relatively low affinity antibodies. Although antibodies have been developed for single carbohydrate conjugates, specificity is often poor and optimal binding usually requires six or more residues.20,22 In spite of these reports, one mAb, 13F9, was able to achieve nanomolar affinity for NATOG. In addition to affinity, obtaining high selectively was crucial for an operational LFIA due to the significant presence of glucuronidated compounds in urine. Thus, even a small off target affinity could obstruct the assay due to the abundance. Surprisingly, many of the mAbs had several orders of magnitude less affinity for the structural analogs than NATOG (Table 1). In addition, the most prevalent analogs in urine, glucose (9.0–143.1 μmol/mmol creatinine), glucuronic acid (3.0–144.7 μmol/mmol creatinine), tyramine (0.33–4.296 μmol/mmol creatinine), and tyrosine (0.72–27.35 μmol/mmol creatinine), exhibited little to no measurable affinity (average creatinine values for healthy adults: 5.9–35 000 μm).19,28
Table 1. Evaluation of Antibodies by ELISA Competition To Investigate Selectivity Towards Structural Analogs of N-Acetyl-tyramine-O-glucuronide (NATOG) Using DG1a.

Antibody Kdapp values presented in μM for NATOG (green) are compared to off target values within one order of magnitude (yellow) and those with a lower Kdapp value (red).
When both the NATOG affinity and specificity results are taken into account, the three most promising mAbs were chosen for large scale production and purification, characterization by SPR, and evaluation in our LFIA system. Antibodies included one high affinity binding mAb, 13F9, and two moderate binding mAbs, 8C7 and 15A3, displaying only minor cross-reactivity. Purified mAbs were reassessed by competition ELISA and provided results consistent with our initial studies (Tables 1 and S1). Further binding analysis was performed using a surface plasmon resonance (SPR)-based competitive assay which closely mirrors our competitive LFIA format. The IC50 values demonstrated several orders of magnitude separation between NATOG and its structural analogs except for p-nitrophenyl glucuronide (Table 2). Although this cross-reactivity may be considered a potential liability, p-nitrophenyl glucuronide is not a natural metabolite and therefore should not impact our LFIA. To explore whether there was an immunodominant response directed toward the linker moiety within the hapten–linker–carrier complex, competition ELISA experiments were conducted with BSA–SQ1.20−22 Remarkably, the mAbs demonstrated marginally higher binding for the SQ1 variant suggesting that the immune response is primarily against the NATOG scaffold (Table S2).
Table 2. Monoclonal Antibody Surface Plasmon Resonance (SPR) Binding Results Using DG1 with N-Acetyl-tyramine-O-glucuronide (NATOG), Structural Analogs, and Urine Metabolitesa.

Antibody IC50 values for NATOG (green) are compared to off target values within one order of magnitude (yellow).
Lateral Flow Immunoassay Dipstick Diagnostic for Onchocerciasis
Due to the relatively small size of the biomarker, the competitive LFIA format was chosen to detect NATOG instead of the more traditional sandwich LFIA (Figure 4).29,30 In a competitive LFIA, a colloidal gold antibody conjugate that is specific for the biomarker is placed on the conjugation pad and a biomarker–protein conjugate is applied to test line. When urine is introduced to the sample pad, it migrates along the nitrocellulose membrane allowing for the colloidal gold antibody to competitively bind between the biomarker in the urine and the biomarker–BSA conjugate on the test line. As a result, a colored test line is indicative of a negative sample, and inversely, a clear test line indicates a positive sample. When this LFIA is incorporated into a reader, the intensity of the test line can be used to determine the concentration of biomarker in a semiquantitative fashion.
Figure 4.
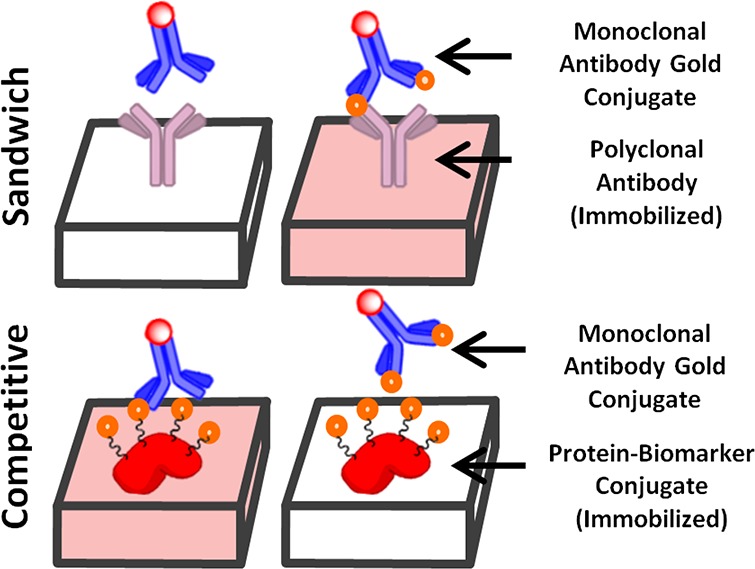
Sandwich assay detects biomarker by the tandem binding of immobilized polyclonal antibodies with a gold labeled mAb across the biomarker. A competitive assay detects biomarker by disrupting the binding between an immobilized protein–biomarker conjugate and a gold labeled mAb.
