

Abstract
BACKGROUND:
Microsatellite instability (MSI) is an emerging, actionable phenotype in oncology that informs tumor response to immune checkpoint pathway immunotherapy. However, there remains a need for MSI diagnostics that are low cost, highly accurate, and generalizable across cancer types. We developed a method for targeted high throughput sequencing of numerous microsatellite loci with pan-cancer informativity for MSI using single-molecule molecular inversion probes (smMIPs).
METHODS:
We designed a smMIP panel targeting 111 loci highly informative for MSI across cancers. We developed an analytical framework taking advantage of smMIP-mediated error correction to specifically and sensitively detect instability events without the need for typing matched normal material.
RESULTS:
Using synthetic DNA mixtures, smMIPs were sensitive to at least 1% MSI positive cells and were highly consistent across replicates. The fraction of identified unstable microsatellites discriminated tumors exhibiting MSI from those lacking MSI with high accuracy across colorectal (100% diagnostic sensitivity and specificity), prostate (100% diagnostic sensitivity and specificity), and endometrial cancers (95.8% diagnostic sensitivity and 100% specificity). MSI-PCR, the current standard-of-care molecular diagnostic for MSI, proved equally robust for colorectal tumors, but evidenced multiple false negative results in prostate (81.8% diagnostic sensitivity and 100% specificity) and endometrial (75.0% diagnostic sensitivity and 100% specificity) tumors.
CONCLUSIONS:
smMIP capture provides an accurate, diagnostically sensitive, and economical means to diagnose MSI across cancer types without reliance on patient-matched normal material. The assay is readily scalable to large numbers of clinical samples, enables automated and quantitative analysis of microsatellite instability, and is readily standardized across clinical laboratories.
Keywords: next-generation DNA sequencing, microsatellite instability, cancer, mismatch repair, screening, immunotherapy
INTRODUCTION
Microsatellite instability (MSI) is a molecular diagnostic finding in oncology that reflects a state of genomic hypermutability. Spontaneous gains or losses of nucleotides from DNA in repetitive tracts are the pathognomonic finding of MSI and serve as the basis for its clinical diagnosis (1). Tumors exhibiting MSI (i.e., MSI-high, or MSI-H cancers) are often marked by disruption of the mismatch repair (MMR) system through mutation or epigenetic silencing (1,2), which impairs the correction of length-altering somatic mutations that occur at high rates within microsatellites (2) and results in the generation of new microsatellite alleles.
MSI was initially discovered and employed diagnostically in colorectal cancers (3,4), but recent developments have elevated the importance of MSI testing as an emerging diagnostic in oncology that is applicable across cancer types (5–7). First, MSI has now been reported in tumors from multiple tissues, including endometrial, ovarian, gastric, prostate, and glioblastoma (2,8,9). Recent genome-scale analyses (10,11,12) have found that MSI is a generalized feature of malignancies and occurs at some frequency in virtually all tumor types. Second, therapeutic agents active against the immune checkpoint pathway (programmed death-ligand 1 and programmed cell death protein 1 inhibitors) have been approved by the United States Food and Drug Administration in the treatment of MSI-H cancers irrespective of their tissue of origin (5,13), where they potentiate immune cell recognition of mutation-associated neoantigens and improve patient outcomes (5).
Standard-of-care molecular diagnosis of MSI is currently achieved using multiplexed PCR-based testing (MSI-PCR), typically directed against five informative microsatellite markers (3,14). Although widely used, MSI-PCR has been validated only for colorectal tumors (1,14) and can have poor performance in malignancies derived from other tissues (15–17), likely reflecting differences in the mutational frequency of microsatellite loci across cancer types (10,11). Moreover, MSI-PCR is susceptible to false negatives (18), owing to artifacts related both to tumor heterogeneity or contamination with non-neoplastic immune infiltrate (19) and to lower mutational burdens in the context of specific MMR pathway deficiencies (15,20). Lastly, MSI-PCR has limited prognostic utility: recent work indicates that functional (21) and genome-scale assays incorporating quantification of microsatellite instability across numerous loci (10) provide superior predictive information regarding patient outcomes than does binary classification of tumors as MSI-H or microsatellite stable (MSS).
Several groups (10,22,23–25) have alternatively used next-generation sequencing (NGS) to assay MSI by examining dozens to hundreds of repetitive loci that are incidentally sequenced along with regions of interest during targeted gene enrichment or exome sequencing. By virtue of their ability to broadly interrogate the genome, these approaches offer substantial advantages over conventional methods in terms of diagnostic accuracy and applicability across cancer types (10,22,24,25). Nevertheless, determination of MSI by NGS currently requires large-scale targeted gene enrichment or exome sequencing data, which is material-limiting for many clinical specimens and results in a high cost of testing. Consequently, such diagnostics are typically reserved for late-stage or widely metastatic disease (26). Moreover, most analytic approaches require sequence data from matched normal material (23,24), increasing costs and workflow demands. NGS methods for determining MSI are consequently impractical as primary screening tests and are presently provided as value-added information for larger, panel-based sequencing tests when they are applied to advanced or otherwise recalcitrant malignancies.
