

Abstract
Activators of Nuclear factor-erythroid 2-related factor 2 (NRF2) could lead to promising therapeutics for prevention and treatment of oxidative stress and inflammatory disorders. Ubiquitination and subsequent degradation of the transcription factor NRF2 is mediated by Kelch-like ECH-associated protein-1 (KEAP1). Inhibition of the KEAP1/NRF2 interaction with small molecules leads to NRF2 activation. Previously, we and others described naphthalene-based NRF2 activators, but the 1,4-diaminonaphthalene scaffold may not represent a drug-like scaffold. Paying particular attention to aqueous solubility, metabolic stability, potency, and mutagenicity, we modified a previously known, naphthalene-based non-electrophilic NRF2 activator to give a series of non-naphthalene and heterocyclic scaffolds. We found that, compared to previously reported naphthalene-based compounds, a 1,4-isoquinoline scaffold provides a better mutagenic profile without sacrificing potency, stability, or solubility.
Table of contents Graphic
Introduction:
Detoxification enzymes act as the cell’s main defense mechanism to counter oxidative and electrophilic stress. Detoxification enzymes include, but are not limited to, NAD(P)H quinone oxidoreductase 1 (NQO1), heme oxygenase 1 (HMOX1), glutamate-cysteine ligase-modifier and—catalytic subunits (GCLM and GCLC), and glutathione S-transferases (GST). The transcription of these and other genes is regulated by the transcription factor, NRF2 (nuclear factor-erythroid2-related factor 2), a member of the cap ‘n’ collar family of basic leucine zipper proteins. Endogenous NRF2 levels are maintained by KEAP1 (Kelch like ECH associated protein 1), which acts as a negative regulator of NRF2. KEAP1 is a 69 kDa protein that exists as a homodimer, and it contains 27 cysteine residues, which act as sensors for oxidative or electrophilic stressors.1, 2, 3 In the absence of oxidative or electrophilic stressors, NRF2 is degraded by the E3 ubiquitin ligase Cullin 3 (CUL3), which binds to KEAP1’s BTB domain (Figure 1A).4 KEAP1 functions as a substrate adaptor protein, by bridging NRF2 to CUL3. A lysine-rich region located between ETGE and DLG motifs of NRF2 is well-positioned for ubiquitination, and NRF2 is subsequently degraded through the ubiquitin/proteasome system.5–9Inhibition of the KEAP1/NRF2 interaction prevents this ubiquitination and degradation, leading to an increase in NRF2 levels. NRF2 then translocates to the nucleus, heterodimerizes with the small MAF (sMAF) transcription factors and binds to antioxidant response elements (AREs) in the promotor regions of many detoxification genes;2, 5, 10–15 thus, inhibitors of the KEAP1/NRF2 interaction serve as NRF2 activators.
Figure 1:

(A) Transcription factor NRF2, when bound to substrate adaptor KEAP1, is ubiquitinated by E3 ligase CUL3, leading to proteosomal degradation. Two mechanisms explain activation of NRF2 by electrophiles. (B) In the CUL3 dissociation mechanism, cysteine modification by an electrophile causes dissociation of CUL3, leading to accumulation of NRF2. (C) In the hinge-and-latch mechanism, cysteine modification causes non-optimal placement of NRF2 for degradation. (D) In the conformational cycling model, cysteine modification locks KEAP1 and NRF2 in a conformation not ideal for ubiquitination, preventing KEAP1 from binding nascent NRF2, leading to accumulation of NRF2 (E) The non-electrophilic mechanism aims to create a non-covalent inhibitor of the protein-protein interaction between KEAP1 and NRF2 to activate NRF2.
Oxidative and electrophilic stressors induce transcription of detoxification enzymes by disrupting the interaction of NRF2 and KEAP1, but they do so indirectly. Three prevailing mechanisms exist to explain this disruption, namely: 1) the CUL3 dissociation model, 2) the hinge and latch model, and 3) the conformational cycling model.6, 8, 9, 16, 17 In all three models, a KEAP1 homodimer binds to a single NRF2 molecule through high affinity ETGE and low affinity DLG motifs within the Neh2 domain of NRF2. Under electrophilic stress, modification of specific cysteine sulfhydryl groups leads to loss of CUL3, displacement of NRF2’s DLG motif, or a non-optimal conformation for ubiquitination. In the CUL3 dissociation model, a cysteine in the BTB domain, Cys151, of KEAP1 is modified, leading to dissociation of CUL3 from KEAP1, preventing NRF2 ubiquitination (Figure 1B). The hinge and latch model is triggered through electrophilic modification of cysteine residues in Keap1’s IVR region, rather than the BTB domain of KEAP1. This modification changes KEAP1’s conformation causing release of the DLG motif, while NRF2 remains bound at the ETGE motif “hinge.” According to this model, release of the DLG motif “latch” results in a binding pose of NRF2 that is inaccessible for ubiquitination (Figure 1C). In the conformational cycling model, all of the partners are still bound to one another, but covalent modification of KEAP1 brings about conformational changes in KEAP1 and NRF2 that disfavor ubiquitination of NRF2 (Figure 1D).17 All of these models result in an inability to ubiquitinate and degrade NRF2 in the cytosol, leading to NRF2 translocation into the nucleus. NRF2 then complexes with small MAF proteins, binds to antioxidant response elements, and transcribes the above mentioned detoxification enzymes.2, 10, 11, 14
Due to the role of NRF2 in cytoprotection, several NRF2 activators have been tested in the clinic against a number of disease states.18–20 All of these clinical candidates have utilized the same mechanism of action: an electrophile is introduced to interact with the sulfhydryl groups of KEAP1 and activate NRF2. Several NRF2 activators have proceeded as far as phase III clinical trials with a few making it into clinical use; however, many of these electrophilic compounds may have selectivity issues. For instance, both sulforaphane (3) and a bardoxolone (1) may have hundreds of molecular targets (Chart 1).21, 22 Similarly, Schulze-Topphoff et al. have shown that the mechanism of action of dimethyl fumarate 2 (Tecfidera®, Chart 1) in multiple sclerosis may be independent of the KEAP1/NRF2 axis.23 A selective, non-electrophilic molecule could be useful to disentangle the contribution of NRF2 activation against a particular pathophysiological state.24
Chart 1:

Electrophilic activators of NRF2
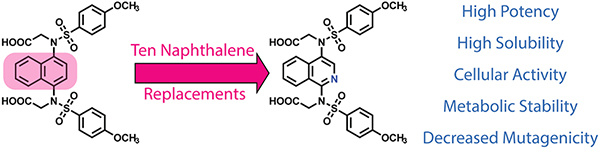
The binding sites of NRF2’s ETGE and DLG motifs on KEAP1 represent “hot spots” that can be targeted with non-electrophilic small molecules and peptides. This route differs from the electrophilic mechanism of NRF2 activation, and may provide advantages in terms of on-target selectivity.19, 25–27 We and others have previously reported several NRF2 activators that use a 1,4-substituted naphthalene core.1, 16, 28–30 These compounds show low nanomolar potencies in vitro and increase transcription of NRF2 target genes in cells; however, the 1,4-diaminonaphthalene core imparts properties that may negatively impact the development of these molecules. For instance, Winkel et al. showed that naphthalene 4 (RA-839) was active in vivo only if Phase 1 metabolism were inhibited (Chart 2).31 Naphthalene itself is metabolically activated to naphthalene oxide and naphthoquinones, and others have shown that it can covalently react with proteins in vitro and in vivo.32–35 Additionally, some aminonaphthalenes show carcinogenic and mutagenic properties.36, 37 Taken together, these findings suggest that the central naphthalene scaffold may not be an optimal core for this series of compounds.
Chart 2:

Naphthalene 4 (RA-839) is active in vivo only if Phase 1 metabolism is inhibited.
To compare novel-scaffold NRF2 activators to extant naphthalene-based activators, we were interested in studying the physical properties of these molecules beyond potency. There have been some recent perspectives that suggest that the emphasis of medicinal chemists, particularly those in academia, on potency often leads to highly potent, but poorly drug-like, molecules.38 With these concerns in mind, we designed and synthesized compounds with scaffolds to replace the naphthalene as a means to study potency, aqueous solubility, metabolic stability and mutagenicity of NRF2 activators. Our results indicate that a 1,4-substituted isoquinoline presents a new scaffold for NRF2 activators that shows similar potency, aqueous solubility, and metabolic stability to the 1,4-substituted naphthalene. Additionally, a mini-Ames assay shows that the 1,4-substituted isoquinoline displays an improved mutagenic profile relative to the 1,4-substituted naphthalene. These results have important implications for the design of future NRF2 activators.
Results:
Analog Design.
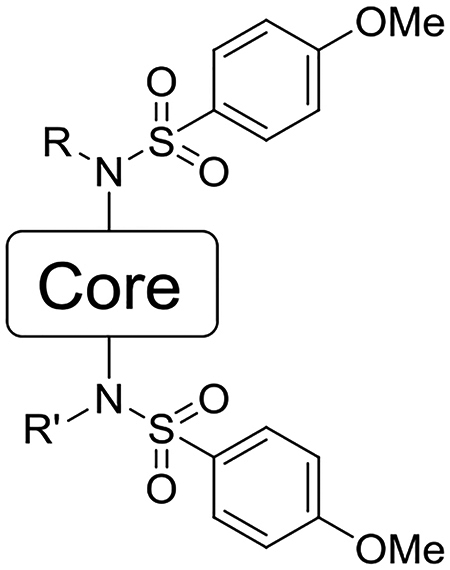
NRF2 activators displaying a large set of different cores were synthesized based off the naphthalene compound 6a. In previous work, we and others reported compounds varying the terminal rings, the sulfonamide R groups, the sulfonamide linker, and possible isosteric replacements for the carboxylates.1, 29, 30 Additionally, we reported that the 1,4-substitution pattern was optimal, as other substitution patterns showed complete loss of activity. In the current work of designing non-naphthalene derivatives, we maintained the sulfonamides, either unsubstituted or with bis-carboxymethyl substitutions, and we varied the connecting scaffold. Previously, we showed that bis-carboxamide 6b showed high potency.1 During the course of the current studies, we determined that the amides of 6b hindered aqueous solubility and caused rapid degradation in liver microsomes. As a result, we did not pursue this substituent in the current study.
We sought to synthesize compounds that maintained a display of substituents that was similar to that of the naphthalenes. To accomplish this, we synthesized molecules that featured a retention of the topology of the naphthalene that displayed different electronics (i.e., 1,4-isoquinoline; 5,8-quinoline; 1,4-phthalazine; 4,7-indole) (Figure 2). We also synthesized compounds that no longer contained the 6,6-fused ring system to examine the distal ring and possibilities of altering it (i.e., 1,4-benzene; 2,5-pyridine; o-xylene; 3,5-biphenyl; phthalamide). Lastly, we synthesized compounds that no longer contained an aromatic scaffold to fully determine what was necessary for activity (i.e., butene).
Figure 2:

Non-fused (red), acyclic (blue) and heterocyclic (magenta) analogs were designed to replace the naphthalene core of non-covalent NRF2 activators.
Analog Synthesis.
Compounds 5, 6a, and 6b were synthesized as previously described.1
Benzenes 9b and 11b and di-substituted pyridine 11a were synthesized (Scheme 1) starting from the reaction of phenylene-1,4-diamine 7b or 2,5-diamino pyridine 7a and p-methoxybenzenesulfonyl chloride 8 in pyridine to obtain the corresponding sulfonamides 9a and 9b. Reaction with ethyl bromoacetate in the presence of K2CO3 followed by saponification gave 11a and 11b.
Scheme 1. Synthesis of compounds 11a and 11ba.

aReagents: (a) pyridine, rt, 18–76% yield; (b) RBr, K2CO3, DMF, rt, 80% yield; (c) NaOH, MeOH, reflux. 67–80% yield; rt: room temperature
Analogs with a 1,4-isoquinoline core were synthesized according to Scheme 2. 1-hydroxyisoquinoline 12 was converted to 1,4-dibromoisoquinoline 13 using PBr5. An Ullmann-type coupling with p-methoxybenzenesulfonamide 14, followed by alkylation and saponification yielded isoquinoline 17. In the synthesis of isoquinoline 17, undesired alkylation of the isoquinoline ring nitrogen, rather than the desired sulfonamide alkylation, could have occurred while installing the carboxymethyl functionalities (i.e. 15 → 16). 2D-NMR experiments confirmed the assigned sites of alkylation (Figure S8).
Scheme 2. Synthesis of 1,4-isoquinolines 15 and 17a.

aReagents: (a) PBr5, 140 °C, 89% yield; (b) CuI, N,N’-dimethylethylenediamine, K2CO3, MeCN, 70 °C, 60% yield; (c) RBr, K2CO3, DMF, rt, 77% yield; (d) NaOH, MeOH, reflux, 36% yield. rt: room temperature
Compounds with a quinoline core were synthesized as shown in Scheme 3. 5-Aminoquinoline 18 was reacted with HONO in the presence of aniline to form the diazo compound 19, which was further reduced in the presence of stannous chloride to yield 5,8-diaminoquinoline 20.39 Sulfonylation followed by alkylation and saponification yielded quinoline 23.
Scheme 3. Synthesis of quinolines 21 and 23a.

aReagents: (a) aniline, NaNO2, HCl, AcOH, NaOAc, H2O, 30% yield; (b) SnCl2, HCl, reflux, 50% yield; (c) p-methoxybenzenesulfonyl chloride, pyridine, rt, 77% yield; (d) RBr, K2CO3, DMF, rt; (e) NaOH, MeOH, reflux, 67% yield over 2 steps. rt: room temperature
Analogs containing an indole core were synthesized as shown in Scheme 4. 2,4-Difluoronitrobenzene 24 was sequentially aminated at the 2- and 4-positions to yield a differentially bis-aminated intermediate 25. Cyclization was carried out in the presence of TFA to yield 4-benzylamino-7-nitroindole. Reduction and hydrogenolysis yielded 4,7-diaminoindole 26,40 which was further sulfonylated, alkylated, and saponified to yield indole 29. HNMR results confirmed that compound 27 was sulfonylated at the 1- and 4-positions. Singlets at 10.69, 9.79, and 9.61, each integrating to 1H, correspond to the indole NH, and two sulfonamide NH groups.
Scheme 4. Synthesis of indoles 27 and 29a.