A BSA–NATOG (DG1 or SQ1) conjugate was used as the test line to compete with NATOG in the urine for the binding of a colloidal gold antibody conjugate. On the basis of affinity/selectivity, three antibodies, 13F9, 8C7, and 15A3, were conjugated to colloidal gold nanoparticles for LFIA investigations. The LFIA strips consisted of a nitrocellulose membrane with an immobilized BSA–NATOG conjugate (DG1 or SQ1) on the test line and a gold nanoparticle labeled antibody on the conjugation pad (13F9, 8C7, 15A3). The strips were run on a flat surface by applying the urine sample (35 μL) to the sample pad and were dried before scanning. The LFIA conditions were screened at two concentrations of NATOG in urine (0, 30 μM) and with the three antibodies (13F9, 8C7, 15A3), at three separate volumes of antibody conjugate (0.5, 1.0, 1.5 μL). In addition, two test line NATOG–BSA conjugates (DG1, SQ1) were screened at one volume (0.5 μL) at four concentrations (0.25, 0.35, 0.5, 1.0 mg/mL). Under these screening conditions, the LFIA produced intensely colored test lines that were not readily discernible between both NATOG concentrations. However, by diluting the urine samples with buffer (PBS, HEPES, or MEPS) at various urine/buffer ratios (1:1, 3:2, 2:1), a clear distinction was achieved. With optimized conditions (0.35 mg/mL DG1, 0.5 mL of 13F9, 3:2 urine/PBS), signal intensity correlated with the NATOG concentration. Furthermore, both the DG1 and SQ1 test lines were indistinguishable during optimization, consistent with the ELISA Kdapp values and further demonstrating mAb selectivity for NATOG rather than the tether (Table S2).
A linear dependency was established by analyzing six spiked urine samples (0, 7.5, 15, 22.5, 30, 37.5, 45 μM) in triplicate, averaging their peak heights, and plotting the results with a linear fit (R2 = 0.91) (Figure 5). However, because of a limited dynamic range (10–50 μM) and limit of quantitation (38 μM), the current state of this LFIA is best described as semiquantitative. To evaluate color stability, tested strips were stored under ambient condition and exhibited no visual variation in color intensity even after several months. This is consistent with other colloidal gold LFIAs that have demonstrated reliability up to one year.31,32 This stability allows for strips used at the point-of-care to undergo reevaluation at a centralized laboratory for quality assurance.
Figure 5.
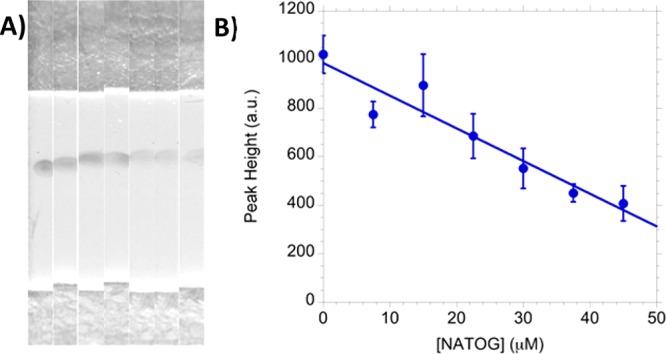
(A) Images of LFIA strips spiked using urine with 0, 7.5, 15, 22.5, 30, 37.5, and 45 μM of NATOG (left to right). (B) The triplicate average of concentration range with a linear fit (R2 = 0.91).
