

Abstract
Water molecules are an important factor in protein-ligand binding. Upon binding of a ligand with a protein’s surface, waters can either be displaced by the ligand or may be conserved and possibly bridge interactions between the protein and ligand. Depending on the specific interactions made by the ligand, displacing waters can yield a gain in binding affinity. The extent to which binding affinity may increase is difficult to predict, as the favorable displacement of a water molecule is dependent on the site-specific interactions made by the water and the potential ligand. Several methods have been developed to predict the location of water sites on a protein’s surface, but the majority of methods are not able to take into account both protein dynamics and the interactions made by specific functional groups. Mixed-solvent molecular dynamics (MixMD) is a cosolvent simulation technique that explicitly accounts for the interaction of both water and small molecule probes with a protein’s surface, allowing for their direct competition. This method has previously been shown to identify both active and allosteric sites on a protein’s surface. Using a test set of eight systems, we have developed a method using MixMD to identify conserved and displaceable water sites. Conserved sites can be determined by an occupancy-based metric to identify sites which are consistently occupied by water even in the presence of probe molecules. Conversely, displaceable water sites can be found by considering the sites which preferentially bind probe molecules. Furthermore, the inclusion of six probe types allows the MixMD method to predict which functional groups are capable of displacing which water sites. The MixMD method consistently identifies sites which are likely to be non-displaceable and predicts the favorable displacement of water sites that are known to be displaced upon ligand binding.
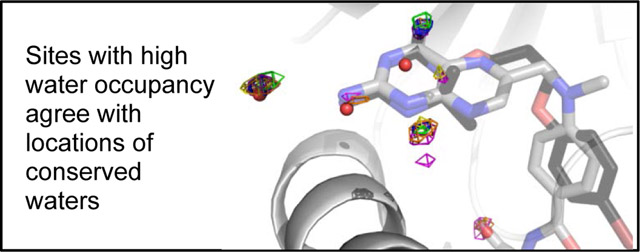
Graphical Abstract
Introduction
Water molecules play an important role in protein-ligand interactions. The specific conservation or displacement of water molecules is a significant factor in molecular recognition1, drug selectivity2, and a ligand’s binding affinity3. Upon ligand binding, waters at the binding interface must be displaced or participate in interactions between the protein and ligand. Waters at the binding interface fall into one of three categories: 1) waters which are always conserved, 2) waters which may be displaced by some ligands but not others, and 3) waters which are always displaced.
In the strategic design of ligands, scientists frequently try to increase a ligand’s affinity by selectively displacing waters. It would be advantageous for researchers attempting this to have a means to predict whether a water site could be displaced, and whether this would lead to an increase in a ligand’s affinity. To this end, a number of computational methods have been developed which attempt to predict the relative ease of displacement of a water site. For example, statistical methods such as AcquaAlta4, Consolv5, HINT/RANK6, and Waterscore7 utilize varying molecular descriptors such as crystallographic B-factors, number of hydrogen bonds, and descriptors of surrounding residues to analyze a hydration site. While these methods are relatively fast, they give predictive rates in the range of 50–70% depending on the method and test set used. Water prediction methods have also been incorporated into docking software. For example, the WaterDock methodology used with AutoDock Vina reported successful prediction of a water molecule’s displacement in 75% of cases8. Alternatively, Monte Carlo simulations of water molecules may be performed to predict their locations and binding affinity, such as in the Just Add Water Molecules (JAWS) method3, 9.
Since the specific interactions that determine whether a water molecule can be displaced or not are inherently site dependent, methods based on static structures may not accurately capture the variability among binding sites of different systems. Molecular dynamics-based methods are a promising alternative, as they are able to account for dynamics and interactions specific to each protein. For example, inhomogeneous fluid solvation theory (IFST) provides a means of calculating binding energies, including enthalpic and entropic components, from molecular dynamics (MD) simulations10, 11. This method has been implemented into the WaterMap tool and successfully applied to a number of targets12–17. McCammon and coworkers used a model system of receptor-ligand binding to understand the thermodynamic effects of water displacement into bulk solvent18. The same authors further characterized the energetics of ligand binding in additional test systems with varying charge properties19 and emphasized the fundamental role of water in mediating these interactions. A later study by Raman and MacKerell used cosolvent simulations to identify binding hotspots on Factor Xa and P38 Map kinase, followed by individual simulations of one probe in each hotspot site. They used those individual simulations to characterize the thermodynamics of protein-ligand interactions in explicit solvent using an IFST-based method20. They also noted the essential role of water and provided an analytical framework for calculating thermodynamic properties of protein-ligand interactions. The SPAM method also utilizes molecular dynamics simulations to calculate the affinity of a water site by considering the probability distribution of the interaction energies of each water site with its surroundings21.
While these methods are useful to analyze the energetics of individual water sites and predict their potential for displacement, they do not test the ability of specific functional groups to displace each site. In recent years, several cosolvent simulation techniques have been developed to map favorable interactions within a protein’s binding site, including the MixMD, SILCS, and MDmix methods22–31. In cosolvent MD simulations, a protein is initially immersed in a solution of small molecule probes and water. Following MD simulations during which the probes and water compete for binding to the protein’s surface, the solvent occupancy can be calculated to identify locations on the protein’s surface which preferentially interact with either the solvent probes or water. Most studies of cosolvent simulations to date have focused on the behavior of probe molecules. A preliminary study of the ability of SILCS to identify displaceable water sites for Factor Xa showed promising results.27 However, few systems were examined and crystallographic waters were retained during system setup which may have biased the observed results. Alvarez-Garcia and Barril examined the ability of the MDmix method to predict displaceable waters25, but only two systems and two probe types were tested which provides a limited view of the potential for diverse functional groups to displace specific water sites. Post-processing of the trajectories allowed for the binding affinity of each water or probe site to be calculated, which was assessed for a linear correlation with the fraction of crystal structures containing a water molecule at each site.25 While these methods are similar in their use of mixed solvents, each has methodological differences, such as the use of different probe molecules, whether individual probe molecules are run alone or in combination, and the use of restraints on protein and solvent atoms. The Mixed-Solvent Molecular Dynamics (MixMD) method has been developed by our group and previously shown to identify both active and allosteric sites32. In the present manuscript, we validate and extend the use of MixMD to map water sites and gauge their potential for displacement.