To address the need for a high diagnostic accuracy, low-cost MSI assay with applicability across cancer types, here we used single molecule molecular inversion probe (smMIP) (27) capture coupled with high throughput sequencing to provide a novel, pan-cancer diagnostic for MSI. smMIPs integrate highly multiplexed targeted sequencing with error-correction schemes based on unique molecular identifiers (UMIDs), allowing the inexpensive and accurate typing of microsatellite loci (28). By using smMIPs to interrogate large numbers of microsatellites that are highly informative for a MSI-H phenotype across cancer types(10), we demonstrate high levels of diagnostic sensitivity and specificity with robust limits of detection across disparate cancer types, without the need for typing matched normal material.
MATERIALS AND METHODS
Samples, MSI diagnosis, and Cell Lines
Residual DNA from clinical samples (fresh frozen tumor or formalin fixed paraffin embedded material, Supplemental Table 1) was obtained retrospectively and de-identified according to University of Washington Institutional Review Board guidelines. This dataset represented a convenience sample of tumor specimens having been submitted for genetic or genomic characterization and for which sufficient residual DNA (at least 100 ng) was available for analysis. Cases testing positive for MSI-H and an approximately equivalent number of MSS specimens were randomly selected for each tumor type. Genomic DNA from reference individual NA12878 was obtained from Coriell Biorepository (Camden, New Jersey), and cell line LS174T was from ATCC (Manassas, Virginia). This project was approved by the University of Washington Human Subjects Division and was conducted in accordance with the Declaration of Helsinki. All data analysis in this study was performed in a blinded fashion.
Diagnosis of MSI status for colon, prostate, and MSI-H endometrial tumors was established by NGS-based analysis for microsatellite instability events using the mSINGS algorithm with a cutoff of 0.2 unstable sites defining MSI-H (22), based on large-panel targeted gene capture (29) or targeted amplification data (30). Where data were available for confirmatory diagnosis, somatic mutations in MMR-pathway genes were enumerated relative to matched germline tissue, as was clinical immunohistochemistry for MMR pathway genes as performed by the University of Washington Medical Center. MSS endometrial specimens were primarily identified using immunohistochemistry for MMR pathway genes, as per clinical diagnostic pathways. Minimum tumor cellularity of 20% was required for all assays.
MSI-PCR (MSI Analysis System, Version 1.2, Promega, which interrogates mononucleotide repeat markers BAT-25, BAT-26, MONO-27, NR-21 and NR-24) was performed for all samples per manufacturer instructions with independent review by two or three molecular pathologists, using two or more positive loci to define an MSI-H diagnosis.
smMIP design, capture, and rebalancing.
We considered microsatellite loci that were previously identified as preferentially unstable in MSI-H rather than MSS tumors originating from colorectal, endometrial, and stomach (10). Loci that were unstable in <5% of a particular cancer type were omitted. The probability of a locus being preferentially unstable in MSI-H tumors across cancer types was assessed after controlling for cancer type using the Cochran-Mantel-Haenszel test. A smMIP capture panel was designed against Human Genome build 19 (hg19) to target the most statistically significant 150 loci using the program MIPgen (31) with the following flags: -max_capture_size 140, -min_capture_size 100, -logistic_priority_score 0.5, -common_snps off, -arm_lengths 18:24,19:23,20:22,21:21, -tag_sizes 4,4. Probes were synthesized by Integrated DNA Technologies (Coralville, Iowa) using standard salt purification.
Probes were 5’ phosphorylated and used in capture reactions as described elsewhere (32). Briefly, 100–500 ng genomic DNA was hybridized with the panel using successive cycles of denaturation and cooling in the presence of DNA polymerase and DNA ligase, exonuclease treated, and PCR amplified to generate sequencing libraries. Initially, smMIP probes were pooled together at equimolar concentrations, used to capture genomic DNA from reference specimen NA12878, and sequenced. The mean relative abundance of unique UMIDs originating from each probe was determined, and probes were empirically rebalanced to achieve better performance uniformity and to remove several probes with consistently poor performance (n=12). We also eliminated probes that empirically demonstrated a high rate of instability in MSS specimens (difference in instability rates < 9% between MSS and MSI-H tumors, n=27). This resulted in a final panel of 111 smMIP probes (Supplemental Table 2).
Sequencing and Data processing
smMIP libraries were sequenced using an Illumina NextSeq 500 operating 300 cycle chemistries. Reads were processed and mapped as described elsewhere (32), except that i) self-assembled reads less than 53 bp in length were discarded, and ii) reads were mapped to a reduced representation of hg19 comprised of the targeted microsatellite regions padded with 30 bp of flanking sequence to reduce alternative read mappings. Mapped reads were then grouped on their UMIDs, and groups with two or more reads were retained. Error correction was performed by discarding UMID groups of two reads that did not match exactly, and by picking a representative read matching the majority rule sequence for groups of three or more. The length of error corrected reads at each locus was calculated and normalized to the most abundant allele length using mSINGS (22) (v3.2, release 26ef15d).