Population Distribution, Cutoff, and Patient Samples
With the linear relationship established in the spiked urine samples, authentic patient samples were analyzed to validate the LFIA. In order to mirror how this diagnostic would function in the field, a NATOG concentration diagnostic cutoff was determined by population distribution analysis. O. volvulus positive patient samples including monoinfections and coinfections with O. volvulus (Ov/Ll, Ov/Mp, and Ov/Ll/Mp) were combined with O. volvulus monoinfection values resulting in a NATOG concentration value of 39.2 ± 2.5 μM (±SEM; N = 293). All samples without detectable O. volvulus (O. volvulus negative control, L. loa monoinfection, M. perstans monoinfection, L. loa/M. perstans coinfection, and Lymphatic filariasis) were combined and resulted in an average concentration of c(NATOG) = 9.29 ± 0.95 μM (±SEM; N = 255). The negative log of NATOG concentration (p[NATOG]) for patient samples (Npos = 293, Nneg = 255) was grouped in 0.5p[NATOG] bins (3.5, 4, 4.5, ...) to obtain frequency (eq S1), and the resulting points were then fit to a normal distribution to evaluate the population overlap (Figure 6).33 Even though this analysis may contain false negatives due to misdiagnosed individuals experiencing low intensity infections, a distinct separation between the groups was observed. On the basis of this distribution, a cutoff of 25 μM was set to ensure a 95% confidence in a positive diagnosis. With a cutoff set, the LFIA was assessed with O. volvulus positive individuals (N = 14) and O. volvulus negative (N = 13) urine samples. To evaluate the LFIA over a wide range of concentrations for both positive and negative samples, these patients were selected to be representative of the binned sample groups (the plotted points) utilized in the distribution analysis (Figure 6). A correct diagnosis was defined as arriving at the same conclusion indicated by mass spectroscopy quantification for the given cutoff (25 μM). Under these conditions, the LFIA assay was able to correctly diagnose 85% of the patient samples (Table S3). The 15% incorrectly diagnosed was likely due to minor errors introduced via variability in urine composition. Although this sample set is limited (N = 27), it demonstrates the feasibility of targeting NATOG in a LFIA.
Figure 6.
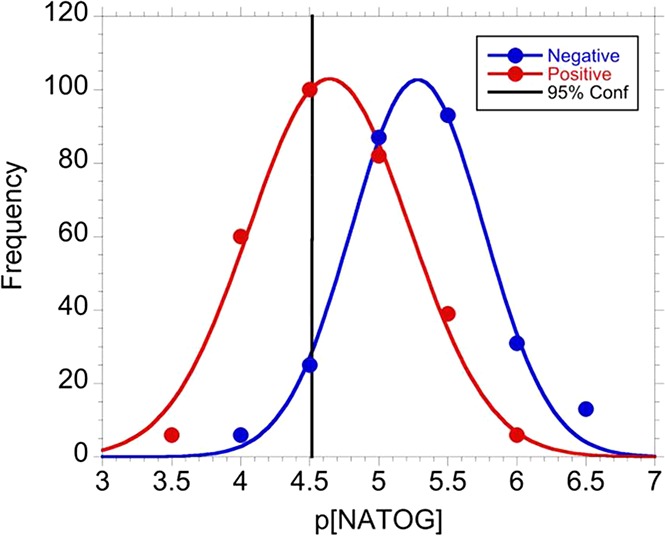
Distribution of pooled p[NATOG] for both positive (red) and negative (blue) patients. The 95% confidence interval is displayed at 25 μM.
Conclusion
In summary, a semiquantitative competitive LFIA for the biomarker NATOG has been developed utilizing mAbs produced from immunization with a KLH–NATOG conjugate. The mAb selected for the LFIA demonstrated superior specificity in the presence of similar glucuronidated metabolites and was implemented in the assay by conjugation with gold nanoparticles. A linear concentration dependency was observed in urine samples spiked with NATOG, and a diagnostic NATOG concentration cutoff for onchocerciasis (25 μM) was determined by the analysis of a large cohort of authentic patient samples (N = 548). Utilizing the linear correlation and cutoff value, the LFIA assay was compared to mass spectrometry data and exhibited 85% accuracy when diagnosing a subset of patients (N = 27). Our present study demonstrates that this LFIA has the potential to be the least invasive diagnostic for onchocerciasis and provide a means to monitor active infection and treatment progression in the field.
Methods
Reagents
The colloidal gold nanoparticle solution (CG-40 Colloidal Gold 40 nm) was obtained from ABIOX. Sucrose (CAS: 57-50-1) and Tween-20 (CAS: 9005-64-5) were purchased from Sigma-Aldrich. KLH (Cat# 77600) and BSA (Cat# 77110) were obtained from ThermoFisher Scientific. PBS buffer (pH 7.4, 137 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4, and 2 mM KH2PO4) and borate (pH 9, 50 mM) were both prepared.
