

SUMMARY
Rhinovirus is a leading cause of acute respiratory infections and asthma attacks, but infections are also frequently cleared from the nasal mucosa without causing symptoms. We sought to better understand host defense against rhinovirus by investigating antiviral defense in primary human nasal and bronchial airway epithelial cells cultured ex vivo. Surprisingly, upon rhinovirus infection or RIG-I stimulation, nasal-derived epithelial cells exhibited much more robust antiviral responses than bronchial-derived cells. Conversely, RIG-I stimulation triggered more robust activation of the NRF2-dependent oxidative stress response in bronchial cells compared to nasal cells. NRF2 activation dampened epithelial antiviral responses, whereas NRF2 knockdown enhanced antiviral responses and was protective during rhinovirus infection. These findings demonstrate a tradeoff in epithelial defense against distinct types of airway damage, namely, viral versus oxidative, and reveal differential calibration of defense responses in cells derived from different airway microenvironments.
In Brief
Airway epithelial cells form the first line of defense against harmful substances that enter the airway. Mihaylova et al. show that viral RNA detection triggers both the NRF2-mediated oxidative stress response and the antiviral interferon response in epithelial cells and demonstrates a tradeoff between these defense responses.
Graphical Abstract
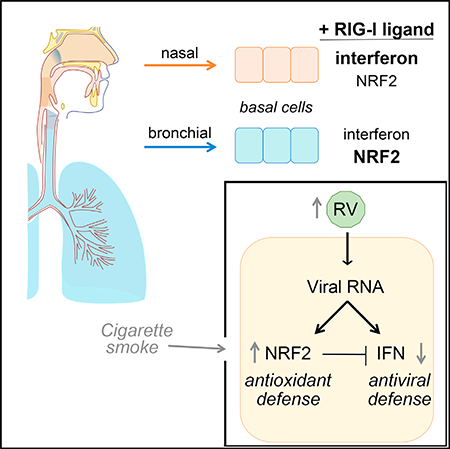
INTRODUCTION
Respiratory virus infections cause an estimated 500 million colds per year in the US and contribute to the roughly 2 million annual hospitalizations for respiratory illness (Fendrick et al., 2003; NIAID, 2001; Pfunter et al., 2013) However, recent evidence suggests that presence of respiratory viruses in the nasal passages is even more common but that viruses are often cleared without causing symptoms (Bosch et al., 2013; Jartti et al., 2008). For example, in a recent family surveillance study, respiratory viruses were detected on average 7.3 weeks per year per person, but almost half of infections were asymptomatic (44%; Byington et al., 2015). These data suggest that, in many cases, airway defense responses enable efficient local viral clearance without engaging defenses that lead to symptoms, such as excessive inflammation and mucus production. To better understand the molecular basis of antiviral defense responses in the airway, we have focused on interactions between epithelial cells and rhinovirus (RV), the most frequent cause of colds, asthma attacks, and exacerbations of chronic airway disease (Gern, 2010). Multiple lines of evidence indicate that innate defenses of airway epithelial cells can efficiently block RV replication and clear infection at its earliest stages. Airway epithelial cells are the target cells within which RV replicates, but RV replication within these cells can trigger powerful innate defense responses, including induction of type I and type III interferons (IFNs) and interferon-stimulated genes (ISGs), programmed cell death, and RNaseL activity, all of which can block RV replication (Foxman et al., 2015, 2016; Slater et al., 2010; Wang et al., 2009). Furthermore, epithelial cell interferon responses triggered by RNA virus infection are attenuated in several patient groups susceptible to severe RV illness, including asthmatics and smokers, further supporting the idea that epithelial cell defenses are critically important for optimal control of RV infection (Contoli et al., 2006; Jaspers et al., 2010; Wu et al., 2016; Wark et al., 2005).
RV first enters the respiratory tract in the nasal passages, but cells and cell lines of bronchial origin are by far more commonly used as experimental tools. Here, we sought to compare antiviral responses of primary epithelial cells cultured from the nasal or bronchial airway mucosa of healthy donors. We were particularly interested in examining nasal epithelial cells because previous work showed that incubating primary airway cells at cool temperature, mimicking the conditions of the nasal passages, dampens antiviral responses triggered by cytoplasmic RNA (Foxman et al., 2015, 2016). This finding suggests that nasal and bronchial epithelial cells might require different calibration of innate responses to maintain effective antiviral defense in distinct in vivo anatomical microenvironments.
Here, we report fundamental differences in the responses of nasal and bronchial primary human epithelial cells to rhinovirus infection or direct stimulation of the viral RNA sensor RIG-I. We studied primary nasal- or bronchial-derived airway epithelial cells using a culture system that models basal cells, the regional progenitor cells of the airway epithelium central to epithelial defense and repair following mucosal injury (Rock et al., 2010). In cells derived from both sites, RIG-I stimulation triggered activation of well-characterized signaling pathways, mediating protective responses against both viral infection and intracellular oxidative stress. Interestingly, however, nasal cells showed a more predominant interferon response, whereas bronchial cells exhibited a more predominant oxidative stress response. Further investigation revealed evidence for antagonism between activity of the NRF2-mediated oxidative stress response and RIG-Idependent interferon and ISG defense in epithelial cells and a surprising cytoprotective effect of NRF2 knockdown during RV infection due to decreased viral replication. Based on these findings, we propose a model in which epithelial cell-intrinsic defense mechanisms are tailored for different airway microenvironments to optimize airway protection.
RESULTS
Antiviral Responses of Primary Airway Epithelial Cells Track with Site of Origin in the Respiratory Tract
Previous work showed that incubating primary airway epithelial cells at cool temperature (33°C), mimicking the conditions of the nasal passages, diminished antiviral responses triggered by cytoplasmic RNA, including induction of type I and type III interferons (Foxman et al., 2015, 2016). These findings suggest that nasal epithelial cells may require adaptations to maintain robust antiviral defense within their naturally cooler local microenvironment in vivo. To examine this hypothesis, we obtained primary nasal or bronchial epithelial cells from healthy donors (commercially; see STAR Methods), cultured them on collagen under conditions that promote a basal cell phenotype, and then infected with rhinovirus 1B (RV1B). All experiments were performed at low passage number (P3 or fewer). Compared to bronchial epithelial cells, nasal-derived epithelial cells displayed much more robust secretion of the type III interferon, IFNλ1, following rhinovirus infection at 37°C, at a time point when viral load was equivalent in both cell types (Figures 1A and S1). Consistent with previous studies, when cells were incubated at 33°C, IFNλ1 secretion was greatly reduced (Figure S1). Interestingly, the low but detectable level of IFNλ1 secreted by nasal cells at nasal temperature (33°C) was comparable to levels observed in bronchial epithelial cells at lung temperature (37°C; Figure S1). Because the levels of virus replication may contribute to the difference in IFNl1 levels between the two cell types, we next performed experiments using a non-replicating RIG-I ligand, stem loop RNA (SLR), SLR14, and a short 5ʹ-triphosphorylated RNA ligand of the cytoplasmic innate immune sensor RIG-I (Linehan et al., 2018). Following transfection of cells with SLR14, we observed more robust IFNλ1 secretion and induction of mRNA for IFNλ1, IFNβ, and the interferon-stimulated gene OAS1 in nasal cells compared to bronchial cells when both cell types were incubated at the same temperature (Figures 1B–1H).