Detection of microsatellite instability by smMIP
To detect instability events without the need for patient-matched normal material, we analyzed 30 residual peripheral blood specimens (which are constitutionally negative for MSI) as above to establish a baseline for sequencing artifacts. At each targeted locus, the distribution of alleles across samples was aligned based on the most abundant allele length, and the mean and standard deviation of relative abundance was calculated for minor alleles at each position.
Instability in clinical specimens was evaluated against these summary statistics after matching the distribution of alleles against the baseline at the most abundant allele length. Loci were considered unstable if they had one or more alleles at greater relative abundance than predicted by the mean and standard deviation of the baseline at p ≤0.001 (z-test). If a given allele length in a clinical specimen was absent from the baseline, a mean relative abundance of zero and standard deviation equal to the next closest allele were used to assess significance.
Data availability
Sequence data have been submitted to the NCBI Sequence Read Archive (SRA) under study accession PRJNA423139. Source code for the data analysis pipeline is available on request.
RESULTS
Error correction, linearity, limit of detection, and assay reproducibility
To enable the diagnosis of MSI across disparate cancer types, we designed a smMIP panel targeting microsatellite loci predicted to be preferentially unstable in MSI-H tumors from multiple tissues of origin (10). smMIP-mediated error correction substantially decreased the number of artefactual microsatellite alleles resulting from slipped strand mispairing during PCR amplification (“stutter” artifact (28)), enabling the identification of true allele lengths (Fig. 1).
Figure 1. smMIP-mediated error correction for microsatellite locus typing.
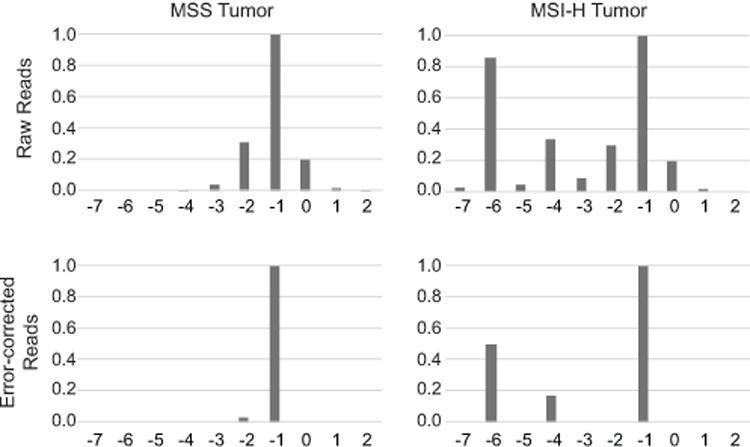
Sequence results from a representative locus (remsi_23) are shown for MSS and MSI-H colorectal cancers before and after smMIP-mediated error correction. In all panels, the X-axis indicates the length of a microsatellite allele with respect to the human reference genome, and the Y-axis displays the relative abundance of signal as normalized to the most abundant microsatellite allele.
We first evaluated the performance characteristics of smMIP capture using defined control material. We combined DNA extracted from MSI-H colorectal cancer cell line LS174T with that from a non-neoplastic cell line having an intact MMR system (NA12878) at various proportions to simulate tumor heterogeneity or admixture with non-neoplastic material. Triplicate preparations of each dilution were generated and separately subjected to smMIP testing to estimate assay variability.
Pure material from LS174T exhibited instability at 79.1% of the loci typed (Fig. 2), whereas only 7.1% of microsatellites in the MSS diluent were unstable. Remarkably, all dilutions containing 1% MSI-H cells or more evidenced numbers of unstable loci that were readily and significantly (p<1X10−5, two-tailed t-test) distinguishable from the MSS control. The fraction of loci identified as unstable was highly consistent across replicates (mean standard deviation of measurement = 0.016 unstable loci), as were instability calls for individual loci, with 81.9% concordance observed. The fraction of unstable loci quantified was significantly associated with the abundance of MSI-H material (R2=0.89). We concluded that smMIP-based determination of MSI was highly reproducible and diagnostically sensitive to detecting at least 1% MSI-H cells in a heterogeneous tumor microenvironment.
Figure 2. Sensitivity and limit of detection.