Protein–Hapten Conjugation
DG1–KLH Immunization Conjugation
Trityl-protected NATOG hapten (7.8 mg, 12.4 μmol) was dissolved in anhydrous dichloromethane (600 μL). Under vigorous stirring at room temperature, first, trifluoroacetic acid (100 μL) and then trimethylsilane (500 μL) were added to the solution. The solution was stirred at room temperature for 15 min. The reaction mixture was transferred into an Eppendorff tube, and the solvent was removed with a stream of nitrogen. The solid obtained was dissolved in PBS buffer (pH 7.4, 10 mM) and mixed well to dissolve all water-soluble components; the mixture was then separated through centrifugation at 3000 rpm for 2 min. The supernatant was then removed and slowly added to the corresponding amount of preactivated BSA–maleimide conjugate protein (1–5 mg of deprotected hapten versus 1 mg of protein). The solution was mixed gently for 12 h at 4 °C. Afterward, the KLH conjugate was purified and/or rebuffered using the Zeba desalting columns according to the manufacturer’s manual.
DG1–BSA Test Line Conjugation
To BSA (100 μL, 6.7 mg/mL) in PBS buffer (pH 7.4) was added N-(γ-maleimidobutyrloxy)succinimide ester (1 mL, 3.82 mg/mL), and the solution was mixed gently at room temperature for 30 min. The mixture was then transferred to a Zeba spin desalting column (7K MWCO, 5 mL) and centrifuged for 2 min (1000g). During this time, the protected hapten (7 mg) was dissolved in anhydrous dichloromethane (500 μL), and trifluoroacetic acid (100 μL) was added followed by trimethylsilane (500 μL). The solution was stirred at room temperature for 20 min before the solvent was removed with a stream of nitrogen. The solid was dissolved in PBS buffer (100 μL) and mixed well; the mixture was centrifuged at 3000 rpm for 2 min. The supernate was then slowly added to the BSA–GMBS solution, and the sample was mixed gently for 12 h at 4 °C. The mixture was then dialyzed into PBS buffer (2 × 45 mL) overnight at 4 °C. Conjugation efficiency (18.5 copies) was calculated by the difference in mass between BSA–GMBS and BSA–GMBS–hapten by MALDI-TOF.
SQ1 Conjugation
To BSA (300 μL, 1.7 mg/mL) in borate buffer (pH 9, 50 mM) was added the square amide hapten (11.91 mg) dissolved in borate buffer (100 μL, pH 9, 50 mM), and the solution was mixed gently at room temperature for 24 h. The mixture was then dialyzed into PBS buffer (2 × 45 mL) overnight at 4 °C. Conjugation efficiency (13.7 copies) was calculated by the difference in mass between BSA and BSA–hapten by MALDI-TOF.
NATOG Monoclonal Antibody Production
A group of four mice (A/J, 6 week old) were injected intraperitoneally with DG1–KLH (100 μg) in PBS which had been premixed with a Sigma Adjuvant System. After 2 weeks, all mice received a booster injection identical to the initial immunization injection. Five weeks later, an injection of DG1–KLH (50 μg) was administered intravenously (tail vein); the animals were then sacrificed 3 days later, and the spleens removed. Hybridoma generation followed from this point as per literature procedure.34
Competition ELISA
The limiting dilution amounts of NATOG–BSA conjugate and mAb were predetermined for each antibody and used for competition ELISAs as described. A 96-well plate (Costar 3690) was coated overnight at 4 °C with NATOG–BSA and then blocked with 5% skim milk in PBS for 1 h at room temperature. The blocking buffer was removed, and an aliquot (15 μL) of serial diluted competing antigen, NATOG or other small molecule, was added followed by an equal amount of mAb. After incubation for 1 h at room temperature, the plate was washed; 30 μL of a 1:10 000 dilution of a donkey-antimouse IgG/horseradish peroxidase conjugate (Jack ImmunoResearch; code number: 715-035-151) in 2% BSA–PBS was added, and the sample was incubated for 30 min at room temperature. Following a washing step, the plate was developed with TMB (3,3′,5,5′-tetramethylbenzidine) as the chromogenic substrate (ThermoFisher Scientific; #34021) and H2SO4 (2 M) as the stopping solution. The Kdapp was defined as the concentration of competing antigen that resulted in one-half of the absorbance measured in the absence of competing antigen.