Figure 1. IFNλ1 and ISG Induction in Nasal and Bronchial Epithelial Cells following Rhinovirus Infection or SLR14 Exposure.

(A) Primary nasal and bronchial epithelial cells were inoculated with RV-1B, MOI 0.1, and incubated at 37C for 48 hr, at which time supernatants were collected for ELISA. Bars show IFNl1 protein in supernatant.
(B–H) Primary nasal and bronchial epithelial cells were transfected with RIG-I ligand SLR14 for 1 hr and then medium was added and cells were incubated at 37C. Supernatants were collected for ELISA, and cells were collected for RNA isolation and qRT-PCR at the time points shown.
(B) IFNλ1 in the supernatant was measured by ELISA at 24 hr.
(C and D) Fold change in mRNA for IFNλ1 (C) or IFNβ (D) at 24 hr.
(E) IFNλ1 in the supernatant was measured by ELISA at 0–8 hr.
(F–H) Fold change in mRNA for IFNλ1 (F), IFNβ (G), and Oas1(H) at time points 0–8 hr post-stimulation. Bars represent mean and SD of 2 or 3 replicate experimental wells per condition. mRNA level is plotted relative to the level in resting bronchial cells (t = 0 hr).
Data are representative of at least 3 independent experiments with airway epithelial cells of each type from two or more different donors. Significant differences between nasal and bronchial cell levels by paired t test are shown with asterisks: ***p < 0.0005; **p < 0.005; and *p < 0.05.
See also Figure S1.
To assess whether differences in nasal and bronchial cell responses to RIG-I ligand were due to differences in transfection efficiency, we created a modified SLR14 labeled with Alexa 488 (SLR14–488). Flow cytometric analysis of nasal and bronchial cells following transfection with SLR14–488 revealed an increase in fluorescence to an equivalent degree for both cell types, indicating equivalent transfection efficiency (Figures S1C and S1D). Consistent with results seen with unlabeled SLR14 (Figure 1), SLR14–488 stimulated greater induction of mRNAs encoding IFNλ1 and the interferon stimulated gene IFIT2 in nasal cells compared to bronchial cells (Figures S1E and S1F). These findings indicate that differences between nasal and bronchial-derived cells in interferon and ISG induction are not due to differences in transfection efficiency.
RIG-I Stimulation Triggers Differential Activation of the Antiviral Interferon Response and the NRF2-Dependent Oxidative Stress Response in Nasal and Bronchial Cells
To better understand the differences in the response to RIG-I stimulation in epithelial cells cultured from nasal or bronchial sites, we performed RNA sequencing (RNA-seq) to compare the transcriptomes of resting and SLR14-stimulated cells. First, we examined resting gene expression in both cell types (Figure S2). We observed many commonalities and some differences in resting gene expression (Figure S2A). Both nasal and bronchial cells expressed lineage markers of airway basal cells, the self-renewing regional progenitor cells of the airway epithelium (Figure S2B). This finding is consistent with previous studies showing that the epithelial cells that proliferate from primary airway mucosa in conventional culture have a basal cell phenotype, with expression of basal cell lineage markers KRT5 and TP63 (Hackett et al., 2011; Rock et al., 2010). Examination of transcripts for RIG-I and other signaling molecules involved in innate immune recognition of RNA viruses revealed a trend toward equal or slightly higher expression levels in bronchial cells at rest compared to nasal cells (Figures S2C and S2D), therefore did not provide an explanation for the phenotype shown in Figure 1. Ingenuity pathway analysis revealed some small differences in pathway activation at rest but did not reveal a clear-cut reason for the greater interferon response in nasal cells following response to RIG-I stimulation (Figure S2E).
To assess whether the low-passage primary cells used in this study retained characteristics specific to the site of origin, we examined region-specific mRNA biomarkers. Previous work showed that regional progenitor cells derived from different airway regions retain gene expression patterns reflecting the site of origin within the respiratory tract, and furthermore that progenitor cells can proliferate and differentiate to form a differentiated airway epithelium with characteristics of the site of origin when reintroduced into a 3D matrix (Kumar et al., 2011). Using published microarray data (GSE32606), we identified the top 10 differentially expressed genes in nasal versus trachealderived airway progenitor cells and examined expression of these transcripts in cells used in this study. The differential expression pattern in our RNA-seq data comparing nasal and bronchial-derived cells largely mirrored the pattern seen previously in nasal versus tracheal-derived regional progenitor cells (Figures S2F and S2G). Next, we performed qRT-PCR on nasal and bronchial cells from different donors used in this study and found enrichment of the nasal-associated biomarker FOXG1 in nasal cells from different donors and higher expression of the tracheal and bronchial-enriched mRNA SERPINF1 in bronchial cells from different donors (Figure S2H). These results indicate that nasal and bronchial-derived cells cultured under the conditions used in this study retain gene expression patterns reflective of the site of origin within the respiratory tract.
Next, we compared the transcriptional changes observed in nasal or bronchial cells following stimulation with the RIG-I ligand SLR14 (Figure 2). Consistent with the known role of RIG-I like receptors (RLRs), stimulation with SLR14 led to enrichment in transcripts associated with the antiviral response, including “interferon signaling,” “activation of IRF by cytosolic PRRs,” and “role of PRR in recognition of bacteria and viruses” (Figures 2A and 2B). Interestingly, the other top pathway enriched by RIG-I stimulation was the NRF2-mediated oxidative stress response (Figures 2A and 2B). NRF2 is a transcription factor that is activated by oxidative stress in the cytosol, leading to transcription of diverse targets involved in neutralizing reactive oxygen species and restoring homeostasis (Suzuki and Yamamoto, 2015). Notably, transcripts related to interferon signaling dominated the response to SLR14 in nasal cells, whereas transcripts related to the NRF2 pathway were more significantly enriched in bronchial cells (Figures 2B–2D). As shown on radar plots representing mean fragments per kilobase mapped (FPKM), overall mRNA levels of canonical ISGs were strongly induced and more highly expressed in nasal cells compared to bronchial cells (Figure 2C), whereas transcripts encoding antioxidant enzymes, a subset of NRF2 targets, were more highly expressed in bronchial cells compared to nasal cells following RIG-I stimulation (Figure 2D). A more complete list of enriched pathways and associated transcripts is shown in Table S1. The effects of SLR14 treatment were not predictable from the resting transcriptomes, which showed a slight enrichment for both pathways in bronchial cells at rest compared to resting nasal cells (Figure S2E).
