

Abstract
Neuroinflammation and mitochondrial dysfunction, key mechanisms in the pathogenesis of Parkinson's disease (PD), are usually explored independently. Loss‐of‐function mutations of PARK2 and PARK6, encoding the E3 ubiquitin protein ligase Parkin and the mitochondrial serine/threonine kinase PINK1, account for a large proportion of cases of autosomal recessive early‐onset PD. PINK1 and Parkin regulate mitochondrial quality control and have been linked to the modulation of innate immunity pathways. We report here an exacerbation of NLRP3 inflammasome activation by specific inducers in microglia and bone marrow‐derived macrophages from Park2−/− and Pink1−/− mice. The caspase 1‐dependent release of IL‐1β and IL‐18 was, therefore, enhanced in Park2−/− and Pink1−/− cells. This defect was confirmed in blood‐derived macrophages from patients with PARK2 mutations and was reversed by MCC950, which specifically inhibits NLRP3 inflammasome complex formation. Enhanced NLRP3 signaling in Parkin‐deficient cells was accompanied by a lack of induction of A20, a well‐known negative regulator of the NF‐κB pathway recently shown to attenuate NLRP3 inflammasome activity. We also found an inverse correlation between A20 abundance and IL‐1β release, in human macrophages challenged with NLRP3 inflammasome inducers. Overall, our observations suggest that the A20/NLRP3‐inflammasome axis participates in the pathogenesis of PARK2‐linked PD, paving the way for the exploration of its potential as a biomarker and treatment target.
Keywords: human macrophages, neuroinflammation, NLRP3‐inflammasome, Parkin, Parkinson's disease, primary microglia
Abbreviations
- 3‐MA
3‐methyladenine
- A20 (=TNFAIP3)
antiapoptotic signaling protein 20
- AD
Alzheimer disease
- AIM2
absent in melanoma 2
- ALS
Amyotrophic lateral sclerosis
- ASC
apoptosis‐associated speck‐like protein containing carboxyl‐terminal CARD
- CARD
Caspase activation and recruitment domain
- DA neurons
dopaminergic neurons
- DAMPS
damage‐associated molecular pattern
- DMEM
Dulbecco's modified Eagle medium
- DREAM
downstream regulatory element antagonist modulator
- FCS
fetal calf serum
- GM‐CSF
granulocyte‐macrophage colony‐stimulating factor
- HMGB1
high‐mobility group box 1
- IBA‐1
ionized calcium‐binding adapter molecule 1
- IKKβ
inhibitor of nuclear factor kappa‐B kinase subunit beta
- IL
interleukin
- LPS
lipopolysaccharide
- MAC‐1 (=CD11B)
macrophage antigen‐1
- MAVS
mitochondrial antiviral signaling
- MCP‐1
monocyte chemotactic protein 1
- MPTP
1‐méthyl‐4‐phényl‐1,2,3,6‐tétrahydropyridine
- MQC
mitochondrial quality control
- MW
molecular weight
- NF‐κB
nuclear factor‐kappa B
- NLRs
nod‐like receptors
- NLRP1/3
NLR family, pyrin domain‐containing 1/3
- NLRC4
NLR family CARD domain‐containing protein 4
- PBMC
peripheral blood mononuclear cell
- PEI
polyethylenimine
- PD
Parkinson disease
- O/AA
oligomycin + antimycin A
- PD
Parkinson's disease
- RLR
RIG‐I like receptors
- TLRs
toll‐like receptors
- TNFα
tumor necrosis factor alpha
- TNFAIP3 (=A20)
tumor necrosis factor alpha‐induced protein 3
- USF1
upstream stimulatory factor 1
1. INTRODUCTION
Parkinson's disease (PD) is a multifactorial disorder involving a complex interplay between various pathogenic mechanisms, including mitochondrial dysfunction and neuroinflammation. The discovery of the function of the PD‐linked genes PARK2/Parkin and PINK1, which act together to regulate various mitochondrial quality control (MQC) mechanisms, including mitophagy, has strengthened the role of mitochondrial dysfunction (McLelland, Soubannier, Chen, McBride, & Fon, 2014; Narendra, Tanaka, Suen, & Youle, 2008; Pickrell & Youle, 2015; Sayre, 1989). The neuroinflammation observed in PD is characterized by excessive microglial cell activation, supported by histological findings from post‐mortem examinations and recent positron emission tomography imaging of the brain (Halliday & Stevens, 2011; Hirsch & Hunot, 2009). Moreover, high levels of proinflammatory cytokines have been observed in the brain and cerebrospinal fluid of patients compared with controls (Hirsch & Hunot, 2009).
Molecular patterns associated with invading organisms or released during degenerative processes (danger‐associated molecular patterns, DAMPs) bind to specific receptors, including those of the NOD‐like receptor (NLR) family, thereby activating signaling cascades that promote pathogen elimination and tissue repair. The NLR family member NLRP3 and its intracellular signaling complex, the inflammasome, play a broad role in various inflammatory conditions (Gross, Thomas, Guarda, & Tschopp, 2011; Lee, Kang, Lee, & Jo, 2013). They have recently been linked to the pathogenesis of Alzheimer's disease (AD), and are suspected to contribute to neurodegeneration in PD (Codolo et al., 2013; Heneka et al., 2013; Sarkar et al., 2017; Yan et al., 2015) The NLRP3 inflammasome is activated, in a two‐step mechanism, in response to DAMP stimuli, including amyloid beta‐oligomers and α‐synuclein (Codolo et al., 2013; Y. Zhou et al., 2016): (1) a priming step mediated by TLR and the nuclear translocation of NF‐κB (nuclear factor‐kappa B), associated with the transcriptional induction of NLRP3 and the immature forms of the proinflammatory cytokines interleukin (IL)‐1β and IL‐18 and (2) an activation step, during which NLRP3 and its adaptor ASC (apoptosis‐associated speck‐like protein containing a CARD) assemble into a complex at the endoplasmic reticulum‐mitochondrion interface (Martinon, Burns, & Tschopp, 2002; Rossol et al., 2012). This complex recruits and activates caspase‐1, leading to the processing and release of IL‐1β and IL‐18 (Latz, Xiao, & Stutz, 2013). Regulatory negative feedback loops operate in immune cells, both at the transcriptional level and at the level of NLRP3 complex formation and stability, to prevent overactivation of the NLRP3 inflammasome (Guarda et al., 2009; Mishra et al., 2013; Walle et al., 2014).
Mitochondria have recently emerged as a platform for the shaping of innate immune responses, including in the context of NLRP3‐dependent signaling (Shimada et al., 2012; R. Zhou, Yazdi, Menu, & Tschopp, 2011). Mitochondrial quality control (MQC) plays a key role in attenuating this pathway, by preserving a cohort of functional mitochondria within the cell (Lazarou, 2015; Zhong et al., 2016). Several studies have linked Parkin and PINK1 to innate immunity (Greene, Whitworth, Andrews, Parker, & Pallanck, 2005; Matheoud et al., 2016; Torres‐Odio et al., 2017). A loss of PARK2 function increases susceptibility to mycobacterial infection and sensitivity to inflammation‐related dopaminergic (DA) neuron degeneration (Chopra et al., 2014; Frank‐Cannon et al., 2008; Lazarou, 2015; Manzanillo et al., 2013; Mira et al., 2004), while the expression of PARK2 and PINK1 is stimulated by hepatitis viruses (Khan, Syed, Kim, & Siddiqui, 2016). Parkin regulates the NF‐κB‐dependent inflammatory pathway (Henn et al., 2007), and Parkin deficiency enhances the production of cytokines, such as TNFα, IL‐6 and monocyte chemoattractant protein‐1 (MCP‐1; de Léséleuc et al., 2013; Tran et al., 2011).
In this study, we explored the link between MQC impairment and neuroinflammation in PD further, by investigating the response of PARK2‐ and PINK1‐deficient primary microglial cells to proinflammatory stimuli and dissecting the impact of Parkin and PINK1 dysfunction on NLRP3 inflammasome signaling. We found that a loss of PARK2 function exacerbated inflammasome activation by attenuating a negative feedback loop restricting NLRP3 inflammasome activity, in both primary murine immune cells and primary macrophages from patients with PARK2 mutations. These results suggest that the NLRP3‐inflammasome participates in the pathogenesis of PD as a result of PARK2 dysfunction, paving the way for the exploration of this complex as a target for disease prevention and treatment.
2. MATERIALS AND METHODS
2.1. Patients
Informed consent was obtained from all participants, and the genetic studies were approved by local ethics committees (INSERM, CCPPRB du Groupe Hospitalier Pitié‐Salpêtrière, Paris, France).
Patients (n = 6, Table 1) and control subjects (5) gave written informed consent for blood assessment. Primary human macrophages were differentiated from peripheral blood mononuclear cells (PBMCs) obtained from fresh blood samples by stimulation with recombinant human GM‐CSF (Peprotech, Neuilly‐sur‐Seine, France), and cultured as previously described (Xue et al., 2014). PBMCs were isolated by Ficoll (Histopaque, Sigma Aldrich, L'Isle d'Abeau Chesnes, France) density gradient centrifugation from freshly collected blood (40 ml). After several centrifugations, the PBMC layer was transferred to a CD14+ magnetic microbeads column (Miltenyi, Bergisch Gladbach, Germany) and purified with the Miltenyi Monocyte Isolation Kit to obtain a suspension highly enriched in monocytes.
Table 1.