aReagents: (a) aminoacetaldehyde dimethyl acetal, i-Pr2NEt, MeOH, reflux, 86% yield; (b) benzylamine, 1,4-dioxane, reflux, 93% yield; (c) F3CCO2H, CH2Cl2, rt, 85% yield; (d) Pd/C, EtOH/HCl, H2, 35 psi, 100% yield; (e) p-methoxybenzenesulfonyl chloride, pyridine, rt, 60% yield; (f) RBr, K2CO3, DMF, rt, 82% yield; (g) TFA, CH2Cl2, rt, 20% yield. rt: room temperature
Phthalazine 34 was prepared beginning from phthalonitrile 30, which was condensed with hydrazine in methanol to give 1,4-diaminophthalazine 31, which yielded the sulfonamide 32. Alkylation of the sulfonamide gave only the mono-alkylated products 33 and 34, regardless of the reaction conditions employed.
Analogs of 6a that did not feature a fused ring system were synthesized using the appropriate electrophilic starting materials (Scheme 6). Substituted sulfonamides were used as nucleophiles to yield xylene 38, phthalamide 40, and butene 42 after saponification.
Scheme 6. Synthesis of compounds 38, 40, and 42a.

aReagents: (a) pyridine, rt 55–58% yield; (b) RBr, K2CO3, DMF, rt, 70–80% yield; (c) NaOH, MeOH, reflux, 70–74% yield; (d) F3CCO2H, CH2Cl2, rt, 55% yield. rt: room temperature
Biphenyl 46 was synthesized as shown in Scheme 7. 1-bromo-3,5-dinitrobenzene 43 was reacted with phenylboronic acid via a Suzuki coupling to create the biphenyl core. The nitro groups were reduced to amines to yield a diamino intermediate 44, which was then sulfonylated, alkylated, and saponified to arrive at the analog 46.
Scheme 7. Synthesis of biphenyl 46.

aReagents: (a) phenylboronic acid, Pd(OAc)2, K3PO4, H2O, reflux, quant. yield; (b) SnCl2, MeOH, reflux, 99% yield; (c) p-methoxybenzenesulfonyl chloride, pyridine, THF, reflux; (d) ethyl bromoacetate, K2CO3, DMF; (e) NaOH, MeOH, reflux, 51% yield over 3 steps. rt: room temperature
In vitro assays.
The IC50 of each compound for inhibiting the interaction of a fluorescent NRF2 peptide and the Kelch domain of KEAP1 was determined using fluorescence anisotropy.41 The results are shown in Table 1. The heterocyclic cores ranged in potency: the 1,4-isoquinoline 17 (IC50 = 60 nM) retained much of the potency of the parent naphthalene 6a, the 5,8-quinoline 23 was well tolerated (IC50 = 101 nM), the phthalazine 34 was tolerated (IC50 = 1000 nM), and the indole 29 was tolerated (IC50 = 1300 nM). Unfortunately, the indole 29 was found to be unstable, which precluded its further consideration. The non-fused systems all showed little to no activity: the phenyl 11b was tolerated (IC50 = 980 nM), whereas the xylene 38, pyridine 11b, phthalamide 40, and biphenyl 46 were all inactive (>25 μM). The removal of the core to give butene 42 also produced an inactive compound.
Table 1: IC50 and Kd values.
IC50 values, shown in nM units with 95% confidence intervals, were determined by fluorescence anisotropy. Kd values, shown in nM units ± standard deviation, were determined by surface plasmon resonance. aFluorescent interference from 15 impeded accurate determination of IC50. b IC50 and Kd values reported in Jain et al.1. ND = Not determined
| Compound | Core | R | R’ | IC50 (nM) (95% CI) | Kd (nM) |
|---|---|---|---|---|---|
| 5 |  |
−H | −H | 940 b (850 – 1300) | 1700 ± 120 b |
| 6a | −CH2COOH | −CH2COOH | 25 b (19–31) | 20 ± 3 b | |
| 6b | −CH2CONH2 | −CH2CONH2 | 63 b (52 – 85) | 44 ± 8 b | |
| 9b |  |
−H | −H | >25000 | ND |
| 11b | −CH2COOH | −CH2COOH | 980 (600–1600) | ND | |
| 11a | 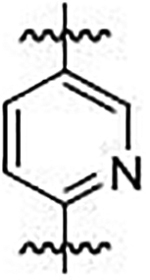 |
−CH2COOH | −CH2COOH | >25000 | ND |
| 15 | 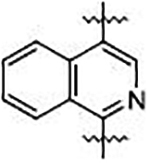 |
−H | −H | NDa | ND |
| 17 | −CH2COOH | −CH2COOH | 60 (30–110) | 102 ± 16 | |
| 21 | 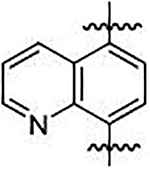 |
−H | −H | >25000 | ND |
| 23 | −CH2COOH | −CH2COOH | 101 (80–130) | 103 ± 2 | |
| 27 | 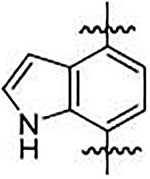 |
−H | −H | >25000 | ND |
| 29 | −CH2COOH | −CH2COOH | 1300 (390–5400) | ND | |
| 32 | 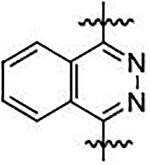 |
−H | −H | >25000 | ND |
| 34 | −CH2COOH | −H | 1100 (435–2500) | > 10 μM | |
| 38 | 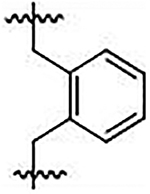 |
−CH2COOH | −CH2COOH | >25000 | ND |
| 40 | 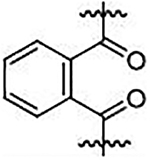 |
−CH2COOH | −CH2COOH | >25000 | ND |
| 42 | 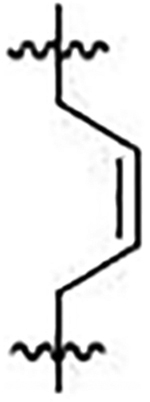 |
−CH2COOH | −CH2COOH | >25000 | ND |
| 46 | 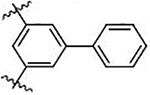 |
−CH2COOH | −CH2COOH | >25000 | ND |
Compounds with low IC50 values in the FP assay were confirmed using an orthogonal Surface Plasmon Resonance (SPR) assay (Table 1). 1,4-Isoquinoline 17 exhibited a Kd of 102 nM, 5,8-quinoline 24 exhibited a Kd of 103 nM, and phthalazine 34 exhibited a Kd of >10 μM, in good agreement with the results of the FP assay.
Aqueous Solubility.
The aqueous solubility of all compounds at pH 7.4 was determined as shown in Table 2. Heterocycles 23 and 17, phthalamide 40, and butane 42 all showed solubility comparable to the naphthalene parent compound 6a. Xylene 38 and benzene 11b showed poor solubility. Pyridine 11b and biphenyl 46 both showed moderate solubility. In all cases except phthalazine 32, unsubstituted sulfonamides were poorly soluble (solubility < 10 μM). The bis-amide 6b that we previously reported as a potent NRF2 activator was shown to have poor solubility, which, in part, led us to focus our attention on the carboxymethyl analogs.
Table 2:
Aqueous solubility, shown in μM units ± standard deviation.
| Compound | Aqueous Solubility (μM) |
|---|---|
| 5 | 5.4 ± 0.9 |
| 6a | 440 ± 3 |
| 6b | 22 ± 9 |
| 9b | 1.2 ± 0.2 |
| 11b | 1.2 ± 0.2 |
| 11a | 190 ± 0.8 |
| 15 | 3.8 ± 0.1 |
| 17 | 380 ± 3 |
| 21 | 6.1 ± 0.1 |
| 23 | 390 ± 4 |
| 27 | 0.86 ± 0.3 |
| 29 | N.D.b |
| 32 | 370 ± 7 |
| 34 | 380 ± 7 |
| 38 | 1.3 ± 0.3 |
| 40 | 490 ± 90 |
| 42 | 420 ± 12 |
| 46 | 270 ± 3 |
NRF2 Stabilization and NRF2 Target Gene Activation.
To see the effects of KEAP1 inhibitors on NRF2 activation, we carried out a screening assay to determine the effects of naphthalene acid 6a, naphthalene amide 6b, isoquinoline 17, quinoline 23, and phthalazine 34 on NRF2 protein stability and its downstream target gene activation in non-transformed immortalized human keratinocytes (HaCat cells). Cells were treated with compounds for 6 h to determine the stabilization of NRF2 and the protein expression levels of NQO1, a well-known target of NRF2 (Figure 3). Treatment of HaCaT cells with 6a, 6b, and 23 for 6 h caused a modest increase in protein levels of NRF2 and NQO1 expression in a manner comparable to CDDO-Im 1, a well-known potent electrophilic activator of NRF2. Phthalazine 34 caused no increase in either NRF2 protein levels or NQO1 protein levels in this assay.
Figure 3:

HaCaT cells were treated with 10 μM compound (6a, 6b, 17, 23; positive control CDDO-Im) or vehicle (DMSO) for 6 h (A) or 24 h (D) as shown in the immunoblots. Cell lysates were prepared, equal amount of protein was probed with antibodies specific for NRF2, NQO1 or GCLM. GAPDH was used as a reference. Quantification of band intensities of NRF2 (B, E), NQO1 (C, F) and GCLM levels (G) are shown. DMSO-treated sample values were used as one arbitrary unit.*:p<0.05; **:p<0.01; ***:p<0.001; if no * is indicated, p>0.05.
We next analyzed NRF2 and NQO1 protein expression levels in HaCat cells treated with compounds for a prolonged period (i.e., 24 h). Increased protein levels of NRF2 and NQO1 were noted in cells treated with naphthalene acid 6a, naphthalene amide 6b, or isoquinoline 17 for 24 h compared to vehicle-treated counterparts (Figure 3). Likewise, both NRF2 and NQO1 protein levels were higher in cells treated with CDDO-Im 1. Quinoline 23 had no effect on either NRF2 stability or NQO1 expression. We also analyzed the expression of GCLM, a well-established target of NRF2. GCLM protein levels were significantly higher in cells treated with naphthalene acid 6a, naphthalene amide 6b, isoquinoline 17, or CDDO 1 compared to vehicle controls. Unlike NRF2 protein stabilization or NQO1 protein expression, GCLM protein expression was modestly increased in cells treated with quinoline 23.
Positive results in the screeening assay seen with isoquinoline 17 and quinoline 23 led us to examine nuclear accumulation of NRF2 (immunoblot) and expression of other NRF2 target genes (qPCR) in HaCaT cells treated with isoquinoline 17 and quinoline 23 at both 6 and 24 hour time points. NRF2 accumulation in the nucleus was increased two-fold when treated with isoquinoline 17 compared to DMSO control at both time points analyzed (Figure 4). A small significant increase in NRF2 nuclear accumulation was observed in cells treated with quinoline 23 at both time points. Analysis of mRNA levels in the HaCaT cells at 6 hours showed isoquinoline 17 produced a significant increase in HMOX1, GCLM, and GCLC mRNA expression (Figure 5). Isoquinoline 17 also produced an increase in NQO1 mRNA expression at 24 hours. Quinoline 23 produced a small increase in NQO1, GCLM, and GCLC mRNA expression at 6 and 24 hours, with no increase observed for HMOX1 mRNA expression. mRNA expression for SOD was also measured. SOD is not an NRF2 target gene, so, as expected, neither isoquinoline 17 nor quinoline 23 produced a significant increase in SOD expression. Western blot analysis of NQO1 protein levels corroborated the mRNA expression levels by showing a modest increase in protein expression in comparison to DMSO for both isoquinoline 17 and quinoline 23 (SI Figure S11).
Figure 4:

The effects of non-electrophilic activators 17 (orange) and 23 (purple) and positive control CDDO-Im on the nuclear accumulation of NRF2 in HaCaT Cells. (A) HaCaT cells were treated with DMSO (vehicle) or indicated compound (10 μM) for 6 hours and 24 hours, Nuclear (N) and cytosolic (C) fractions were prepared, equal amount of protein was immunoblotted using antibodies specific for NRF2. β-actin / Lamin A/C was used as loading control. (B) Quantification of Nrf2 band intensity using β-actin as reference. DMSO-treated sample value was used as one arbitrary unit. Error bars represent the standard error from four independent experiments. *:p<0.05; **:p<0.01; ***:p<0.001; if no * is indicated, p>0.05.
Figure 5:

The effects of non-electrophilic activators 17 (orange) and 23 (purple) on NRF2 target gene expression in HaCaT Cells. (A) HaCaT cells were treated with DMSO (vehicle) or indicated compound (10 μM) for 6 hours and 24 hours. cDNA were prepared from mRNA and analyzed for the target gene expression (A: HMOX1; B: NQO1; C: GCLC; D: GCLM; E: SOD) using qPCR with specific primers. Relative fold change was calculated with β-actin as reference. DMSO-treated sample value was used as one unit. Error bars represent the standard error from 3–4 independent samples. *:p<0.05; **:p<0.01; ***:p<0.001; if no * is indicated, p>0.05.
Metabolic Stability in Liver Microsomes.
Active and moderately active compounds that showed good solubility were subjected to human and mouse liver microsomes for 20 minutes to ascertain the metabolic stability of the compounds. 1,4-Isoquinoline 17, naphthalene 6a, and 5,8-quinoline 23 were stable at 20 minutes in both human and mouse liver microsomes (Figure 6). Phthalazine 34 and 5,8-quinoline 23 showed reduced stability in human liver microsomes, but were comparable to the other compounds in mouse liver microsomes. The naphthalene amide 6b was metabolically unstable, showing 0% compound remaining after 20 minutes in both mouse and human liver microsomes, providing further evidence that amide analogs may be rapidly hydrolyzed. The unsubstituted naphthalene 5 was metabolically unstable in mouse liver microsomes, showing only 15% remaining after 20 minutes.
Figure 6: Stability to Liver Microsomes.

Compounds were incubated with mouse liver microsomes (A) and human liver microsomes (B) for 20 min. Percentage of compound remaining after incubation was determined by LC-MS.
Mutagenicity in a Mini-Ames Assay.
To probe our hypothesis that the naphthalene may prove to be mutagenic, we submitted naphthalene 6a and isoquinoline 17 to a mini-Ames assay in Salmonella typhimurium and Escherichia coli. The compounds were tested for their ability to induce reverse mutations at the histidine locus of strains of Salmonella typhimurium (TA98, TA100, TA1535, TA1537) and Escherichia coli (WP2 uvrA (pKM101)) in the presence and absence of exogenous metabolic activation (Aroclor 1254 induced rat liver S9). Both naphthalene 6a and isoquinoline 17 were screened at concentrations of 1.5, 4, 10, 25, 64, 160, 400 and 1000 μg/well. No cytotoxicity was observed in either naphthalene 6a or isoquinoline 17 at any dose level. Naphthalene 6a induced > 3-fold increases (for TA1535) and > 2-fold increases (for TA98 and TA100) that were statistically significant at higher dose levels in the presence of S9 mix, compared to vehicle control (Figure 7). Dose response was also observed in 6a for TA98 and TA100; therefore, naphthalene 6a was judged to be positive for mutagenicity. On the other hand, isoquinoline 17 induced a > 2-fold increase in only one strain (for TA98) at the highest dose level compared to the vehicle control. No dose response was observed for isoquinoline 17 (Figure 7). Taken together, these data lead us to conclude that isoquinoline 17 has a better mutagenic profile than naphthalene 6a.
Figure 7: Mini-Ames Assay Results.