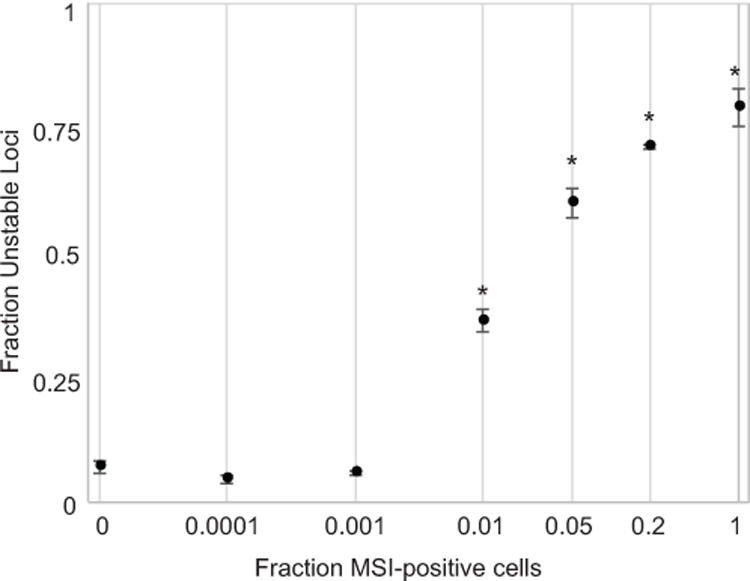
Linearity and reproducibility of the smMIP panel for identifying MSI in synthetic material comprised of a MSI-H cell line combined with a MSS cell line at differing levels of relative abundance. Error bars indicate the standard deviation of three independent replicates for each dilution. Asterisks indicate significance at p<1X10−5.
Diagnosis of MSI across cancer types by smMIP
We next investigated the ability of smMIP capture to diagnose MSI status in cohorts of tumors known to exhibit different patterns of genomic MSI (10): colorectal (n=33 MSI-H, n=35 MSS), endometrial (n=24 MSI-H, n=19 MSS), and prostate (n=11 MSI-H, n=22 MSS). Clinical MSI diagnosis was established in most specimens using targeted gene capture for causative MMR mutations with integrated genome-scale MSI testing by NGS (22,29), enabling more comprehensive phenotypic and genotypic characterization of MSI status (Supplemental Table 1).
MSI-H specimens displayed, on average, significantly higher burdens of unstable sites than MSS tumors (p = 6.27X10−36, 2-tailed t-test, Fig. 3, Supplemental Table 1). Specimens were readily stratified into two groups that were consistent with their clinical diagnosis as MSS or MSI-H using any breakpoint between 0.14 and 0.22 unstable loci, determined as the optimal decision threshold by receiver operating characteristic analysis. The background mutation rates of MSS specimens were not markedly different across cancer types, with a mean fraction of 0.057 mutated loci per tumor. MSI-H specimens had nearly an order of magnitude greater numbers of unstable loci (0.56) but spanned a range of instability values within individual cancer types. The mean total load of unstable loci per MSI-H tumor was comparable across cancer types, supporting the pan-cancer instability of the targeted microsatellite markers. Colorectal tumors exhibited the highest mutation rates (mean 0.61 unstable loci) which was significantly greater than MSI-H endometrial cancers (0.50 unstable loci, p = 0.014, 2-tailed t-test) but not distinguishable from MSI-H prostate cancers (0.53 unstable loci, p = 0.13, 2-tailed t-test). Analysis of the mutational profiles in each of the three types of malignancy (Supplemental Table 3) indicated that a subset of loci were unstable at greater frequency in one or more specific cancer types, consistent with earlier reports (10) and suggesting that this finding reflects subtle, lineage-specific mutational profiles at the selected loci rather than greater mutation frequency overall.
Figure 3. Diagnosis of MSI status across cancer types.
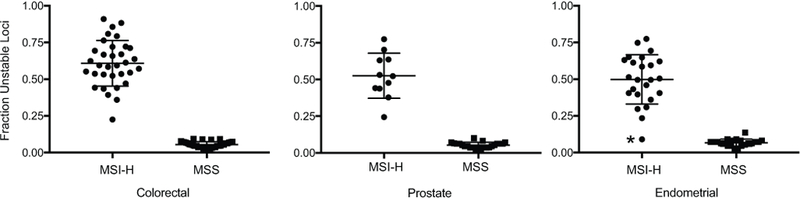
The Y-axis reports the fraction of microsatellite loci identified as unstable in tumors that are positive or negative for the MSI-H phenotype. Tumor type is indicated. The horizontal line for each category indicates the mean, and error bars show standard deviation. The single false negative classification is indicated with an asterisk.
Of the 144 specimens tested, one MSI-H endometrial tumor (ENDO-POS-17), which was confirmed to harbor double somatic mutations in the MMR gene MSH6 and loss of MMR gene expression by IHC, was falsely diagnosed as MSS by our assay. We obtained 100% diagnostic sensitivity and 100% diagnostic specificity for both colorectal and prostate tumors, whereas diagnostic sensitivity of 95.8% and diagnostic specificity of 100% was obtained for endometrial cancers. The smMIP assay was 98.5% sensitive and 100% specific for diagnosing MSI-H when considering results across cancer types altogether (Table 1).
Table 1.