Age, sex and PARK2 genotype of the donors of the blood macrophages used in this study
| Age (years) | Sex | PARK2 mutation | |
|---|---|---|---|
| Control subject 1 | 44 | M | No mutation |
| Control subject 2 | 40 | M | No mutation |
| Control subject 3 | 30 | M | No mutation |
| Control subject 4 | 39 | F | No mutation |
| Control subject 5 | 49 | F | No mutation |
| PARK2 PD Patient 1 | 31 | M | c.[(412 + 1_413‐1)_(734 + 1_735‐1)del];[633A>T] |
| PARK2 PD Patient 2 | 41 | M | c.[673delG];[673delG] |
| PARK2 PD Patient 3 | 44 | M | c.[673delG];[673delG] |
| PARK2 PD Patient 4 | 33 | M | c.[(534 + 1_535‐1)_(618 + 1_619‐1)del];[155delA] |
| PARK2 PD Patient 5 | 30 | F | c.[(7 + 1_8‐1)_(171 + 1_172‐1)del];[827delA] |
| PARK2 PD Patient 6 | 37 | F | c.[(171 + 1_172‐1)_(534 + 1_535‐1)del]; c.[(171 + 1_172‐1)_(534 + 1_535‐1)del] |
2.2. Mice
Animals were housed in an accredited animal facility. They were handled, and cared for in accordance with French and European regulations on animal research and the recommendations of the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The experiments were approved by the animal facility board of directors, the Institute's Welfare Committee and Comité d'Ethique pour l'expérimentation animale Charles Darwin. Park2−/− mice were generated and brought into the C56Bl/6j genetic background as previously described (Damiano et al., 2014; Itier et al., 2003). The Pink1−/− mouse line was established at the MCI/ICS (Mouse Clinical Institute – Institut Clinique de la Souris‐, Illkirch, France; http://www-mci.u-strasbg.fr). The targeting vector was constructed as follows: a 0.95 kb fragment encompassing exons 6 and 7 was amplified by PCR (from 129S2/SvPas ES cells genomic DNA) and subcloned in an MCI proprietary vector; this MCI vector contains a LoxP site as well as a floxed and flipped Neomycin resistance cassette; two 4.3 kb fragments (corresponding to the 5′ and 3′ homology arms) were PCR amplified and further subcloned into step1 plasmid to generate the final targeting construct. The linearized construct was electroporated in 129S2/SvPas mouse embryonic stem (ES) cells. After selection, targeted clones were identified by long‐range PCR using internal and external primers and further confirmed by Southern blot with an internal (Neo) probe and 2 external (5′ and 3′) probes. Two positive ES clones were injected into C57BL/6J blastocysts. Male chimaeras were bred with Flp deleter females (Rodríguez et al., 2000) and gave germline transmission generating the conditional allele. The Flp transgene was segregated in the new breeding step. Heterozygous conditional knock‐out mice were bred with Cre deleter animals (Dupe et al., 1997) in order to obtain the full knock‐out allele. Experimental groups of age‐matched littermate Park2−/− (or Pink1−/−) and WT mice were generated by intercross of heterozygous Park2+/− (or Pink1+/−) mice.
2.3. Isolation of microglial cells
Microglial cultures were prepared from the brains of postnatal day 1 mouse pups, as previously described (Sepulveda‐Diaz et al., 2016). Whole brains were harvested, the meninges were stripped away and brain tissue pieces were placed in 4 ml of Leibovitz's L‐15 medium (Invitrogen Life Technologies, Saint Aubin, France). Cells were then mechanically dissociated in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat‐inactivated fetal calf serum (FCS) (Biowest LLC/Eurobio, Les Ulis, France) and 1% penicillin/streptomycin (both from Invitrogen Life Technologies). The final supernatant was centrifuged at 220g for 5 min and the cells were plated on polyethyleneimine (PEI; Sigma Aldrich)‐coated culture flasks and incubated at 37°C in a humidified atmosphere containing 95% air and 5% CO2. Some of the culture medium was removed after 48 hr, but no additional medium was added until total detachment of astrocytes, after 16–18 days of culture.
2.4. Production of bone marrow‐derived macrophages
Suspensions of bone marrow cells were generated from the tibias and femurs of 8‐ to 12‐week‐old mice, as previously described (Weischenfeldt & Porse, 2008). The cell mixture was suspended in DMEM medium (Life Technologies) supplemented with 15% heat‐inactivated FCS (Invitrogen Life Technologies), streptomycin (50 µg/ml; Invitrogen Life Technologies) and penicillin (50 IU/ml; Invitrogen Life Technologies) and allowed to differentiate in the presence of recombinant mouse CSF‐1 (rmCSF‐1, 75 ng/ml; ImmunoTools, Friesoythe, Germany). BMDMs were then dispensed into Petri dishes and incubated at 37°C under an atmosphere containing 8% CO2 in air. After six days of culture, BMDMs were detached and used to seed the same medium, supplemented with rmCSF‐1 (20 ng/ml).
2.5. In vitro stimulation of immune cells
Cells were primed with ultrapure LPS (10 ng/ml; from Escherichia coli strain 0111:B4; Sigma‐Aldrich) for 3 or 24 hr and stimulated with ATP (1 mM; Sigma‐Aldrich) for 40 min, or with nigericin (2 µM; Sigma‐Aldrich) for 2 hr. The NLRP3 inflammasome was inhibited by incubating the cells in the presence of MCC950 (10 µM) for 15 min before LPS exposure (Bertin Pharma, Montigny le Bretonneux, France). 3‐MA was prepared immediately before use and added to the culture medium at a final concentration of 10 mM, 15 min before cell priming with LPS.
2.6. Reverse transcription and real‐time PCR
Total RNA was purified from microglial cell with the RNeasy plus midi kit (QIAGEN, Hilden, Germany) and quantified on a Nanodrop 8000 spectrometer (Thermo Scientific, Cergy‐Pontoise, France). Reverse transcription was performed with the iScript Reverse Transcription Supermix for RT‐qPCR kit (Biorad, Hercules, CA) and 1 µg of total RNA per sample. Real‐time PCR was carried out in 384‐well plates (Roche, Penzberg, Germany), with a LightCycler 480 instrument (Roche). A mixture of SoAdvanced Universal SYBR Green Supermix (Biorad), primers (0.5 mM) and cDNA (1 µl) was used for the reaction. Fold‐change differences in expression were calculated with REST software.1 The data for each gene listed in Table 2 were normalized relative to the expression of the housekeeping gene, PPIA.
Table 2.
List of primers used in RT‐PCR analyses and their sequences
| Gene | Forward | Reverse |
|---|---|---|
| Il1a | CAAACTGATGAAGCTCGTCA | TCTCCTTGAGCGCTCACGAA |
| Il1b | CTGTGTCTTTCCCGTGGACC | CAGCTCATATGGGTCCGACA |
| Il6 | ATGAAGTTCCTCTCTGCAAGA | GGTTTGCCGAGTAGATCTCAA |
| Nlrp3 | CCTTGGACCAGGTTCAGTG | TCCGGTTGGTGCTTAGACT |
| Ppia | AGCATACAGGTCCTGGCATC | CATGCCTTCTTTCACCTTCC |
| Tnf | TCTTCTCATTCCTGCTTGTGG | GGTCTGGGCCATAGAACTGA |
| Tnfaip3 | TTTGTGGAAACAGGACTTTGC | TGGATTTCTTCCAGGGAATTG |
2.7. Immunoblot analyses and ELISA assays
Cell extracts were homogenized in radioimmunoprecipitation assay (RIPA) buffer containing 25 mM β‐glycerophosphate, 50 mM sodium fluoride, 2 mM sodium pyrophosphate, protease inhibitor cocktail (Roche), 1× anti‐phosphatase cocktail mix (100×, Thermo Scientific), and 1× anti‐protease cocktail mix (100×, Thermo Scientific). They were kept on ice for 30 min and centrifuged at 10,000g for 30 min at 4°C. Protein concentrations were determined with the Micro BCA Protein Assay Reagent Kit (Thermo Scientific), according to the manufacturer's protocol. Protein samples (25–40 μg) were separated on gradient NuPAGE Bis‐Tris gels (Biorad) and electroblotted onto nitrocellulose membranes (GE Healthcare, Chalfont St. Giles, UK) in 25 mM Tris buffer (pH 8.3) containing 200 mM glycine and 20% ethanol. After protein transfer, membranes were blocked in 5% nonfat dry milk in PBS and incubated with primary antibody. Immunoblots were probed with primary antibodies against the following proteins: A20 (sc‐166692, Santa Cruz, Danvers, MA), caspase 1 (AG‐20B‐0042‐C100, Adipogen Life Sciences, San Diego, CA), NLRP3 (AG‐20B‐0014, Adipogen Life Sciences), and tubulin‐α (ab7291, Abcam, Cambridge, UK).
Proteins were visualized by incubation with fluorescent secondary antibodies: IR Dye 700DX‐conjugated anti‐mouse IgG, IR Dye 800CW‐conjugated anti‐goat IgG and IR Dye 800CW‐conjugated anti‐rabbit IgG (Rockland Immunochemical Inc., Gilbertsville, PA), or by enhanced chemiluminescence (Thermo Scientific). Chemiluminescence/fluorescence signals were detected by placing the blot against film (ECL, Amersham Hyperfilm) or by capture with an Odyssey imaging system (Li‐Cor Biosciences, Lincoln, NE), and quantified with Multigauge software (Fuji film, Tokyo, Japan). Cell culture supernatants were assayed, at the indicated times, for the presence of pro‐inflammatory cytokines, by mouse and human IL‐1β, TNFα, CCL2/MCP1, and IL‐6 DuoSet ELISA (R&D Systems, Minneapolis, MN) or IL‐18 ELISA (MBL/R&D Systems), according to the manufacturer's instructions. Absorbance was read at 450 nm, with a 96‐well plate reader, and cytokine levels were calculated from a standard curve.
2.8. Immunofluorescence staining
Cells were washed in PBS, fixed by incubation with 4% paraformaldehyde (Sigma Aldrich) for 20 min and permeabilized by incubation in 0.2% Triton X‐100 (Sigma Aldrich) in PBS for 10 min. Non‐specific antibody binding was blocked by incubation in PBS containing 10% normal goat serum (NGS; Thermo Fisher Scientific, Waltham, MA) for 1 hr. The cells were incubated overnight at 4°C with the primary antibody diluted in PBS containing 2% NGS, and then at room temperature for 45 min with the secondary antibody diluted in the same solution. We used primary antibodies against the following proteins: A20 (sc‐166692, Santa Cruz), CD11B/MAC‐1 (MCA711, AbD Serotec/Biorad), IBA1 (019–19741, Wako, Richmond, VA), NLRP3 (AG‐20B‐0014, Adipogen Life Sciences), TOM20 (ab56783, Santa Cruz), and VDAC (ab15895, Abcam). The secondary antibodies used were Alexa Fluor 488‐conjugated anti‐mouse/rabbit (A11070/A11029, Invitrogen), and Alexa Fluor 568‐conjugated anti‐mouse/rabbit (A11036/A11031, Invitrogen) antibodies. Cells were incubated with a 1/5,000 dilution of Hoechst stain (ImmunoChemistry Technology, Bloomington, MN) in PBS, for 5 min.