Figure 2. Transcriptome Changes in Response to RIG-I Ligand in Human Nasal and Bronchial Airway Epithelial Cells.

RNA-seq was performed on RNA isolated from two replicate wells of stimulated and unstimulated nasal or bronchial epithelial cells (total of eight samples) following SLR14 stimulation (for 1 hr) followed by incubation for 7 hr at 37°C. Libraries were prepared for paired-end RNA sequencing from two replicate samples per condition.
(A) Dot plot depicts change in expression levels for all transcripts in stimulated versus unstimulated cells. Dots outside the gray box represent tran-scripts significantly increased (235 for bronchial; 533 for nasal) or decreased (47 for bronchial; 217 for nasal) in response to SLR14 stimulation (Log2FC > 1 or Log2FC < −1; p-adj < 0.05).
(B) Bar graph shows p values associated with top four ingenuity pathways enriched in response to SLR14 stimulation in both cell types (Z score > 1; p < 0.01).
(C and D) Radar plots show average fragments per kilobase mapped (FPKM) for transcripts contributing to the “interferon signaling” ingenuity pathway (C) or for antioxidant enzymes associated with the NRF2-mediated oxidative stress response ingenuity pathway (D) in SLR14-stimulated (solid line) and unstimulated (dashed line) cells. Gridlines in (C) range from 0 to 500 FPKM (intervals of 100) and gridlines in (D) range from 0 to 200 FPKM (intervals of 50). Throughout, orange lines represent nasal cells and black lines represent bronchial cells.
NRF2-Mediated Oxidative Stress Response Antagonizes Epithelial Antiviral Defense and Promotes Rhinovirus Replication
Previous studies have reported that NRF2 activation during viral infection in macrophages and dendritic cells is associated with decreased inflammatory and antiviral responses (Ahmed et al., 2017; Olagnier et al., 2014; Yageta et al., 2011). This work, combined with the observed relatively lower antiviral response and greater NRF2-mediated response in bronchial cells compared to nasal cells (Figure 2), suggested that NRF2 activation in airway epithelial cells might be antagonizing activation of RIG-I-dependent antiviral responses. To test this hypothesis, we asked whether NRF2 knockdown in bronchial epithelial cells increased RIG-I-dependent interferon and ISG induction. NRF2 knockdown enhanced induction of mRNA encoding IFNl1 and the ISG IFIT2 following SLR14 exposure (Figures 3A–3C). As expected, knockdown of mitochondrial antiviral signaling protein (MAVS), an essential signaling adaptor downstream of RIG-I, abrogated SLR14-dependent antiviral responses (Figures 3A–3C). Next, we tested the effect of NRF2 activation on the robust SLR14-triggered interferon and ISG responses observed in nasal epithelial cells. To do this, we pretreated cells overnight with the well-characterized NRF2 activator sulforaphane prior to RIG-I stimulation and then stimulated cells with SLR14 and assessed gene expression after 5 hr. Sulforaphane (SULF) pretreatment significantly reduced interferon and ISG induction upon subsequent stimulation with SLR14 (Figures 3D and 3E). Sulforaphane pretreatment led to sustained NRF2 activation, as indicated by induction of an NRF2-regulated gene, glutamate-cysteine ligase catalytic subunit (GCLC), whereas SLR14 did not induce GCLC mRNA under these conditions (Figure 3F). These results indicate that NRF2 activity antagonizes interferon induction in both types of airway cells.
Figure 3. Effect of NRF2 Knockdown or NRF2 Activation on Interferon and ISG Response to RIG-I Ligand SLR14.

(A and B) Primary human bronchial epithelial cells were transfected with siRNA targeting NRF2 or MAVS or with control siRNA (RNA-induced silencing complex [RISC]-free). After recovery for four days, cells were transfected with the RIG-I ligand SLR14 and then incubated for 6 hr at 37°C, followed by RNA isolation and qRT-PCR for mRNA encoding IFNλ1 (A) and IFIT2 (B). Significant difference between transcript levels in control siRNA-treated and NRF2-siRNA-treated cells by unpaired t test is indicated with asterisks (*p = 0.02; **p = 0.003).
(C) qRT-PCR was also performed on unstimulated cells following NRF2 or MAVS knockdown to check knockdown efficiency.
(D–F) Primary human nasal epithelial cells were pretreated with 10 μM sulforaphane (SULF) for 18 hr. After 3 hr recovery in medium only, cells were transfected with SLR14. After 5 hr incubation at 37°C, cells were collected for RNA isolation and qRT-PCR for mRNA encoding IFNλ1 (D), IFIT2 (E), or GCLC
(F). Graph shows untreated cells with no stimulation (Ctrl) or SLR14 exposure (SLR14) or sulforaphane-pretreated cells with no stimulation (SULF/Ctrl) or SLR14 exposure (SULF/SLR14). Significant differences between control and SULF pretreated cells by unpaired t test are indicated (#p < 0.0001). Bars show mean and SD of 2–4 replicate experimental wells per condition. Graph titles indicate mRNA assessed by qRT-PCR. Results are representative of at least three independent experiments.
See also Figure S4.
NRF2 Knockdown within Host Cells Decreases Rhinovirus Replication
The observations reported here indicate that, within airway epithelial cells, NRF2 activity antagonizes RIG-I-mediated antiviral signaling. Previous work has shown that alteration in signaling by RIG-I-like receptors can have profound effects on the outcome of RV infection in host cells with robust interferon responses (Foxman et al., 2015). Therefore, we sought to probe the effect of modulating NRF2 activity within host cells on RV replication. First, we targeted NRF2 activity using small interfering RNA (siRNA) knockdown in nasal epithelial cells and then examined rhinovirus amplification from a low MOI. Strikingly, at 40 hr post-infection, viral titer was >10-fold higher in supernatants from control cells than NRF2 knockdown cells and virus-induced cytopathic effect was significantly more advanced in control cells than knockdown cells (Figures 4A–4C). NRF2 knockdown cells exhibited significantly higher expression of the ISG IFIT2 and significantly lower levels of mRNAs encoding NRF2 and the NRF2 target NQO1 compared to control cells (Figures 4D–4F). These findings indicate that NRF2 knockdown cells are protected from RV replication, consistent with the observed enhancement in ISG induction (Figure 4D).
Figure 4. Effect of NRF2 Activity on RV1B Replication in Primary Human Nasal Epithelial Cells.

For a Figure360 author presentation of Figure 4, see https://doi.org/10.1016/j.celrep.2018.08.033.
(A–F) Primary human nasal epithelial cells (HNECs) were transfected with siRNA targeting NRF2 or RISC-free negative control siRNA and allowed to recover for 48 hr and then infected with RV-1B, MOI 0.05 at 33°C.