Compounds 6a (turquoise) and 17 (orange) and vehicle control DMSO (white) were tested for their ability to induce reverse mutations at the histidine locus of strains TA98 (A), TA100 (B), TA1535 (C), and TA1537 (D) of Salmonella typhimurium and strain WP2 uvrA(pKM101) (E) of Escherichia coli in the presence of exogenous rat liver S9 fractions. Individual revertant colonies were counted and graphed as a function of compound concentration (400 and 1000 μg/well). Fold-change for those conditions that were >2.0× greater than DMSO vehicle control are shown above each bar, along with p-values determined from a two-tailed t-test.
Discussion:
Previously, our lab investigated the structure-activity relationship of the distal substitutions on the naphthalene-based NRF2 activator 5. We found that an electron-donating group on the terminal phenyl groups resulted in the most potent inhibitors. We also found that the sulfonamide linkages were superior to similar linkages and that substituting anything other than a carboxymethyl group or carboxamidomethyl group off the sulfonamide also resulted in an inactive compound.
Having explored the rest of the molecule, we turned our attention to the naphthalene scaffold. While there are drugs, including propranolol, that contain a naphthalene core, naphthalenes have been identified as a minor mutagenic scaffold in the Ames Mutagenicity Tree.36 A related naphthalene 4, RA-839, showed in vivo activity only if Phase 1 metabolism were blocked. Although the identity of the RA-839 metabolites are unknown, they may include reactive intermediates, as naphthalenes are known to be metabolically activated to reactive naphthalene oxides and naphthoquinones. 32–35 With these points in mind, we explored alternative scaffolds.31
Our previous results suggested that compounds with amide-containing sidechains (e.g., 6b) were comparable in potency to those containing carboxylic acid-containing sidechains.1 During the course of the present study, however, we found that the amides are both relatively insoluble in water and metabolically unstable in liver microsomes, compared to the acids. This is in good agreement with work reported by Lu et al., who showed that amide-containing compounds were less soluble than acid-containing compounds at pH 7.4.30 Because we were interested in compounds that would show good solubility and metabolic stability, we synthesized scaffolds that were decorated with N-carboxymethyl groups.
To probe the necessity of a 6,6-fused ring system, we synthesized non-fused rings and substitutions that might afford a disposition of substituents similar to the naphthalene scaffold. These substitutions included butene 42, 1,4-substituted pyridine 11a, 1,4-substituted benzene 11b, 1,3,5-substituted benzene 46, o-xylene 38, and phthalamide 40. Of these, only the benzene 11b showed activity in the fluorescence anisotropy assay, albeit at values ca. 40-fold higher than the parent naphthalene 6a. These compounds displayed varying levels of solubility that did not divulge any discernible pattern. The butene 42 and phthalamide 40 showed solubilities similar to the parent naphthalene 6a, the pyridine 11a and 1,3,5-substituted benzene 11a showed slight solubility, and the o-xylene and 1,4-substituted benzene 11b proved insoluble. Because these compounds showed poor potency and solubility comparable to other scaffolds, we did not carry them into liver microsome stability studies.
To probe the necessity of the all-carbon framework of the naphthalene scaffold, we replaced the naphthalene core with nitrogen-containing heterocycles, including 1,4-phthalazine 34, 1,4-isoquinoline 17, 5,8-quinoline 23, and 4,7-indole 29. Of these, 5,8-quinoline 23 and 1,4-isoquinoline 17 retained good potency in both FP (IC50 = 101 and 60 nM, respectively) and SPR assays (Kd = 103 and 102 nM, respectively). The 1,4-phthalazine 34 scaffold was less potent (IC50 = 1000 nM; Kd >10 μM). Previously, we reported that a naphthalene mono-acid showed good affinity for KEAP1 (IC50 = 61 nM).1Phthalazine 34, which is also a mono-acid, is less potent than the naphthalene mono-acid, but this is in agreement with the affinity of the unsubstituted cores: unsubstituted naphthalene 5 has an IC50 of 0.96 μM, while the unsubstituted phthalazine 32 has an IC50 of > 25μM. Taken together, these data suggest that the all-carbon framework of the naphthalene is not necessary for binding to KEAP1, and other 6,6-fused systems can be used to replace the naphthalene scaffold.
The binding of this class of compounds is dictated, in part, by a π-cation interaction between the π-system of the inhibitor and Arg415, first observed by Marcotte et al.27Dougherty and coworkers have comprehensively studied the binding energy of cations to a number of different π-systems. Their work suggests that electron-rich systems, like indole, should be better cation binders than naphthalene, which should be better than electron-poor systems like quinoline and isoquinoline.42–44 In our work, we see that substituted and unsubstituted naphthalenes 5 and 6a show a slightly higher affinity than the corresponding quinolines (21 and 23) and isoquinolines (17), which is consistent with this ordering (Table 1). On the other hand, indoles 27 and 29 show an IC50 of >25 μM and 1300 nM, while naphthalene 5 shows an IC50 of 960 nM. While the instability of the indole-containing di-acid may complicate this analysis, these data may suggest that the contribution of the π-cation interaction to binding may be offset by other factors. Additional analogs and future experiments may shed more light on these intriguing observations.
With two charged carboxylates, naphthalene 6a shows high aqueous solubility at pH 7.4 (440 μM). These high solubility values are maintained across most of the heterocyclic scaffolds, as well. The carboxylates clearly contribute to the solubility, as the unsubstituted compounds 5, 9b, and 15 show only modest aqueous solubilities.
The stabilities of these compounds in mouse and human liver microsomes suggest that the most metabolically stable compounds are naphthalene 6a, 1,4-isoquinoline 17, and 5,8-isoquinoline 23. After 20 minutes of incubation with mouse or human liver microsomes, there was little loss of compound. This is in contrast to the bis-amide 6b and the unsubstituted compound 5, which were both rapidly degraded in this time frame. This is in good agreement with a previous report that naphthalene 4 (Chart 2), which is alkylated on only one aromatic nitrogen, is metabolized quickly in vivo.31 These data suggest that several 1,4-diamino scaffolds are stable, as long as both nitrogen atoms are alkylated. Further evidence for this point comes in the metabolic stability data for phthalazine 34, with approximately 50% of the compound remaining after 20 minutes of incubation with human liver microsomes. The mono-carboxylate system presents its own advantages over the bis-carboxylate system; the overall charge of the molecule is lessened which may assist in cell permeability. The current results suggest, however, that the substitution pattern may play a major role in the metabolic stability of the molecules, which may imply that a balance is needed between charge state and substitution.
Our results obtained from immunoblot analysis demonstrate NRF2 stabilization and transactivation of its target gene expression by compounds 6a, 6b, or 17 in human keratinocytes in a manner comparable to CDDO 1, a potent activator of NRF2. The effects of these compounds were more pronounced at 24 h, although they showed a modest effect on NRF2 stabilization and NQO1 expression when treated for a shorter period (6 h). CDDO 1 had a similar effect on NRF2 and its target gene activation in HaCat cells. Previously, we have shown increased protein levels of NRF2 stabilization and its target gene activation by NRF2 activator compounds in lung epithelial cells at 6h-post treatment.1 A robust increase in NRF2 stabilization and the induction of NRF2 target gene expression in HaCat cells by the activators at 24 h but not at 6 h post-treatment could be attributable to cell-specific effects, as CDDO 1, a potent NRF2 activator, also showed a similar activation pattern in HaCat cells. Nonetheless, our studies showed 6a, 6b, and 17 activate NRF2 target gene expression in human keratinocytes in a manner comparable to CDDO 1.
Our immunoblot analysis led us to measure nuclear accumulation of NRF2 and NQO1 in conjunction with mRNA expression of NRF2 target genes in HaCaT cells treated with isoquinoline 17 and quinoline 23 (Figure 5). The results showed that treatment with isoquinoline 17 produced a 2-fold increase of NRF2 accumulation in the nucleus at 6 and 24 hours along with a corresponding increase in NRF2 target genes NQO1, HMOX1, GCLM, and GCLC. Treatment with quinoline 23 showed a modest increase of NRF2 accumulation in the nucleus at 6 and 24 hours with a small increase in NRF2 target genes NQO1, GCLM, and GCLC. No increase in HMOX1 was observed in cells treated with quinoline 23.
Even though quinoline 23 showed good affinity for KEAP1 and activated NRF2 target genes, it was not completely stable to human liver microsomes, with 50% remaining after 20 minutes. We are uncertain of the identities of the metabolites, but, taken together with the potential for an 8-aminoquinoline scaffold to chelate metals, these data suggest that this scaffold should be deprioritized.
A mini-Ames assay conducted on naphthalene acid 6a and isoquinoline 17 showed that neither compound is cytotoxic, but the data collected led us to conclude that isqouinoline 17 showed a better mutagenic profile than naphthalene 6a. Naphthalene 6a induced > 2-fold increases and > 3-fold increases of reverse mutations at the histidine locus of three strains of Salmonella typhimurium (TA98, TA100, TA1535) in a dose-dependent manner. Isoquinoline 17 induced a > 2-fold increase of reverse mutations at the histidine locus of Salmonella typhimurium strain TA98 at only the highest dose, with no dose-response observed. These data demonstrated that isoquinoline 17 had a better mutagenic profile than naphthalene 6a and should be studied more closely.
Generally, the compounds’ abilities to activate NRF2 target genes correlated with in vitro potency, solubility, and metabolic stability, as naphthalene 6a, isoquinoline 17, and quinoline 23 all showed good in vitro potency and solubility, as well as varying degrees of metabolic stability. An exception is the bis-amide 6b, which was metabolically unstable and poorly soluble but able to activate NRF2 target genes. It is possible that one or both of the amides are hydrolyzed to acids (e.g., 6a) in cell culture, which may produce its activation of NRF2 target genes. The neutrally charged amides may support cell permeability, similar to the effect we have previously reported for an ester,1 but the poor aqueous solubility of amide 6b suggests that a primary amide is a poor mask of the acid for optimization of these compounds. You and coworkers have previously reported that isosteres of carboxylic acids are soluble, cell-permeable, and supportive of NRF2-regulated gene activation.30 Taken together with our data, these results suggest that an isostere or prodrug approach may be a good strategy for multi-parameter optimization of this class of molecules.
Indeed, it is likely that the bis-acid substitution pattern is not optimal for development of a preclinical candidate.45 We are working to reduce the number of carboxylic acids contained in the 1,4-isoquinoline scaffold. These compounds need further optimization and verification if their effects are to be translated to clinical trials, but they present a promising strategy for developing Nrf2 activators.