2.9. Automated cell imaging and confocal microscopy
We used the ArrayScan high‐content screening reader (XTI Live High Content Platform, Thermo Fisher Scientific) for quantitative protein localization and for the determination of fluorescence levels, or morphological changes in cells, in accordance with published procedures (Fetz, Knauer, Bier, von Kries, & Stauber, 2009). One common step in our analyses was the adjustment of the assay protocol, through the modification of several parameters, to ensure optimal object identification, including background correction, the setting of a threshold of pixel definition derived from the Hoechst signal and object segmentation parameters. This step optimized object identification, making it possible to exclude irregular “non‐cellular” objects automatically and to quantify various cell parameters rapidly in thousands of cells. We then used the following arrayscan protocols: “Spot detector” (mitochondrial marker, A20 aggresomes), “General measurement intensity” (IBA1, NLRP3 and A20 fluorescence levels). Confocal microscopy was performed with a Leica Sp7 confocal microscope (Leica, Wetzlar, Germany). MetaMorph software (Roper Scientific, Ottobrun, Germany) was used for image acquisition
2.10. Statistical analyses
Statistical analyses were performed with Prism 6 (Graph Pad Software). Pooled results are expressed as means ± SEM. Two‐way ANOVA was used to analyze the effect of treatments (LPS, LPS + nigericin/ATP, 3‐MA). Unpaired Student's t tests (unless the data were shown not to be normally distributed) or Mann and Whitney tests were used for other comparisons. The correlation between IL‐1β release and A20 levels was assessed by calculating Spearman's correlation coefficient. For all tests, p values <.05 were considered statistically significant.
3. RESULTS
3.1. Loss of Park2 function leads to the overactivation of microglial cells in response to proinflammatory stimuli
Lipopolysaccharide (LPS), a bacterial endotoxin, is a powerful activator of innate immune responses in microglial cells. Its binding to TLRs induces morphological changes and the release of a broad range of pro‐inflammatory cytokines (Horvath, Nutile‐McMenemy, Alkaitis, & Deleo, 2008). We studied the response of primary microglial cells from WT and Park2−/− mice challenged with LPS for 24 hr. We observed a strong increase in the expression of the macrophage/microglia activation marker Iba‐1 (ionized calcium‐binding adapter molecule 1) in WT cells (Figure 1a,d). This increase in Iba‐1 expression was associated with for the morphological changes typical of activated microglia, such as hypertrophic/ameboid cell bodies with ramified projections, as shown by staining for the membrane marker MAC‐1/CD11b (Figure 1a–c). Microglia from Park2−/− mice appeared to be more responsive to LPS than WT microglia, as shown by their significantly greater Iba‐1 staining intensity, MAC‐1‐positive cell area and proportion of hypertrophic cells, on automated immunofluorescence analysis. We also assessed the impact of LPS exposure on the release of pro‐inflammatory cytokines, focusing specifically on TNFα, IL‐1β, IL‐6, IL‐18, and MCP‐1, which are normally induced by LPS challenge. As expected, LPS exposure strongly increased the release of the cytokines tested (Figure 1e). A similar effect was observed in Park2−/− microglia, but with a greater release of the inflammasome‐related cytokines IL‐1β and IL‐18.
Figure 1.
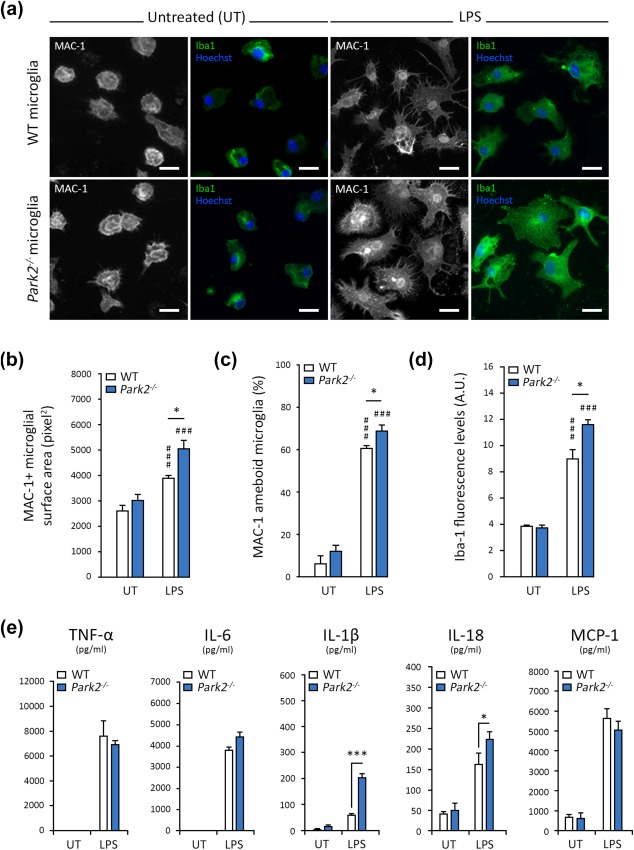
Park2−/− microglia are more strongly activated than WT cells following exposure to the bacterial endotoxin LPS. (a–d) Representative images illustrating the activation of microglial cells exposed to LPS, as explored by automated microscopy assessing the cell surface area (stained for MAC‐1) and Iba1 intensity (a), with the corresponding quantifications (b–d). The microglial surface area (in pixels) of LPS‐treated Park2−/− microglia (n = 10) is larger than that of WT cells (n = 10; b). This modification is associated with a higher percentage of MAC‐1+ microglia with an ameboid morphology (c) and stronger Iba1 staining intensity (d). (e) LPS triggers release of TNFα, IL‐6, IL‐1β, IL‐18, and MCP‐1, quantified by ELISA with normalization according to cell numbers (AU), into the supernatant of WT microglia (n = 10). Larger amounts of IL‐1β and IL‐18 are released by Park2−/− cells (n = 10). n is the number of independent experiments. Error bars indicate the SEM. *p < .05, **p < .01, ***p < .001. # versus the corresponding untreated control (UT) unless otherwise indicated. Scale bars: 10 µm
3.2. Loss of Park2/Pink1 function exacerbates NLRP3 inflammasome activation by specific inducers
We investigated whether the increase in IL‐1β and IL‐18 production in Park2−/− microglia following LPS exposure was due to overactivation of the NLRP3 inflammasome. We used the small molecule MCC950 to inhibit NLRP3‐dependent responses selectively, without affecting inflammasome complexes dependent on other innate immune receptors, such as AIM2, NLRC4 and NLRP1 (Coll et al., 2015). We also used a classical two‐signal model for direct activation of the NLRP3 inflammasome, based on exposure to LPS for 3 hr (signal I) followed by exposure to microbial toxin, nigericin, or ATP for 2 hr (signal II), to promote K+ efflux from the cell, leading to caspase 1 activation and pro‐IL‐1β/IL‐18 processing and release into the extracellular space(Cullen, Kearney, Clancy, & Martin, 2015; Munoz‐Planillo et al., 2013).
Microglial cells exposed to LPS‐nigericin or LPS‐ATP released massive amounts of IL‐1β and IL‐18, 10 times and twice higher, respectively, than the amounts released in response to LPS alone (Figure 2a,b). Parkin deficiency exacerbated these responses, whereas MCC950 abolished them in both WT and Park2−/− microglia. MCC950 also abolished the release of excessive amounts of IL‐1β and IL‐18 from Park2−/− cells treated with LPS only. The overproduction of IL‐1β and IL‐18 in Park2−/− microglia was accompanied by an increase in NLRP3 levels, as revealed by immunofluorescence assays (Figure 2c,d) and immunoblotting (Figure 2e,f). Similarly, the immunoblotting of cellular supernatants for caspase 1 revealed that PARK2−/− microglial cells released larger amounts of the p20 proteolytic fragment, a subunit of the active caspase 1 complex, than WT cells (Figure 2e,g). We also analyzed high‐mobility group box 1 (HMGB1), a ubiquitous, mostly nuclear chromatin‐binding protein that is translocated to the cytosol and released into the extracellular space by immune cells during inflammation, where it acts as a proinflammatory cytokine (Scaffidi, Misteli, & Bianchi, 2002). In cells treated with ATP or nigericin, HMGB1 release is dependent on activation of the NLRP3 inflammasome and caspase 1 (Lu et al., 2012). We therefore performed an automated immunofluorescence‐based quantitative analysis of the nuclear localization of HMGB1 in different conditions, to obtain an indirect estimate of the nucleocytoplasmic translocation and release of this molecule. As expected, following LPS‐nigericin/‐ATP treatment, HMGB1 was translocated to the cytoplasm in both WT and Park2−/− microglial cells. (Supporting Information Figure S1). Consistent with our previous observations suggesting that the NLRP3 inflammasome is overactivated in the absence of Parkin, translocation levels were higher in Park2−/− cells than in WT cells.
Figure 2.