Methods
MD simulations
Eight systems were selected for the present test: Heat Shock Protein 90 (HSP90, PDB:1AH6)33, Bromodomain Containing Protein 4 (BRD4, PDB:2OSS)34, Dihydrofolate Reductase (DHFR, PDB:1DG8)35, TEM-1 β-Lactamase (PDB:1ZG4)36, Neuraminidase (PDB:4HZV)37, β-Secretase (BACE, PDB:1W50)38, Thrombin (PDB:3U69)39, and Penicillopepsin (PDB:3APP)40. These proteins were selected based on the criteria that they had apo crystal structures with better than 2 Å resolution and that each had multiple comparable ligand-bound structures in which water molecules were conserved, displaced, or selectively displaced relative to the apo structure. All crystallographic waters were removed prior to system setup, so there was no pre-biasing of the simulations to place waters in any particular locations. Hydrogens were added and side-chain positions were optimized using MolProbity41. Using a layered cosolvent approach, each protein was surrounded with a layer of probes (acetonitrile, isopropyl alcohol, N-methylacetamide, pyrimidine, or a methylammonium/acetate mix) followed by a layer of TIP3P water in a 5%/95% v/v ratio29. Input files were prepared with tleap using the AMBER FF99SB force field and parameters developed by Ryde for NADP and NADPH42–44. Sodium or chloride ions were used to neutralize the systems, and ACE/NME caps were added to protein chains when appropriate. The systems were initially minimized with restraints on the protein for 5000 steps, followed by 2500 steps of minimization on the entire system. The systems were gradually heated at constant volume over 40,000 steps with a 2 fs timestep and restraints of 10 kcal/mol-Å2 on the protein. After the systems had reached 300K, they were equilibrated at constant pressure for 1.75 ns as the restraints were gradually removed. Production runs were carried out for each system for 20 ns with the Andersen thermostat45. Previous work by our group has found that 20 ns is sufficient for convergence of calculated occupancy values. In total, 50 simulations were performed in AMBER12 for each protein; ten independent runs of 20 ns each per probe type44. This provided a total of 1 μs of total MD production for each protein.
Probe and Water Occupancy Calculation
The resulting trajectories were aligned using the AmberTools ptraj utility, and the occupancy of the probes and water during the last 10 ns of each simulation were calculated using a 0.5 Å grid over the entire solvent box44. To simplify further analysis, the resulting occupancies were normalized into σ units, using the equation:
| (1) |
where xi is the raw count at grid point i, μ is the mean occupancy over all grid points, and σ is the standard deviation across all grid occupancies. The occupancies can then be visualized in σ units, corresponding to the number of standard deviations above the mean occupancy (much like viewing electron density from crystal structures). Water and probe occupancy was visualized in PyMOL46. The maximum water occupancy within 1.4 Å of each water site in the apo structure was calculated using in-house perl and python scripts to parse the ptraj-generated occupancy files.
Water-site Conservation
To assess the ability of the MixMD method to find conserved water sites on a system-wide scale, we compared the water sites identified in the simulation with those found in comparable crystal structures. Comparable structures were identified using the Sequence Clusters from the Protein Data Bank at 95% sequence identity. This returns a list of crystal structures in the PDB at the specified similarity ranked by quality factor (based on resolution and R-value). The entries from this list (up to the top 99) with resolution better than 2.5 Å were selected for comparison. To identify hydration sites in each crystal structure, the structures were aligned using the wRMSD tool47 and PyMOL46, and clusters of waters were identified using WatCH48. WatCH clusters water molecules using a 2.4 Å threshold to identify water molecules occupying the same region. Each cluster was considered to be a water site. The experimental conservation of each water site was then calculated as the percentage of structures which had a water molecule within the cluster relative to the total number of structures. Waters conserved in the great majority of the crystal structures were visually inspected to determine if they were displaced by a ligand at any time (note that electron density was examined when available to determine whether the placement of ligand atoms were justified). If no ligand was found to displace these sites, they were considered to be conserved. In essence, a water could be absent from a structure or two and still be considered conserved as long as it was not actually displaced by a ligand. This was to allow for the subjectivity of water placement in crystallography. All reported percent conservation values in this manuscript refer to the percent conservation calculated from this analysis. It is important to note that observed experimental conservation will not necessarily be correlated with the displaceability of a water site. For example, multiple structures of a protein are frequently solved containing a series of related ligands, which may displace the same water molecule in every case. In addition, water molecules may be capable of being displaced, but ligands targeting that site may not yet have been developed (eg. waters on the edge of a binding site may be displaceable but current ligands do not extend that far).
Receiver Operator Characteristic (ROC) Curves
Each water molecule in the apo structure that was within 3.5 Å of any ligand was specifically examined, and the occupancies of water at each site in each of the simulations was calculated. The minimum occupancy across all simulations (see supplemental information) was used as a score. Traditional ROC plots were used to compare the scores of conserved water to displaced waters and free waters (defined below). Enrichments, Matthews correlation coefficients (MCCs), and areas under the curve (AUCs) were calculated using JMP Pro 1049.
Results and Discussion
Predicting Water Displaceability
It is important for us to acknowledge that there is an inherent difficulty in an assessment of displaceable and non-displaceable water. Displaced water are easily identified from comparing apo and bound crystal structures. However, a water that is not displaced by known ligands could still be displaced by a new ligand design. In essence, all conserved waters in crystal structures should be considered “potentially non-displaceable.” Our definition of conserved waters requires that no ligand displace them and they be present in the overwhelming majority of available structures. Waters that are not displaced by ligands but are also highly variable in crystal structures are considered to be “free” water in our analysis. We systematically analyzed all the waters within 3.5Å of all bound ligands in the crystal structures, and compared the water occupancies for the same positions in the MixMD simulations.
If we score waters based on their occupancies, the ROC plot in Figure 1 shows that our simulations can clearly distinguish conserved waters over free waters and displaced waters. Excellent AUC are seen for both conserved vs free (0.86) and vs displaced water (0.77). Optimal points on the ROC plots can be determined by a few different metrics. The points of maximal enrichment are given in Figure 1 as are the maximum MCC values. A very strict cutoff can be established by the maximum enrichment value of 23σ on the conserved vs displaced curve, which sacrifices a number of conserved waters in order to eliminate the displaced waters. A softer limit of 5σ is obtained from the maximal MCC point. This value aims to maximize the number of conserved water by allowing a small number of displaced waters to be misclassified as non-displaceable. Below, we discuss systems based on the strict 23σ cutoff, and the results for the soft limit are given in the supplemental information.
Figure 1.