Surface Plasmon Resonance
A surface plasmon resonance (SPR)-based competition assay was performed for the three lead antibodies, 13F9, 8C7, and 15A3, to assess their binding affinities toward NATOG and other small molecules. Measurements were conducted on a Biacore 3000 instrument (Biacore AB, Uppsala, Sweden) in HBS-EP buffer at room temperature. The ligand, NATOG–BSA (DG1), was immobilized onto a CM5 sensor chip (GE Healthcare Life Sciences) through amine coupling using standard NHS/EDC chemistry; unconjugated BSA was immobilized on the reference flow cell. The dilution factor for each mAb was standardized such that a similar response measured in response units (RU) was achieved with DG1–BSA immobilized on the chip surface. Samples containing varying concentrations of compound and a constant concentration of mAb were preincubated at room temperature prior to injection for 2 min at 30 μL/min. After a 2 min dissociation phase, the chip surface was regenerated with glycine–HCl (10 mM, pH 1.5). The binding data displayed as a sensogram plot (RU vs time) for each sample was analyzed, and an IC50 value for all mAb–compound combinations was calculated. The RU values in the presence of inhibitor were normalized to the RU values in the absence of inhibitor, and the resulting data was fit to the equation log(inhibitor) vs response–variable slope using GraphPad Prism. The resulting competitive IC50 value is representative of the antibody Kd binding constant for small molecule binding.35
Conjugation of Gold Nanoparticles to Antibodies
To a colloidal gold nanoparticle solution (1.5 mL) was added aqueous potassium carbonate (30 μL, 0.2 M) followed by the antibody solution (5 μL, 1.2 mg/mL), and the solution was mixed for 15 min. A PBS buffered (pH 7.4) 10% BSA solution (50 μL) was added, and the solution mixed for an additional 15 min. The mixture was centrifuged for 5 min at 12 000 rpm; supernate was removed, and the pellet was suspended in conjugate buffer (0.3 mL, 5 mM borate, pH 8, 0.1% BSA, 0.15% Tween-20, and 5% sucrose). The homogeneous red solution was stored at 5 °C and warmed to room temperature prior to application.
Clinical Specimens
Human urine from O. volvulus infected and uninfected volunteers was collected as part of a European Union Seventh Framework Programme Research grant, contract 131242 “Enhanced Protective Immunity Against Filariasis”. Samples consisted of preexisting, deidentified diagnostic specimens collected with written informed consent; in cases of illiteracy, a literate witness signed and a thumbprint was made by the participant. Infected individuals were diagnosed by palpable nodules or microfilaridermia by skin snip.5,6 Uninfected individuals were classified as persons with no palpable nodules and microfilariae negative skin snips. Both positive and negative samples were obtained from regions endemic with onchocerciasis. Human urine from volunteers infected with L. loa, M. perstans, and dual or triple infections with O. volvulus was collected as part of the RADIO Consortium supported by a grant to the Institute of Microbiology, Immunology and Parasitology, University Hospital of Bonn from the Bill & Melinda Gates Foundation (OPP1083888). Infections were confirmed by microscopic examination of blood-borne microfilariae as previously described.3,4,36
Preparation of the Test Strips
The LFIA strip was assembled from a sample pad (Millipore GFCP203080), the membrane (CN95 Sartorius), the wick pad (EMI 30250), and the backing card (GL-57312). These components were applied to the backing card and cut to the dimensions of 3 × 60 mm. Once assembled, BSA–NATOG conjugate (0.5 μL of 0.35 mg/mL) was applied to position A and Ab–gold conjugate (0.6 μL) was applied to position B by pipetting; the solutions dried for an hour at room temperature (Figure 7). The strips were stored in a desiccator at room temperature prior to use.
Figure 7.

Lateral flow immunoassay highlighting the sample pad (A), the conjugation pad (B), and the test line (C).
General Test Procedure
Urine samples (30 μL) were diluted 3:2 with PBS buffer (pH 7.4, 137 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4, and 2 mM KH2PO4) and applied to position C by pipetting and run horizontally (Figure 7). The strips were dried at room temperature prior to scanning. Quantitation of test line spot intensities was performed on a Lumos Leelu Benchtop Reader, Lumos Diagnostics, Carlsbad, CA, USA. The Lumos Leelu reader was set to scan the entire strip, illuminating with red and green light, with a 4 ms exposure. Peak identification was centered at 610 pixels with 50 pixel tolerance, and background values were set on the left side (475–525 pixels) and the right side (700–750 pixels) of the peak. The peak heights were recorded to approximate NATOG concentration.