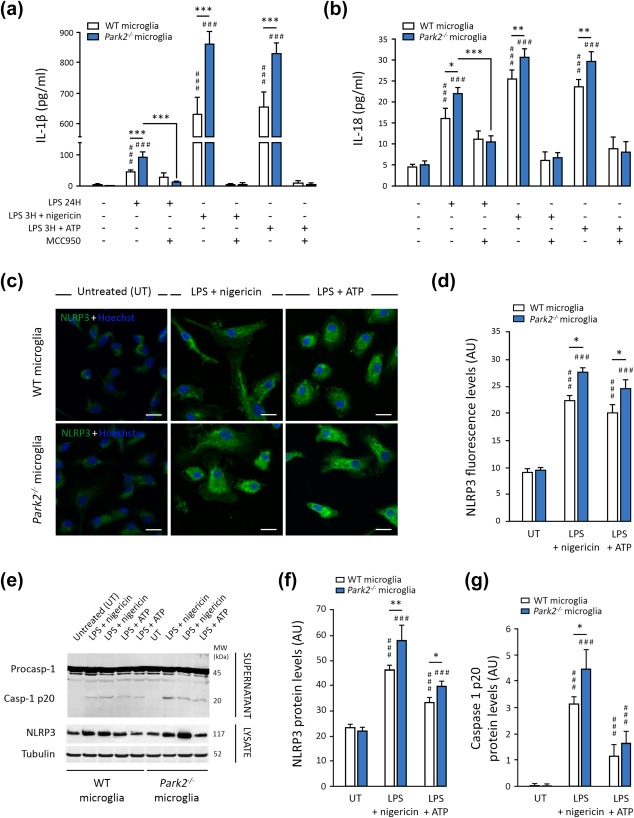
Parkin deficiency exacerbates NLRP3 inflammasome pathway activation in microglial cells. (a and b) WT microglial cells (n = 10) were treated with LPS alone or together with an inflammasome inducer: nigericin or ATP. Nigericin and ATP increase the release of IL‐1β (a) and IL‐18 (b) relative to LPS treatment alone. These effects are exacerbated in Park2−/− cells (n = 10) and completely abolished by treatment with a specific inhibitor of NLRP3‐ASC inflammasome oligomerization, MCC950. (c–g) Nigericin and ATP also strongly increase NLRP3 levels and caspase 1 cleavage in WT cells, as revealed by immunocytochemistry (c and d) and immunoblotting (e–g; n = 6). All these parameters are significantly exacerbated in Park2−/− microglia (n = 6). n is the number of independent experiments. Error bars indicate the SEM. *p < .05, **p < .01, ***p < .001. # versus corresponding untreated control (UT) unless otherwise indicated. Scale bars: 10 µm
We investigated whether the loss of PINK1 function affected microglial responses in a similar way to PARK2 dysfunction, by repeating these analyses in PINK1‐deficient microglial cells. We again observed an overactivation of the NLRP3 inflammasome after exposure to LPS‐nigericin, as illustrated by caspase 1 cleavage (Supporting Information Figure S2a,b) and the levels of IL‐1β release (Supporting Information Figure S2d). However, these changes were not accompanied by a significant increase in NLRP3 levels (Supporting Information Figure S2a,c,e,f).
3.3. The loss of PARK2/PINK1‐dependent MQC abolishes the attenuation of NLRP3 inflammasome activation in microglial cells
The Parkin‐dependent mitophagy induced by NF‐κB in response to NLRP3‐activating stimuli was recently shown to restrict NLRP3 activation (Zhong et al., 2016). We therefore reinvestigated the possibility of a link between NLRP3 inflammasome overactivation in microglial cells from Park2−/− and Pink1−/− mice and a loss of PINK1/Parkin‐dependent MQC regulation. We evaluated the release of IL‐1β and IL‐18 triggered by LPS‐nigericin in cells in the presence and absence of the autophagy inhibitor 3‐methyladenine (3‐MA), which blocks autophagosome formation by inhibiting class III phosphatidylinositol 3‐kinases. Treatment with 3‐MA mimicked the effect of PARK2 and PINK1 dysfunctions, leading to the overproduction of inflammasome‐related cytokines in WT cells (Figure 3a,b); 3‐MA did not further increase the release of IL‐1β/IL‐18 triggered by LPS‐nigericin in Park2−/− microglia, consistent with a role in the same pathway (Figure 3a,b).
Figure 3.
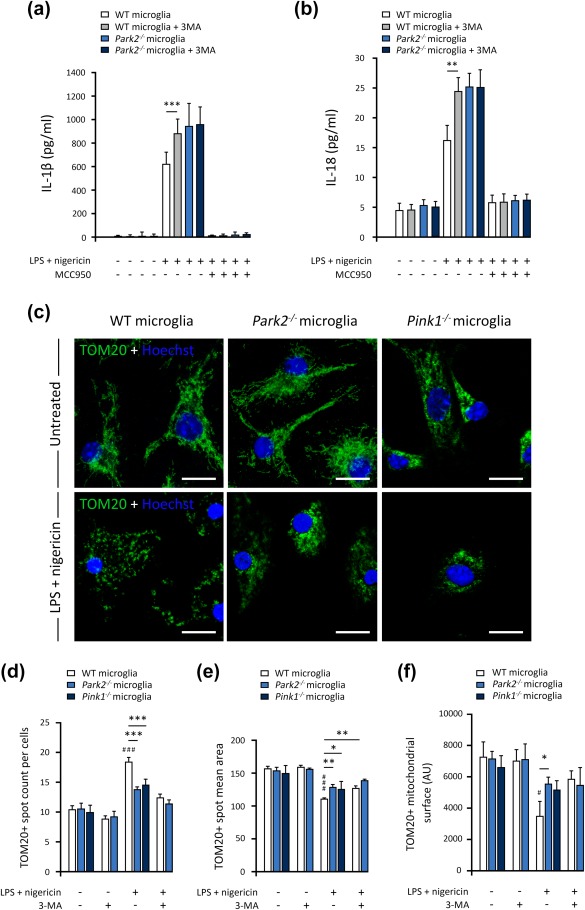
The changes to the mitochondrial network in microglial cells induced by inflammasome activators are attenuated by Parkin and PINK1 deficiencies. (a and b) 3‐MA (3‐methyladenine) treatment of WT microglia mimics the effect of Parkin loss on the release of IL‐1β (a) and IL‐18 (b). (c) Confocal imaging, illustrating the mitochondrial network stained for the mitochondrial outer membrane protein TOM20, in WT, Park2−/− and Pink1−/− microglial cells, after treatment with LPS and nigericin. (d–f) Arrayscan quantification of mitochondrial spot number (d), size (e), and mitochondrial surface area (f), showing mitochondrial fragmentation and loss in WT microglia exposed to LPS and nigericin. These changes are not observed in Park2−/− and Pink1−/− cells or following treatment with 3‐MA. n is the number of independent experiments. The error bars indicate the ±SEM. *p < .05, **p < .01, ***p < .001. # versus corresponding untreated control (UT) unless otherwise indicated. Scale bars: 10 µm
We then explored the impact of PARK2 and PINK1 dysfunction or 3‐MA on the changes to mitochondrial morphology triggered by LPS‐nigericin in microglial cells. As reported for other immune cell types (Park et al., 2015), exposure to LPS‐nigericin triggered mitochondrial fragmentation in WT cells (Figure 3c). Consistent with these qualitative observations, automated immunofluorescence quantification revealed an increase of up to 80% in the number of spots positive for the mitochondrial markers TOM20 and VDAC1 in WT cells exposed to LPS‐nigericin relative to untreated cells; this increase was accompanied by a significant decrease in mean spot area (Figure 3d,e and Supporting Information Figure S3b,c). No such changes were observed in Park2−/− and Pink1−/− cells or after treatment with 3‐MA (Figure 3 and Supporting Information Figure S3). In these cells, the mitochondrial network appeared more compact and aggregated in the perinuclear region, suggesting a defect of mitochondrial degradation. Consistent with this hypothesis, the exposure of WT microglial cells to LPS‐nigericin led to a significant decrease in the overall mean mitochondrial area per cell, whereas no such decrease was observed in Park2−/− and Pink1−/− microglia, or following treatment with 3‐MA, supporting the link to defective mitophagy (Figure 3f). Accordingly, the effects of 3‐MA and PARK2 dysfunction on the changes to the mitochondrial network triggered by LPS‐nigericin were not additive, indicating that both sets of effects were linked to the inhibition of mitophagy.
3.4. Loss of Park2 function interferes with the attenuation of NLRP3 inflammasome priming mediated by A20
Overactivation of the NLRP3 inflammasome in Park2−/− microglia was accompanied by a strong increase in NLRP3 levels (Figure 2) that was not observed in Pink1−/− cells. This suggests that, in addition to mitophagy, Parkin deficiency impairs a second regulatory loop that attenuates NLRP3 inflammasome activation, possibly linked to NF‐κB‐dependent priming. We tested this hypothesis, by exploring the levels of mRNA for NLRP3 and related cytokines by reverse transcription and quantitative PCR (RT‐qPCR) in Park2−/−, Pink1−/− and WT microglia (Figure 4a). As expected, LPS exposure in WT microglia led to a large increase in the levels of Il1b and Nlrp3 mRNAs, directly linked to activation of the NF‐κB pathway (Horvath et al., 2008). The addition of nigericin attenuated this effect, although the mRNA levels of the studied cytokines remained higher than in untreated cells (O'Connor, Harton, Zhu, Linhoff, & Ting, 2003). The lowering of mRNA levels by nigericin was less marked in Park2‐deficient cells, resulting in higher levels of Il1b and Nlrp3 than in control cells (Figure 4a).
Figure 4.