ROC plots are given for scoring active-site waters based on occupancies from the MixMD simulations. The x-axis plots the percentage of conserved waters and the y-axis plots the percentage of free (red line) or displaced (blue line) waters. The points of maximum enrichment are marked with circles, and maximum MCC values are marked with triangles. Occupancies (in σ units) are noted next to the points on the graphs.
MixMD water occupancy can be visualized directly to identify sites which may be non-displaceable. Since the MixMD simulations are performed with both small molecule probes and water, sites that favorably bind water over probe molecules (non-displaceable sites) have greater levels of water occupancy than sites which more favorably bind probes (displaceable sites). As shown in Figure 2, when water occupancy is visualized at high σ values, only a few sites are observed. These sites are locations that are very frequently occupied by water despite the presence of probe molecules and are therefore considered to be non-displaceable. As σ values are decreased, sites that are less frequently occupied by water molecules are identified. Since these sites are not as frequently occupied by water when probe molecules are present, they can be considered to be potentially displaceable. As distinct water sites will inherently have higher levels of expected occupancy than bulk water, we sought to describe the distribution of occupancies for only the local maxima within the active-site region (defined as within 3.5 Å of any ligand). In the sections that follow, results of this method for eight systems are shown in order to demonstrate its ability to predict displaceable and non-displaceable water molecules. Tables with the water occupancy values for each system are given in the supplementary information.
Figure 2.

Water occupancies are shown as colored mesh from the MixMD simulations (colored by the probe type from each simulation). At high occupancy levels, few water sites are identified. These are sites which are repeatedly occupied by water molecules even in the presence of probe molecules. Water sites that first appear at lower sigma values are less frequently occupied by water when probe molecules are present. Note that these isocontours are for waters, not probes in the simulations.
Comparing Site-Specific Binding Preferences
HSP90
HSP90 has been well studied, and many potent inhibitors exist50. Site A, as shown in Figure 3, is found in 100% of homologous structures and is predicted by MixMD simulations to be favorably conserved. Studies focused on the structure-activity relationship of HSP90 have noted the tightly coordinated nature of this water molecule, leading researchers to avoid displacing this site51. Site B, on the other hand, is displaced by ligands containing either a hydroxyl group or a carbonyl group, as shown with geldanamycin bound to HSP90 in Figure 352. This is consistent with the MixMD prediction that this site is displaceable by N-methyl acetamide, with the hydrogen-bond donor and acceptor regions of N-methylacetamide occupying similar orientations to those found in ligand bound structures33, 53. HSP90 has previously been studied by Alvarez-Garcia and Barril using their cosolvent simulation method MDmix25, and by Haider and Huggins using IFST with MCSS54. IFST with MCSS predicted site B to be conserved based on predicted ΔG values, whereas MDmix predicts site B (1AH6:393, 1YER:336) as displaceable, consistent with ligand-bound structures. In Barril’s MDmix, a water site is classified as displaceable if one of the tested probe molecules binds to the site with higher affinity. However, in MDmix only ethanol and acetamide solvent mixtures were used, which limits the applicability of the data. For example, water 391 (PDB:1AH6) in the crystal structure of HSP90 is displaced by the phosphate groups of ATP (PDB:1BYQ)55. Barril’s MDmix predicts this water to be conserved (water site 325 in 1YER numbering), as none of their probes are capable of displacing this site, while our method predicts this site as displaceable. Thus, the use of multiple probe molecules in MixMD offers a greater predictive power over alternative methods that utilize a more limited set of probes.
Figure 3.

Heat Shock Protein 90. Water density is shown at the 23 σ level, colored according to the probe type included in the simulation. Crystallographic waters (PDB: 1AH633) with 3.5 Å of ligands are shown for reference. Site A is a water site found in 100% of homologous structures, predicted to be conserved by MixMD. Site B is a water displaced by the carbonyl of geldanamycin, predicted to be displaced by N-methylacetamide.
BRD4
Apo BRD4 contains several water molecules which are displaced upon interaction with ligands. For example, site A in Figure 4 is predicted to be displaced by all of the probe types tested. This is consistent with crystal structures of bound ligands, in which many functional groups displace this site, including triazole (PDB:2YEL, WSH), the carbonyl of acetylated histone proteins (PDB:3JVK, peptide), and the oxygen of isoxazole (PDB:3SVF, WDR)56–58. Within the binding pocket, there are a number of water molecules which are found in the majority of crystal structures. For example, site B in Figure 4 is found in 97% of all comparable crystal structures. Interestingly, MixMD predicts that several of these sites can be selectively displaced. A recent crystal structure has been solved verifying this, in which an inhibitor extends deeper into the binding pocket and displaces these sites (PDB:4O7F, 2RQ59), as shown in Figure 4. This highlights the need for water prediction methods. Based on conservation alone, it would appear that these sites are not easily displaced. However, MixMD simulations predict that they can be selectively displaced, in agreement with experimental data.
Figure 4.

BRD4. Water density is shown at the 23 σ level, colored according to the probe type included in the simulation. Crystallographic waters from the apo structure (PDB:2OSS34) that are within 3.5 Å of a ligand are shown for reference. Site A is predicted by MixMD to be displaced, shown with an example ligand (PDB:3UVW, peptide34) displacing the site. Site B is a water site found in 97% of comparable structures, predicted by MixMD to be displaceable is shown with an inhibitor displacing this site (PDB:4O7F, 2RQ59).
DHFR
The results for DHFR provide another good example of MixMD’s ability to discriminate between waters that are always conserved, always displaced, and those that are selectively displaced. In DHFR, 100% of homologous structures contain a water at site A (Figure 5) which is predicted by MixMD to be always conserved. Site B is often displaced, with only 18% of structures containing a water molecule at this location. (Note that this water is not occupied in apo structure PDB:1DG8 used to make the table for DHFR in the supplemental information.) For example, the amino group of methotrexate (PDB:1DF7, MTX) displaces this site, which is in agreement with the MixMD prediction that this site will be displaced by N-methylacetamide35. The inability of other groups to displace this site is illustrated in the binding mode of folic acid to DHFR. Folic acid is almost identical in composition to methotrexate, with differing substituents at two sites, but binds with a different orientation60. In methotrexate, a nitrogen (which occupies site B on the crystal structure) substitutes for an oxygen in folic acid. However, folic acid binds to DHFR with the pteridine ring flipped 180°, which results in the oxygen pointing in the opposite direction. This specificity is captured by the behavior of the probes in the simulations. Visualizing the N-methylacetamide occupancy by atom shows that the nitrogen is oriented in the direction known to be preferred from ligand-bound structures with the oxygen always positioned away from this site. Site C is another example of nitrogen displacing a water molecule. In this case, MixMD predicts that this site can also be displaced by N-methylacetamide, as well as acetate/methylammonium and isopropyl alcohol. Thus, not only can MixMD identify displaceable water sites, but can also identify specific functional groups capable of displacing a site.