The two compounds that show high affinity, stability and solubility are naphthalene 6a and 1,4-isoquinoline 17. These compounds show stabilization of NRF2 and activation of NRF2 target genes in western blots as well. While much work has been done by us1 and others28, 30, 46 on the substituents on the naphthalene scaffold, the current work has identified 1,4-isoquinoline as a new scaffold for optimizing noncovalent NRF2 activators, a topic of considerable current research activity. We foresee that the 1,4-isoquinoline core may provide a number of advantages relative to naphthalene.
First, the naphthalene scaffold may be more toxic and/or mutagenic than the isoquinoline scaffold. Oral toxicity as predicted by PROTOX, an online prediction tool that works through analysis of a structure and comparison to known toxicophores and toxic fragments,47 predicted the naphthalene 6a to be slightly toxic in the class of 4/6 whereas the 1,4-isoquinoline 17 was predicted to be non-toxic in the class 6/6. Lazar toxicity predictions, which generate QSAR models for compounds to predict toxicity,48 mirrored this by predicting the 1,4-isoquinoline 17 to be less toxic than the naphthalene 6a across all categories calculated. Beyond these computational studies, we have conducted a mini-Ames assay on naphthalene 6a and isoquinoline 17, the latter of which was shown to be less mutagenic than naphthalene 6a.
Second, the unsymmetrical nature of the 1,4-isoquinoline serves to desymmetrize the molecule. The binding site on KEAP1 is not symmetrical, and we and others have shown that symmetrical ligands are not required for binding to KEAP1;24, 46 the current work further emphasizes that point.
Lastly, the pharmacokinetics of a negatively charged NRF2 activator may be enhanced by the presence of a basic nitrogen. In support of this, we experimentally determined the LogD7.4 values of naphthalene 6a (−1.9) and 1,4-isoquinoline 17 (−1.7). The slight improvement in LogD7.4 seen with isoquinoline 17 suggests this scaffold may provide a better starting point for further optimization of these molecules.
Overall, there have been a number of medicinal chemistry and biological studies that have been carried out using naphthalene-based NRF2 activators.1, 28, 30, 31, 49, 50 Our data suggest that it may be prudent to consider other scaffolds going forward.
Conclusion
In conclusion, this work demonstrates that the naphthalene core of non-electrophilic NRF2 activators may be replaced with 1,4-isoquinoline, with little to no disadvantage in potency, cellular activity, solubility or metabolic stability. Importantly, isoquinoline 17 displays a better mutagenic profile than naphthalene 17. Other heterocycles and scaffolds were examined, as well, but each of these had at least one liability that isoquinoline 17 did not have. We have also shown that alkylation of the sulfonamide nitrogens in this series is necessary for metabolic stability in liver microsomes. Taken together, these data suggest that, relative to the often-used naphthalene scaffold, isoquinoline 17 represents a preferable core for new NRF2 activators.
Experimental
Chemistry:
General considerations.
1H and 13C NMR spectra were recorded on a Bruker AVIII and AVIII HD 400 spectrometers equipped with a room temperature 5 mm broadband observe (BBO) probes with z-gradient. Chemical shifts were reported in parts per million (δ) using either tetramethylsilane (TMS) or the undeuterated residual solvent peak as an internal standard (CDCl3: 1H δ = 7.26 and 13C δ = 77.2; CD3COCD3: 1H δ = 2.05 and 13C δ = 29.2; CD3OD: 1H δ = 3.31 and 13C = 49.2; CD3CN: 1H δ = 1.96and 13C δ = 118.3; DMSO-d6: 1H δ = 2.50 and 13C δ = 39.5). Purity of each of the final, tested compounds was determined by HPLC on a Shimadzu LC-20AB (Solvent system; MeCN/H2O, 0.1% Formic Acid; Column: Shimadzu C18, 50 μm, 50 × 4.6 mm) and was ≥95% (HPLC Method: 15:85 MeCN:H2O, 0–2 min; 15:85 – 95:5 MeCN:H2O, linear gradient, 2 – 9 min; 95:5 MeCN:H2O, 9–10.7 min; 95:5 – 15:85 MeCN:H2O, linear gradient, 10.7 – 11 min; 15:85 MeCN:H2O, 11 – 13 min. UV, 254 nm). The HRMS spectra were recorded on a Shimadzu LCMS-IT-TOF. Flash chromatography was performed on a Teledyne-Isco Combiflash Rf+ system using preloaded silica gel cartridges (230–400 mesh). Reactions were monitored by thin-layer chromatography (TLC) on silica gel GHLF plates (250 μm, Macherey-Nagel, Inc.).
N,N’-(pyridine-2,5-diyl)bis(4-methoxybenzenesulfonamide) (9a)
This previously unreported compound was synthesized in a manner similar to a published procedure.1 2,5-diaminopyridine dihydrochloride 7a (3.0 g, 1.0 eq, 16.47 mmol) was dissolved in a mixture of THF (18 mL) and pyridine (6 mL). The mixture was heated to reflux and allowed to stir for a half hour. 4-methoxybenzenesulfonyl chloride 8 (8.16 g, 2.4 eq, 39.52 mmol) was added, and the reaction mixture stirred for 18 hours at reflux. The reaction was monitored by TLC (3:1, ethyl acetate:hexanes) and, upon completion, was quenched with ice water. The precipitate was filtered and washed with methanol. The product 9a was obtained as a white solid (1.5 g 18% yield). 1H NMR (400 MHz, DMSO-d6, δ): 10.8197 (br.s, 1H), 10.04 (s, 1H), 7.68–7.83 (m, 3H), 7.58 (d, J = 8.93 Hz, 2H), 7.39 (dd, J = 2.71, 8.85 Hz, 1H), 6.95–7.13 (m, 5H), 3.81 (s, 3H), 3.80 (s, 3H)
N,N’-(1,4-phenylene)bis(4-methoxybenzenesulfonamide) (9b)
This previously unreported compound was synthesized in a manner similar to a published procedure.51 A solution of p-aminoaniline (0.5 g, 4.6 mmol) and p-methoxybenzenesulfonyl chloride (2.4 g, 11.6 mmol) in pyridine (3–5 mL) was allowed stir at room temperature for 24 h. On completion, the reaction mixture was diluted with EtOAc (40 mL), washed with H2O (2 × 50 mL) and HCl (2N, 2 × 50 mL), dried over Na2SO4, and evaporated to yield the crude product, which, upon recrystallization from EtOH, yielded 1.6 g (76% yield) of (9b) as white needles. 1H NMR (400 MHz, DMSO-d6, δ): 9.93 (s, 2H), 7.49–7.66 (AA’XX’, J = 8.8 Hz, 4H), 6.96–7.09 (AA’XX’, J = 8.8 Hz, 4H), 6.90 (s, 4H), 3.79 (s, 6H); 13C NMR (100 MHz, DMSO-d6, δ): 162.4, 134.1, 131.1, 128.9, 121.5, 114.3, 55.7; HRMS-ESI (−) (m/z): [M-H]− calcd for C20H20N2O6S2, 447.0685; found, 447.0696.
Diethyl 2,2’-(pyridine-2,5-diylbis((4-methoxyphenyl)sulfonyl)azanediyl))diacetate (10a)
This previously unreported compound was synthesized in a manner similar to a published procedure.1 9a (0.700 g, 1.0 eq, 1.40 mmol) was dissolved in DMF (10 mL). Potassium carbonate (K2CO3, 0.445 g, 2.3 eq, 3.22 mmol) was added, and the mixture was stirred for a 0.5 hour at room temperature. Ethyl bromoacetate (340 μL, 2.2 eq, 3.08 mmol) was added, and the reaction mixture stirred for 16 hours at room temperature. The reaction was monitored by TLC (3:1, ethyl acetate:hexanes) and, upon completion, ice water was added to the mixture, whereupon a precipitate formed. The precipitate was filtered and recrystallized from ethanol to yield 0.702 g (80% yield). 1H NMR (400 MHz, DMSO-d6, δ): 8.09 (d, J = 2.5 Hz, 1H), 7.70 (dd, J = 2.0, 6.8 Hz, 2H), 7.62 (dd, J = 2.0, 6.9 Hz, 2H), 7.54 (dd, J = 2.6, 9.8 Hz, 1H), 7.32 (d, J = 9.8 Hz, 1H), 7.10 (dd, J = 2.0, 7.0 Hz, 2H), 7.03 (dd, J = 2.0, 6.9 Hz, 2H), 4.91 (s, 2H), 4.37 (s, 2H), 4.09 (m, 4H), 3.86 (s, 3H), 3.81 (s, 3H), 1.14 (m, 6H).
2,2’-(pyridine-2,5-diylbis(((4-methoxyphenyl)sulfonyl)azanediyl))diacetic acid (11a)
This previously unreported compound was synthesized in a manner similar to a published procedure.1 10a (0.200 g, 1.00 eq, 0.319 mmol) was dissolved in a solution of 15% sodium hydroxide (NaOH, 20 mL), and methanol (MeOH, 20 mL) was added to this. The reaction mixture was stirred at reflux for 8 hours. The reaction was monitored by TLC (3:1, ethyl acetate:hexanes) and, upon completion, the mixture was concentrated in vacuo. The aqueous mixture was acidified to a pH of ~6.0. On formation of precipitate, the flask was placed in a 4 °C refrigerator. The precipitate was then filtered and washed with diethyl ether to yield 0.180 g of a white solid (66% yield). 1H NMR (400 MHz, DMSO-d6, δ): 13.07 (b.s., 2H), 8.05 (d, J = 2.3 Hz, 1H), 7.71 (d, J = 8.8 Hz, 2H), 7.59 (d, J = 8.9 Hz, 2H), 7.55 (dd, J = 2.4, 9.7 Hz, 1H), 7.28 (d, J = 9.8 Hz, 1H), 7.07 (d, J = 8.9, 2H), 7.00 (d, J = 8.8 Hz, 2H), 4.84 (s, 2H), 4.25 (s, 2H), 3.85 (s, 3H), 3.81 (s, 3H). 13C NMR (400 MHz, DMSO-d6, δ): 169.9, 168.4, 163.5, 161.9, 154.6, 143.0, 142.2, 135.7, 130.1, 129.4, 128.4, 124.3, 116.2, 115.1, 114.3, 56.2, 55.9, 54.2, 51.8 HRMS–ESI (+) (m/z): [M+H]+ calcd for C23H23N3O10S2; 565.0892, found: 565.0825.
2,2’-(1,4-phenylene)bis(((4-methoxyphenyl)sulfonyl)azanediyl))diacetic acid (11b)
This previously unreported compound was synthesized in a manner similar to a published procedure.28 Potassium carbonate (0.28 g, 2.01 mmol) and ethyl bromoacetate (0.23 g, 1.4 mmol) were added to a solution of 9b (0.3 g, 0.67 mmol) in anhydrous DMF (2 mL), and the reaction was stirred at room temperature for 3 h. On completion the reaction was quenched with H2O (25 mL) and acidified with HCl (2N, to pH 3). The precipitate was collected by filtration and dried. A fraction of the dried product 10b (0.1 g) was suspended in NaOH (10%, 5 mL) and MeOH (10 mL) and allowed to reflux for 2 h. On completion the reaction mixture was cooled, the organic portion was evaporated, and diluted with H2O (20 mL) and HCl (2N, to pH 3) to yield a white-colored precipitate. The crude was recrystallized from MeOH to yield 0.06 g (67% yield) of 11b as a white-colored solid. 1H NMR (400 MHz, DMSO-d6, δ): 12.86 (br s, 2H), 7.53 (d, J = 8.8 Hz, 4H), 6.97–7.19 (m, 8H), 4.36 (s, 4H), 3.84 (s, 6H); 13C NMR (100 MHz, DMSO-d6, δ): 170.3, 163.2, 139.1, 130.2, 129.9, 128.1, 114.7, 56.2, 52.2; HRMS-ESI (−) (m/z): [M-H]− calcd for C24H24N2O10S2, 563.0794; found, 563.0596.
1,4-dibromoisoquinoline (13)
This previously reported compound was synthesized according to a published procedure.52 In a 50 mL round bottom flash, isocarbostyril (0.50 g, 3.4 mmol) and phosphorus tribromide (2.7 g, 9.8 mmol) were heated at 140–145 °C for 10 min. The solid melted and lightened in color. The liquid solidified, and the reaction was stirred for an additional 10 min. On completion, the reaction mixture was cooled to room temperature, and ice/H2O (10 mL) was poured into the flask. The buff-colored solid was collected by filtration and washed with H2O (2 × 50 mL) and dried under vacuum to obtain 13 (0.86 g, 89% yield) as a buff-colored solid. 1H NMR (400 MHz, CDCl3, δ) 8.45 (s, 1H), 8.33 (d, J = 8.8 Hz, 1H), 8.19 (d, J = 8.4 Hz, 1H), 7.88 (t, 1H), 7.78 (t, 1H).
N,N’-(isoquinoline-1,4-diyl)bis(4-methoxybenzenesulfonamide) (15)
This previously unreported compound was synthesized in a manner similar to a published procedure.53,54 A solution of 1,4-dibromoisoquinoline 13 (230 mg, 0.8 mmol), 4-methoxybenzenesulfonamide 14 (300 mg, 1.6 mmol), CuI (60 mg, 0.3 mmol), K2CO3 (1.18 g, 8.5 mmol), and N1,N2-dimethylethane-1,2-diamine (142 mg, 1.6 mmol) in MeCN (6 mL) was heated at 70 °C for 7 days. On completion the reaction mixture was quenched with H2O (20 mL), and the yellow-colored solid was collected by filtration. The collected solid was washed with Et2O (10 mL) and dried to yield 240 mg (60% yield) of 15 as a yellow-colored solid. 1H NMR (400 MHz, DMSO-d6) δ 8.62 (d, J = 8.3 Hz, 1H), 8.28 (d, J = 8.4 Hz, 1H), 7.86 (t, J = 7.6 Hz, 1H), 7.77 (t, J = 7.7 Hz, 1H), 7.66 (s, 1H), 7.64 – 7.58 (m, 2H), 7.48 – 7.42 (AA’XX’, 2H), 7.14 – 7.08 (AA’XX’, 4H), 3.88 (d, 6H). 13C NMR (CD3CN) δ 163.4, 162.6, 152.5, 135.5, 133.5, 132.5, 130.5, 129.5, 129.3, 128.6, 128.3, 128.0, 126.8, 122.7, 114.3, 114.1, 55.6, 55.4. HRMS-ESI (+) (m/z): [M+H]+ calcd for C23H21N3O6S2, 500.0945; found, 500.0940.
2,2’-(isoquinoline-1,4-diylbis(((4-methoxyphenyl)sulfonyl)azanediyl))diacetic acid (17)