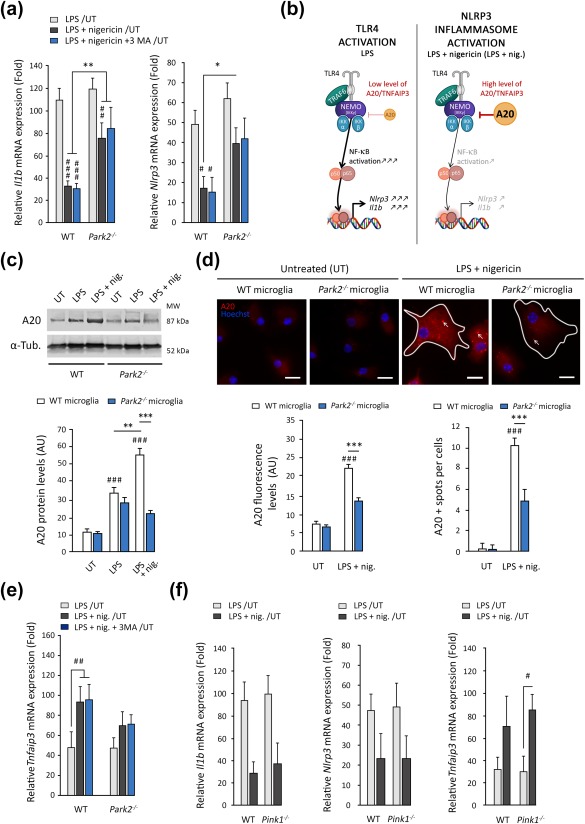
Parkin deficiency abolishes the negative feedback regulation of NLRP3 inflammasome priming by A20. (a) Expression of Il1b and Nlrp3 quantified by RT‐PCR and normalized relative to Pp1a mRNA levels. The expression of all the cytokines and inflammasome components tested increased following LPS exposure. The addition of nigericin decreased the expression of Il1b and Nlrp3 significantly in WT microglia and, to a lesser extent, in Park2−/− microglial cells (n = 5). These changes in expression were not affected by the autophagy inhibitor 3‐MA. (b) Schematic diagram illustrating the downregulation of the NF‐κB‐dependent expression of NLRP3 components driven by the A20/TNFAIP3 protein. In the presence of LPS, A20 is weakly expressed, leading to strong induction of the IKKβ‐NEMO‐NF‐κB pathway and the production of large amounts of inflammasome components. Nigericin treatment triggers an increase in A20 levels, leading to downregulation of the IKKβ‐NEMO‐NF‐κB pathway and a decrease in the expression of Nlrp3 and Il1b. (c–e) A20 protein levels are significantly lower in Park2−/− microglia (n = 4) treated with LPS and nigericin than in WT microglia (n = 4), as illustrated by immunoblot analysis (c) and immunocytochemistry (d). A20 immunofluorescence shows this protein to have a punctate distribution following inflammasome activation in WT cells, which is partly abolished in Parkin‐deficient cells. (e) mRNA levels for Tnfaip3 are also lower following exposure to LPS + nigericin. (f) No difference in expression of Il1b, Nlrp3 and Tnfaip3 was found between WT and Pink1−/− microglia (n = 4). n is the number of independent experiments. Error bars indicate the SEM. *p < .05, **p < .01, ***p < .001. # versus corresponding untreated control (UT) unless otherwise indicated. Scale bars: 10 µm
The ubiquitin‐modifying enzyme A20 (antiapoptotic signaling protein 20), encoded by the rheumatoid arthritis susceptibility gene TNFAIP3, is strongly upregulated in the presence of NLRP3 agonists, and plays a key role in attenuating NLRP3 inflammasome activity (Duong et al., 2015; Walle et al., 2014). A well‐characterized mechanism underlying the negative feedback regulation of the NLRP3 inflammasome by A20 involves the inhibition of NF‐κB activation and NF‐κB‐dependent gene expression (Beyaert, Heyninck, & Van Huffel, 2000; Figure 4b). In our experimental conditions, A20 protein levels in WT cells increased by a factor of more than four following exposure to LPS‐nigericin, as revealed by immunoblotting and confirmed by immunofluorescence approaches (Figure 4c,d). This increase was associated with the appearance of intracellular A20‐positive punctate bodies, previously reported to correspond to insoluble aggresomes in which A20 sequesters inflammatory intermediates required for NF‐κB signaling (Enesa, Moll, Luong, Ferran, & Evans, 2015). The upregulation of A20 protein levels was accompanied by an upregulation of TNFAIP3 expression following exposure to LPS‐nigericin, as indicated by the quantitative analysis of transcript levels (Figure 4e). A20 induction was significantly attenuated, at both the transcript and protein levels, in the absence of Parkin (Figure 4c–e), suggesting that PARK2 dysfunction impairs the transcriptional regulation of A20. PINK1 deficiency or the treatment of cells with 3‐MA had no effect on Tnfaip3 mRNA levels or inflammasome components in response to treatment with LPS‐nigericin, indicating that this regulation does not involve mitophagy (Figure 4a,e,f).
3.5. The NLRP3‐inflammasome is overactivated in immune cells from PD patients with PARK2 mutations
We investigated the relevance of the NLRP3 inflammasome deregulation associated with PARK2 dysfunction to human immune cells in the context of Parkinson's disease. The complexity of the procedures for obtaining living microglial cells from the human brain (Mizee et al., 2017), and the lack of availability of post mortem tissue from familial cases of PD severely limit the exploration of disease‐relevant mechanisms in these cells. A valuable alternative to overcome this limitation is the use of macrophages isolated from circulating blood cells. As a preliminary step, we validated key findings obtained with primary mouse microglial cells in bone marrow‐derived macrophages (BMDMs) from WT and Park2−/− mice. BMDMs are highly responsive to inflammasome activators (Cullen et al., 2015; Gurung et al., 2015). We observed strong activation of the NLRP3 inflammasome after exposure to LPS‐nigericin, as illustrated by increases in the extracellular release of IL‐1β and IL‐18 (Supporting Information Figure S4a,b) and intracellular levels of NLRP3 (Supporting Information Figure 4c,d) relative to treatment with LPS alone and untreated control conditions. As in microglial cells, these responses were exacerbated by Parkin deficiency. Similarly, the increase in A20 levels observed in WT BMDMs subjected to proinflammatory stimuli was attenuated in Park2−/− cells (Supporting Information Figure S4c,e).
We then analyzed these responses in primary human blood‐derived macrophages obtained from control individuals (n = 5) and PD patients with PARK2 mutations (n = 6). Macrophages from control subjects displayed significant activation following exposure to LPS‐nigericin or LPS‐ATP, which strongly induced the NLRP3 inflammasome, as shown by the analysis of intracellular NLRP3 levels and IL‐1β production (Figure 5a,b,d). This activation was exacerbated in macrophages from patients with PARK2 mutations, as shown by the higher levels of NLRP3 immunofluorescence and IL‐1β and the tendency towards higher levels of IL18 release than for cells from control individuals (Figure 5a,b,d,e). It was also associated with a lower abundance of the A20 protein (Figure 5c). Linear regression analysis revealed a significant inverse correlation between A20 abundance and the amounts of IL‐1β released, in human macrophages treated with LPS‐nigericin or LPS‐ATP (Figure 5f,g).
Figure 5.
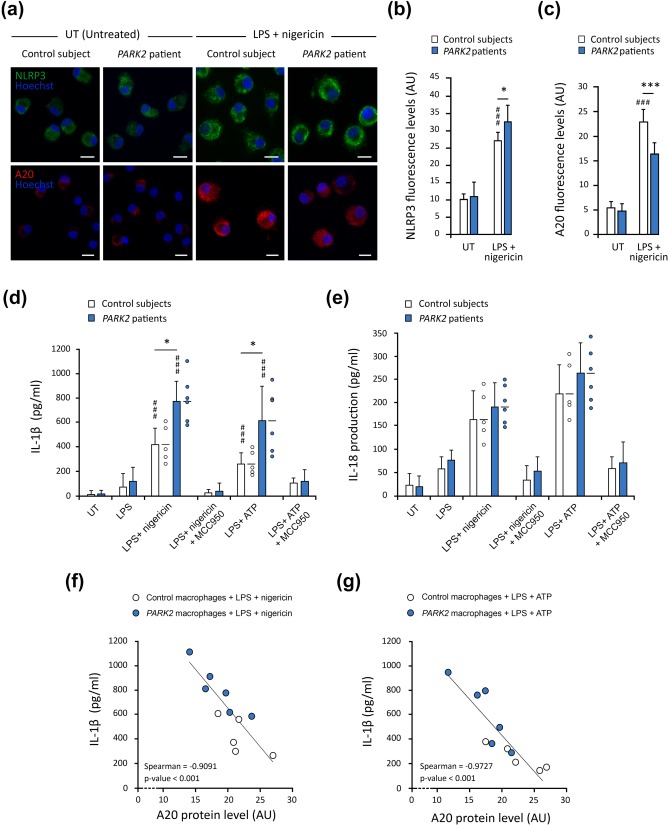
Parkin deficiency leads to NLRP3 inflammasome overactivation in macrophages from PD patients. (a–c) Immunofluorescence staining for NLRP3 and A20 in primary macrophages from control subjects (n = 5) and PD patients with PARK2 mutations (n = 6; a), with the corresponding quantifications. NLRP3 levels are significantly higher in PARK2 macrophages treated with LPS + nigericin than in control cells (b), whereas the induction of A20 is weaker (c). (d and e) ELISA revealed significantly higher levels of IL‐1β release from PARK2 macrophages (n = 6) than from control cells (n = 5). (f and g) Graphic representation illustrating the significant inverse correlation between A20 levels and levels of IL‐1β (d) and IL‐18 (e) released from macrophages of controls and patients, treated with LPS‐nigericin (f) or LPS‐ATP (g). n is the number of independent experiments. The error bars indicate the SEM. *p < .05, **p < .01, ***p < .001. # versus corresponding untreated control (UT) unless otherwise indicated. Scale bars: 10 µm
4. DISCUSSION
The cooperation between PINK1 and Parkin in MQC mechanisms, ranging from the removal of damaged mitochondrial components to the degradation of whole dysfunctional organelles by mitophagy, has been studied in detail (McLelland et al., 2014; Mouton‐Liger, Jacoupy, Corvol, & Corti, 2017; Narendra et al., 2008; Pickrell & Youle, 2015). However, little is known about the impact of PINK1/Parkin‐dependent mitochondrial surveillance mechanisms and their dysfunction on the biology of the different cell types in the central nervous system, including glial cells in particular. Innate immune system abnormalities and CNS inflammation, which have long been thought to be involved in PD pathogenesis, mostly involve microglial cells, the macrophages resident in the brain. We show here that deficiencies of PINK1 and Parkin exacerbate the response of mouse microglial cell and BMDMs to proinflammatory conditions, leading to an excessive release of cytokines associated with the NLRP3 inflammasome. Similarly, the response to specific NLRP3 inflammasome activators was stronger in macrophages from the blood of patients with PARK2 mutations than in those from control subjects. These changes were abolished by treatment with MCC950, a selective inhibitor of the interaction between NLRP3 and its adaptor, ASC (Coll et al., 2015), providing the first evidence for NLRP3 inflammasome deregulation in PD. These findings echo recent observations highlighting a role of dopamine and D1‐receptor signaling in the attenuation of the NLRP3 inflammasome in immune cells, and demonstrating that this pathway protects against the DA neurodegeneration induced by the mitochondrial neurotoxin MPTP (Yan et al., 2015). These observations highlight the potential relevance of the NLRP3 inflammasome to PD pathogenesis. Together with the acknowledged role of this complex in AD (Heneka et al., 2013) or ALS (Amyotrophic lateral sclerosis; Johann et al., 2015), these findings support a more general role, extending beyond inflammatory disorders in neurodegenerative diseases. Remarkably, overactivation of the NLRP3 inflammasome was recently identified as the cause of the greater susceptibility to polymicrobial sepsis‐induced organ failure and death in Pink1−/− and Park2−/− mice, in which it has also been linked to lower DA levels (Kang et al., 2016).