(A) Forty hours post-infection, supernatants were collected for viral titer measurement by plaque assay as shown.
(B and C) Micrographs show appearance of cells treated with control siRNA (B) or siRNA targeting NRF2 (C) at 40 hr post-infection.
(D–F) Relative expression of mRNA encoding IFIT2
(D), NQO1 (E), and NRF2 (F) in uninfected or RV1B infected cells, as assessed by qRT-PCR.
(G) Viral titer in supernatants of nasal epithelial cells 40 hr post-infection with RV1B MOI 0.05 at 33°C, with or without exposure 2% CSE.
(H and I) Nasal epithelial cells were stimulated with SLR14 and then incubated for 3 hr with medium only or medium containing 2% or 4% CSE. RNA was isolated and levels of mRNA encoding IFIT2
(H) and NQO1 (I) were assessed by qRT-PCR. In all panels, bars show mean and SEM of 3–6 replicate experimental wells per condition. Graphs show mRNA level relative to level in control, mocktreated cells. Significant differences by unpaired t test are indicated with asterisks: *p < 0.05; **p < 0.005; and ***p < 0.0001. The scale bar represents 50 μm.
See also Figures S3 and S4.
Next, we sought to test the effect of enhancing NRF2 activity in primary nasal epithelial cells by mimicking a physiological source of NRF2 activation in the airway, exposure to cigarette smoke. Airway epithelial cells from smokers show enhanced expression of NRF2 target genes compared to non-smokers (Spira et al., 2004), and enrichment of NRF2 targets is also observed upon stimulation of cultured bronchial epithelial cells in vitro with cigarette smoke extract (Pickett et al., 2010; Proud et al., 2012). To test the effect of NRF2 activation in primary human nasal epithelial cells on RV1B replication, we exposed cells to cigarette smoke extract (CSE) and assessed RV amplification from a low MOI following incubation at 33C for 40 hr. We observed a significant effect on RV1B replication, which doubled in cells exposed to CSE (Figure 4G). CSE exposure was toxic to NRF2 knockdown (KD) cells (Figure S3), consistent with the known importance of NRF2-dependent responses for cell survival of oxidative stress and cigarette smoke exposure in particular (Ma, 2013). Therefore, although we could not directly test the role of NRF2 in CSE-dependent increase in RV replication using knockdown, we explored the hypothesis that CSE exposure activates NRF2 and concomitantly suppresses antiviral interferon responses by testing the effect of CSE exposure on the basal and SLR14-induced expression of IFIT2 and NQO1 in these cells. We observed that CSE exposure increases expression of the NRF2 target NQO1 and decreases SLR14-dependent IFIT2 expression (Figures 4H and 4I). These findings are consistent with previous studies showing that CSE exposure leads to a decrease in ISG induction and a modest increase in viral replication following RV infection of human bronchial epithelial cells (Eddleston et al., 2011; Proud et al., 2012). Together, these findings suggest that one mechanism whereby CSE exposure may promote RV replication is through antagonism of the interferon response by NRF2 activation. Overall, our findings support a model in which tissue-specific set points and environmental factors decrease or increase the level of NRF2 activation in RV host cells, which in turn promotes or antagonizes the antiviral response of airway epithelial cells (Figure S4).
DISCUSSION
Airway epithelial cells provide frontline defense against a variety of potentially harmful substances that enter the airway from the environment, including respiratory viruses and diverse substances that can cause oxidative damage. We found that stimulation of the innate immune sensor RIG-I within airway epithelial cells activates two central mechanisms that protect against these sources of damage: the antiviral interferon response (greater activation in nasal cells than in bronchial cells) and the NRF2-mediated response to oxidative stress (greater activation in bronchial cells compared to nasal cells). Here, we present evidence that NRF2 activation antagonizes the antiviral interferon response in the airway epithelium and evidence for cell intrinsic regulation (i.e., innate differences between nasal and bronchial cells) and environmental regulation (i.e., exposure to cigarette smoke that triggers NRF2 activation) of the balance between these two defense mechanisms in airway epithelial cells.
The antagonism we observed between NRF2 activity and interferon and ISG induction in airway epithelial cells fits with antagonism of immune defense by NRF2 observed in other models. NRF2 activity was observed to suppress the interferon response and virus-induced apoptosis in a study of Dengue virus infection in dendritic cells (Olagnier et al., 2014), and NRF2 knockout led to exaggerated IRF3 and nuclear factor kB (NF-kB) activation in mouse models of sepsis (Thimmulappa et al., 2006). NRF2 activation has been shown to dampen production of NF-kB-dependent pro-inflammatory cytokines in diverse settings, and in fact, NRF2 activators are currently used clinically as anti-inflammatory agents (Suzuki and Yamamoto, 2017). Reactive oxygen species (ROS) promote cellintrinsic innate antiviral signaling, including RIG-I signaling (Koarai et al., 2010; Tal et al., 2009), and neutralization of ROS has been proposed to be the mechanism whereby NRF2 activity suppresses antiviral and pro-inflammatory cytokine production (Ma, 2013). Interestingly, ROS may enhance innate immune signaling in part by effects on nucleic acid ligands. For example, oxidation of cytosolic DNA was shown to enhance stimulator of interferon genes (STING)-dependent innate immune signaling due to decreased degradation of oxidized DNA by TREX1 (Gehrke et al., 2013). It will be interesting to explore how oxidation of RNA ligands or other components of the RIG-I signaling pathway might influence innate immune signaling in future studies. In addition to effects on neutralization of ROS, an additional mechanism of NRF2-dependent suppression of innate immune signaling was proposed recently, in which chromatin immunoprecipitation (ChIP)-seq studies in lipopolysaccharide (LPS)-stimulated macrophages showed suppression of cytokine transcription by binding of NRF2 upstream of cytokine promoters (Kobayashi et al., 2016). The mechanism(s) underlying the antagonism between RIG-I signaling and NRF2 activation in airway epithelial cells will be an important avenue for future investigation. Our findings also emphasize the importance of considering how experimental conditions impact oxidative stress in studies of innate immune signaling.
Despite antagonism between antiviral innate immune defense and NRF2 activity, NRF2 activation can have either protective or deleterious effects for the host during viral infection. We observed a cytoprotective effect of NRF2 knockdown during rhinovirus infection in nasal epithelial cells, consistent with enhanced ISG induction and diminished viral replication (Figure 4). However, NRF2 activity was essential for cell survival during CSE exposure (Figure S3). Therefore, any suppressive effects of NRF2 activation on antiviral defense represent a necessary compromise in the presence of both CSE and viral infection. Likewise, Olagnier et al. (2014) observed a protective effect of NRF2 knockdown during Dengue virus infection of dendritic cells. In contrast, during influenza infection of cigarettesmoke-exposed mice, NRF2 knockout diminished host survival (Yageta et al., 2011). These results likely reflect the fact that host resistance and clearance of infection is the best pathway to health in some disease settings, whereas the antioxidant response takes precedence to promote host survival in others, particularly when multiple stressors are encountered at once.