This previously unreported compound was synthesized in a manner similar to a published procedure.1 Potassium carbonate (83 mg, 0.6 mmol) and ethyl bromoacetate (70 mg, 0.4 mmol) were added to a solution of N,N’-(isoquinoline-1,4-diyl)bis(4-methoxybenzenesulfonamide) 15, (100 mg, 0.20 mmol) in anhydrous DMF (2 mL), and the reaction was stirred overnight at room temperature. On completion the reaction was quenched with H2O (10 mL) and the solid was collected by filtration to yield a brown-colored solid which was subjected to saponification using NaOH (10%, 0.5 mL) and MeOH (2 mL) and allowed to reflux for 1.5 h. On completion the reaction mixture was cooled, and the organic portion was evaporated and diluted with H2O (10 mL). The solid was removed by filtration, and the filtrate was acidified with HCl (2N, to pH 2) and extracted with EtOAc (2 × 5 mL). The combined organic portions were washed with H2O (2 × 10 mL), dried (Na2SO4), and evaporated to yield a yellow solid as crude product (77% yield). 15 mg of the crude product was further purified by preperative HPLC (C18, 20–95% MeCN with 0.1% formic acid, 205 mL/min) to yield 5 mg of 17 as a buff-colored solid. 1H NMR (400 MHz, DMSO-d6, δ) 8.62 (d, J = 8.4 Hz, 1H), 8.28 (d, J = 8.4 Hz, 1H), 7.86 (t, J = 7.6 Hz, 1H), 7.77 (t, J = 7.6 Hz, 1H), 7.66 (s, 1H), 7.64 – 7.56 (AA’XX’, 2H), 7.49 – 7.41 (AA’XX’, 2H), 7.15 – 7.07 (AA’XX’, 4H), 4.42 (s, 2H), 4.30 (d, J = 31.7 Hz, 2H), 3.88 (s, 3H), 3.87 (s, 3H). 1H NMR (CD3COCD3, 400 MHz, δ) 8.81 (d, J =8.6 Hz, 1 H), 8.31 (d, J =8.3 Hz, 1 H), 7.93 (s, 1 H), 7.84 – 7.91 (m, 1 H), 7.77 – 7.83 (m, 1 H), 7.70 – 7.75 (m, 2 H), 7.52 – 7.57 (m, 2 H), 7.09 – 7.18 (m, 4 H), 4.67 – 4.78 (m, 1 H), 4.48 – 4.64 (m, 3 H), 3.98 (s, 3 H), 3.96 ppm (s, 3 H) 13C NMR (CD3CN, δ): 170.4, 169.7, 164.7, 164.6, 153.5, 142.7, 138.2, 133.6, 132.5, 131.6, 131.1, 130.4, 129.6, 128.9, 128.8, 128.8, 124.2, 115.3, 115.2, 56.6, 56.6, 53.6, 52.1. 13C NMR (CD3COCD3, 101 MHz, δ): 171.0, 170.1, 165.3, 165.2, 154.4, 143.7, 138.8, 134.1, 132.8, 132.3, 131.8, 131.8, 130.5, 130.0, 129.8, 129.0, 124.8, 115.8, 115.8, 57.0, 57.0, 54.3, 52.6 ppm; HRMS-ESI (+) (m/z): [M+H]+ calcd for C27H25N3O10S2, 616.1054; found, 616.1036.
(E/Z)-8-(phenyldiazenyl)quinolin-5-amine (19)
This previously unreported compound was synthesized in a manner similar to a published procedure.55 In a 250 mL round-bottomed flask, aniline (1.0 g) was dissolved in HCl (1N, 30 mL), and the solution was chilled in an ice-bath (solution 1). NaNO2 (0.76 g) was added and the reaction mixture was allowed to stir for 15 min. In a separate 250 mL round-bottomed flask, quinoline-5-amine 18 (1.5 g) was dissolved in AcOH (1N, 20 mL), NaOAc (saturated, 20 mL) and H2O (200 mL) and chilled in an ice-bath below 10 °C (solution 2). Solution 1 was slowly added to solution 2 while maintaining the temperature below 10 °C, during which time the solution changed color from red to deep-red. After complete addition the reaction was allowed to stir for an additional 15 min and poured into NH4OH (100 mL) and ice to obtain a red precipitate. The solid was collected by filtration and washed further with H2O (300 mL) and recrystallized from EtOH/H2O (50/50) to yield 0.8 g (30% yield) of 19 as deep-red needles. The product was utilized for the synthesis of 20 without further purification or characterization.
Quinoline-5,8-diamine (20)
This previously unreported compound was synthesized in a manner similar to a published procedure.40 Tin chloride dihydrate (5.1 g, 22.6 mmol) was added to a solution of (E/Z)-8-(phenyldiazenyl)quinolin-5-amine 19 (0.7 g, 2.8 mmol) in conc. HCl (20 mL), and the reaction mixture was allowed to stir at reflux for 4 h. On completion the reaction was cooled in an ice-bath, and KOH was slowly added until a pH of 8–9 was attained. The solid was filtered and washed with EtOH (2 × 100 mL), and the organic portion was kept aside for slow evaporation to obtain 0.22 g (50% yield) of 20 as needles. 1H NMR (400 MHz, DMSO-d6, δ): 8.61–8.73 (m, 1H), 8.38 (d, J = 8.6 Hz, 1H), 7.27–7.42 (m, 1H), 6.72 (d, J = 7.8 Hz, 1H), 6.59 (d, J = 7.8 Hz, 1H), 5.03 (br. s, 2H), 4.96 (br. s, 2H).
N,N’-(quinoline-5,8-diyl)bis(4-methoxybenzenesulfonamide) (21)
This previously unreported compound was synthesized in a manner similar to a published procedure.51 A solution of quinoline-5,8-diamine 20 (0.1 g, 0.6 mmol) and p-methoxybenzenesulfonyl chloride (0.27 g, 1.3 mmol) in pyridine (4 mL) was allowed to stir overnight at room temperature. On completion, the reaction mixture was diluted with EtOAc (40 mL); washed with H2O (2 × 50 mL) and brine (50 mL); dried (Na2SO4); and evaporated to yield the crude product, which, upon recrystallization from i-PrOH, yielded 0.24 g (77% yield) of 21 as buff-colored needles. 1H NMR (400 MHz, DMSO-d6, δ): 10.05 (s, 1H), 9.81 (s, 1H), 8.80 (d, J = 4.0 Hz, 1H), 8.29 (d, J = 8.6 Hz, 1H), 7.80 (d, J = 8.6 Hz, 2H), 7.39 – 7.62 (m, 4H), 7.09 (d, J = 8.1 Hz, 1H), 6.96 (t, J = 9.4 Hz, 4H), 3.74 (s, 3H), 3.77 (s, 3H); 13C NMR (100 MHz, DMSO-d6, δ): 163.1, 162.9, 149.8, 139.3, 133.0, 132.9, 131.3, 131.2, 129.7, 129.4, 128.1, 125.7, 124.9, 122.5, 116.2, 114.7, 56.1; HRMS-ESI (+) (m/z): [M+H]+ calcd for C23H21N3O6S2, 500.0905; found, 500.0946.
2,2’-(quinoline-5,8-diyl)bis(((4-methoxyphenyl)sulfonyl)azanediyl))diacetic acid (23)
This previously unreported compound was synthesized in a manner similar to a published procedure.28 Potassium carbonate (0.07 g, 0.48 mmol) and ethyl bromoacetate (0.06 g, 0.35 mmol) were added to a solution of 21 (0.08 g, 0.16 mmol) in anhydrous DMF (2 mL), and the reaction was stirred overnight at room temperature. On completion the reaction was quenched with H2O (25 mL), and the solid was collected by filtration, suspended in NaOH (10%, 5 mL) and MeOH (10 mL), and allowed to reflux for 2 h. On completion the reaction mixture was cooled, and the organic portion was evaporated and diluted with H2O (20 mL). The solid was removed by filtration, and the filtrate was acidified with HCl (2N, to pH 6) and extracted with EtOAc (2 × 25 mL). The combined organic portions were washed with H2O (3 × 50 mL) and brine (20 mL), dried (Na2SO4), and evaporated to yield a pastel green precipitate. The crude product was further recrystallized from MeOH to yield 0.06 g (67% yield) of 23 as a white solid. 1H NMR (400 MHz, CD3OD, δ): 12.86 (s, 1H), 8.51–8.74 (m, 2H), 7.69 (d, J = 8.1 Hz, 1H), 7.51 (d, J = 8.8 Hz, 2H), 7.56 (d, J = 8.6 Hz, 3H), 7.26 (d, J = 8.1 Hz, 1H), 7.11 (d, J = 8.8 Hz, 2H), 6.94 (d, J = 8.8 Hz, 2H), 4.63 (br s, 2H), 4.48 (s, 1H), 4.36 (d, 1H), 3.87 (s, 3H), 3.80 (s, 3H); 13C NMR (100 MHz, DMSO-d6, δ): 170.7, 169.0, 163.5, 163.0, 147.7, 138.3, 136.3, 135.8, 131.2, 130.6, 130.3, 129.4, 128.7, 114.8, 111.6, 56.2; HRMS-ESI (+) (m/z): [M+H]+ calcd for C27H26N3O10S2, 616.1015; found, 616.1060.
N-(2,2-dimethoxyethyl)-5-fluoro-2-nitroaniline
This previously reported compound was synthesized according to a published procedure.56 Diisopropylethylamine (4.4 mL, 13.2 mmol) and aminoacetaldehyde dimethyl acetal (3.3 mL, 30.2 mmol) were added to a solution of 2,4-difluoro-1-nitrobenzene (2.8 mL, 13.2 mmol) in MeOH (100 mL), and the reaction mixture was stirred at reflux for 15 h. On completion, the organic portions were evaporated, and the residue was dissolved in EtOAc (100 mL). The organic portion was washed with H2O (2 × 100 mL) and brine (100 mL), dried (Na2SO4) and evaporated to yield 5.3 g (86% yield) of an orange oil. 1H NMR (400 MHz, CDCl3, δ): 8.28 (br s, 1H), 8.24 (dd, J = 9.5, 6.2, Hz, 1H), 6.53 (dd, J = 11.4, 2.5 Hz, 1H), 6.40 (ddd, J = 9.5, 7.1, 2.8 Hz, 1H), 4.65 (t, J = 5.4 Hz, 1H), 3.46–3.50 (m, 6H), 3.45 (d, 1H), 3.41 (t, J = 5.4 Hz, 2H).
N1-benzyl-N3-(2,2-dimethoxyethyl)-4-nitrobenzene-1,3-diamine (25)
This previously reported compound was synthesized according to a published procedure.56 A solution of N-(2,2-dimethoxyethyl)-5-fluoro-2-nitroaniline (3 g, 12.3 mmol) and benzylamine (8 mL, 72.7 mmol) in 1,4-dioxane (15 mL) was stirred at reflux for 3 days. On completion, the reaction mixture was cooled to room temperature and diluted with EtOAc (80 mL). The mixture was washed with H2O (3 × 100 mL) and HCl (2N, 2 × 75 mL), dried (Na2SO4), and evaporated to yield 3.8 g (93% yield) of 25 as an orange oil which solidified on standing. The product was taken directly to the next step without purification or characterization.
N-benzyl-7-nitro-1H-indol-4-amine
This previously reported compound was synthesized according to a published procedure.56 A solution of N1-benzyl-N3-(2,2-dimethoxyethyl)-4-nitrobenzene-1,3-diamine (25, 3.7 g, 11.4 mmol) and TFA (6 mL) in CH2Cl2 (40 mL) was stirred overnight at room temperature. On completion, the reaction was quenched with H2O (50 mL), and the aqueous layer was further extracted with CH2Cl2 (3 × 30 mL). The combined organic portions were washed with H2O (3 × 100 mL), dried (Na2SO4) and evaporated to yield a crude residue which was purified by column chromatography (silica gel; hexanes/EtOAc; 90:10 to 70:30) to afford 2.5 g (85% yield) of a dark orange solid. 1H NMR (400 MHz, DMSO-d6, δ): 11.61 (br. s, 1H), 8.19 (t, J = 6.3 Hz, 1H), 7.89 (d, J = 9.1 Hz, 1H), 7.28–7.41 (m, 4H), 7.16–7.28 (m, 2H), 6.94 (d, J = 2.02 Hz, 1H), 6.23 (d, J = 9.1 Hz, 1H), 4.61 (d, 2H).
N,N’-(1H-indole-4,7-diyl)bis(4-methoxybenzenesulfonamide) (27)
This previously unreported compound was synthesized in a manner similar to a published procedure.40 Pd/C (10%, 0.1 g) was added to a suspension of N-benzyl-7-nitro-1H-indol-4-amine (1.0 g, 3.7 mmol) in HCl/EtOH (1.25 M, 15 mL) and EtOH (10 mL), and the reaction mixture was hydrogenated using a Parr shaker apparatus (35 psi H2) at room temperature for 24 h. On completion the reaction was filtered over Celite®, and the filter bed was washed with EtOH (20 mL). The combined organic portions were evaporated and dried to yield 0.83 g (quant. yield) of 26 as a brown solid, of which 0.3 g (0.8 mmol) was added to a solution of p-methoxybenzenesulfonyl chloride (0.4 g, 1.9 mmol) in pyridine (4 mL). The mixture was allowed to stir overnight at room temperature. On completion, the reaction mixture was diluted with EtOAc (50 mL) and washed with H2O (2 × 100 mL), HCl (2N, 50 mL) and brine (30 mL); dried (Na2SO4); and evaporated to yield a crude product which, upon washing with Et2O (100 mL), yielded 0.4 g (60% yield) of 27 as a buff-colored solid. 1H NMR (400 MHz, DMSO-d6, δ): 10.69 (s, 1H), 9.79 (s, 1H), 9.61 (s, 1H), 7.59 (d, J = 8.3 Hz, 4H), 6.96 (dd, J = 8.4, 14.5 Hz, 4H), 6.66 (d, J = 7.8 Hz, 1H), 6.45–6.61 (m, 2H), 3.78 (s, 3H), 3.75 (s, 3H); 13C NMR (100 MHz, DMSO-d6 δ): 162.4, 162.2, 131.9, 131.2, 130.9, 129.1, 128.9, 126.8, 125.1, 123.7, 118.9, 115.1, 114.2, 114.1, 112.1, 100.1, 55.6, 55.6; HRMS-ESI (+) (m/z): [M+H]+ calcd for C22H22N3O6S2, 488.0905; found, 488.0937.
2,2’-((1H-indole-4,7-diyl)bis(((4-methoxyphenyl)sulfonyl)azanediyl))diacetic acid (29)
This previously unreported compound was synthesized in a manner similar to a published procedure.28 Potassium carbonate (0.08 g, 0.6 mmol) and tert-butylbromoacetate (0.08 g, 0.4 mmol) were added to a solution of 27 (0.1 g, 0.2 mmol) in anhydrous DMF (2 mL), and the reaction was stirred for 6 h at room temperature. On completion the reaction was quenched with H2O (25 mL) and extracted with EtOAc (2 × 40 mL). The combined organic portion was washed with H2O (2 × 100 mL) and brine (50 mL), dried (Na2SO4), and evaporated to yield a crude brown solid which was further washed with hexanes (30 mL) to yield 0.12 g (82% yield) of the tert-butyl ester 28. Trifluoroacetic acid (0.5 mL) was added to a solution of the tert-butyl ester (0.06 g) in anhydrous CH2Cl2 (4 mL), and the reaction was stirred at room temperature for 5 h. On completion the solvent was evaporated and the crude residue (0.04 g) was purified using preparative HPLC (C18, 50% MeCN with 0.1% formic acid, retention time = 8.8 min, run length = 32 min, 4 mL/min) to yield 10 mg of 29 as a buff-colored solid. 1H NMR (400 MHz, DMSO-d6, δ): 12.85 (br. s, 1H), 11.19 (br. s, 1H), 7.47–7.68 (m, 4H), 7.25 (br s, 1H), 7.00 (d, J = 8.8 Hz, 2H), 7.03 (d, J = 8.8 Hz, 2H), 6.64 (d, J = 8.1 Hz, 1H), 6.55 (d, J = 7.8 Hz, 1H), 6.11 (s, 1H), 4.34 (s, 4H), 3.84 (s, 3H), 3.82 (s, 3H); 13C NMR (100 MHz, DMSO-d6 δ): 162.7, 162.6, 135.0, 131.4, 130.7, 129.9, 129.6, 128.0, 126.6, 124.2, 120.2, 114.2, 114.1, 100.1, 55.7, 55.7; HRMS-ESI (+) (m/z): [M+Na]+ calcd for C26H25N3O10S2, 626.0879; found, 626.0867.