Diagnostic sensitivity and specificity of MSI diagnosis by smMIP and MSI-PCR
| Sensitivity (number measured positive / true positive (% [95% CI*])) |
Specificity (number measured negative / true negatives (% [95% CI*])) |
|||||||
|---|---|---|---|---|---|---|---|---|
| Testing method |
Colorectal | Prostate | Endometrial | All samples | Colorectal | Prostate | Endometrial | All samples |
| MSI-PCR | 32/32 (100% [89.1%– 100%]) |
9/11 ( 81.8% [48.2%– 97.7%]) |
18/24 (75.0% [53.3%– 90.2%]) |
59/67 (88% [77.8%– 94.7%]) |
34/34 (100% [89.7–100%]) |
22/22 (100% [84.6%– 100%]) |
19/19(100% [82.4%– 100%]) |
75/75 (100% [95.2%– 100%]) |
| smMIP | 33/33 (100% [89.4%– 100%]) |
11/11 (100% [71.5%– 100%]) |
23/24 (95.8% [78.9%– 99.9%]) |
67/68 (98.5% [92.1%– 100%]) |
35/35 (100% [90.0%−100%]) |
22/22 (100% [84.6%– 100%]) |
19/19 (100% [82.4%– 100%]) |
76/76 (100% [95.3%– 100%]) |
All CI’s calculated by the Clopper-Pearson (exact) method
Diagnostic comparison and quantitative correlation with MSI-PCR
To compare smMIP capture against conventional “gold-standard” clinical molecular MSI diagnosis, commercially available MSI-PCR (3,14) was performed for all specimens. Two tumors that were successfully typed by smMIP failed MSI-PCR amplification and were subsequently excluded (Supplemental Table 1).
With one exception, discrepancies between MSI-PCR and clinical MSI determinations represented MSI-H cases that were diagnosed as MSS by MSI-PCR. These discrepancies were further examined. Two prostate tumors (Prostate-POS-08 and Prostate-POS-11) evidenced pathogenic inactivating lesions in MMR genes by targeted gene sequencing, indicating false negative MSI-PCR results. Four of the six discrepant endometrial tumors (Endo-POS-09, Endo-POS-17, Endo-POS-20, Endo-POS-22) demonstrated loss of MMR expression by immunohistochemsitry and carried double somatic pathogenic mutations in MMR genes by targeted sequencing, similarly indicating that the MSS diagnosis by MSI-PCR was inaccurate. One endometrial tumor (Endo-POS-14) had equivocal immunohistochemistry results but demonstrated homozygous pathogenic mutations in MSH6 which were consistent with a MSI-H phenotype. The final endometrial tumor (Endo-POS-12), carried only a single somatic mutation in MSH2, but the diagnosis of MSI-H was secondarily supported by loss of MSH2 and MSH6 expression by immunohistochemistry, again supporting an inappropriate MSS diagnosis by MSI-PCR. Of note, MSI-H endometrial tumor ENDO-POS-17 was falsely diagnosed as MSS by both MSI-PCR and smMIP.
Unlike smMIP, the performance of MSI-PCR varied markedly across cancer types (Table 1). MSI-PCR achieved 100% specificity across tumor types and 100% sensitivity in colorectal tumors but only 81.8% sensitivity for prostate and 75.0% sensitivity for endometrial tumors. Considering all specimens, the overall diagnostic sensitivity of MSI-PCR for detecting MSI-H was 88.0%.
Quantification of MSI as a continuous score correlates with patient outcomes better than binary categorization of tumors as either MSI-H or MSS (10). We therefore examined whether the fraction of unstable markers measured by MSI-PCR correlated with those quantified by smMIP (Fig. 4). Although a weak positive correlation between these two measurements was observed for both MSI-H and MSS tumors, the statistical support was not strong (R2 = 0.22 for MSI-H, 0.076 for MSS), indicating that the two assays were dissimilar in their quantification of instability burden.
Figure 4. Quantitative correlation of instability quantified by smMIP and MSI-PCR.
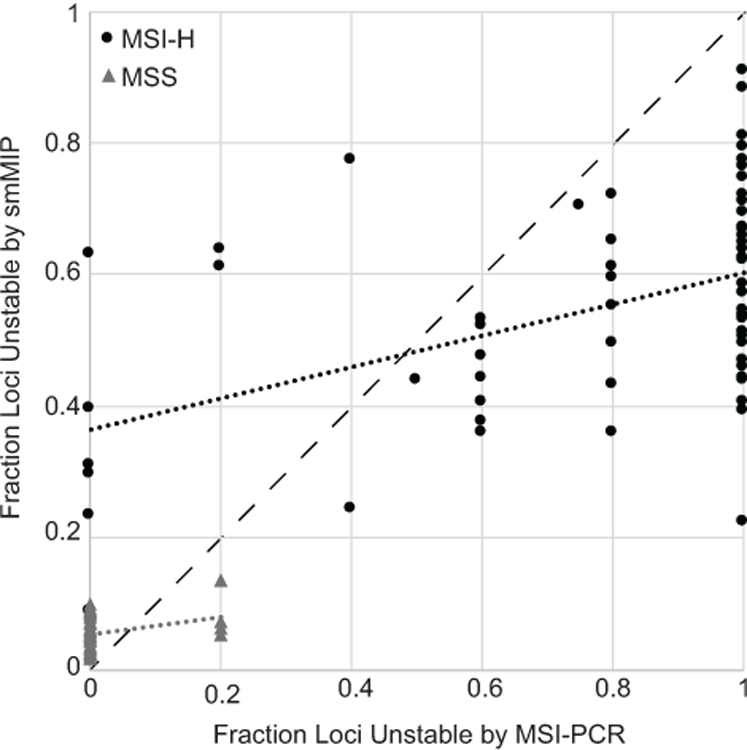
The fraction of unstable loci quantified by smMIP (Y-axis) is shown separately for MSS and MSI-H tumors as a function of the fraction of unstable loci detected by MSI-PCR (X-axis). The dashed black line indicates a theoretical perfect correlation between the two measurements. Linear trend lines are displayed separately for MSS and MSI-H populations.