Acknowledgments
We gratefully acknowledge Diane Kubitz, Beverly Ellis, and the TSRI Antibody Production Core Facility for the preparation of monoclonal antibodies. We thank Kenneth M. Pfarr, Samuel Wanji, Alexander Y. Debrah, Achim Hoerauf, Sara Lustigman, Peter Enyong, Nidia Rizzo, Nancy Cruz-Ortiz, Mauricio Sauerbrey, Eduardo Catú, and Frank O. Richards for their assistance with sample collection. Finally, we thank Lumos Diagnostics for providing advice on LFIA development and granting us access to the Lumos Leelu Benchtop Reader. This work was supported by The Worm Institute of Research and Medicine (WIRM) and The Skaggs Institute for Chemical Biology (D.G., L.M.E., R.J.S., and K.D.J.) and by the Institute for Medical Microbiology, Immunology and Parasitology, University Hospital of Bonn, Germany as part of a consortium grant funded by the Bill and Melinda Gates Foundation (OPP1083888).
Glossary
Abbreviations
- Ab
antibody
- ACN
acetonitrile
- BSA
bovine serum albumin
- ELISA
enzyme-linked immunosorbent assay
- KLH
keyhole limpet hemocyanin
- LFIA
lateral flow immunoassay
- mAb
monoclonal antibody
- NATOG
N-acetyl-tyramine-O-glucuronide
- SPR
surface plasmon resonance
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsinfecdis.8b00163.
DG1 and SQ1 conjugate chemical structures; synthesis of NATOG; competition ELISA on bulk mAbs batches and comparing BSA–hapten conjugates (DG1 and SQ1); LFIA diagnostic determinations and the measured NATOG concentrations; Gaussian formula for NATOG concentration distributions; ethics statement; compound synthesis experimental information (PDF)
Author Present Address
∥ D.G.: Associate Professor, Department of Medicinal Chemistry, Science for Life Laboratory, Uppsala University, Box 574, SE-75123 Uppsala, Sweden.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- WHO (2018) Onchocerciasis - river blindness, http://www.who.int/mediacentre/factsheets/fs095/en/, accessed June 15, 2018.
- WHO (2018) Ivermectin, http://www.who.int/apoc/cdti/ivermectin/en/, accessed June 15, 2018.
- Hopkins A. D. (2016) Neglected tropical diseases in Africa: A new paradigm. Int. Health 8, i28–i33. 10.1093/inthealth/ihv077. [DOI] [PubMed] [Google Scholar]
- Bundy D. A. P.; Dhomun B.; Daney X.; Schultz L. B.; Tembon A. (2015) Investing in Onchocerciasis Control: Financial Management of the African Programme for Onchocerciasis Control (APOC). PLoS Neglected Trop. Dis. 9 (5), e0003508. 10.1371/journal.pntd.0003508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence J.; Sodahlon Y. K.; Ogoussan K. T.; Hopkins A. D. (2015) Growth, Challenges, and Solutions over 25 Years of Mectizan and the Impact on Onchocerciasis Control. PLoS Neglected Trop. Dis. 9 (5), e0003507. 10.1371/journal.pntd.0003507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn C.; Callahan K.; Katabarwa M.; Richards F.; Hopkins D.; Withers P. C. Jr.; Buyon L. E.; McFarland D. (2015) The Contributions of Onchocerciasis Control and Elimination Programs toward the Achievement of the Millennium Development Goals. PLoS Neglected Trop. Dis. 9 (5), e0003703. 10.1371/journal.pntd.0003703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moloo A. (2017) River blindness: shifting from prevention to surveillance and elimination, WHO, Geneva, http://www.who.int/neglected_diseases/news/shifting_from_prevention_to_surveillance_elimination/en/, accessed June 15, 2018.