Recent studies have demonstrated a role for Parkin‐dependent mitophagy in limiting NLRP3 activation in peripheral macrophages and primary fibroblasts (Sumpter et al., 2016; Zhong et al., 2016). Our results are consistent with such a role and indicate that this mechanism is also relevant to microglial cells. Impaired PINK1/Parkin‐dependent MQC therefore probably contributes to the exacerbation of NLRP3 inflammasome activity in cells from Pink1−/−/Park2−/− mice and PARK2‐deficient patients.
However, our study highlights another mechanism underlying NLRP3 inflammasome deregulation in Parkin‐deficient cells. This mechanism involves the well‐known negative regulator of the NF‐κB pathway, A20 (Beyaert et al., 2000), the loss of which was recently shown to lead to NLRP3 inflammasome overactivation in vitro and in vivo, in a model of chronic autoinflammatory rheumatoid arthritis (Duong et al., 2015; Walle et al., 2014). We show here that A20 mRNA and protein levels are very low in immune cells from Park2−/− mice and PARK2 patients treated with NLRP3 agonists. In human macrophages challenged with NLRP3 activators, A20 abundance was inversely correlated with the amount of IL‐1β released, linking A20 deregulation to the exacerbation of NLRP3 inflammasome activation in PD associated with PARK2 dysfunction.
Future studies should investigate the mechanisms underlying this deregulation. A20 expression in immune cells is regulated by two transcription factors with antagonistic functions: the transcriptional activator upstream transcription factor 1 (USF1), and the repressor downstream regulatory element antagonist modulator (DREAM; Amir‐Zilberstein & Dikstein, 2008; Tiruppathi et al., 2014). Analysis of the impact of Parkin deficiency on the abundance of these transcription factors and their nuclear translocation kinetics will pave the way for explorations of their potential regulation by Parkin. A20 induction following treatment with NLRP3 activators was not deregulated in Pink1−/− cells, excluding the possibility of A20 regulation by PINK1‐driven phosphorylation events, and raising the question of the mechanisms underlying Parkin activation in this context. The phosphorylation, by PINK1, of ubiquitin and the N‐terminal ubiquitin‐like domain of Parkin at the common serine 65 residue, has been recognized as a central step in the activation of the E3 ubiquitin ligase activity of Parkin. Interestingly, an alternative route of Parkin activation by ubiquitin phosphorylated on serine 57 has recently been proposed supporting the existence of PINK1‐independent mechanisms associated with multiple ubiquitin phosphorylation sites (George et al., 2017). This is consistent with other observations highlighting PINK1‐independent functions of Parkin (Johnson, Charan, & LaVoie, 2012): (1) Parkin can rescue phenotypes linked to loss‐of‐function mutations in pink1 in Drosophila models (Dodson & Guo, 2007); (2) Parkin inactivates Bax, regulating cytochrome c release and stress‐induced apoptosis independently of PINK1 (Berger et al., 2009; Johnson, Berger, Cortese, & Lavoie, 2012); and (3) Parkin targets phagosomes containing mycobacteria for ubiquitin‐mediated autophagic removal, but there is no evidence for the presence of PINK1 on such phagosomes (Manzanillo et al., 2013).
Overall, our results place Parkin at the crossroads of innate immune pathways relating to the three major families of pattern recognition receptors. The roles of Parkin in Toll‐like receptors (TLR)/NF‐κB (de Leseleuc et al., 2013; Frank‐Cannon et al., 2008; Tran et al., 2011) and RIG‐I‐like receptors (RLR)/MAVS signaling have already been reported (Khan et al., 2016), but we highlight here the role of this protein in the NLR inflammasome pathway. Our study also raises questions about the more general role of the A20 regulatory pathway and its role in modulating the strength of NLRP3 inflammasome activity in patients with sporadic PD. Analyses of these pathways may help to identify subgroups of PD patients with an inflammasome overactivation profile, an essential step towards the development of targeted treatments. One recent study reported a decrease in A20 mRNA levels in the blood of PD patients (Perga et al., 2017) similar to that reported in chronic inflammatory diseases (Gilli et al., 2011), supporting the potential use of A20 as a biomarker. During the last decade, the NLRP3 inflammasome has emerged as a promising pharmacological target for the prevention and treatment of a number of immune‐related diseases, including cancer, type II diabetes, rheumatoid arthritis and AD (Çimen et al., 2016; Coll et al., 2015; Lamkanfi & Dixit, 2012; White, Lawrence, Brough, & Rivers‐Auty, 2017). Strategies for controlling inflammasome activity are already being considered in clinical trials for some of these disorders, and might, if successful, be extended to subgroups of PD patients presenting aberrant NLRP3 signaling.
COMPETING INTERESTS
The authors declare that they have nothing to report.
AUTHOR CONTRIBUTIONS
F. M. L and O. C. designed the study and wrote the paper. F. M. L, T. R., J. S., A. I., S. M. H., and E. I. performed the experiments and analyzed the data. Specifically, F. M. L., T. R., J. S., S. M. H., and A. I. performed the cellular experiments. F. M. L., T. R., and E. I. carried out biochemistry studies. T. R. and A. I. performed RT‐PCR experiments. P. P. M. designed primary culture experiment. G. M. and J. C. C. included and managed patients. J. C. C. and A. B. corrected the paper. All coauthors have reviewed and approve the contents of the manuscript and agree on the order in which their names will be listed in the manuscript.
Supporting information
Additional Supporting Information may be found online in the supporting information tab for this article.
Supporting Information Figure 1
Supporting Information Figure 2
Supporting Information Figure 3
Supporting Information Figure 4
Supporting Information
ACKNOWLEDGMENT
This work received support from the Innovative Medicines Initiative Joint Undertaking under grant agreement n°115568, resources of which are composed of financial contribution from the European Union's Seventh Framework Programme (FP7/2007–2013) and EFPIA companies' in kind contribution. We thank Martin Hofmann‐Apitius, coordinator of the IMI AETIONOMY project. This work was also supported by Institut National de la Santé et de la Recherche Médicale (INSERM), Fondation Institut du Cerveau et de la Moelle Epinière and Agence Nationale pour la Recherche (‘Investissements d'avenir’, grant ANR‐10‐IAIHU‐06), Fondation de France (Grant ID Engt 2016 00066513 ‐ Crosstalk between mitochondrial dysfunction and inflammasome activation) and the Michael J. Fox Foundation (Target Validation Awards Spring 2016 Program, Grant ID 12095, Role of the PINK1/PARKIN pathway in innate immunity). The PINK1 mouse mutant line was established at the Mouse Clinical Institute (Institut Clinique de la Souris, MCI/ICS) in the Genetic Engineering and Model Validation Department with funds from the GIS‐Institut de maladies rares to A. Brice. We thank Sabah Hamadat, David Akbar, Jennifer Fransson, Jeremy Rocca, Valérie Drouet, Erwan Moua, Doriane Foret, Yannick Martinez, Carole Dongmo, Vanessa Brochard, Florence Cormier Maxime Jacoupy, Zoï Erpapazoglou, Fiona Bonello, Emeline Hamon, Fabrice Danjou and Florence Deknuydt for their technical help.