DISCUSSION
Here, we adapted smMIP technology as a novel molecular diagnostic platform for MSI testing that provides several noteworthy advantages over existing conventional and NGS approaches. First, unlike MSI-PCR(14) or amplicon-based NGS MSI panels (30), smMIPs are readily and highly mutiplexable (27), such that thousands of microsatellites may be interrogated in the same assay. This scale enables sufficient numbers of loci to be examined such that mutational biases occurring in any individual cancer type (10) can be effectively avoided, providing a targeted assay that is valid for pan-cancer MSI determination. Although by subsampling data we have found that as few as 40 highly informative loci were able to properly discriminate MSS from MSI-H tumors in this study, the functional redundancy of the larger panel provides increased robustness. Second, due to their capacity for error correction and ability to detect low prevalence alleles (32), smMIP probes are able to identify instability events even in the background of substantial tumor heterogeneity or non-neoplastic cell contamination, which limits both conventional diagnostics and existing NGS approaches. Third, smMIP technology is rooted in massively parallel sequencing, providing a widely adopted digital detection platform that supports automated, algorithmic interpretation (22). Fourth, the modular construction of smMIP panels allows for focused and ultrasensitive detection of other relevant cancer-associated mutations (32) in parallel with MSI. Fifth, our analytic strategy enables the robust detection of MSI status without the need for sequencing patient-matched normal material (22). Lastly, the costs of library construction and required sequencing coverage by this approach are low, enabling economic testing of patient samples at scale (33); here, specimens were prepared and sequenced for <$80 USD in material costs each.
In contrast to MSI-PCR, the current clinical standard-of-care for MSI molecular diagnosis, smMIPs demonstrated superior performance across cancer types (Table 1). MSI-PCR showed substantial numbers of false negative results for both prostate and endometrial tumors, whereas smMIP-based diagnosis improperly classified only a single endometrial specimen. Notably, this discordant endometrial tumor was also classified as MSS by MSI-PCR, and the consistency between these two assays raises the possibility that the tumor’s molecular signature may legitimately reflect a mutational process that is distinct from MSI (10,23). Regardless of this possibility, our findings reinforce the narrow validity of MSI-PCR only within colorectal cancers (15–17) and demonstrate that informed selection of larger numbers of informative markers by smMIP can recapitulate the pan-cancer performance of exome or genome-scale NGS diagnostic approaches for MSI (10,22,24,25).
Although most MSI-H tumors were readily discriminated from MSS, we noted substantial variability in the overall fraction of unstable loci identified (Fig. 3). This finding may partially reflect tumor purity (Fig. 2), but it is important to acknowledge that MSI is better represented as a continuum than a binary classification (10,12). Tumors may span a range of overall MSI burden (10). A continuous instability metric has been shown to be a more meaningful predictor of clinical outcomes than conventional MSI-H and MSS classifications, even when applied among tumors considered MSS (10). The large number of microsatellites that can be interrogated by our smMIP assay aids in achieving quantification of MSI load and its interpretation as a prognostic variable (Fig. 4).
There exist alternative approaches to both MSI-PCR and NGS-based MSI testing; however, none represent an ideal solution. Immunohistochemical detection of MMR-pathway genes in fixed tumor tissue using antibody staining is an established approach utilizing commonly available, but poorly-standardized pathology methods (34). Although inexpensive and currently used in clinical practice, it is less sensitive than molecular methods (34), requires interpretation by trained pathologists, is lower throughput, prone to technical artifacts, and can yields false negatives when point mutations (rather than decreased expression) have affected MMR genes (35) or when MMR dysregulation does not underlie MSI (10,23). Immunoscore (21) quantifies the density of tumor-infiltrating lymphocytes, providing a functional measure of neoantigens resulting from MSI. Immunoscore has been shown to have superior predictive power for tumor recurrence and patient survival than conventional MSI assays (21); however, it is costly to implement, requiring supporting analytic infrastructure, and is low-throughput and labor intensive. As a functional measurement, Immunoscore does not directly assess microsatellite mutations or other underlying genomic alterations. Finally, tumor mutational burden is a disparate, and currently unstandardized, NGS analysis that has recently been used to quantify the DNA lesions present in a cancer (36). However, tumor mutational burden has an unpredictable relationship with MSI, and its prognostic significance for immunotherapy response remains of questionable clinical utility (36–39). The accurate prediction of tumor mutational burden also requires sequencing large numbers of genes, either whole exome sequence data or panels of at least 400 genes in size (40), which is both cost and material limiting for many tumors.