- Vlaminck J.; Fischer P. U.; Weil G. J. (2015) Diagnostic Tools for Onchocerciasis Elimination Programs. Trends Parasitol. 31 (11), 571–582. 10.1016/j.pt.2015.06.007. [DOI] [PubMed] [Google Scholar]
- Boatin B. A.; Toé L.; Alley E. S.; Nagelkerke N. J.; Borsboom G.; Habbema J. D. (2002) Detection of Onchocerca volvulus infection in low prevalence areas: a comparison of three diagnostic methods. Parasitology 125 (6), 545–552. [PubMed] [Google Scholar]
- Duerr H. P.; Raddatz G.; Eichner M. (2008) Diagnostic value of nodule palpation in onchocerciasis. Trans. R. Soc. Trop. Med. Hyg. 102 (2), 148–154. 10.1016/j.trstmh.2007.10.009. [DOI] [PubMed] [Google Scholar]
- Golden A.; Steel C.; Yokobe L.; Jackson E.; Barney R.; Kubofcik J.; Peck R.; Unnasch T. R.; Nutman T. B.; Santos T. D. L.; Domingo G. J. (2013) Extended Result Reading Window in Lateral Flow Tests Detecting Exposure to Onchocerca volvulus: A New Technology to Improve Epidemiological Surveillance Tools. PLoS One 8 (7), e69231. 10.1371/journal.pone.0069231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denery J. R.; Nunes A. A. K.; Hixon M. S.; Dickerson T. J.; Janda K. D. (2010) Metabolomics-based discovery of diagnostic biomarkers for onchocerciasis. PLoS Neglected Trop. Dis. 4 (10), e834. 10.1371/journal.pntd.0000834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Moa M. J.; Van Dorst B.; Lagatie O.; Verheyen A.; Stuyver L.; Biamonte M. A. (2018) Proof-of-Concept Rapid Diagnostic Test for Onchocerciasis: Exploring Peptide Biomarkers and the Use of Gold Nanoshells as Reporter Nanoparticles. ACS Infect. Dis. 4 (6), 912–917. 10.1021/acsinfecdis.8b00031. [DOI] [PubMed] [Google Scholar]
- Lagatie O.; Verheyen A.; Nijs E.; Van Dorst B.; Debrah L. B.; Debrah A.; Supali T.; Sartono E.; Stuyver L. J. (2018) Evaluation of the diagnostic performance of Onchocerca volvulus linear epitopes in a peptide enzyme-linked immunosorbent assay. Am. J. Trop. Med. Hyg. 98 (3), 779–785. 10.4269/ajtmh.17-0756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Globisch D.; Moreno A. Y.; Hixon M. S.; Nunes A. A. K.; Denery J. R.; Specht S.; Hoerauf A.; Janda K. D. (2013) Onchocerca volvulus-neurotransmitter tyramine is a biomarker for river blindness. Proc. Natl. Acad. Sci. U. S. A. 110 (11), 4218–4223. 10.1073/pnas.1221969110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Globisch D.; Eubanks L. M.; Shirey R. J.; Pfarr K. M.; Wanji S.; Debrah A. Y.; Hoerauf A.; Janda K. D. (2017) Bioorganic & Medicinal Chemistry Letters Validation of onchocerciasis biomarker N-acetyltyramine-O-glucuronide. Bioorg. Med. Chem. Lett. 27 (15), 3436–3440. 10.1016/j.bmcl.2017.05.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Globisch D.; Specht S.; Pfarr K. M.; Eubanks L. M.; Hoerauf A.; Janda K. D. (2015) Litomosoides Sigmodontis: A Jird Urine Metabolome Study. Bioorg. Med. Chem. Lett. 25 (24), 5804–5807. 10.1016/j.bmcl.2015.10.037. [DOI] [PubMed] [Google Scholar]
- Lagatie O.; Ediage E. N.; Debrah L. B.; Diels L.; Nolten C.; Vinken P.; Debrah A.; Dillen L.; Silber S.; Stuyver L. J. (2016) Evaluation of the diagnostic potential of urinary N-Acetyltyramine-O,β-glucuronide (NATOG) as diagnostic biomarker for Onchocerca volvulus infection. Parasites Vectors 9 (1), 1–10. 10.1186/s13071-016-1582-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouatra S.; Aziat F.; Mandal R.; Guo A. C.; Wilson M. R.; Knox C.; Bjorndahl T. C.; Krishnamurthy R.; Saleem F.; Liu P.; Dame Z. T.; Poelzer J.; Huynh J.; Yallou F. S.; Psychogios N.; Dong E.; Bogumil R.; Roehring C.; Wishart D. S. (2013) The Human Urine Metabolome. PLoS One 8 (9), e73076. 10.1371/journal.pone.0073076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimburg-Molinaro J.; Rittenhouse-Olson K. (2009) Development and Characterization of Antibodies to Carbohydrate Antigens. Glycomics: Methods and Protocols 341–357. 10.1007/978-1-59745-022-5_24. [DOI] [PubMed] [Google Scholar]
- Bay S.; Huteau V.; Zarantonelli M. L.; Pires R.; Ughetto-Monfrin J.; Taha M. K.; England P.; Lafaye P. (2004) Phosphorylcholine-carbohydrate-protein conjugates efficiently induce hapten-specific antibodies which recognize both Streptococcus pneumoniae and Neisseria meningitidis: A potential multitarget vaccine against respiratory infections. J. Med. Chem. 47 (16), 3916–3919. 10.1021/jm040783p. [DOI] [PubMed] [Google Scholar]