We propose a model in which airway epithelial cells’ responses to airway injury may be differentially regulated to optimize defense responses in distinct microenvironments and can vary depending on the recent exposures of the cell. As shown in Figure S4, in our model, virus-induced RIG-I signaling leads to activation of both the antiviral interferon response and the NRF2-dependent oxidative stress response. In nasal epithelial cells, there is an inherent bias toward less NRF2 activation and greater activation of the interferon and ISG response compared to bronchial cells (Figure S4A). This bias may serve to enable nasal epithelial cells to maintain robust antiviral defense in the relatively cool temperatures of the nasal passages (Foxman et al., 2015; Figure S1). This bias may also reflect the fact that the nasal epithelium is also the initial site of entry for many respiratory viruses and a site in which the consequences of local epithelial damage and inflammation are relatively low. In contrast, in the bronchi, maintaining tissue integrity and suppressing inflammatory responses to keep the large airways open is of paramount importance, fitting with the observed difference in set point favoring the NRF2 response in bronchial epithelial cells. It is also important to note that this study models defense responses of airway basal cells from different airway microenvironments, but in vivo, the relative proportions of differentiated epithelial cell types likely also play an important role in airway region-specific defense, such as the increased proportion of mucus-producing cells in the nasal passages compared to the bronchi (Iwasaki et al., 2017).
In our model, in addition to inherent region-specific adaptations of progenitor cells from different airway sites, recent environmental exposures can further influence the balance between defense responses in the airway epithelium. For example, the NRF2 response would be expected to be activated in cells exposed to sources of oxidative stress in the airway, which could include environmental pollutants, such as diesel exhaust, cigarette smoke, microbial metabolites, or the byproducts of oxidative metabolism by resident cells or infiltrating leukocytes. Although NRF2 activation would move cells to a new state of adaptation that enhances cell survival during oxidative stress, such cells would be maladapted to viral infection due to dampening of the antiviral interferon response (Figure S4B). We modeled this scenario in this study by exposing nasal epithelial cells to cigarette smoke extract, resulting in increased NRF2 activity and decreased RIG-I signaling (Figures 4G–4I). Previous studies have shown diminished antiviral responses in bronchial epithelial cells following cigarette smoke exposure (Eddleston et al., 2011; Proud et al., 2012); our results indicate that a possible underlying mechanism for this finding is antagonism between NRF2 activation and antiviral signaling.
Importantly, there is considerable evidence that epithelial cells from patient groups susceptible to severe rhinovirus infection have a depressed antiviral interferon and ISG response. Our findings suggest that activation of the NRF2-mediated oxidative stress response in these cells could be the mechanistic basis for this phenotype. There is evidence supporting this model in one such patient group: smokers. Published data from smokers reveal a striking transcriptional signature of NRF2 activation (Spira et al., 2004). Chronic obstructive pulmonary disease (COPD) and asthma patients are also highly susceptible to serious illness following RV infection, and in fact, RV is the top trigger of childhood asthma exacerbations (Gern, 2010). Ex vivo experiments have shown evidence for defects in cell-intrinsic innate immunity in airway epithelial cells from asthmatics and smokers compared to healthy controls (Contoli et al., 2006; Wu et al., 2016; Wark et al., 2005; Jaspers et al., 2010). Our findings suggest that it will be important to investigate whether an aberrant shift toward NRF2-mediated antioxidant defense could underlie the observed defect in epithelial antiviral defense, leading to increased RV susceptibility in these patients.
In summary, the findings reported here demonstrate antagonism between two key defense mechanisms in airway epithelial cells and demonstrate how the activity levels of these responses are tailored to different set points in cells derived from different airway regions (nasal versus bronchial). We also demonstrate that NRF2 activation by an environmental oxidative stress can shift this balance and create vulnerability to rhinovirus infection. These results compel further investigation of the role of NRF2 activation in RV-susceptible patient groups and indicate that finding ways to protect the airway epithelium from intracellular oxidative stress, and thereby avert NRF2 activation, may lead to effective strategies to enhance natural defense against rhinovirus infection.
STAR⋆METHODS
Detailed methods are provided in the online version of this paper and include the following:
- EXPERIMENTAL MODEL AND SUBJECT DETAILS
-
○Primary human airway epithelial cells
-
○H1-HeLa cells
-
○Rhinovirus 1B
-
○
- METHOD DETAILS
-
○Viral infection or RIG-I stimulation of primary cells
-
○ELISA
-
○Plaque assay to determine viral titer
-
○siRNA knockdown
-
○RNA-Seq sample preparation and sequencing
-
○Synthesis, purification, and labeling of SLR14–488 nucleotide
-
○
- QUANTIFICATION AND STATISTICAL ANALYSIS
-
○Statistical comparisons of viral titers, protein expression, and mRNA expression
-
○RNASeq analysis
-
○
STAR⋆METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| Bacterial and Virus Strains | ||
| RV-1B | ATCC | VR-1645 |
| Biological Samples | ||
| Primary human bronchial epithelial cells | LONZA | Cat# CC-2540S |
| Primary human nasal epithelial cells | Promocell, Germany | Cat# C-12620 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| SLR14 RIG-I ligand | provided by Anna Pyle, Yale University | N/A |
| SLR14–488 RIG-I ligand | provided by Anna Pyle, Yale University | N/A |
| Cigarette Smoke Extract | provided by Charles Dela Cruz, Yale University | N/A |
| Critical Commercial Assays | ||
| iScript cDNA synthesis Kit | Bio-Rad | Cat# 1708891 |
| iTaq Universal SYBR green | Bio-Rad | Cat# 1725120 |
| RNeasy Mini Kit | QIAGEN | Cat# 74106 |
| Interferon lambda 1 ELISA kit | eBiosciences | Cat# 88–7296-22 |
| Deposited Data | ||
| RNASeq data, resting and SLR14 stimulated nasal and bronchial-derived epithelial cells | This paper | GEO: GSE117884 |
| Experimental Models: Cell Lines | ||
| Human: H1-HeLa cells | Provided by W.M. Lee | Lee et al., 2015 |
| Oligonucleotides | ||
| siRNA negative control (RISC-free) | Dharmacon | Cat# D-001220–01 |
| siRNA targeting NRF2 | Dharmacon | Cat# D-003755–01 |
| siRNA targeting MAVS | Dharmacon | Cat# D-024237–02 |
| Primers used for RT-qPCR, see Table S2 | This paper | N/A |
| Software and Algorithms | ||
| GraphPad Prism 7.0 | GraphPad Software | https://www.graphpad.com |
| Ingenuity Pathway Analysis software | QIAGEN | https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis |
| btrim | Kong, 2011 | N/A |
| tophat2 | Kim et al., 2013 | N/A |
| DEseq2 | Love et al., 2014 | N/A |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ellen Foxman (ellen.foxman@yale.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Primary human airway epithelial cells
Primary human nasal and bronchial cells were purchased from commercial vendors Lonza (bronchial cells, cat# CC2450S) and Promocell (nasal cells, cat# C-12620). Companies did not provide donor-specific information apart from age, gender, and healthy status, therefore our institution did not require a protocol for human subjects research. Cells from adult donors of both genders were included and no gender differences were noted for the phenotypes assessed in this study. For RNASeq, one donor was used for each cell type, with 2 biological replicates per condition. Other experimental results are representative of results from multiple different bronchial or nasal cell donors as indicated in the figure legends. Primary human nasal and bronchial epithelial cells were cultured on type I collagen coated tissue culture plates using BEGM growth media and supplements (Lonza, Cat# CC-3171 and CC-4175) using Lonza protocols. Cells were used at passage 3 or lower. For SLR14 stimulation experiments and RV infection experiments, hydrocortisone and epinephrine were omitted from the standard media supplements.