Figure 5.

Dihydrofolate Reductase. Water density is shown at the 23 σ level, colored according to the probe type included in the simulation. Crystallographic waters from the apo structure (PDB:1DG835) that are within 3.5 Å of a ligand are shown for reference. Site A is a water that is found in 100% of comparable crystal structures, predicted to be conserved by MixMD. In site B, this water is seen in structures PDB:4KL9 and 4KLX. It is known to be displaced by nitrogen, and it is predicted by MixMD to be displaced by N-methylacetamide. Site C is a water site known to be displaced by nitrogen, predicted by MixMD to be displaced by N-methylacetamide, acetate/methylammonium, and isopropyl alcohol. (PDB:1DF735(MTX) and 1DG735(WRB))
β-lactamase
Apo β-lactamase contains a number of water sites which are experimentally known to be displaced in ligand-bound structures61, 62. This is consistent with MixMD predictions that these sites will be displaced by probe molecules. In addition to the displaceable water sites in β-lactamase, the MixMD simulations also predict the location of conserved waters, including the cluster of water molecules known to be important in stabilizing the Ω-loop63. However, there is one exception, as shown in Figure 6. Classic inhibitors of β-lactamase form a covalent attachment to the enzyme following nucleophilic attack of the β-lactam ring by a deprotonated serine. The carbonyl oxygen of the β-lactam ring displaces a water molecule, while a nearby conserved water molecule coordinated to Glu-166 is involved in hydrolysis of the β-lactam ring61. MixMD simulations predict both of these sites to be conserved. This discrepancy is likely due to the fact that our molecular dynamics simulations cannot account for the formation of covalent bonds between the protein and inhibitor, which therefore mimics the apo structure in which the serine is free to coordinate with the water molecule.
Figure 6.

β-lactamase. Water density is shown at the 23 σ level, colored according to the probe type included in the simulation. Crystallographic waters from the apo structure (PDB:1ZG436) that are within 3.5 Å of a ligand are shown for reference. Site A is known to be conserved and is well mapped by waters in our simulations. MixMD also correctly predicts many of the waters in the active site of β-lactamase as being displaced, but there is one known discrepancy. Site B is predicted to be conserved when it is actually displaced by a covalent bond between the ligand and protein. This is attributed to the inability to account for potential covalent modifications within an MD simulation. (PDB:1BT5-IM261, 1ERM-BJI62)
Neuraminidase
Upon ligand binding to neuraminidase, a few water sites are conserved near the binding site. For example, a cluster of water molecules, shown in Figure 7 site A, are found in 100% of homologous crystal structures. MixMD simulations predict the conservation of these sites in the presence of all probes tested, consistent with their high experimental conservation. On the other hand, a number of waters are displaced upon ligand binding, for instance by the carboxyl group of the ligand, as shown in Figure 7 site B37. MixMD predicts the displacement of these sites, as indicated by the lack of water density at this location and the presence of acetate density. Although other methods have been applied to neuraminidase, they require additional steps to generate comparable information. For example, neuraminidase has been previously studied by the JAWS method9. While the JAWS method was able to identify favorable and unfavorable hydration sites in the active site, the method requires the use of ligand-bound structures to identify water sites that would be displaced upon ligand binding. Our MixMD method does not require ligand-bound structures, and all of these simulations were initiated from apo structures. MixMD simulations could be easily extended to study sequence level changes. For example, neuraminidase variants are common, and show differing susceptibilities to inhibitors64. Interestingly, the number of water sites contained in the active site has been shown to vary depending on the mutant studied, and it has been suggested as one factor influencing the observed variations in binding affinity of inhibitors65. MixMD could potentially be used for further study of neuraminidase variants, to yield insight into the specific factors that mediate the observed water occupancy and variable binding affinities of inhibitors.
Figure 7.

Neuraminidase. Crystallographic waters (PDB:4HZV37) within 3.5 Å of any ligand are shown, along with water density from the MixMD simulations shown at the 23 σ level, colored according to the probe type included in the simulation. Site A shows a cluster of conserved water sites found in 100% of homologous structures, predicted by MixMD to be conserved. Site B shows water sites displaced by carboxyl of ligand (Zanamivir shown, PDB:4I00, ZMR37) and predicted by MixMD to be displaced. The inset figure shows the occupancy of the acetate probe which displaces these sites.
β-secretase
In the structure of β-secretase, the MixMD method identifies a conserved water site which is found in greater than 95% of crystal structures. The method also predicts the displacement of several water molecules known to be displaced in the majority of crystal structures. Interestingly, MixMD is able to predict the presence of a water molecule which bridges interactions between the ligand and protein in some cases, but is displaced by a ligand in others. In the apo-structure of BACE, this water molecule interacts with the two catalytic aspartates. As shown in Figure 8A, the amino group of inhibitors can displace this water site by mimicking this interaction, or this water site can be conserved to bridge interactions between the protein and ligand, as shown in Figure 8B. The MixMD simulations predict that this water site can be displaced by the acetate/methylammonium, N-methylacetamide, and pyrimidine probes (as evidenced by the lack of water occupancy from these simulations). This is consistent with known ligands that use an amino group (PDB:4RCD, 3LL) to displace the water site by interacting with both aspartates66. Our simulations modeled the two aspartates as deprotonated, which has been shown to be the preferred protonation states for a subset of BACE inhibitors67. Other inhibitors, including those which place a hydroxy group at this site, preferentially interact with BACE when one of the aspartates is protonated67, 68. The MixMD results generated from the simulations with doubly deprotonated aspartates are consistent with this, which predicted this site to be favorably conserved in the presence of isopropyl alcohol. This water site has also been previously analyzed with the WaterMap method to guide synthesis efforts69. One of the goals of that study was to develop BACE inhibitors that did not displace the catalytic water in order to reduce the number of hydrogen bonds present in the inhibitor and yield a ligand with more desirable drug-like properties. While the WaterMap method was successfully applied to explain SAR results, complementary structure-based drug design efforts were required. Alternatively, the MixMD method can be used, which allows users to predict the ease of displacement of a water site, while simultaneously predicting the location of favorable interactions of the probe molecules within the binding site. This information can then be used to identify favorable interactions that may be targeted with future ligands. Thus, cosolvent simulation methods, including MixMD, yield additional information compared to other methods that rely solely on water for predictions.