- Lipinski T.; Luu T.; Kitov P. I.; Szpacenko A.; Bundle D. R. (2011) A structurally diversified linker enhances the immune response to a small carbohydrate hapten. Glycoconjugate J. 28 (3–4), 149–164. 10.1007/s10719-011-9331-8. [DOI] [PubMed] [Google Scholar]
- Yin Z.; Chowdhury S.; McKay C.; Baniel C.; Wright W. S.; Bentley P.; Kaczanowska K.; Gildersleeve J. C.; Finn M. G.; BenMohamed L.; Huang X. (2015) Significant Impact of Immunogen Design on the Diversity of Antibodies Generated by Carbohydrate-Based Anticancer Vaccine. ACS Chem. Biol. 10 (10), 2364–2372. 10.1021/acschembio.5b00406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurm F. R.; Klok H. A. (2013) Be squared: Expanding the horizon of squaric acid-mediated conjugations. Chem. Soc. Rev. 42 (21), 8220–8236. 10.1039/c3cs60153f. [DOI] [PubMed] [Google Scholar]
- Hou S.; Saksena R.; Kováč P. (2008) Preparation of glycoconjugates by dialkyl squarate chemistry revisited. Carbohydr. Res. 343 (2), 196–210. 10.1016/j.carres.2007.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima R.; Ono M.; Aiso S.; Akita H. (2005) Synthesis of methyl 1-O-(4-hydroxymethamphetaminyl)-alpha-d-glucopyranouronate. Chem. Pharm. Bull. 53 (6), 684–687. 10.1248/cpb.53.684. [DOI] [PubMed] [Google Scholar]
- Anand A.; Kulkarni M. V. (2017) Click chemistry approach for the regioselective synthesis of iso-indoline-1,3-dione-linked 1,4 and 1,5 coumarinyl 1,2,3-triazoles and their photophysical properties. Synth. Commun. 47 (7), 722–733. 10.1080/00397911.2017.1283524. [DOI] [Google Scholar]
- Sugita O.; Uchiyama K.; Yamada T.; Sato T.; Okada M.; Takeuchi K. (1992) Reference values of serum and urine creatinine, and of creatinine clearance by a new enzymatic method. Ann. Clin. Biochem. 29 (5), 523–528. 10.1177/000456329202900506. [DOI] [PubMed] [Google Scholar]
- Posthuma-Trumpie G. A.; Korf J.; Van Amerongen A. (2009) Lateral flow (immuno)assay: Its strengths, weaknesses, opportunities and threats. A literature survey. Anal. Bioanal. Chem. 393 (2), 569–582. 10.1007/s00216-008-2287-2. [DOI] [PubMed] [Google Scholar]
- Sajid M.; Kawde A. N.; Daud M. (2015) Designs, formats and applications of lateral flow assay: A literature review. J. Saudi Chem. Soc. 19 (6), 689–705. 10.1016/j.jscs.2014.09.001. [DOI] [Google Scholar]
- Wang Z.; Zhi D.; Zhao Y.; Zhang H.; Wang X.; Ru Y.; Li H. (2014) Lateral flow test strip based on colloidal selenium immunoassay for rapid detection of melamine in milk, milk powder, and animal feed. Int. J. Nanomed. 9 (1), 1699–1707. 10.2147/IJN.S58942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng X. Z.; Song J. L.; Zhan W. B. (2012) Development of a colloidal gold immunochromatographic test strip for detection of lymphocystis disease virus in fish. J. Appl. Microbiol. 113 (4), 737–744. 10.1111/j.1365-2672.2012.05389.x. [DOI] [PubMed] [Google Scholar]
- Koo J. W.; Parham F.; Kohn M. C.; Masten S. A.; Brock J. W.; Needham L. L.; Portier C. J. (2002) The association between biomarker-based exposure estimates for phthalates and demographic factors in a human reference population. Environ. Health Perspect 110 (4), 405–410. 10.1289/ehp.02110405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubitz K., and Keinan E. (2005) Production of Monoclonal Catalytic Antibodies: Principles and Practice. In Catalytic Antibodies, pp 491–504, Wiley-Blackwell, Hoboken, NJ, 10.1002/3527603662.ch18. [DOI] [Google Scholar]
- Bremer P. T.; Kimishima A.; Schlosburg J. E.; Zhou B.; Collins K. C.; Janda K. D. (2016) Combatting Synthetic Designer Opioids: A Conjugate Vaccine Ablates Lethal Doses of Fentanyl Class Drugs. Angew. Chem., Int. Ed. 55 (11), 3772–3775. 10.1002/anie.201511654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P. K.; Ajay A.; Kushwaha S.; Tripathi R. P.; Misra-Bhattacharya S. (2010) Towards novel antifilarial drugs: challenges and recent developments. Future Med. Chem. 2 (2), 251–283. 10.4155/fmc.09.170. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.