1,4-diaminophthalazine (31)
This previously reported compound was synthesized according to a published procedure.57 A solution of phthalonitrile (2.6 g, 20 mmol) and hydrazine hydrate (55%, 16.4 mL), in MeOH (100 mL) was stirred overnight at room temperature and refluxed for 2 h. On completion, the organic solvent was partially evaporated until a precipitate formed, which was dissolved using hot MeOH to yield 2 g (65% yield) of 31 as yellow-colored needles. 1H NMR (400 MHz, DMSO-d6, δ) 8.13–8.08 (m, 2H), 7.83–7.77 (m, 2H), 5.94 (br. s, 4H).
N,N’-(phthalazine-1,4-diyl)bis(4-methoxybenzenesulfonamide) (32)
This previously unreported compound was synthesized in a manner similar to a published procedure.51 A solution of 1,4-diaminophthalazine 31 (0.3 g, 1.9 mmol) and p-methoxybenzenesulfonyl chloride (0.85 g, 4.1 mmol) in pyridine (3 mL) and 1,4-dioxane (10 mL) was allowed to stir overnight at reflux. On completion, the reaction mixture was quenched with H2O (100 mL), and the precipitate was collected by filtration. The collected solid was further washed with Et2O (3 × 30 mL), and the crude product was recrystallized with HCl in EtOH (1.2 M) to obtain 32 (0.3 g, 31% yield) as a buff-colored solid. 1H NMR (400 MHz, DMSO-d6, δ) 11.50 (br. s, 1H), 8.14–8.28 (m, 2H), 7.82 (d, J = 8.8 Hz, 6H), 6.96 (d, J = 8.6 Hz, 4H). 3.78 (s, 3H); 13C NMR (100 MHz, DMSO-d6, δ) 163.0, 148.7, 143.2, 133.0, 131.4, 131.2, 129.4, 127.4, 124.8, 124.3, 116.1, 114.7, 56.1; HRMS-ESI (+) (m/z): [M+H]+ calcd for C22H20N4O6S2, 501.0858; found, 501.0908.
Ethyl N-(4-((4-methoxyphenyl)sulfonamido)phthalazin-1-yl)-N-((4-methoxyphenyl)sulfonyl)glycinate (33)
This previously unreported compound was synthesized in a manner similar to a published procedure.1 Lithium tert-butoxide (0.06 g, 0.8 mmol) and ethyl bromoacetate (0.1 g, 0.6 mmol) were added to a solution of 32 (0.05g, 0.1 mmol) in anhydrous DMF (3 mL), and the reaction was stirred for 24 h at room temperature. On completion the reaction was quenched with 2N HCl (25 mL), and the buff-colored solid was collected by filtration to yield 0.06 g (quant. yield) of a brown solid. 30 mg of this crude material was subjected to purification using preparative HPLC to yield 10 mg of 33 as a white solid. 1H NMR (400 MHz, CD3CN, δ) 8.45–8.55 (m, 1H), 8.41 (dd, J = 8.2, 0.6 Hz, 1H), 7.99–8.13 (m, 1H), 7.89–7.99 (m, 3H), 7.68–7.83 (m, 2H), 7.09–7.15 (m, 2H), 7.02–7.09 (m, 2H), 4.03 (q, J = 7.1 Hz, 2H), 3.94 (s, 3H), 3.86 (s, 3H), 1.06 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, CD3CN, δ) 167.6, 164.1, 162.8, 134.4, 134.4, 133.4, 130.9, 129.3, 129.2, 128.2, 127.5, 125.6, 114.5, 114.1, 61.4, 55.7, 55.4, 51.7, 13.2; HRMS-ESI (+) (m/z): [M+H]+ calcd for C26H26N4O8S2, 587.1226; found, 587.1220.
N-(4-((4-methoxyphenyl)sulfonamido)phthalazin-1-yl)-N-((4-methoxyphenyl)sulfonyl)glycine (34)
This previously unreported compound was synthesized in a manner similar to a published procedure.1 35 mg of ester 33 was suspended in NaOH (10%, 2 mL) and MeOH (10 mL) and allowed to reflux for 3 h. On completion the reaction mixture was cooled, and the organic solvent was evaporated, diluted with H2O (10 mL), and washed with Et2O (20 mL). The aqueous portion was acidified with HCl (2N, to pH 2), and the precipitate was collected by filtration to yield 18 mg of crude product, which was purified using preparative HPLC to yield 10 mg of 34 as a white solid. 1H NMR (400 MHz, CD3CN, δ) 8.49 (d, J = 7.8 Hz, 1H), 8.38 (d, J = 8.1 Hz, 1H), 7.98–8.08 (m, 1H), 7.94 (d, J = 9.1 Hz, 2H), 7.85–7.92 (m, 1H), 7.72 (d, J = 9.1 Hz, 2H), 7.00–7.16 (m, 4H), 4.24 (br s, 2H), 3.92 (s, 3H), 3.86 (s, 3H); 13C NMR (100 MHz, CD3CN, δ) 164.1, 163.0, 146.6, 134.4, 133.4, 131.0, 129.7, 128.4, 127.6, 125.6, 117.5, 114.6, 114.3, 55.8, 55.6; HRMS-ESI (+) (m/z): [M+H]+ calcd for C24H22N4O8S2, 559.0913; found, 559.0915.
Methyl ((4-methoxyphenyl)sulfonyl)glycinate (36a)
This previously reported compound was synthesized according to a published procedure.58 A solution of glycine methyl ester hydrochloride (1.0 g, 7.9 mmol) and p-methoxybenzenesulfonyl chloride (1.8 g, 8.7 mmol) in pyridine (7 mL) was allowed stir at room temperature for 24 h. On completion, the reaction mixture was quenched with HCl (2N, 30 mL) and ice. The white precipitate was collected by filtration and washed with H2O (2 × 50 mL) to yield the crude product, which upon recrystallization from i-PrOH yielded 1.2 g (58% yield) of 36a as white needles. 1H NMR (400 MHz, DMSO-d6, δ) 8.02 (t, J = 6.2 Hz, 1H), 7.67–7.77 (m, J = 8.6 Hz, 2H), 7.05–7.17 (m, J = 8.8 Hz, 2H), 3.84 (s, 3H), 3.65 (d, J = 6.1 Hz, 2H), 3.53 (s, 3H).
Tert-butyl ((4-methoxyphenyl)sulfonyl)glycinate (36b)
This previously reported compound was synthesized according to a published procedure.58 A solution of glycine tert-butyl ester hydrochloride (0.31 g, 1.8 mmol) and p-methoxybenzenesulfonyl chloride (0.4 g, 1.9 mmol) in pyridine (5 mL) was allowed to stir at room temperature for 4 h. On completion, the reaction mixture was quenched with HCl (2N, 30 mL) and ice. The yellow-colored precipitate was collected by filtration and washed with H2O (2 × 50 mL) to yield 0.3 g (54% yield) of 36b as yellow-colored solid. 1H NMR (400 MHz, DMSO-d6, δ) 7.73–7.86 (m, 2H), 6.91–7.05 (m, 2H), 3.87 (s, 3H), 3.66 (d, 2H), 1.36 (s, 9H).
2,2’-((1,2-phenylenebis(methylene))bis(((4-methoxyphenyl)sulfonyl)azanediyl))diacetic acid (38)
This previously unreported compound was synthesized in a manner similar to a published procedure.1 Potassium carbonate (0.16 g, 1.14 mmol) and methyl ((4-methoxyphenyl)sulfonyl)glycinate 36a (0.19 g, 0.76 mmol) were added to a solution of α,α’-dibromoxylene (0.1 g, 0.38 mmol) in anhydrous DMF (2 mL), and the reaction was stirred overnight at room temperature. On completion the reaction was quenched with H2O (25 mL), and the solid was collected by filtration and recrystallized with MeOH to yield 0.17 g (70% yield) of methyl ester, of which 0.1 g was suspended in NaOH (10%, 2 mL) and MeOH (10 mL) and allowed to reflux for 4 h. On completion the reaction mixture was cooled, and the organic portion was evaporated and diluted with H2O (10 mL). The solid was removed by filtration, and the filtrate was acidified with HCl (2N, to pH 2). The white-colored precipitate was collected by filtration. The crude solid was washed with Et2O (2 × 20 mL) and dried to yield 0.07 g (74% yield) of 38 as a white-colored solid. 1H NMR (400 MHz, DMSO-d6, δ) 12.60 (br. s, 2H), 7.76 (d, J = 8.6 Hz, 4H), 7.26–7.31 (m, 2H), 7.20–7.26 (m, 2H), 7.09 (d, J = 8.8 Hz, 4H), 4.44 (s, 4H), 3.85 (s, 6H); 13C NMR (100 MHz, DMSO-d6, δ) 170.3, 163.0, 134.8, 130.9, 129.8, 129.1, 128.0, 114.8, 56.1, 49.5, 48.7; HRMS-ESI (+) (m/z): [M+Na]+ calcd for C26H28N2O10S2, 615.1083; found, 615.1068.
2,2’-(phthaloylbis(((4-methoxyphenyl)sulfonyl)azanediyl))diacetate (40)
This previously unreported compound was synthesized in a manner similar to a published procedure.59 tert-Butyl ((4-methoxyphenyl)sulfonyl)glycinate 36b (0.15 g, 0.5 mmol) was added to a solution of phthaloyl chloride (0.05 g, 0.3 mmol) in pyridine (4 mL), and the reaction was stirred overnight at room temperature. On completion the reaction was quenched with H2O (25 mL) and extracted with EtOAc (2 × 40 mL). The combined organic portions were washed with HCl (2N, 2 × 50 mL), H2O (2 × 100 mL), and brine (50 mL); dried (Na2SO4); and evaporated to yield a brown oil as crude product which was further purified by column chromatography (silica gel; Hexanes/EtOAc; 100:10 to 100:45) to afford 0.1 g (55% yield) of the tert-butyl ester of 39. Trifluoroacetic acid (0.5 mL) was added to a solution of the tert-butyl ester (0.05 g) in anhydrous CH2Cl2 (4 mL), and the reaction was stirred at room temperature overnight. On completion the solvent was evaporated, and the crude residue was recrystallized from i-PrOH to yield 0.02 g (50% yield) 40 as a white solid. 1H NMR (400 MHz, DMSO-d6, δ) 7.87 (d, J = 8.3 Hz, 4H), 7.58 (br s, 2H), 7.44 (br s, 2H), 7.11 (d, J = 8.5 Hz, 4H), 4.18 (s, 4H), 3.86 (s, 6H); 13C NMR (100 MHz, DMSO-d6 δ) 170.7, 164.0, 162.5, 136.7, 136.3, 130.5, 129.1, 125.8, 115.1, 114.6, 56.1; HRMS-ESI (+) (m/z): [M+Na]+ calcd for C26H24N2O12S2, 643.0668; found, 643.0634.
(E/Z)-2,2’-(but-2-ene-1,4-diyl)bis(((4-methoxyphenyl)sulfonyl)azanediyl))diacetate (42)
This previously unreported compound was synthesized in a manner similar to a published procedure.1 Potassium carbonate (0.19 g, 1.4 mmol) and methyl ((4-methoxyphenyl)sulfonyl)glycinate 36a (0.24 g, 0.94 mmol) were added to a solution of 1,4-dibromobutene (0.1 g, 0.47 mmol) in anhydrous DMF (2 mL), and the reaction was stirred overnight at room temperature. On completion the reaction was quenched with H2O (25 mL), and the solid was collected by filtration and recrystallized with acetone/i-PrOH to yield 0.22 g (80% yield) of methyl ester of which 0.15 g was suspended in NaOH (10%, 2 mL) and MeOH (10 mL) and allowed to reflux for 4 h. On completion the reaction mixture was cooled, the organic portion was evaporated and diluted with H2O (10 mL). The solid was removed by filtration, and the filtrate was acidified with HCl (2N, to pH 2). The white-colored precipitate was collected by filtration. The crude solid was washed with Et2O (2 × 20 mL) and dried to yield 0.1 g (70% yield) of 42 as a white-colored solid. 12.68 (br. s, 2H), 7.72 (d, J = 8.8 Hz, 4H), 7.09 (d, J = 8.8 Hz, 4H), 5.40 (br. s, 2H), 3.84 (s, 7H), 3.79 (s, 4H), 3.73 (br. s, 4H); 13C NMR (100 MHz, DMSO-d6, δ) 170.5, 162.9, 131.6, 129.6, 129.0, 114.8, 56.1, 49.5, 47.7; HRMS-ESI (+) (m/z): [M+Na]+ calcd for C22H26N2O10S2, 565.0927; found, 565.0932.
3,5-dinitro-1,1’-biphenyl
This previously unreported compound was synthesized in a manner similar to a published procedure.60 1-Bromo-3,5-dinitrobenzene (100 mg, 0.404 mmol), phenylboronic acid (54.2 mg, 0.444 mmol), Pd(OAc)2 (5%, 4.9 mg) and potassium phosphate (86.0 mg, 0.405 mmol) were added to 1 mL of H2O and allowed to reflux overnight. On completion, the reaction mixture was cooled, quenched with HCl (1N, 2 × 10mL) and extracted using ethyl acetate (10 mL) and brine (10 mL) and H2O (2 × 10 mL), dried over Na2SO4, and evaporated to yield 0.176 g of a yellow-white solid. 1H NMR (400 MHz, CDCl3, δ) 9.04 (s, 1H), 8.80 (s, 2H), 7.74–7.69 (d, J = 6.88 Hz, 2H), 7.62–7.52 (m, 3H).
1,1’-biphenyl-3,5-diamine (44)
Dihydrate tin (II) chloride (0.87 g, 3.8 mmol) was added to 3,5-dinitro-1,1’-biphenyl (67 mg, 0.27 mmol) in methanol (3.9 mL) under argon and allowed to reflux for 1 hour. The reaction was quenched with excess saturated NaHCO3; extracted using ethyl acetate (10 mL), brine (10 mL), and H2O (2 × 10 mL); dried over Na2SO4; and evaporated to yield (0.050 g, 98.7%) of a red-brown oil. 1H NMR (400 MHz, CDCl3, δ) 7.60–7.53 (d, J = 7.48 Hz, 2H) 7.45–7.30 (m, 3H), 6.41–6.34 (s, 2H), 6.07 (s, 1H), 3.69 (s, 1H).
3N,N’-(1,1’-biphenyl]-3,5-diyl)bis(4-methoxybenzenesulfonamide) (45)
The biphenyl 44 (80.0 mg, 0.43 mmol) was added to 4-methoxybenzenesulfonylchloride (396 mg, 1.92 mmol) in pyridine (0.75 mL) and THF (0.5 mL) and refluxed under argon for 24 hours. The reaction was quenched with HCl (1N, 2 × 10mL); extracted using ethyl acetate (10 mL), brine (10 mL) and H2O (2 × 10 mL); dried over Na2SO4; and taken to the next step. 1H NMR (400 MHz, CDCl3, δ) 7.76–7.71 (d, J = 8.72 Hz, 4H), 7.44–7.35 (m, 5H), 6.99–6.90 (m, 7H), 6.49 (s, 2H), 3.86 (s, 6H).