H1-HeLa cells
H1-HeLa cells used for generating rhinovirus stock and performing plaque assay were a generous gift from Wai-Ming Lee (Lee et al., 2015). The following media were used for HeLa cell maintenance and RV infection as in previous studies (Foxman et al., 2015): growth media, MEM (11095, GIBCO), 10%NCS, non-essential amino acids, penicillin-streptomycin; infection media for virus amplification: MEM, 5% NCS, non-essential amino acids, penicillin-streptomycin, and 30mM MgCl2.
Rhinovirus 1B
RV-1B was obtained from ATCC (VR-1645) and amplified in H1-HeLa cells. To concentrate and purify virus, virus in H1-HeLa lysates was pelleted by ultracentrifugation through a 30% sucrose cushion (SW28, 25,000 rpm, 5 hr) and virus was resuspended in PBS. Titer was determined by plaque assay.
METHOD DETAILS
Viral infection or RIG-I stimulation of primary cells
Primary nasal and bronchial epithelial cells were grown in collagen-coated flasks and cultured identically at 37°C, then plated in 12-well or 24- well collagen-coated plates for experiments. For viral infection, subconfluent cells were inoculated with RV-1B diluted in PBS+0.1% BSA for 1 hr at 33°C to achieve indicated multiplicity of infection (MOI). At t = 1hr, inoculum was removed, medium was added, and plates were replaced in the 33°C incubator or shifted to 37°C. To stimulate RIG-I, a 5′-triphosphorylated stem-loop RNA containing 14 base-pairs (SLR14) was used (Linehan et al., 2018). Medium was removed and cells were stimulated with SLR14 complexed with Lipofectamine 2000 (Invitrogen 1168–019) in Optimem for 1 hr, then the stimulus was removed and medium re-added prior to incubation for the indicated time period(s). For cigarette smoke extract experiments, nasal cells were pre-incubated with medium ± 2% CSE (v/v) for 6 hr prior to inoculation, then medium was removed during inoculation, and re-added prior to incubation for 40hr at 33C. For CSE/SLR14 stimulation experiments, cells were incubated o/n with medium ± 2% or 4% CSE, then medium was removed during SLR14 transfection, and re-added following stimulation for 3 hr incubation prior to RNA isolation and RT-qPCR.
ELISA
IFNλ1 protein concentration in cell supernatants was measured using a commercial IFNλ1 ELISA kit (eBiosciences cat#88–7296-22). In brief, primary airway epithelial cells were infected with RV1B or stimulated with SLR14, then fresh medium was added to cells and cells were incubated ay 33°C or 37°C for the indicated time points. Cell supernatants were then collected and stored at –80°C. ELISA assay was performed on neat and diluted supernatants per manufacturer’s instructions to obtain results in the linear range of the assay.
Plaque assay to determine viral titer
Supernatants were collected at the indicated time points and titer was determined by plaque assay on H1-HeLa cells as reported previously (Foxman et al., 2015). Briefly, H1-HeLa cells in 6 well plates were inoculated with 200 μL of cell lysate, serially diluted in PBS+0.1% BSA. Plates were incubated at 33°C for 1 hour with rocking, then overlaid with plaque assay medium (1X MEM, 5% FBS, 0.3% agarose, 30mM MgCl2, 30 μg/ml DEAE-Dextran) and incubated at 33°C for 3 days prior to staining with crystal violet.
siRNA knockdown
siRNAs targeting NRF2 or MAVS, or non-targeting control siRNA, were obtained from GE-Dharmacon and were transfected into primary human airway epithelial cells following the manufacturer’s protocol for the Dharmafect transfection reagent (GE Dharmacon, cat#T-2001–02). Cells were transfected when < 30% confluent. Transfection was performed for 6 hr at 37°C, after which the medium was changed to fresh complete BEGM. Cells were allowed to incubate for additional 2–4 days at 37°C with daily medium changes to fresh complete BEGM prior to initiating SLR14 stimulation or RV1B infection experiments. Medium was changed to BEGM without hydrocortisone or epinephrine for experiments.
RNA-Seq sample preparation and sequencing
RNA was prepared from primary cells using RNEasy QIAGEN kit and library preparation was performed at the Yale Center for Genomic Analysis as follows. mRNA was purified from approximately 500ng of total RNA with oligo-dT beads and sheared by incubation at 94C and cDNA library was prepared using standard protocols. Indexed libraries that met appropriate cut-offs were quantified by qRT-PCR using a commercially available kit (KAPA Biosystems) and insert size distribution determined with the LabChip GX or Agilent Bioanalyzer. Samples with a yield of ≥ 0.5 ng/ul were used for sequencing. Sample concentrations were normalized to 10 nM and loaded onto Illumina flow cells and sequenced using 75 bp paired-end sequencing on an Illumina HiSeq 2500 per Illumina protocols. Signal intensities were converted to individual base calls during a run using the system’s Real Time Analysis (RTA) software.