Figure 8.

β-Secretase. Crystallographic waters from the apo structure of BACE (PDB: 1W5038) and the bridging water in the ligand bound structure (PDB:4FM7, WAT90969) are shown for reference. A) MixMD predicts the displacement of the circled water site by acetate/methylammonium, N-methylacetamide, and pyrimidine probes. The ligand from PDB:4RCD66(3LL) is shown for comparison. Water density is shown at the 23 σ level, colored according to the probe type included in the simulation. The inset figure shows the methylammonium density at 150 σ. B) This site may also be conserved and bridge interactions between the ligand and protein, as predicted by the simulations with acetonitrile and isopropyl alcohol. The ligand from PDB:4FM769(OUP) is shown for comparison.
Thrombin
Upon ligand binding to thrombin, several water sites are displaced, as shown in Figure 939. MixMD simulations predict that these sites can be displaced, as indicated by the lack of water density in the figure. Interestingly, a number of water sites are observed in the MixMD simulations which are known to be involved in thrombin’s activity. Thrombin is allosterically regulated by a Sodium ion, whose binding site is connected to the active site via a water channel70, 71. One of the benefits of the MixMD methodology is the ability to contour occupancy at different levels, corresponding to a range of very high to moderate to low occupancy. While high σ levels in the presence of probe molecules were used as a cutoff for classifying water conservation, lower σ values still identify discrete water sites with occupancy greater than that of bulk water. When the water occupancy is visualized at lower occupancy levels, such as 5σ, several additional sites within the water channel of thrombin are identified, pointing to MixMD’s ability to identify not only absolutely conserved sites, but also water sites that will be occupied in the absence of bound ligands.
Figure 9.

Thrombin. Crystallographic waters within the active site (PDB:3U6939) are shown, along with the water density from the MixMD simulations at the 23 σ level, colored according to the probe type included in the simulation. Site A is a water site that is predicted to be always displaced, shown with a peptide-inhibitor. (PDB:3U8O39).
Penicillopepsin
Ligands of penicillopepsin displace a number of water sites, as shown in Figure 10. Near the active-site region, only one water site is predicted as being conserved (Figure 10 site B), while all other sites are predicted to be potentially displaceable. Aspartic proteases, such as penicillopepsin, unvaryingly have a water molecule that interacts with the two active aspartates and is involved in catalysis72. This location is shown at site A in Figure 10. However, this water may be displaced by inhibitors that interact with these aspartates. For instance, this water is displaced by the phosphonate portion of the ligand shown in Figure 1073. This is consistent with the MixMD predictions that this site will be displaced. As MixMD incorporates both charged and uncharged probe molecules, the method is able to predict the displacement of water sites that commonly bind charged ligands, as shown in the case of site A. Additionally, MixMD predicts the displacement of several other water sites, consistent with ligand-bound crystal structures which show the majority of water sites in this region to be displaced upon binding. MixMD also predicts the location of water sites which are known to be conserved, including the water located at site B. This water molecule is buried and participates in a network of interactions that are essential to stabilize the active site72. It is conserved in 100% of related structures of penicillopepsin as well as in structures of related aspartic proteases. Furthermore, disruption of this stabilizing network of interactions has been show to disrupt the active-site geometry in related enzymes72, illustrating the biological importance in conserving this water site.
Figure 10.

Penicillopepsin. Crystallographic waters (PDB:3APP40) within the active site are shown, along with the water density from the MixMD simulations at the 23 σ level, colored according to the probe type included in the simulation. Site A is a water displaced by phosphonate-containing ligand (PDB:1BXO, PP773), and it is predicted as displaceable by the MixMD simulations. Site B is an important water site found in 100% of related structures which participates in a network of stabilizing interactions. It is predicted as being conserved.
Conclusions
As shown in the examples above, the MixMD method consistently identifies water molecules that may be displaced by ligands as well as those that are conserved in crystal structures. Although ligands may be designed to displace a water site, this is not necessarily accompanied by a corresponding increase in binding affinity if the ligand does not adequately mimic the specific contacts previously made by the water molecule. Using the MixMD method, favorable binding sites on the protein’s surface are determined for multiple functional groups. This in turn allows for the prediction of conserved and displaceable water sites, while simultaneously determining which groups can successfully displace them. If GPU adapted molecular dynamics programs are used, MixMD simulations for a single system can be completed in as little as one day.
MixMD is able to identify specific groups that can displace a site and identify conserved water sites that play important roles and are involved in protein function. In addition, MixMD successfully identifies a displaceable water site that is predicted by other methods as being conserved, as shown in the results of HSP90. MixMD had one shortcoming in the β-lactamase case where a covalently attached ligand can displace a water molecule that was predicted to be non-displaceable. Efforts are currently underway to expand the available probe set to include additional groups, which is expected to extend MixMD’s predictive power. Overall, the MixMD method successfully classifies the displacement of water sites by common functional groups. These results may be used in the strategic design of ligands to determine which water sites should be conserved and which sites can be favorably displaced. Furthermore, MixMD results can also give insight into pockets that ligands may be most favorably extended into, by predicting sites that are favorably desolvated.
Supplementary Material
Acknowledgement
This work has been supported in part by the National Institutes of Health (R01 GM65372) and the University of Michigan’s MCubed program. SG thanks the Rackham Graduate School at the University of Michigan, Ann Arbor for a Rackham Research Grant to purchase computational resources.
Footnotes
Supporting Information
Figures labeled with the residue number from the apo crystal structure and tables of the MixMD occupancy values are given for each system.