Mouton‐Liger F, Rosazza T, Sepulveda‐Diaz J, et al. Parkin deficiency modulates NLRP3 inflammasome activation by attenuating an A20‐dependent negative feedback loop. Glia. 2018;66:1736–1751. 10.1002/glia.23337
Funding information Grant Sponsor: Innovative Medicines Initiative Joint Undertaking under grant agreement, Grant/Award Number: 115568; Grant Sponsor: European Union's Seventh Framework Program, Grant/Award Number: FP7/2007‐2013; Grant Sponsor: EFPIA company; Institut National de la Santé et de la Recherche Médicale (INSERM); Grant Sponsor: Fondation Institut du Cerveau et de la Moelle Epinière; Grant Sponsor: Agence Nationale pour la Recherche “Investissements d'avenir”, Grant/Award Number: ANR‐10‐IAIHU‐06; Grant Sponsor: Fondation de France, Grant/Award Number: ID Engt 2016 00066513; Grant Sponsor: Michael J. Fox Foundation (Target Validation Awards Spring 2016 Program), Grant/Award Number: ID 12095; Grant Sponsor: GIS‐Institut de maladies rares
REFERENCES
- Amir‐Zilberstein, L. , & Dikstein, R. (2008). Interplay between E‐box and NF‐kappaB in regulation of A20 gene by DRB sensitivity‐inducing factor (DSIF). Journal of Biological Chemistry, 283(3), 1317–1323. 10.1074/jbc.M706767200 [DOI] [PubMed] [Google Scholar]
- Berger, A. K. , Cortese, G. P. , Amodeo, K. D. , Weihofen, A. , Letai, A. , & LaVoie, M. J. (2009). Parkin selectively alters the intrinsic threshold for mitochondrial cytochrome c release. Human Molecuar Genetics, 18(22), 4317–4328. 10.1093/hmg/ddp384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyaert, R. , Heyninck, K. , & Van Huffel, S. (2000). A20 and A20‐binding proteins as cellular inhibitors of nuclear factor‐kappa B‐dependent gene expression and apoptosis. Biochemical Pharmacology, 60(8), 1143–1151. [DOI] [PubMed] [Google Scholar]
- Chopra, R. , Kalaiarasan, P. , Ali, S. , Srivastava, A. K. , Aggarwal, S. , Garg, V. K. , … Bamezai, R. N. K. (2014). PARK2 and proinflammatory/anti‐inflammatory cytokine gene interactions contribute to the susceptibility to leprosy: A case‐control study of North Indian population. BMJ Open, 4(2), e004239 10.1136/bmjopen-2013-004239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Çimen, I. , Kocatürk, B. , Koyuncu, S. , Tufanlı, Ö. , Onat, U. I. , Yıldırım, A. D. , … Erbay, E. (2016). Prevention of atherosclerosis by bioactive palmitoleate through suppression of organelle stress and inflammasome activation. Science Translational Medicine, 8(358), 358ra126 10.1126/scitranslmed.aaf9087 [DOI] [PubMed] [Google Scholar]
- Codolo, G. , Plotegher, N. , Pozzobon, T. , Brucale, M. , Tessari, I. , Bubacco, L. , & de Bernard, M. (2013). Triggering of inflammasome by aggregated alpha‐synuclein, an inflammatory response in synucleinopathies. PLoS One, 8(1), e55375 10.1371/journal.pone.0055375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coll, R. C. , Robertson, A. A. B. , Chae, J. J. , Higgins, S. C. , Muñoz‐Planillo, R. , Inserra, M. C. , … O'Neill, L. A. J. (2015). A small‐molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nature Medicine, 21(3), 248–255. 10.1038/nm.3806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen, S. P. , Kearney, C. J. , Clancy, D. M. , & Martin, S. J. (2015). Diverse activators of the NLRP3 inflammasome promote IL‐1beta secretion by triggering necrosis. Cell Reports, 11(10), 1535–1548. 10.1016/j.celrep.2015.05.003 [DOI] [PubMed] [Google Scholar]
- Damiano, M. , Gautier, C. A. , Bulteau, A.‐L. , Ferrando‐Miguel, R. , Gouarne, C. , Paoli, M. G. , … Lombès, A. (2014). Tissue‐ and cell‐specific mitochondrial defect in Parkin‐deficient mice. PLoS One, 9(6), e99898 10.1371/journal.pone.0099898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Léséleuc, L. , Orlova, M. , Cobat, A. , Girard, M. , Huong, N. T. , Ba, N. N. , … Schurr, E. (2013). PARK2 mediates interleukin 6 and monocyte chemoattractant protein 1 production by human macrophages. PLoS Neglected Tropical Diseases, 7(1), e2015 10.1371/journal.pntd.0002015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson, M. W. , & Guo, M. (2007). Pink1, Parkin, DJ‐1 and mitochondrial dysfunction in Parkinson's disease. Current Opinion in Neurobiology, 17(3), 331–337. 10.1016/j.conb.2007.04.010 [DOI] [PubMed] [Google Scholar]
- Duong, B. H. , Onizawa, M. , Oses‐Prieto, J. A. , Advincula, R. , Burlingame, A. , Malynn, B. A. , & Ma, A. (2015). A20 restricts ubiquitination of pro‐interleukin‐1beta protein complexes and suppresses NLRP3 inflammasome activity. Immunity, 42(1), 55–67. 10.1016/j.immuni.2014.12.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupe, V. , Davenne, M. , Brocard, J. , Dolle, P. , Mark, M. , Dierich, A. , … Rijli, F. M. (1997). In vivo functional analysis of the Hoxa‐1 3' retinoic acid response element (3'RARE). Development, 124(2), 399–410. [DOI] [PubMed] [Google Scholar]
- Enesa, K. , Moll, H. P. , Luong, L. , Ferran, C. , & Evans, P. C. (2015). A20 suppresses vascular inflammation by recruiting proinflammatory signaling molecules to intracellular aggresomes. Faseb J, 29(5), 1869–1878. 10.1096/fj.14-258533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fetz, V. , Knauer, S. K. , Bier, C. , von Kries, J. P. , & Stauber, R. H. (2009). Translocation biosensors – Cellular system integrators to dissect CRM1‐dependent nuclear export by chemicogenomics. Sensors (Basel), 9(7), 5423–5445. 10.3390/s90705423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank‐Cannon, T. C. , Tran, T. , Ruhn, K. A. , Martinez, T. N. , Hong, J. , Marvin, M. , … Tansey, M. G. (2008). Parkin deficiency increases vulnerability to inflammation‐related nigral degeneration. Journal of Neuroscience, 28(43), 10825–10834. 10.1523/JNEUROSCI.3001-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- George, S. , Wang, S. M. , Bi, Y. , Treidlinger, M. , Barber, K. R. , Shaw, G. S. , & O'Donoghue, P. (2017). Ubiquitin phosphorylated at Ser57 hyper‐activates parkin. Biochimica et Biophysica Acta, 10.1016/j.bbagen.2017.06.023 [DOI] [PubMed] [Google Scholar]
- Gilli, F. , Navone, N. D. , Perga, S. , Marnetto, F. , Caldano, M. , Capobianco, M. , … Bertolotto, A. (2011). Loss of braking signals during inflammation: A factor affecting the development and disease course of multiple sclerosis. Archives of Neurology, 68(7), 879–888. 10.1001/archneurol.2011.32 [DOI] [PubMed] [Google Scholar]
- Greene, J. C. , Whitworth, A. J. , Andrews, L. A. , Parker, T. J. , & Pallanck, L. J. (2005). Genetic and genomic studies of Drosophila parkin mutants implicate oxidative stress and innate immune responses in pathogenesis. Human Molecular Genetics, 14(6), 799–811. 10.1093/hmg/ddi074 [DOI] [PubMed] [Google Scholar]
- Gross, O. , Thomas, C. J. , Guarda, G. , & Tschopp, J. (2011). The inflammasome: An integrated view. Immunological Reviews, 243(1), 136–151. 10.1111/j.1600-065X.2011.01046.x [DOI] [PubMed] [Google Scholar]
- Guarda, G. , Dostert, C. , Staehli, F. , Cabalzar, K. , Castillo, R. , Tardivel, A. , … Tschopp, J. (2009). T cells dampen innate immune responses through inhibition of NLRP1 and NLRP3 inflammasomes. Nature, 460(7252), 269–273. 10.1038/nature08100 [DOI] [PubMed] [Google Scholar]
- Gurung, P. , Li, B. , Subbarao Malireddi, R. K. , Lamkanfi, M. , Geiger, T. L. , & Kanneganti, T. D. (2015). Chronic TLR stimulation controls NLRP3 inflammasome activation through IL‐10 mediated regulation of NLRP3 expression and caspase‐8 activation. Scientific Reports, 5(1), 14488 10.1038/srep14488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliday, G. M. , & Stevens, C. H. (2011). Glia: Initiators and progressors of pathology in Parkinson's disease. Movement Disorders, 26(1), 6–17. 10.1002/mds.23455 [DOI] [PubMed] [Google Scholar]
- Heneka, M. T. , Kummer, M. P. , Stutz, A. , Delekate, A. , Schwartz, S. , Vieira‐Saecker, A. , … Golenbock, D. T. (2013). NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature, 493(7434), 674–678. 10.1038/nature11729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henn, I. H. , Bouman, L. , Schlehe, J. S. , Schlierf, A. , Schramm, J. E. , Wegener, E. , … Winklhofer, K. F. (2007). Parkin mediates neuroprotection through activation of IkappaB kinase/nuclear factor‐kappaB signaling. Journal of Neuroscience, 27(8), 1868–1878. 10.1523/JNEUROSCI.5537-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch, E. C. , & Hunot, S. (2009). Neuroinflammation in Parkinson's disease: A target for neuroprotection? The Lancet. Neurology, 8(4), 382–397. 10.1016/S1474-4422(09)70062-6 [DOI] [PubMed] [Google Scholar]
- Horvath, R. J. , Nutile‐McMenemy, N. , Alkaitis, M. S. , & Deleo, J. A. (2008). Differential migration, LPS‐induced cytokine, chemokine, and NO expression in immortalized BV‐2 and HAPI cell lines and primary microglial cultures. Journal of Neurochemistry, 107(2), 557–569. 10.1111/j.1471-4159.2008.05633.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itier, J. M. , Ibanez, P. , Mena, M. A. , Abbas, N. , Cohen‐Salmon, C. , Bohme, G. A. , … Garcia de Yebenes, J. (2003). Parkin gene inactivation alters behaviour and dopamine neurotransmission in the mouse. Human Molecular Genetics, 12(18), 2277–2291. 10.1093/hmg/ddg239 [DOI] [PubMed] [Google Scholar]
- Johann, S. , Heitzer, M. , Kanagaratnam, M. , Goswami, A. , Rizo, T. , Weis, J. , … Beyer, C. (2015). NLRP3 inflammasome is expressed by astrocytes in the SOD1 mouse model of ALS and in human sporadic ALS patients. Glia, 63(12), 2260–2273. 10.1002/glia.22891 [DOI] [PubMed] [Google Scholar]
- Johnson, B. N. , Berger, A. K. , Cortese, G. P. , & Lavoie, M. J. (2012). The ubiquitin E3 ligase parkin regulates the proapoptotic function of Bax. Proceedings of the National Academy of Sciences of the United States of America, 109(16), 6283–6288. 10.1073/pnas.1113248109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, B. N. , Charan, R. A. , & LaVoie, M. J. (2012). Recognizing the cooperative and independent mitochondrial functions of Parkin and PINK1. Cell Cycle, 11(15), 2775–2776. 10.4161/cc.21261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, R. , Zeng, L. , Xie, Y. , Yan, Z. , Zhou, B. , Cao, L. , … Tang, D. (2016). A novel PINK1‐ and PARK2‐dependent protective neuroimmune pathway in lethal sepsis. Autophagy, 12(12), 2374–2385. 10.1080/15548627.2016.1239678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, M. , Syed, G. H. , Kim, S. J. , & Siddiqui, A. (2016). Hepatitis B virus‐induced Parkin‐dependent recruitment of linear ubiquitin assembly complex (LUBAC) to mitochondria and attenuation of innate immunity. PLoS Pathogens, 12(6), e1005693 10.1371/journal.ppat.1005693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamkanfi, M. , & Dixit, V. M. (2012). Inflammasomes and their roles in health and disease. Annual Review of Cell and Developmental Biology, 28, 137–161. 10.1146/annurev-cellbio-101011-155745 [DOI] [PubMed] [Google Scholar]