Synthesis, purification, and labeling of SLR14–488 nucleotide
The RNA oligonucleotide SLR-14Am (5′ppp GGA UCG AUC GAU CG UAmCG CGA UCG AUC GAU CC (Am - aminomodifier C6dT (Glen Research) was synthesized on a MerMade 12 RNA-DNA synthesizer (BioAutomation) using TBDMS-protected phosphoramidites (BioAutomation) as previously described (Zlatev et al., 2012). Base deprotection was carried out in a 1:1 solution of 30% ammonium hydroxide (JT Baker) and 40% methylamine (Sigma) at 65°C for 10 min as described (Wincott et al., 1995). The supernatant was cooled on ice, transferred to a new vial and evaporated to dryness. Then 0.5 mL of ethanol was added and the mixture was evaporated again. In order to deprotect 2′-hydroxyl groups, the dry oligonucleotide residue was incubated with 0.5 mL of 1 M solution of tetrabutylammonium fluoride (TBAF) in THF (Sigma) at room temperature for 36 h. Then 0.5 mL of 2M sodium acetate (pH 6.0) was added, the mixture was evaporated to a 0.5 mL volume, extracted with 4× 0.8 mL of ethyl acetate and ethanol precipitated. RNA oligonucleotide was then purified on a 20% denaturing polyacrylamide gel as described (Fedorova et al., 2005). Purified SLR-14Am oligonucleotide was dissolved in 200 μL of 0.25 M sodium bicarbonate buffer (pH 9.2). Then a solution containing 0.5 mg of AlexaFluor 488 NHS ester (Life Technologies Corp.) in 200 μL formamide was added and the reaction mixture was incubated at room temperature for 2 hours. The labeled oligonucleotide (SLR14–488) was ethanol precipitated and purified on a 20% denaturing polyacrylamide gel as described (Turunen et al., 2014).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical comparisons of viral titers, protein expression, and mRNA expression
For comparison of viral titers or gene or protein expression, GraphPad Prism 7.0 was used to determine mean and SD or mean and SEM of replicates which are graphed as indicated, and p values were determined using an unpaired t test.
RNASeq analysis
The raw reads of RNA-seq experiments were trimmed off sequencing adaptors and low quality regions by btrim (Kong, 2011). The trimmed reads were mapped to human genome (GRCh37) by tophat2 (Kim et al., 2013). The counts of reads for each gene were based on Ensembl annotation (release 70). After the counts were collected, the differential expression analysis was done by DEseq2 (Love et al., 2014), which calculated the adjusted p values. Pathway analysis was performed using Ingenuity Pathway Analysis software (QIAGEN).
DATA AND SOFTWARE AVAILABILITY
The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (Edgar et al., 2002). The accession number for the nasal and bronchial RNA-seq experiments, which were performed at the same time, is GEO: GSE117884. Nasal data was also cited previously as GEO: GSE107898 (Landry and Foxman, 2018).
Supplementary Material
Highlights.
Activating RIG-I triggers host defense against viruses and oxidative stress
These defenses are calibrated differently in nasal and bronchial epithelial cells
Defense against oxidative stress antagonizes the antiviral interferon response
ACKNOWLEDGMENTS
The authors would like to thank Peter Tattersall and the Tattersall lab for helpful discussions and support. This work was supported by a K08 award from the NIH (5K08AI119139 to E.F.F.) and was also supported in part by the Howard Hughes Medical Institute and awards from the NIH (R01AI054359 and R42AI120269 to A.I. and R01HL126094 to C.D.). O.F. is a Research Scientist, and A.M.P. and A.I. are Investigators of the Howard Hughes Medical Institute.
Footnotes
DECLARATION OF INTERESTS
A.M.P. has a patent pending for RIG-I stimulation agent SLR-14.
SUPPLEMENTAL INFORMATION
Supplemental Information includes four figures and two tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.08.033.
REFERENCES
- Ahmed SM, Luo L, Namani A, Wang XJ, and Tang X (2017). Nrf2 signaling pathway: pivotal roles in inflammation. Biochim. Biophys. Acta 1863, 585–597. [DOI] [PubMed] [Google Scholar]
- Bosch AA, Biesbroek G, Trzcinski K, Sanders EA, and Bogaert D (2013). Viral and bacterial interactions in the upper respiratory tract. PLoS Pathog. 9, e1003057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byington CL, Ampofo K, Stockmann C, Adler FR, Herbener A, Miller T, Sheng X, Blaschke AJ, Crisp R, and Pavia AT (2015). Community surveillance of respiratory viruses among families in the Utah Better Identification of Germs-Longitudinal Viral Epidemiology (BIG-LoVE) study. Clin. Infect. Dis 61, 1217–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contoli M, Message SD, Laza-Stanca V, Edwards MR, Wark PA, Bartlett NW, Kebadze T, Mallia P, Stanciu LA, Parker HL, et al. (2006). Role of deficient type III interferon-lambda production in asthma exacerbations. Nat. Med 12, 1023–1026. [DOI] [PubMed] [Google Scholar]
- Eddleston J, Lee RU, Doerner AM, Herschbach J, and Zuraw BL (2011). Cigarette smoke decreases innate responses of epithelial cells to rhinovirus infection. Am. J. Respir. Cell Mol. Biol 44, 118–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, and Lash AE (2002). Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorova O, Boudvillain M, Kawaoka J, and Pyle AM (2005). Nucleotide analog interference mapping and suppression: specific applications in studies of RNA tertiary structure, dynamic helicase mechanism and RNA-protein interactions. In Handbook of RNA Biochemistry, Hartmann RK, ed. (Weinheim, Germany: Wiley-VCH; ), pp. 259–293. [Google Scholar]
- Fendrick AM, Monto AS, Nightengale B, and Sarnes M (2003). The economic burden of non-influenza-related viral respiratory tract infection in the United States. Arch. Intern. Med 163, 487–494. [DOI] [PubMed] [Google Scholar]
- Foxman EF, Storer JA, Fitzgerald ME, Wasik BR, Hou L, Zhao H, Turner PE, Pyle AM, and Iwasaki A (2015). Temperature-dependent innate defense against the common cold virus limits viral replication at warm temperature in mouse airway cells. Proc. Natl. Acad. Sci. USA 112, 827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foxman EF, Storer JA, Vanaja K, Levchenko A, and Iwasaki A (2016). Two interferon-independent double-stranded RNA-induced host defense strategies suppress the common cold virus at warm temperature. Proc. Natl. Acad. Sci. USA 113, 8496–8501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehrke N, Mertens C, Zillinger T, Wenzel J, Bald T, Zahn S, Tüting T, Hartmann G, and Barchet W (2013). Oxidative damage of DNA confers resistance to cytosolic nuclease TREX1 degradation and potentiates STINGdependent immune sensing. Immunity 39, 482–495. [DOI] [PubMed] [Google Scholar]