References
- 1.Snyder P; Mecinovic J; Moustakas D; Thomas S; Harder M; Mack E; Lockett M; Héroux A; Sherman W; Whitesides G, Mechanism of the hydrophobic effect in the biomolecular recognition of arylsulfonamides by carbonic anhydrase. Proceedings of the National Academy of Sciences of the United States of America 2011, 108, 17889–17894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Levinson NM; Boxer SG, A conserved water-mediated hydrogen bond network defines bosutinib’s kinase selectivity. Nat Chem Biol 2014, 10, 127–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Michel J; Tirado-Rives J; Jorgensen W, Energetics of displacing water molecules from protein binding sites: consequences for ligand optimization. Journal of the American Chemical Society 2009, 131, 15403–15411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rossato G; Ernst B; Vedani A; Smiesko M, AcquaAlta: a directional approach to the solvation of ligand-protein complexes. Journal of chemical information and modeling 2011, 51, 1867–1881. [DOI] [PubMed] [Google Scholar]
- 5.Raymer ML; Sanschagrin PC; Punch WF; Venkataraman S; Goodman ED; Kuhn LA, Predicting conserved water-mediated and polar ligand interactions in proteins using a K-nearest-neighbors genetic algorithm. Journal of molecular biology 1997, 265, 445–64. [DOI] [PubMed] [Google Scholar]
- 6.Amadasi A; Spyrakis F; Cozzini P; Abraham D; Kellogg G; Mozzarelli A, Mapping the energetics of water-protein and water-ligand interactions with the “natural” HINT forcefield: predictive tools for characterizing the roles of water in biomolecules. Journal of molecular biology 2006, 358, 289–309. [DOI] [PubMed] [Google Scholar]
- 7.Garcia-Sosa AT; Mancera RL; Dean PM, WaterScore: a novel method for distinguishing between bound and displaceable water molecules in the crystal structure of the binding site of protein-ligand complexes. Journal of molecular modeling 2003, 9, 172–82. [DOI] [PubMed] [Google Scholar]
- 8.Ross G; Morris G; Biggin P, Rapid and accurate prediction and scoring of water molecules in protein binding sites. PloS one; 2012, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Michel J; Tirado-Rives J; Jorgensen W, Prediction of the water content in protein binding sites. The journal of physical chemistry. B 2009, 113, 13337–13346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Themis L, Inhomogeneous Fluid Approach to Solvation Thermodynamics. 1. Theory. The Journal of Physical Chemistry B 1998, 102. [Google Scholar]
- 11.Lazaridis T, Inhomogeneous Fluid Approach to Solvation Thermodynamics. 2. Applications to Simple Fluids. The Journal of Physical Chemistry B 1998, 102, 3542–3550. [Google Scholar]
- 12.Young T; Abel R; Kim B; Berne B; Friesner R, Motifs for molecular recognition exploiting hydrophobic enclosure in protein-ligand binding. Proceedings of the National Academy of Sciences of the United States of America 2007, 104, 808–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abel R; Young T; Farid R; Berne B; Friesner R, Role of the active-site solvent in the thermodynamics of factor Xa ligand binding. Journal of the American Chemical Society 2008, 130, 2817–2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beuming T; Farid R; Sherman W, High-energy water sites determine peptide binding affinity and specificity of PDZ domains. Protein science : a publication of the Protein Society 2009, 18, 1609–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pearlstein R; Hu Q-Y; Zhou J; Yowe D; Levell J; Dale B; Kaushik V; Daniels D; Hanrahan S; Sherman W; Abel R, New hypotheses about the structure-function of proprotein convertase subtilisin/kexin type 9: analysis of the epidermal growth factor-like repeat A docking site using WaterMap. Proteins 2010, 78, 2571–2586. [DOI] [PubMed] [Google Scholar]
- 16.Christopher H; Thijs B; Woody S, Hydration Site Thermodynamics Explain SARs for Triazolylpurines Analogues Binding to the A2A Receptor. ACS Medicinal Chemistry Letters 2010, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Robinson D; Sherman W; Farid R, Understanding kinase selectivity through energetic analysis of binding site waters. ChemMedChem 2010, 5, 618–627. [DOI] [PubMed] [Google Scholar]
- 18.Setny P; Baron R; McCammon JA, How Can Hydrophobic Association Be Enthalpy Driven? Journal of Chemical Theory and Computation 2010, 6, 2866–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baron R; Setny P; McCammon JA, Water in Cavity−Ligand Recognition. Journal of the American Chemical Society 2010, 132, 12091–12097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raman EP; MacKerell AD Jr., Spatial analysis and quantification of the thermodynamic driving forces in protein-ligand binding: binding site variability. Journal of the American Chemical Society 2015, 137, 2608–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guanglei C; Jason MS; Eric SM, SPAM: A Simple Approach for Profiling Bound Water Molecules. Journal of Chemical Theory and Computation 2013, 9. [DOI] [PubMed] [Google Scholar]
- 22.Seco J; Luque F; Barril X, Binding site detection and druggability index from first principles. Journal of medicinal chemistry 2009, 52, 2363–2371. [DOI] [PubMed] [Google Scholar]
- 23.Guvench O; MacKerell A, Computational fragment-based binding site identification by ligand competitive saturation. PLoS computational biology 2009, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alvarez-Garcia D; Barril X, Relationship between Protein Flexibility and Binding: Lessons for Structure-Based Drug Design. Journal of Chemical Theory and Computation 2014, 10, 2608–2614. [DOI] [PubMed] [Google Scholar]
- 25.Alvarez-Garcia D; Barril X, Molecular Simulations with Solvent Competition Quantify Water Displaceability and Provide Accurate Interaction Maps of Protein Binding Sites. Journal of medicinal chemistry 2014, 57, 8530–8539. [DOI] [PubMed] [Google Scholar]