Diethyl 2,2’-(1,1’-biphenyl]-3,5-diylbis(((4-methoxyphenyl)sulfonyl)azanediyl)diacetate
Ethyl-2-bromoacetate (83 μL, 0.75 mmol) and the previously formed sulfonamide 45 were added to potassium carbonate (0.130 g, 0.93 mmol) and DMF (2 mL) and stirred at room temperature for 2 hours. The reaction was extracted using ethyl acetate (10 mL), brine (10 mL), and H2O (2 × 10mL); dried over Na2SO4; evaporated; and taken to the next step. 1H NMR (400 MHz, CDCl3, δ) 7.66–7.62 (d, J =7.68 Hz, 2H), 7.45–7.36 (m, 5H), 7.05–7.01 (m, 1H), 6.99–6.95 (d, J =9.1 Hz, 2H), 4.35 (s, 2H), 4.20–4.13 (q, 4H), 3.89 (s, 4H), 2.07 (s, 1H), 1.56 (s, 6H), 1.31–1.21 (t, 6 H).
2,2’-(1,1’-biphenyl]-3,5-diylbis(((4-methoxyphenyl)sulfonyl)azanediyl) diacetic acid (46)
NaOH (15%) and methanol (2.4 mL) were added to the previously formed ester and allowed to reflux for 1 hour. On completion, the reaction mixture was cooled; quenched with HCl (1N, 20 mL); extracted using ethyl acetate (10 mL), brine (10 mL), and H2O (2 × 10mL); dried over Na2SO4; and purified using preparative HPLC (C18, 30–95% MeCN with 0.1% formic acid) to yield a white powder (0.024 g, 51%). 1H NMR (400 MHz, CD3CN, δ) 7.64–7.57 (d, 4H), 7.50–7.37 (m, 5H) 7.37–7.32 (s, 2H), 7.05–7.00 (d, 4 H) 4.39–4.33 (s, 4H), 3.91–3.84 (s, 6H); 13C NMR (100 MHz, CD3CN, δ): 163.4, 142.2, 141.2, 138.8, 129.9, 129.0, 128.2, 126.8, 126.0, 125.8, 114.3, 55.6, 52.0; HRMS-ESI (−) (m/z): [M-H]− calcd for C30H28N2O10S2, 639.1191, found 639.1149
Kelch domain expression and purification:
KEAP1 Kelch domain was expressed and purified as previously described 1
Fluorescence anisotropy assays:
Fluorescence anisotropy assays were performed as previously described.1 Briefly, small molecules ability to inhibit the KEAP1-NRF2 interaction were assayed using the Kelch domain of KEAP1 and a fluorescein-labeled 9-mer peptide containing the ETGE motif of the Neh2 domain of NRF2 41. The experiments were performed in triplicate and sigmoidal concentration-response curves were fitted to the data using Graphpad Prism 6.1 software
Surface Plasmon Resonance Assay:
Surface Plasmon Resonance assays were performed as previously described.1 Briefly, the Kelch domain of KEAP1 was immobilized on a CM5 sensor chip. Increasing concentrations of compound solutions were applied to the protein-bound chip, and response units were measured. The experiments were conducted in triplicate with a technical repeat. Approximately 40% of captured KEAP1 was active based on the comparison between calculated RUmax and achieved RUmax:
1-site binding saturation curves were fitted to the data using GraphPad Prism 6.1.
Gene expression analysis:
Spontaneously immortalized human keratinocytes (HaCaT cell line, kindly provided by Dr. Luisa DiPietro, University of Illinois) were grown in Dubecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum and 1% antibiotics. Cells at 70–80% confluence were treated with compounds as indicated for 6 h or 24 h in the presence of complete medium. Cells were lysed in a 20 mM Tris (pH 7.5) buffer containing 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM Na3VO4, 5 mM β-glycerophosphate, and 1 μg/mL leupeptin. Equal amount of protein (~40 μg) was separated on a 10% SDS-PAGE, blotted on the membrane, and probed with anti-NRF2 (sc-13032, Santa Cruz Biotech, Santa Cruz, CA), anti-NQO1 (A1518, Neo Scientific, Cambridge, MA) or anti-GCLM (14241–1-AP, Proteintech, Rosemont, IL) antibodies. Membranes were stripped and probed with anti-GAPDH antibodies (SC-25778, Santa Cruz Biotech, Santa Cruz, CA). The blots were developed using HyGlo-Chemi antibody detection kit (Denville Scientific Inc, Metuchen, NJ) and images were visualized on ChemiDoc (Biorad). Band intensities were quantified using ImageJ software with GAPDH intensity as a reference. The values of vehicle treated samples were considered as one arbitrary unit.
Mini-Ames Assay:
A Mini-Ames Assay was used to evaluate the compounds 17 and 6a, for their ability to induce reverse mutations at the histidine locus of strains of Salmonella typhimurium (TA98, TA100, TA1535, TA1537) and Escherichia coli (WP2 uvrA (pKM101)) in the presence and absence of exogenous metabolic activation (Aroclor 1254 induced rat liver S9). 17 and 6a were soluble in DMSO up to 50 mg/mL; therefore DMSO was chosen as the solvent. The assay was conducted in the presence or absence of the S9 mix along with concurrent negative/solvent control and positive controls. Dose levels tested in the Mini-Ames assay were 1.5, 4, 10, 25, 64, 160, 400 and 1000 μg/well. Compound dilutions were prepared immediately prior to use.
A top agar consisting of 0.6% (w/v) agar and 0.5% (w/v) NaCl was melted. 10 mL of 0.5 mM l-histidine/biotin solution or 5 mL of 0.5 mM Tryptophan solution was added to the melted top agar per 100 mL top agar. This solution was aliquoted at 1.6 mL per tube and held at approximately 45 °C. 80 μL of the compound solution, 400 μL of S9 mix (for S9-activated) or phosphate buffered saline (for non-activated) and 80 μL of the bacterial culture were added to the 1.6 mL of top agar. After mixing the top agar mix, every 540 μL of the resulting mix was added into one well of the 6-well plates (34.8-mm dishes) containing solidified approximately 5 mL of minimal glucose agar media (1.5% agar, 2% glucose, in Vogel-Bonner medium E). Each treatment was plated in triplicate (three wells) except vehicle control which was plated in sextuple. As soon as the soft agar solidified, the 6-well plates were incubated at 37±2 °C for 48 to 72 hours. Plates were counted immediately following the incubation period.
The number of revertant colonies in each well was counted manually and recorded. Pinpoint colonies appearing on a well in conjunction with an absence of background lawn were not revertants and were not scored (Mortelmans and Zeiger, 2000).
The condition of the bacterial background lawn was evaluated for evidence of cytotoxicity. Evidence of cytotoxicity was scored relative to the negative control plate and recorded along with the revertant count for that plate. Cytotoxicity was evaluated as a decrease in the number of revertant colonies per well and/or a thinning or disappearance of the bacterial background lawn. Precipitation was evaluated after the incubation period by visual examination without magnification. Precipitation could be confirmed microscopically.
Kinetic Solubility Studies:
A variation of a previously reported method was employed to determine the compounds’ solubility in buffer at pH 7.4.61Briefly, 10 mM DMSO stock solutions of compounds were diluted to 400 μM in buffer (200 mM HEPES, 150 mM NaCl, pH 7.4). The mixture was stirred for 1 hr, filtered, and then injected onto a Shimadzu LC-20AB HPLC system (Solvent system: gradient from 15% MeCN/85% H2O to 95% MeCN/5% H2O (+0.1% formic acid) over 13 minutes; Column: Shimadzu C18, 50 μm, 50 × 4.6 mm; (UV, 254 nm). A 10 mM stock solution was prepared by completely dissolving test compound in DMSO. This solution was diluted to six known concentration solutions (1200 μM, 400 μM, 120 μM, 40 μM, 12 μM, and 4 μM) in MeCN. Each solution was analyzed by HPLC, and a calibration curve was plotted using the peak areas from the standard concentrations. The equilibrium solubility of test compounds was determined by quantifying the concentration of test solutions against the calibration curve. (SI Figure S10). Experiments were run in triplicate and results were tabulated and reported as mean ± standard deviation (Table 2).
2-D NMR Studies:
Standard pulse programs found in the Bruker TopSpin™ 3.x software library were applied for detecting direct (HSQC) and long-range heteronuclear correlations (HMBC), as well as through-space correlations (2D NOESY and selective 1D NOESY). Phase sensitive HSQC experiment was acquired with the heteronuclear one bond coupling constant of 145 Hz, 32 scans per increment, relaxation delay of 1.5 s, 2048 and 128 data points in direct and indirect dimension, respectively. The HMBC experiments were acquired with the heteronuclear long range coupling constants of 2 and 8 Hz, with the number of data points set to 2048 × 512 or 2048 × 384, respectively. Carbon spectra obtained with DEPTQ135 experiment served as an external reference. For the 2D NOESY using purge pulses and gradients, the number of data points were set to 2048 × 192, with the relaxation time of 2 s and mixing time of 0.8 s. The data were processed using square sine-bell weighting functions in both dimensions. One dimensional gradient-selective NOESY experiments were performed with the Gaussian pulse of 2 ms and the relaxation mixing time of 0.6 s.
Metabolism Studies:
Materials.
LC-MS grade acetonitrile (MeCN), methanol (MeOH) and formic acid, biology grade DMSO were purchased from Fisher Scientific (Pittsburgh, PA). LC-MS grade water was generated using a Barnstead™ Nanopure Diamond water system (Thermo Scientific, Pittsburgh, PA). All other reagents, if not specified, were purchased from Sigma Aldrich (Saint Louis, MO). Pooled human liver microsomes and mouse liver microsomes were purchased from Sekisui XenoTech (Kansas City, KS).
Preparation of standard.
The initial stock solutions of test compounds were prepared in DMSO at 10 mM, and then diluted with MeOH to 200 μM before use. All stock solutions were stored at −20 °C. Liver microsomes were aliquoted upon arrival and stored at −80 °C.
Liquid chromatographic and mass spectrometric conditions.
Samples were analyzed on a Shimadzu 8040 triple-quadrupole mass spectrometer (Shimadzu, Addison, IL) equipped with electrospray ion source and Shimadzu Nexera XR UHPLC system. Mobile phase A contained water with 0.025% (v/v) FA, mobile phase B consisting of MeCN with 0.1% (v/v) FA. 5 μL of sample was injected. Study compounds were eluted on a Kinetex C18 column (3.0 × 50 mm, 2.6 μm; Phenomenex, Torrance, CA) at 500 μL/min using a gradient program: mobile phase B started and maintained at 15% for 1 min, then increased to 95% in 4 min.
Mass spectrometric analysis was performed in positive mode for all study compounds except 6a using multiple reaction monitoring (MRM). Nebulizing gas flow and drying gas flow were set to 2 L/min and 15 L/min. DL temperature, heat block temperature and ion spray voltage were maintained at 300 °C, 450 °C and 4500 V, respectively. The precursor ion and product ion of each compound and corresponding Q1 Pre Bias, collision energy (CE) and Q3 Pre Bias are listed in Table 3.
Table 3.
MRM transitions (precursor ion Q1, product ion Q3) and compound related parameters (Q1 Pre Bias, CE, Q3 Pre Bias)
| Compound Name | Q1 | Q3 | Q1 Pre Bias | CE | Q3 Pre Bias |
|---|---|---|---|---|---|
| 34 | 559.0 | 107.0 | −26 | −53 | −16 |
| 6a | 613.1 | 170.9 | 30 | 40 | 18 |
| 17 | 616.1 | 428.1 | −30 | −20 | −19 |
| 23 | 616.0 | 274.0 | −30 | −28 | −27 |
| 6b | 581.1 | 426.1 | −20 | −13 | −28 |
Microsomal stability.
The microsomal stability of study compounds was assessed using human liver microsomes and mouse liver microsomes individually. Briefly, pooled liver microsomes (0.5 mg/mL) were pre-incubated with 1 μM of the study compound in potassium phosphate buffer (100 mM, pH 7.4) at 37 °C for 5 min. The reaction was initiated by adding freshly prepared NADPH (1.5 mM) and quenched by adding equal volume of cold MeCN at 20 min after incubation. Samples were kept at −20 °C for 10 min, followed by centrifugation at 14000 rpm at 4 °C for 15 min. The supernatant was extracted and diluted two times with a mixture of MeCN : water (50:50, v/v). The resulting solution was analyzed by LC-MS. Peak area of each target compound was used for quantitation. Each compound was also incubated in buffer only, and without NADPH. The metabolic stability of the study compound was determined by the percentage of compound remaining after the incubation.
Supplementary Material
Scheme 5. Synthesis of phthalazines 32 and 34a.