Pan-cancer MSI determination by smMIPs provides enhanced testing compared with existing testing modalities with respect to generalizability, performance, cost, workflow, and interpretation. Use of this technology and subsequent data analysis are scalable and amenable to standardization across laboratories, offering a means to accurately and economically screen large numbers of tumors for MSI, regardless of their stage or tissue of origin. Although here we have demonstrated robust performance across three anatomically and physiologically distinct tumor types, future work will seek to expand the range of cancers for which the assay has been empirically evaluated and to better characterize quantitative metrics of MSI and their clinical correlates.
Supplementary Material
ACKNOWLEDGEMENTS:
This work was supported by grants CA192980 and CA222344 from the National Cancer Institute (to SJS), a Prostate Cancer Foundation Award (to CCP), and Congressional Designated Medical Research Program (CDMRP) award PC131820 (to CCP).
List of Abbreviations:
- MSI
microsatellite instability
- MSI-H
microsatellite instability high
- MSS
microsatellite stable
- smMIP
single-molecule molecular inversion probe
- MMR
mismatch repair
- NGS
Next-Generation DNA sequencing
- UMID
unique molecular identifier
List of Human Genes:
- MSH6
mutS homolog 6
REFERENCES:
- 1.Murphy KM, Zhang S, Geiger T, Hafez MJ, Bacher J, Berg KD, et al. Comparison of the microsatellite instability analysis system and the Bethesda panel for the determination of microsatellite instability in colorectal cancers. J Mol Diagn 2006;8:305–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vilar E, Gruber SB. Microsatellite instability in colorectal cancer-the stable evidence. Nat Rev Clin Oncol 2010;7:153–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de la Chapelle A, Hampel H. Clinical relevance of microsatellite instability in colorectal cancer. J Clin Oncol 2010;28:3380–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beamer LC, Grant ML, Espenschied CR, Blazer KR, Hampel HL, Weitzel JN, et al. Reflex immunohistochemistry and microsatellite instability testing of colorectal tumors for Lynch syndrome among US cancer programs and follow-up of abnormal results. J Clin Oncol 2012;30:1058–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med 2015;372:2509–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dudley JC, Lin M-T, Le DT, Eshleman JR. Microsatellite Instability as a Biomarker for PD-1 Blockade. Clin Cancer Res Off J Am Assoc Cancer Res 2016;22:813–20. [DOI] [PubMed] [Google Scholar]
- 7.Xiao Y, Freeman GJ. The microsatellite instable subset of colorectal cancer is a particularly good candidate for checkpoint blockade immunotherapy. Cancer Discov 2015;5:16–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res 1998;58:5248–57. [PubMed] [Google Scholar]
- 9.Pritchard CC, Morrissey C, Kumar A, Zhang X, Smith C, Coleman I, et al. Complex MSH2 and MSH6 mutations in hypermutated microsatellite unstable advanced prostate cancer. Nat Commun 2014;5:4988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hause RJ, Pritchard CC, Shendure J, Salipante SJ. Classification and characterization of microsatellite instability across 18 cancer types. Nat Med 2016;22:1342–50. [DOI] [PubMed] [Google Scholar]
- 11.Cortes-Ciriano Isidro Lee Sejoon, Park Woong-Yang, Kim Tae-Min, Park Peter. A molecular portrait of microsatellite instability across multiple cancers. bioRxiv [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bonneville R, Krook MA, Kautto EA, Miya J, Wing MR, Chen H-Z, et al. Landscape of Microsatellite Instability Across 39 Cancer Types. JCO Precis Oncol 2017;1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017;357:409–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bacher JW, Flanagan LA, Smalley RL, Nassif NA, Burgart LJ, Halberg RB, et al. Development of a fluorescent multiplex assay for detection of MSI-High tumors. Markers 2004;20:237–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hampel H, Frankel W, Panescu J, Lockman J, Sotamaa K, Fix D, et al. Screening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res 2006;66:7810–7. [DOI] [PubMed] [Google Scholar]
- 16.Faulkner RD, Seedhouse CH, Das-Gupta EP, Russell NH. BAT-25 and BAT-26, two mononucleotide microsatellites, are not sensitive markers of microsatellite instability in acute myeloid leukaemia. Br J Haematol 2004;124:160–5. [DOI] [PubMed] [Google Scholar]