- 26.Raman E; Yu W; Guvench O; Mackerell A, Reproducing crystal binding modes of ligand functional groups using Site-Identification by Ligand Competitive Saturation (SILCS) simulations. Journal of chemical information and modeling 2011, 51, 877–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raman E; Yu W; Lakkaraju S; Mackerell A, Inclusion of Multiple Fragment Types in the Site Identification by Ligand Competitive Saturation (SILCS) Approach. Journal of chemical information and modeling 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lexa K; Carlson H, Improving protocols for protein mapping through proper comparison to crystallography data. Journal of chemical information and modeling 2013, 53, 391–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lexa KW; Goh GB; Carlson HA, Parameter Choice Matters: Validating Probe Parameters for Use in Mixed-Solvent Simulations. Journal of chemical information and modeling 2014, 54, 2190–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ung PMU; Ghanakota P; Graham SE; Lexa KW; Carlson HA, Identifying binding hot spots on protein surfaces by mixed-solvent molecular dynamics: HIV-1 protease as a test case. Biopolymers 2016, 105, 21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lexa KW; Carlson HA, Full Protein Flexibility is Essential for Proper Hot-Spot Mapping. Journal of the American Chemical Society 2011, 133, 200–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ghanakota P; Carlson HA, Moving Beyond Active-Site Detection: MixMD Applied to Allosteric Systems. J Phys Chem B 2016, 120, 8685–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prodromou C; Roe SM; Piper PW; Pearl LH, A molecular clamp in the crystal structure of the N-terminal domain of the yeast Hsp90 chaperone. Nature structural biology 1997, 4, 477–82. [DOI] [PubMed] [Google Scholar]
- 34.Filippakopoulos P; Picaud S; Mangos M; Keates T; Lambert JP; Barsyte-Lovejoy D; Felletar I; Volkmer R; Muller S; Pawson T; Gingras AC; Arrowsmith CH; Knapp S, Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li R; Sirawaraporn R; Chitnumsub P; Sirawaraporn W; Wooden J; Athappilly F; Turley S; Hol WG, Three-dimensional structure of M tuberculosis dihydrofolate reductase reveals opportunities for the design of novel tuberculosis drugs. Journal of molecular biology 2000, 295, 307–23. [DOI] [PubMed] [Google Scholar]
- 36.Stec B; Holtz KM; Wojciechowski CL; Kantrowitz ER, Structure of the wild-type TEM-1 beta-lactamase at 1.55 A and the mutant enzyme Ser70Ala at 2.1 A suggest the mode of noncovalent catalysis for the mutant enzyme. Acta Crystallogr D Biol Crystallogr 2005, 61, 1072–9. [DOI] [PubMed] [Google Scholar]
- 37.Li Q; Qi J; Wu Y; Kiyota H; Tanaka K; Suhara Y; Ohrui H; Suzuki Y; Vavricka CJ; Gao GF, Functional and structural analysis of influenza virus neuraminidase N3 offers further insight into the mechanisms of oseltamivir resistance. Journal of virology 2013, 87, 10016–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Patel S; Vuillard L; Cleasby A; Murray CW; Yon J, Apo and inhibitor complex structures of BACE (beta-secretase). Journal of molecular biology 2004, 343, 407–16. [DOI] [PubMed] [Google Scholar]
- 39.Figueiredo AC; Clement CC; Zakia S; Gingold J; Philipp M; Pereira PJ, Rational design and characterization of D-Phe-Pro-D-Arg-derived direct thrombin inhibitors. PloS one 2012, 7, e34354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.James MN; Sielecki AR, Structure and refinement of penicillopepsin at 1.8 A resolution. Journal of molecular biology 1983, 163, 299–361. [DOI] [PubMed] [Google Scholar]
- 41.Vincent BC; Arendall WB; Jeffrey JH; Daniel AK; Robert MI; Gary JK; Laura WM; Jane SR; David CR, MolProbity: all-atom structure validation for macromolecular crystallography. Acta crystallographica. Section D, Biological crystallography 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ryde U, Molecular dynamics simulations of alcohol dehydrogenase with a four- or five-coordinate catalytic zinc ion. Proteins: Structure, Function, and Bioinformatics 1995, 21, 40–56. [DOI] [PubMed] [Google Scholar]
- 43.Holmberg N; Ryde U; Bulow L, Redesign of the coenzyme specificity in L-lactate dehydrogenase from bacillus stearothermophilus using site-directed mutagenesis and media engineering. Protein engineering 1999, 12, 851–6. [DOI] [PubMed] [Google Scholar]
- 44.Case DA, Darden TA, Cheatham TE, Simmerling CL, Wang J, Duke RE, Luo R, Walker RC, Zhang W, Merz KM, Roberts B, Hayik S, Roitberg A, Seabra G, Swails J, Goetz AW, Kollossvary I, Wong KF, Paesani F, Vanicek J, Wolf RM, Liu J, Wu X, Brozell SR, Steinbrecher T, Gohlke H, Cai Q, Ye X, Wang J, Hsieh MJ, Cui G, Roe DR, Mathews DH, Seetin MG, Salomon-Ferrer R, Sagui C, Babin V, Luchko T, Gusarov S, Kovalenko A, and Kollman PA AMBER 12, University of California, San Francisco: 2012. [Google Scholar]
- 45.Andrea TA; Swope WC; Andersen HC, The role of long ranged forces in determining the structure and properties of liquid water. The Journal of Chemical Physics 1983, 79, 4576–4584. [Google Scholar]
- 46.PyMOL 1.8.4.0, Schrodinger: 2016.
- 47.Damm KL; Carlson HA, Gaussian-weighted RMSD superposition of proteins: a structural comparison for flexible proteins and predicted protein structures. Biophys J 2006, 90, 4558–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sanschagrin PC; Kuhn LA, Cluster analysis of consensus water sites in thrombin and trypsin shows conservation between serine proteases and contributions to ligand specificity. Protein Sci 1998, 7, 2054–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Institute SAS, Inc.: Cary, N.C. [Google Scholar]
- 50.Trepel J; Mollapour M; Giaccone G; Neckers L, Targeting the dynamic HSP90 complex in cancer. Nature reviews. Cancer 2010, 10, 537–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kung PP; Sinnema PJ; Richardson P; Hickey MJ; Gajiwala KS; Wang F; Huang B; McClellan G; Wang J; Maegley K; Bergqvist S; Mehta PP; Kania R, Design strategies to target crystallographic waters applied to the Hsp90 molecular chaperone. Bioorganic & medicinal chemistry letters 2011, 21, 3557–62. [DOI] [PubMed] [Google Scholar]