- Gern JE (2010). The ABCs of rhinoviruses, wheezing, and asthma. J. Virol 84, 7418–7426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett NR, Shaykhiev R, Walters MS, Wang R, Zwick RK, Ferris B, Witover B, Salit J, and Crystal RG (2011). The human airway epithelial basal cell transcriptome. PLoS ONE 6, e18378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki A, Foxman EF, and Molony RD (2017). Early local immune defences in the respiratory tract. Nat. Rev. Immunol 17, 7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jartti T, Jartti L, Peltola V, Waris M, and Ruuskanen O (2008). Identification of respiratory viruses in asymptomatic subjects: asymptomatic respiratory viral infections. Pediatr. Infect. Dis. J 27, 1103–1107. [DOI] [PubMed] [Google Scholar]
- Jaspers I, Horvath KM, Zhang W, Brighton LE, Carson JL, and Noah TL (2010). Reduced expression of IRF7 in nasal epithelial cells from smokers after infection with influenza. Am. J. Respir. Cell Mol. Biol 43, 368–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koarai A, Sugiura H, Yanagisawa S, Ichikawa T, Minakata Y, Matsunaga K, Hirano T, Akamatsu K, and Ichinose M (2010). Oxidative stress enhances toll-like receptor 3 response to double-stranded RNA in airway epithelial cells. Am. J. Respir. Cell Mol. Biol 42, 651–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi EH, Suzuki T, Funayama R, Nagashima T, Hayashi M, Sekine H, Tanaka N, Moriguchi T, Motohashi H, Nakayama K, and Yamamoto M (2016). Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun 7, 11624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong Y (2011). Btrim: a fast, lightweight adapter and quality trimming program for next-generation sequencing technologies. Genomics 98, 152–153. [DOI] [PubMed] [Google Scholar]
- Kumar PA, Hu Y, Yamamoto Y, Hoe NB, Wei TS, Mu D, Sun Y, Joo LS, Dagher R, Zielonka EM, et al. (2011). Distal airway stem cells yield alveoli in vitro and during lung regeneration following H1N1 influenza infection. Cell 147, 525–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry ML, and Foxman EF (2018). Antiviral response in the nasopharynx identifies patients with respiratory virus infection. J. Infect. Dis 217, 897–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WM, Chen Y, Wang W, and Mosser A (2015). Growth of human rhinovirus in H1-HeLa cell suspension culture and purification of virions. Methods Mol. Biol 1221, 49–61. [DOI] [PubMed] [Google Scholar]
- Linehan MM, Dickey TH, Molinari ES, Fitzgerald ME, Potapova O, Iwasaki A, and Pyle AM (2018). A minimal RNA ligand for potent RIG-I activation in living mice. Sci. Adv 4, e1701854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q (2013). Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol 53, 401–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NIAID (2001). The Common Cold, NIAID Fact Sheet (NIAID).
- Olagnier D, Peri S, Steel C, van Montfoort N, Chiang C, Beljanski V, Slifker M, He Z, Nichols CN, Lin R, et al. (2014). Cellular oxidative stress response controls the antiviral and apoptotic programs in dengue virusinfected dendritic cells. PLoS Pathog. 10, e1004566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfunter A, Wier LM, and Stocks C (2013). Most Frequent Conditions in U.S. Hospitals, 2011 HCUP Statistical Brief #162 (Rockville, MD: Agency for Healthcare Research and Quality; ). [PubMed] [Google Scholar]
- Pickett G, Seagrave J, Boggs S, Polzin G, Richter P, and Tesfaigzi Y (2010). Effects of 10 cigarette smoke condensates on primary human airway epithelial cells by comparative gene and cytokine expression studies. Toxicol. Sci 114, 79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proud D, Hudy MH, Wiehler S, Zaheer RS, Amin MA, Pelikan JB, Tacon CE, Tonsaker TO, Walker BL, Kooi C, et al. (2012). Cigarette smoke modulates expression of human rhinovirus-induced airway epithelial host defense genes. PLoS ONE 7, e40762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock JR, Randell SH, and Hogan BL (2010). Airway basal stem cells: a perspective on their roles in epithelial homeostasis and remodeling. Dis. Model. Mech 3, 545–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater L, Bartlett NW, Haas JJ, Zhu J, Message SD, Walton RP, Sykes A, Dahdaleh S, Clarke DL, Belvisi MG, et al. (2010). Co-ordinated role of TLR3, RIG-I and MDA5 in the innate response to rhinovirus in bronchial epithelium. PLoS Pathog. 6, e1001178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spira A, Beane J, Shah V, Liu G, Schembri F, Yang X, Palma J, and Brody JS (2004). Effects of cigarette smoke on the human airway epithelial cell transcriptome. Proc. Natl. Acad. Sci. USA 101, 10143–10148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, and Yamamoto M (2015). Molecular basis of the Keap1-Nrf2 system. Free Radic. Biol. Med 88 (Pt B), 93–100. [DOI] [PubMed] [Google Scholar]
- Suzuki T, and Yamamoto M (2017). Stress-sensing mechanisms and the physiological roles of the Keap1-Nrf2 system during cellular stress. J. Biol. Chem 292, 16817–16824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tal MC, Sasai M, Lee HK, Yordy B, Shadel GS, and Iwasaki A (2009). Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc. Natl. Acad. Sci. USA 106, 2770–2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimmulappa RK,Lee H,Rangasamy T,Reddy SP,Yamamoto M,Kensler TW, and Biswal S (2006). Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J. Clin. Invest 116, 984–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turunen JJ, Pavlova LV, Hengesbach M, Helm M, Mueller S, Hartmann RK, and Frilander MJ (2014). RNA ligation In Handbook of RNA Biochemistry, Hartmann RK, ed. (Weinheim, Germany: Wiley-VCH; ), pp. 45–88. [Google Scholar]
- Wang Q, Nagarkar DR, Bowman ER, Schneider D, Gosangi B, Lei J, Zhao Y, McHenry CL, Burgens RV, Miller DJ, et al. (2009). Role of double-stranded RNA pattern recognition receptors in rhinovirus-induced airway epithelial cell responses. J. Immunol 183, 6989–6997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wark PA, Johnston SL, Bucchieri F, Powell R, Puddicombe S, LazaStanca V, Holgate ST, and Davies DE (2005). Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J. Exp. Med 201, 937–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wincott F, DiRenzo A, Shaffer C, Grimm S, Tracz D, Workman C, Sweedler D, Gonzalez C, Scaringe S, and Usman N (1995). Synthesis, deprotection, analysis and purification of RNA and ribozymes. Nucleic Acids Res. 23, 2677–2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Zhang W, Booth JL, Hutchings DC, Wang X, White VL, Youness H, Cross CD, Zou MH, Burian D, et al. (2016). Human primary airway epithelial cells isolated from active smokers have epigenetically impaired antiviral responses. Respir. Res 17, 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yageta Y, Ishii Y, Morishima Y, Masuko H, Ano S, Yamadori T, Itoh K, Takeuchi K, Yamamoto M, and Hizawa N (2011). Role of Nrf2 in host defense against influenza virus in cigarette smoke-exposed mice. J. Virol. 85, 4679–4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlatev I, Manoharan M, Vasseur JJ, and Morvan F (2012). Solid-phase chemical synthesis of 5ʹ-triphosphate DNA, RNA, and chemically modified oligonucleotides. Curr. Protoc. Nucleic Acid Chem, Chapter 1, Unit1.28. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.