aReagents: (a) NH2NH2, MeOH, rt, reflux, 65% yield; (b) pyridine, 1,4-dioxane, reflux, 31% yield; (c) ethyl bromoacetate, LitBuO, DMF, rt, 100% yield; (d) NaOH, MeOH/H2O, 54% yield. rt: room temperature
Acknowledgments:
The authors would like to thank the following funders for their support of this work: National Institute of Arthritis, Musculoskeletal and Skin Diseases to TWM (1R01 AR069541-01A1), National Heart, Lung, and Blood Institute to SPR (1R01 HL136946-01), the Chancellor’s Discovery Fund at UIC (to TWM and SPR), and the Michael Reese Research and Education Foundation (to TWM and SPR). The authors are grateful to Dr. Dejan Nikolic for his assistance with some of the mass spectrometry experiments.
Abbreviations Used:
- ARE
antioxidant response element
- BTB
Tramtrack and Bric-à-Brac
- CUL3
cullin 3
- DMEM
Dulbecco’s Modified Eagle’s Medium
- DMF
N,N-dimethylformamide
- DMSO
dimethyl sulfoxide
- EtOAc
ethyl acetate
- GAPDH
Glyceraldehyde 3-phosphate dehydrogenase
- GCLM
Glutamate-Cysteine Ligase Modifier
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HMBC
heteronuclear multiple bond correlation
- HMOX1
heme oxygenase 1
- HPLC
high-performance liquid chromatography
- HRMS
high resolution mass spectrometry
- HSQC
heteronuclear single quantum correlation
- IVR
intervening region
- KEAP1
kelch-like ECH associated protein 1
- LC-MS
liquid chromatography mass spectrometry
- NADPH
nicotinamide adenine dinucleotide phosphate
- NOESY
Nuclear Overhauser Effect Spectroscopy
- NQO1
NAD(P)H quinone dehydrogenase 1
- NRF2
nuclear factor (erythroid-derived 2)-like 2
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- UHPLC
ultra-high performance liquid chromatography
Footnotes
The authors declare no competing financial interest.
Supporting Information
2D HNMR structure validation of 17; HMBC, HSQC, DEPTQ, and 2D Noesy data for 17; solubility calibration curves; immunoblot data for Nrf2 target gene expression; results of mini ames assay; HPLC retention times; protocols for HPLC methods
Molecular formula strings (CSV)
References
- 1.Jain AD; Potteti H; Richardson BG; Kingsley L; Luciano JP; Ryuzoji AF; Lee H; Krunic A; Mesecar AD; Reddy SP; Moore TW Probing the structural requirements of non-electrophilic naphthalene-based Nrf2 activators. Eur. J. Med. Chem 2015, 103, 252–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Itoh K; Chiba T; Takahashi S; Ishii T; Igarashi K; Katoh Y; Oyake T; Hayashi N; Satoh K; Hatayama I; Yamamoto M; Nabeshima Y An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun 1997, 236, 313–322. [DOI] [PubMed] [Google Scholar]
- 3.Itoh K; Wakabayashi N; Katoh Y; Ishii T; Igarashi K; Engel JD; Yamamoto M Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cullinan SB; Gordan JD; Jin J; Harper JW; Diehl JA The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: oxidative stress sensing by a Cul3-Keap1 ligase. Mol. Cell. Biol 2004, 24, 8477–8486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Furukawa M; Xiong Y BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol. Cell. Biol 2005, 25, 162–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lo SC; Li X; Henzl MT; Beamer LJ; Hannink M Structure of the Keap1:Nrf2 interface provides mechanistic insight into Nrf2 signaling. EMBO J. 2006, 25, 3605–3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McMahon M; Thomas N; Itoh K; Yamamoto M; Hayes JD Dimerization of substrate adaptors can facilitate cullin-mediated ubiquitylation of proteins by a “tethering” mechanism: a two-site interaction model for the Nrf2-Keap1 complex. J. Biol. Chem 2006, 281, 24756–24768. [DOI] [PubMed] [Google Scholar]
- 8.Tong KI; Katoh Y; Kusunoki H; Itoh K; Tanaka T; Yamamoto M Keap1 recruits Neh2 through binding to ETGE and DLG motifs: characterization of the two-site molecular recognition model. Mol. Cell. Biol 2006, 26, 2887–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tong KI; Kobayashi A; Katsuoka F; Yamamoto M Two-site substrate recognition model for the Keap1-Nrf2 system: a hinge and latch mechanism. Biol. Chem 2006, 387, 1311–1320. [DOI] [PubMed] [Google Scholar]
- 10.Ohtsubo T; Kamada S; Mikami T; Murakami H; Tsujimoto Y Identification of NRF2, a member of the NF-E2 family of transcription factors, as a substrate for caspase-3(-like) proteases. Cell Death Differ. 1999, 6, 865–872. [DOI] [PubMed] [Google Scholar]
- 11.Hayes JD; Chanas SA; Henderson CJ; McMahon M; Sun C; Moffat GJ; Wolf CR; Yamamoto M The Nrf2 transcription factor contributes both to the basal expression of glutathione S-transferases in mouse liver and to their induction by the chemopreventive synthetic antioxidants, butylated hydroxyanisole and ethoxyquin. Biochem. Soc. Trans 2000, 28, 33–41. [DOI] [PubMed] [Google Scholar]
- 12.Dinkova-Kostova AT; Holtzclaw WD; Cole RN; Itoh K; Wakabayashi N; Katoh Y; Yamamoto M; Talalay P Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. U S A 2002, 99, 11908–11913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kwak MK; Wakabayashi N; Greenlaw JL; Yamamoto M; Kensler TW Antioxidants enhance mammalian proteasome expression through the Keap1-Nrf2 signaling pathway. Mol. Cell. Biol 2003, 23, 8786–8794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McMahon M; Itoh K; Yamamoto M; Hayes JD Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J. Biol. Chem 2003, 278, 21592–21600. [DOI] [PubMed] [Google Scholar]
- 15.Nguyen T; Sherratt PJ; Huang HC; Yang CS; Pickett CB Increased protein stability as a mechanism that enhances Nrf2-mediated transcriptional activation of the antioxidant response element. Degradation of Nrf2 by the 26 S proteasome. J. Biol. Chem 2003, 278, 4536–4541. [DOI] [PubMed] [Google Scholar]
- 16.Richardson BG; Jain AD; Speltz TE; Moore TW Non-electrophilic modulators of the canonical Keap1/Nrf2 pathway. Bioorg. Med. Chem. Lett 2015, 25, 2261–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baird L; Swift S; Lleres D; Dinkova-Kostova AT Monitoring Keap1-Nrf2 interactions in single live cells. Biotechnol. Adv 2014, 32, 1133–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crunkhorn S Deal watch: Abbott boosts investment in NRF2 activators for reducing oxidative stress. Nat. Rev. Drug. Discov 2012, 11, 96. [DOI] [PubMed] [Google Scholar]
- 19.Magesh S; Chen Y; Hu L Small molecule modulators of Keap1-Nrf2-ARE pathway as potential preventive and therapeutic agents. Med. Res. Rev 2012, 32, 687–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mitsuishi Y; Motohashi H; Yamamoto M The Keap1-Nrf2 system in cancers: stress response and anabolic metabolism. Front. Oncol 2012, 2, 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yore MM; Kettenbach AN; Sporn MB; Gerber SA; Liby KT Proteomic analysis shows synthetic oleanane triterpenoid binds to mTOR. PLoS One 2011, 6, e22862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clulow JA; Storck EM; Lanyon-Hogg T; Kalesh KA; Jones LH; Tate EW Competition-based, quantitative chemical proteomics in breast cancer cells identifies new target profiles for sulforaphane. Chem. Commun 2017, 53, 5182–5185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schulze-Topphoff U; Varrin-Doyer M; Pekarek K; Spencer CM; Shetty A; Sagan SA; Cree BA; Sobel RA; Wipke BT; Steinman L; Scannevin RH; Zamvil SS Dimethyl fumarate treatment induces adaptive and innate immune modulation independent of Nrf2. Proc. Natl. Acad. Sci. U S A 2016, 113, 4777–4782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davies TG; Wixted WE; Coyle JE; Griffiths-Jones C; Hearn K; McMenamin R; Norton D; Rich SJ; Richardson C; Saxty G; Willems HM; Woolford AJ; Cottom JE; Kou JP; Yonchuk JG; Feldser HG; Sanchez Y; Foley JP; Bolognese BJ; Logan G; Podolin PL; Yan H; Callahan JF; Heightman TD; Kerns JK Monoacidic Inhibitors of the Kelch-like ECH-Associated Protein 1: Nuclear Factor Erythroid 2-Related Factor 2 (KEAP1:NRF2) Protein-Protein Interaction with High Cell Potency Identified by Fragment-Based Discovery. J. Med. Chem 2016, 59, 3991–4006. [DOI] [PubMed] [Google Scholar]
- 25.Hu L; Magesh S; Chen L; Wang L; Lewis TA; Chen Y; Khodier C; Inoyama D; Beamer LJ; Emge TJ; Shen J; Kerrigan JE; Kong AN; Dandapani S; Palmer M; Schreiber SL; Munoz B Discovery of a small-molecule inhibitor and cellular probe of Keap1-Nrf2 protein-protein interaction. Bioorg. Med. Chem. Lett 2013, 23, 3039–3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.London N; Raveh B; Schueler-Furman O Druggable protein–protein interactions – from hot spots to hot segments. Curr. Opin. Chem. Biol 2013, 17, 952–959. [DOI] [PubMed] [Google Scholar]
- 27.Marcotte D; Zeng W; Hus JC; McKenzie A; Hession C; Jin P; Bergeron C; Lugovskoy A; Enyedy I; Cuervo H; Wang D; Atmanene C; Roecklin D; Vecchi M; Vivat V; Kraemer J; Winkler D; Hong V; Chao J; Lukashev M; Silvian L Small molecules inhibit the interaction of Nrf2 and the Keap1 Kelch domain through a non-covalent mechanism. Bioorg. Med. Chem 2013, 21, 4011–4019. [DOI] [PubMed] [Google Scholar]
- 28.Jiang ZY; Lu MC; Xu LL; Yang TT; Xi MY; Xu XL; Guo XK; Zhang XJ; You QD; Sun HP Discovery of potent Keap1-Nrf2 protein-protein interaction inhibitor based on molecular binding determinants analysis. J. Med. Chem 2014, 57, 2736–2745. [DOI] [PubMed] [Google Scholar]
- 29.Jiang ZY; Xu LL; Lu MC; Pan Y; Huang HZ; Zhang XJ; Sun HP; You QD Investigation of the intermolecular recognition mechanism between the E3 ubiquitin ligase Keap1 and substrate based on multiple substrates analysis. J. Comput. Aided. Mol. Des 2014, 28, 1233–1245. [DOI] [PubMed] [Google Scholar]
- 30.Lu MC; Tan SJ; Ji JA; Chen ZY; Yuan ZW; You QD; Jiang ZY Polar recognition group study of Keap1-Nrf2 protein-protein interaction inhibitors. ACS Med. Chem. Lett 2016, 7, 835–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Winkel AF; Engel CK; Margerie D; Kannt A; Szillat H; Glombik H; Kallus C; Ruf S; Gussregen S; Riedel J; Herling AW; von Knethen A; Weigert A; Brune B; Schmoll D Characterization of RA839, a noncovalent small molecule binder to Keap1 and selective activator of Nrf2 signaling. J. Biol. Chem 2015, 290, 28446–28455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schwarz LR; Mezger M; Hesse S Effect of decreased glucuronidation and sulfation on covalent binding of naphthalene in isolated rat hepatocytes. Toxicol. 1980, 17, 119–122. [DOI] [PubMed] [Google Scholar]
- 33.Buckpitt A; Boland B; Isbell M; Morin D; Shultz M; Baldwin R; Chan K; Karlsson A; Lin C; Taff A; West J; Fanucchi M; Van Winkle L; Plopper C Naphthalene-induced respiratory tract toxicity: metabolic mechanisms of toxicity. Drug Metab. Rev 2002, 34, 791–820. [DOI] [PubMed] [Google Scholar]
- 34.Tsuruda LS; Lame MW; Jones AD Formation of epoxide and quinone protein adducts in B6C3F1 mice treated with naphthalene, sulfate conjugate of 1,4-dihydroxynaphthalene and 1,4-naphthoquinone. Arch. Toxicol 1995, 69, 362–367. [DOI] [PubMed] [Google Scholar]
- 35.Buckpitt AR; Warren DL Evidence for hepatic formation, export and covalent binding of reactive naphthalene metabolites in extrahepatic tissues in vivo. J. Pharmacol. Exp. Ther 1983, 225, 8–16. [PubMed] [Google Scholar]
- 36.Hsu KH; Su BH; Tu YS; Lin OA; Tseng YJ Mutagenicity in a molecule: Identification of core structural features of mutagenicity using a scaffold analysis. PLoS One 2016, 11, e0148900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bulbulyan MA; Figgs LW; Zahm SH; Savitskaya T; Goldfarb A; Astashevsky S; Zaridze D Cancer incidence and mortality among beta-naphthylamine and benzidine dye workers in Moscow. Int. J. Epidemiol 1995, 24, 266–275. [DOI] [PubMed] [Google Scholar]
- 38.Rafferty MF No denying it: Medicinal chemistry training is in big trouble. J. Med. Chem 2016, 59, 10859–10864. [DOI] [PubMed] [Google Scholar]
- 39.Garst ME; Lukton D Phenyldiazenes from phenyl diazonium fluoborate and carbanions. Synth. Commun 1980, 10, 155–160. [Google Scholar]
- 40.Wuts PGM; Greene TW Protection for the amino group In Greene’s Protective Groups in Organic Synthesis, John Wiley & Sons, Inc., Hoboken, New Jersey: 2006; pp 696–926. [Google Scholar]
- 41.Inoyama D; Chen Y; Huang X; Beamer LJ; Kong AN; Hu L Optimization of fluorescently labeled Nrf2 peptide probes and the development of a fluorescence polarization assay for the discovery of inhibitors of Keap1-Nrf2 interaction. J. Biomol. Screen 2012, 17, 435–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dougherty DA Cation-pi interactions in chemistry and biology: a new view of benzene, Phe, Tyr, and Trp. Science 1996, 271, 163–168. [DOI] [PubMed] [Google Scholar]
- 43.Dougherty DA Cation-pi interactions involving aromatic amino acids. J. Nutr 2007, 137, 1504S–1508S; discussion 1516S-1517S. [DOI] [PubMed] [Google Scholar]
- 44.Dougherty DA The cation-pi interaction. Acc. Chem. Res 2013, 46, 885–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pallesen JS; Tran KT; Bach A Non-covalent small-molecule Kelch-like ECH-Associated Protein 1-Nuclear Factor Erythroid 2-Related Factor 2 (Keap1-Nrf2) inhibitors and their potential for targeting central nervous system diseases. J. Med. Chem 10.1021/acs.jmedchem.8b00358. Published Online: May 11, 2018. [DOI] [PubMed] [Google Scholar]
- 46.Yasuda D; Yuasa A; Obata R; Nakajima M; Takahashi K; Ohe T; Ichimura Y; Komatsu M; Yamamoto M; Imamura R; Kojima H; Okabe T; Nagano T; Mashino T Discovery of benzo[g]indoles as a novel class of non-covalent Keap1-Nrf2 protein-protein interaction inhibitor. Bioorg. Med. Chem. Lett 2017, 27, 5006–5009. [DOI] [PubMed] [Google Scholar]
- 47.Drwal MN; Banerjee P; Dunkel M; Wettig MR; Preissner R ProTox: a web server for the in silico prediction of rodent oral toxicity. Nucleic Acids Res. 2014, 42, W53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maunz A; Gutlein M; Rautenberg M; Vorgrimmler D; Gebele D; Helma C Lazar: a modular predictive toxicology framework. Front. Pharmacol 2013, 4, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lu M; Zhou HS; You QD; Jiang Z Design, synthesis, and initial evaluation of affinity-based small-molecule probes for fluorescent visualization and specific detection of Keap1. J. Med. Chem 2016, 59, 7305–7310. [DOI] [PubMed] [Google Scholar]
- 50.Jiang ZY; Xu LL; Lu MC; Chen ZY; Yuan ZW; Xu XL; Guo XK; Zhang XJ; Sun HP; You QD Structure-activity and structure-property relationship and exploratory in vivo evaluation of the nanomolar Keap1-Nrf2 protein-protein interaction inhibitor. J. Med. Chem 2015, 58, 6410–6421. [DOI] [PubMed] [Google Scholar]
- 51.Briffett NE; Hibbert F Alkaline hydrolysis of dibenzoylaminonaphthalenes in 70%(v/v) Me2SO-H2O and the effect of a neighbouring amide group. J. Chem. Soc., Perkin Trans. 2 1989, 7, 765–768. [Google Scholar]
- 52.Dumas JP; Joe TK; Kluender HCE; Lee W; Nagarathnam D; Sibley RN; Su N; Boyer SJ; Dixon JA Preparation of Substituted Pyridines and Pyridazines with Angiogenesis Inhibiting Activity for Pharmaceutical use as Antitumor Agents. WO2001023375A2, 2001. [Google Scholar]
- 53.Qin H-L; Panek JS Total synthesis of the Hsp90 inhibitor geldanamycin. Org. Lett 2008, 10, 2477–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wrona IE; Gozman A; Taldone T; Chiosis G; Panek JS Synthesis of reblastatin, autolytimycin, and non-benzoquinone analogues: potent inhibitors of Heat Shock Protein 90. J. Org. Chem 2010, 75, 2820–2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Price CC; Voong S-T 4-Nitro-1-naphthylamine. Org. Synth 1948, 28, 80–81. [Google Scholar]
- 56.Liu K; Yin D Efficient method for the synthesis of 2,3-unsubstituted nitro containing indoles from o-fluoronitrobenzenes. Org. Lett 2009, 11, 637–639. [DOI] [PubMed] [Google Scholar]
- 57.Haider N; Holzer W Product class 10: Phthalazines. Sci. Synth 2004, 16, 315–372. [Google Scholar]
- 58.Attolino E; Calderone V; Dragoni E; Fragai M; Richichi B; Luchinat C; Nativi C Structure-based approach to nanomolar, water soluble matrix metalloproteinases inhibitors (MMPIs). Eur. J. Med. Chem 2010, 45, 5919–5925. [DOI] [PubMed] [Google Scholar]
- 59.Thompson QE The diacylation of amides by acyl chloride—pyridine compounds. J. Am. Chem. Soc 1951, 73, 5841–5846. [Google Scholar]
- 60.Li Z; Gelbaum C; Fisk JS; Holden B; Jaganathan A; Whiteker GT; Pollet P; Liotta CL Aqueous Suzuki coupling reactions of basic nitrogen-containing substrates in the absence of added base and ligand: Observation of high yields under acidic conditions. J. Org. Chem 2016, 81, 8520–8529. [DOI] [PubMed] [Google Scholar]
- 61.Zhou L; Yang L; Tilton S; Wang J Development of a high throughput equilibrium solubility assay using miniaturized shake-flask method in early drug discovery. J. Pharm. Sci 2007, 96, 3052–3071. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.