- 17.Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Working Group. Recommendations from the EGAPP Working Group: genetic testing strategies in newly diagnosed individuals with colorectal cancer aimed at reducing morbidity and mortality from Lynch syndrome in relatives. Genet Med Off J Am Coll Med Genet 2009;11:35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bacher JW, Sievers CK, Albrecht DM, Grimes IC, Weiss JM, Matkowskyj KA, et al. Improved Detection of Microsatellite Instability in Early Colorectal Lesions. PloS One 2015;10:e0132727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Müller A, Giuffre G, Edmonston TB, Mathiak M, Roggendorf B, Heinmöller E, et al. Challenges and pitfalls in HNPCC screening by microsatellite analysis and immunohistochemistry. J Mol Diagn JMD 2004;6:308–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang L Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. Part II. The utility of microsatellite instability testing. J Mol Diagn 2008;10:301–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mlecnik B, Bindea G, Angell HK, Maby P, Angelova M, Tougeron D, et al. Integrative Analyses of Colorectal Cancer Show Immunoscore Is a Stronger Predictor of Patient Survival Than Microsatellite Instability. Immunity 2016;44:698–711. [DOI] [PubMed] [Google Scholar]
- 22.Salipante SJ, Scroggins SM, Hampel HL, Turner EH, Pritchard CC. Microsatellite instability detection by next generation sequencing. Clin Chem 2014;60:1192–9. [DOI] [PubMed] [Google Scholar]
- 23.Huang MN, McPherson JR, Cutcutache I, Teh BT, Tan P, Rozen SG. MSIseq: Software for Assessing Microsatellite Instability from Catalogs of Somatic Mutations. Sci Rep 2015;5:13321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kautto EA, Bonneville R, Miya J, Yu L, Krook MA, Reeser JW, et al. Performance evaluation for rapid detection of pan-cancer microsatellite instability with MANTIS. Oncotarget 2017;8:7452–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Middha S, Zhang L, Nafa K, Jayakumaran G, Wong D, Kim HR, et al. Reliable Pan-Cancer Microsatellite Instability Assessment by Using Targeted Next-Generation Sequencing Data. JCO Precis Oncol 2017;1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller FA, Hayeems RZ, Bytautas JP, Bedard PL, Ernst S, Hirte H, et al. Testing personalized medicine: patient and physician expectations of next-generation genomic sequencing in late-stage cancer care. Eur J Hum Genet EJHG 2014;22:391–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hiatt JB, Pritchard CC, Salipante SJ, O’Roak BJ, Shendure J. Single molecule molecular inversion probes for targeted, high-accuracy detection of low-frequency variation. Genome Res 2013;23:843–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carlson KD, Sudmant PH, Press MO, Eichler EE, Shendure J, Queitsch C. MIPSTR: a method for multiplex genotyping of germline and somatic STR variation across many individuals. Genome Res 2015;25:750–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pritchard CC, Salipante SJ, Koehler K, Smith C, Scroggins S, Wood B, et al. Validation and implementation of targeted capture and sequencing for the detection of actionable mutation, copy number variation, and gene rearrangement in clinical cancer specimens. J Mol Diagn 2014;16:56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hempelmann JA, Scroggins SM, Pritchard CC, Salipante SJ. MSIplus: Integrated Colorectal Cancer Molecular Testing by Next-Generation Sequencing. J Mol Diagn JMD 2015; [DOI] [PubMed] [Google Scholar]
- 31.Boyle EA, O’Roak BJ, Martin BK, Kumar A, Shendure J. MIPgen: optimized modeling and design of molecular inversion probes for targeted resequencing. Bioinforma Oxf Engl 2014;30:2670–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Waalkes A, Penewit K, Wood BL, Wu D, Salipante SJ. Ultrasensitive detection of acute myeloid leukemia minimal residual disease using single molecule molecular inversion probes. Haematologica 2017;102:1549–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neveling K, Mensenkamp AR, Derks R, Kwint M, Ouchene H, Steehouwer M, et al. BRCA Testing by Single-Molecule Molecular Inversion Probes. Clin Chem 2017;63:503–12. [DOI] [PubMed] [Google Scholar]
- 34.Lindor NM, Burgart LJ, Leontovich O, Goldberg RM, Cunningham JM, Sargent DJ, et al. Immunohistochemistry versus microsatellite instability testing in phenotyping colorectal tumors. J Clin Oncol Off J Am Soc Clin Oncol 2002;20:1043–8. [DOI] [PubMed] [Google Scholar]
- 35.Shia J Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. Part I. The utility of immunohistochemistry. J Mol Diagn JMD 2008;10:293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med 2017;9:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015;350:207–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Danilova L, Wang H, Sunshine J, Kaunitz GJ, Cottrell TR, Xu H, et al. Association of PD-1/PD-L axis expression with cytolytic activity, mutational load, and prognosis in melanoma and other solid tumors. Proc Natl Acad Sci U S A 2016;113:E7769–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brown SD, Warren RL, Gibb EA, Martin SD, Spinelli JJ, Nelson BH, et al. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res 2014;24:743–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nagahashi M, Wakai T, Shimada Y, Ichikawa H, Kameyama H, Kobayashi T, et al. Genomic landscape of colorectal cancer in Japan: clinical implications of comprehensive genomic sequencing for precision medicine. Genome Med 2016;8:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequence data have been submitted to the NCBI Sequence Read Archive (SRA) under study accession PRJNA423139. Source code for the data analysis pipeline is available on request.