- 52.Millson SH; Chua CS; Roe SM; Polier S; Solovieva S; Pearl LH; Sim TS; Prodromou C; Piper PW, Features of the Streptomyces hygroscopicus HtpG reveal how partial geldanamycin resistance can arise with mutation to the ATP binding pocket of a eukaryotic Hsp90. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 2011, 25, 3828–37. [DOI] [PubMed] [Google Scholar]
- 53.Sharp SY; Roe SM; Kazlauskas E; Cikotiene I; Workman P; Matulis D; Prodromou C, Co-crystalization and in vitro biological characterization of 5-aryl-4-(5-substituted-2–4-dihydroxyphenyl)-1,2,3-thiadiazole Hsp90 inhibitors. PloS one 2012, 7, e44642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Haider K; Huggins D, Combining solvent thermodynamic profiles with functionality maps of the Hsp90 binding site to predict the displacement of water molecules. Journal of chemical information and modeling 2013, 53, 2571–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Obermann WM; Sondermann H; Russo AA; Pavletich NP; Hartl FU, In vivo function of Hsp90 is dependent on ATP binding and ATP hydrolysis. The Journal of cell biology 1998, 143, 901–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chung CW; Coste H; White JH; Mirguet O; Wilde J; Gosmini RL; Delves C; Magny SM; Woodward R; Hughes SA; Boursier EV; Flynn H; Bouillot AM; Bamborough P; Brusq JM; Gellibert FJ; Jones EJ; Riou AM; Homes P; Martin SL; Uings IJ; Toum J; Clement CA; Boullay AB; Grimley RL; Blandel FM; Prinjha RK; Lee K; Kirilovsky J; Nicodeme E, Discovery and characterization of small molecule inhibitors of the BET family bromodomains. Journal of medicinal chemistry 2011, 54, 3827–38. [DOI] [PubMed] [Google Scholar]
- 57.Vollmuth F; Blankenfeldt W; Geyer M, Structures of the dual bromodomains of the P-TEFb-activating protein Brd4 at atomic resolution. J Biol Chem 2009, 284, 36547–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhao L; Cao D; Chen T; Wang Y; Miao Z; Xu Y; Chen W; Wang X; Li Y; Du Z; Xiong B; Li J; Xu C; Zhang N; He J; Shen J, Fragment-based drug discovery of 2-thiazolidinones as inhibitors of the histone reader BRD4 bromodomain. Journal of medicinal chemistry 2013, 56, 3833–51. [DOI] [PubMed] [Google Scholar]
- 59.Ember SW; Zhu JY; Olesen SH; Martin MP; Becker A; Berndt N; Georg GI; Schonbrunn E, Acetyl-lysine binding site of bromodomain-containing protein 4 (BRD4) interacts with diverse kinase inhibitors. ACS Chem Biol 2014, 9, 1160–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mastropaolo D; Camerman A; Camerman N, Folic Acid: Crystal Structure and Implications for Enzyme Binding. Science 1980, 210, 334–336. [DOI] [PubMed] [Google Scholar]
- 61.Structural Basis for Clinical Longevity of Carbapenem Antibiotics in the Face of Challenge by the Common Class A Beta-Lactamases from Antibiotic-Resistant Bacteria. TO BE PUBLISHED.
- 62.Ness S; Martin R; Kindler AM; Paetzel M; Gold M; Jensen SE; Jones JB; Strynadka NCJ, Structure-Based Design Guides the Improved Efficacy of Deacylation Transition State Analogue Inhibitors of TEM-1 β-Lactamase. Biochemistry 2000, 39, 5312–5321. [DOI] [PubMed] [Google Scholar]
- 63.Bös F; Pleiss J, Conserved Water Molecules Stabilize the Ω-Loop in Class A β-Lactamases. Antimicrobial agents and chemotherapy 2008, 52, 1072–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Samson M; Pizzorno A; Abed Y; Boivin G, Influenza virus resistance to neuraminidase inhibitors. Antiviral Research 2013, 98, 174–185. [DOI] [PubMed] [Google Scholar]
- 65.Vergara-Jaque A; Poblete H; Lee EH; Schulten K; González-Nilo F; Chipot C, Molecular Basis of Drug Resistance in A/H1N1 Virus. Journal of chemical information and modeling 2012, 52, 2650–2656. [DOI] [PubMed] [Google Scholar]
- 66.Dineen TA; Chen K; Cheng AC; Derakhchan K; Epstein O; Esmay J; Hickman D; Kreiman CE; Marx IE; Wahl RC; Wen PH; Weiss MM; Whittington DA; Wood S; Fremeau RT Jr.; White RD; Patel VF, Inhibitors of beta-Site Amyloid Precursor Protein Cleaving Enzyme (BACE1): Identification of (S)-7-(2-Fluoropyridin-3-yl)-3-((3-methyloxetan-3-yl)ethynyl)-5’H-spiro[chromeno[ 2,3-b]pyridine-5,4’-oxazol]-2’-amine (AMG-8718). Journal of medicinal chemistry 2014, 57, 9811–31. [DOI] [PubMed] [Google Scholar]
- 67.Barman A; Prabhakar R, Protonation States of the Catalytic Dyad of β-Secretase (BACE1) in the Presence of Chemically Diverse Inhibitors: A Molecular Docking Study. Journal of chemical information and modeling 2012, 52, 1275–1287. [DOI] [PubMed] [Google Scholar]
- 68.Ghosh AK; Kumaragurubaran N; Hong L; Kulkarni SS; Xu X; Chang W; Weerasena V; Turner R; Koelsch G; Bilcer G; Tang J, Design, synthesis, and X-ray structure of potent memapsin 2 (beta-secretase) inhibitors with isophthalamide derivatives as the P2-P3-ligands. Journal of medicinal chemistry 2007, 50, 2399–407. [DOI] [PubMed] [Google Scholar]
- 69.Brodney M; Barreiro G; Ogilvie K; Hajos-Korcsok E; Murray J; Vajdos F; Ambroise C; Christoffersen C; Fisher K; Lanyon L; Liu J; Nolan C; Withka J; Borzilleri K; Efremov I; Oborski C; Varghese A; O’Neill B, Spirocyclic sulfamides as β-secretase 1 (BACE-1) inhibitors for the treatment of Alzheimer’s disease: utilization of structure based drug design, WaterMap, and CNS penetration studies to identify centrally efficacious inhibitors. Journal of medicinal chemistry 2012, 55, 9224–9239. [DOI] [PubMed] [Google Scholar]
- 70.Wells CM; Di Cera E, Thrombin is a sodium ion activated enzyme. Biochemistry 1992, 31, 11721–11730. [DOI] [PubMed] [Google Scholar]
- 71.Di Cera E; Guinto ER; Vindigni A; Dang QD; Ayala YM; Wuyi M; Tulinsky A, The Na+ Binding Site of Thrombin. Journal of Biological Chemistry 1995, 270, 22089–22092. [DOI] [PubMed] [Google Scholar]
- 72.Prasad BVLS; Suguna K, Role of water molecules in the structure and function of aspartic proteinases. Acta Crystallographica Section D 2002, 58, 250–259. [DOI] [PubMed] [Google Scholar]
- 73.Khan AR; Parrish JC; Fraser ME; Smith WW; Bartlett PA; James MNG, Lowering the Entropic Barrier for Binding Conformationally Flexible Inhibitors to Enzymes. Biochemistry 1998, 37, 16839–16845. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.