- Latz, E. , Xiao, T. S. , & Stutz, A. (2013). Activation and regulation of the inflammasomes. Nature Reviews. Immunology, 13(6), 397–411. 10.1038/nri3452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarou, M. (2015). Keeping the immune system in check: A role for mitophagy. Immunology and Cell Biology, 93(1), 3–10. 10.1038/icb.2014.75 [DOI] [PubMed] [Google Scholar]
- Lee, H. M. , Kang, J. , Lee, S. J. , & Jo, E. K. (2013). Microglial activation of the NLRP3 inflammasome by the priming signals derived from macrophages infected with mycobacteria. Glia, 61(3), 441–452. 10.1002/glia.22448 [DOI] [PubMed] [Google Scholar]
- Lu, B. , Nakamura, T. , Inouye, K. , Li, J. , Tang, Y. , Lundbäck, P. , … Tracey, K. J. (2012). Novel role of PKR in inflammasome activation and HMGB1 release. Nature, 488(7413), 670–674. 10.1038/nature11290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzanillo, P. S. , Ayres, J. S. , Watson, R. O. , Collins, A. C. , Souza, G. , Rae, C. S. , … Cox, J. S. (2013). The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature, 501(7468), 512–516. 10.1038/nature12566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon, F. , Burns, K. , & Tschopp, J. (2002). The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL‐beta. Molecular Cell, 10(2), 417–426. [DOI] [PubMed] [Google Scholar]
- Matheoud, D. , Sugiura, A. , Bellemare‐Pelletier, A. , Laplante, A. , Rondeau, C. , Chemali, M. , … Desjardins, M. (2016). Parkinson's disease‐related proteins PINK1 and Parkin repress mitochondrial antigen presentation. Cell, 166(2), 314–327. 10.1016/j.cell.2016.05.039 [DOI] [PubMed] [Google Scholar]
- McLelland, G. L. , Soubannier, V. , Chen, C. X. , McBride, H. M. , & Fon, E. A. (2014). Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. Embo J, 33(4), 282–295. 10.1002/embj.201385902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mira, M. T. , Alcaïs, A. , Nguyen, V. T. , Moraes, M. O. , Di Flumeri, C. , Vu, H. T. , … Schurr, E. (2004). Susceptibility to leprosy is associated with PARK2 and PACRG. Nature, 427(6975), 636–640. 10.1038/nature02326 [DOI] [PubMed] [Google Scholar]
- Mishra, B. B. , Rathinam, V. A. , Martens, G. W. , Martinot, A. J. , Kornfeld, H. , Fitzgerald, K. A. , & Sassetti, C. M. (2013). Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome‐dependent processing of IL‐1beta. Nature Immunology, 14(1), 52–60. 10.1038/ni.2474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizee, M. R. , Miedema, S. S. , van der Poel, M. , Adelia, Schuurman, K. G. , van Strien, M. E. , … Huitinga, I. (2017). Isolation of primary microglia from the human post‐mortem brain: Effects of ante‐ and post‐mortem variables. Acta Neuropathologica Communications, 5(1), 16 10.1186/s40478-017-0418-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouton‐Liger, F. , Jacoupy, M. , Corvol, J. C. , & Corti, O. (2017). PINK1/Parkin‐dependent mitochondrial surveillance: From pleiotropy to Parkinson's disease. Frontiers in Molecular Neuroscience, 10, 120 10.3389/fnmol.2017.00120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz‐Planillo, R. , Kuffa, P. , Martinez‐Colon, G. , Smith, B. L. , Rajendiran, T. M. , & Nunez, G. (2013). K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity, 38(6), 1142–1153. 10.1016/j.immuni.2013.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra, D. , Tanaka, A. , Suen, D. F. , & Youle, R. J. (2008). Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. The Journal of Cell Biology, 183(5), 795–803. 10.1083/jcb.200809125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor, W., Jr. , Harton, J. A. , Zhu, X. , Linhoff, M. W. , & Ting, J. P. (2003). Cutting edge: CIAS1/cryopyrin/PYPAF1/NALP3/CATERPILLER 1.1 is an inducible inflammatory mediator with NF‐kappa B suppressive properties. Journal of Immunology, 171(12), 6329–6333. [DOI] [PubMed] [Google Scholar]
- Park, S. , Won, J. H. , Hwang, I. , Hong, S. , Lee, H. K. , & Yu, J. W. (2015). Defective mitochondrial fission augments NLRP3 inflammasome activation. Scientific Reports, 5(1), 15489 10.1038/srep15489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perga, S. , Martire, S. , Montarolo, F. , Navone, N. D. , Calvo, A. , Fuda, G. , … Bertolotto, A. (2017). A20 in multiple sclerosis and Parkinson's disease: Clue to a common dysregulation of anti‐inflammatory pathways? Neurotoxicity Research, 32(1), 1–7. 10.1007/s12640-017-9724-y [DOI] [PubMed] [Google Scholar]
- Pickrell, A. M. , & Youle, R. J. (2015). The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease. Neuron, 85(2), 257–273. 10.1016/j.neuron.2014.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez, C. I. , Buchholz, F. , Galloway, J. , Sequerra, R. , Kasper, J. , Ayala, R. , … Dymecki, S. M. (2000). High‐efficiency deleter mice show that FLPe is an alternative to Cre‐loxP. Nature Genetics, 25(2), 139–140. 10.1038/75973 [DOI] [PubMed] [Google Scholar]
- Rossol, M. , Pierer, M. , Raulien, N. , Quandt, D. , Meusch, U. , Rothe, K. , … Wagner, U. (2012). Extracellular Ca2+ is a danger signal activating the NLRP3 inflammasome through G protein‐coupled calcium sensing receptors. Nature Communications, 3(1), 1329 10.1038/ncomms2339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar, S. , Malovic, E. , Harishchandra, D. S. , Ghaisas, S. , Panicker, N. , Charli, A. , … Kanthasamy, A. G. (2017). Mitochondrial impairment in microglia amplifies NLRP3 inflammasome proinflammatory signaling in cell culture and animal models of Parkinson's disease. NPJ Parkinsons Disease, 3(1), 30 10.1038/s41531-017-0032-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayre, L. M. (1989). Biochemical mechanism of action of the dopaminergic neurotoxin 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP). Toxicology Letters, 48(2), 121–149. [DOI] [PubMed] [Google Scholar]
- Scaffidi, P. , Misteli, T. , & Bianchi, M. E. (2002). Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature, 418(6894), 191–195. 10.1038/nature00858 [DOI] [PubMed] [Google Scholar]
- Sepulveda‐Diaz, J. E. , Ouidja, M. O. , Socias, S. B. , Hamadat, S. , Guerreiro, S. , Raisman‐Vozari, R. , & Michel, P. P. (2016). A simplified approach for efficient isolation of functional microglial cells: Application for modeling neuroinflammatory responses in vitro. Glia, 64(11), 1912–1924. 10.1002/glia.23032 [DOI] [PubMed] [Google Scholar]
- Shimada, K. , Crother, T. R. , Karlin, J. , Dagvadorj, J. , Chiba, N. , Chen, S. , … Arditi, M. (2012). Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity, 36(3), 401–414. 10.1016/j.immuni.2012.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumpter, R. , Sirasanagandla, S. , Fernández, Á. , Wei, Y. , Dong, X. , Franco, L. , … Levine, B. (2016). Fanconi anemia proteins function in mitophagy and immunity. Cell, 165(4), 867–881. 10.1016/j.cell.2016.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiruppathi, C. , Soni, D. , Wang, D. E. , Xue, J. , Singh, V. , Thippegowda, P. B. , … Malik, A. B. (2014). The transcription factor DREAM represses the deubiquitinase A20 and mediates inflammation. Nature Immunology, 15(3), 239–247. 10.1038/ni.2823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres‐Odio, S. , Key, J. , Hoepken, H. E. , Canet‐Pons, J. , Valek, L. , Roller, B. , … Auburger, G. (2017). Progression of pathology in PINK1‐deficient mouse brain from splicing via ubiquitination, ER stress, and mitophagy changes to neuroinflammation. Journal of Neuroinflammation, 14(1), 154 10.1186/s12974-017-0928-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran, T. A. , Nguyen, A. D. , Chang, J. , Goldberg, M. S. , Lee, J. K. , & Tansey, M. G. (2011). Lipopolysaccharide and tumor necrosis factor regulate Parkin expression via nuclear factor‐kappa B. PLoS One, 6(8), e23660 10.1371/journal.pone.0023660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walle, L. V. , Van Opdenbosch, N. , Jacques, P. , Fossoul, A. , Verheugen, E. , Vogel, P. , … Lamkanfi, M. (2014). Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature, 512(7512), 69–73. 10.1038/nature13322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weischenfeldt, J. , & Porse, B. (2008). Bone marrow‐derived macrophages (BMM): Isolation and applications. CSH Protocol, 2008(12), pdb.prot5080 pdb prot5080. 10.1101/pdb.prot5080 [DOI] [PubMed] [Google Scholar]
- White, C. S. , Lawrence, C. B. , Brough, D. , & Rivers‐Auty, J. (2017). Inflammasomes as therapeutic targets for Alzheimer's disease. Brain Pathology, 27(2), 223–234. 10.1111/bpa.12478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue, J. , Schmidt, S. V. , Sander, J. , Draffehn, A. , Krebs, W. , Quester, I. , … Schultze, J. L. (2014). Transcriptome‐based network analysis reveals a spectrum model of human macrophage activation. Immunity, 40(2), 274–288. 10.1016/j.immuni.2014.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, Y. , Jiang, W. , Liu, L. , Wang, X. , Ding, C. , Tian, Z. , & Zhou, R. (2015). Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell, 160(1–2), 62–73. 10.1016/j.cell.2014.11.047 [DOI] [PubMed] [Google Scholar]
- Zhong, Z. , Umemura, A. , Sanchez‐Lopez, E. , Liang, S. , Shalapour, S. , Wong, J. , … Karin, M. (2016). NF‐kappaB restricts inflammasome activation via elimination of damaged mitochondria. Cell, 164(5), 896–910. 10.1016/j.cell.2015.12.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, R. , Yazdi, A. S. , Menu, P. , & Tschopp, J. (2011). A role for mitochondria in NLRP3 inflammasome activation. Nature, 469(7329), 221–225. 10.1038/nature09663 [DOI] [PubMed] [Google Scholar]
- Zhou, Y. , Lu, M. , Du, R. ‐H. , Qiao, C. , Jiang, C. ‐Y. , Zhang, K. ‐Z. , … Hu, G. (2016). MicroRNA‐7 targets Nod‐like receptor protein 3 inflammasome to modulate neuroinflammation in the pathogenesis of Parkinson's disease. Molecular Neurodegeneration, 11(1), 28 10.1186/s13024-016-0094-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found online in the supporting information tab for this article.
Supporting Information Figure 1
Supporting Information Figure 2
Supporting Information Figure 3
Supporting Information Figure 4
Supporting Information