

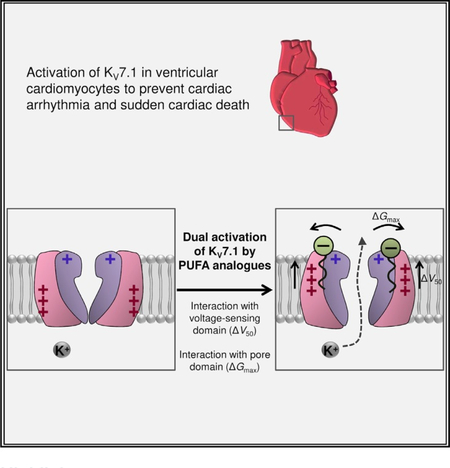
In Brief
Polyunsaturated fatty acid (PUFA) analogs are potentially anti-arrhythmic compounds, and understanding their functional mechanisms can aid in rational drug design. Liin et al. find that negatively charged PUFA analogs activate the cardiac potassium channel KV7.1 by dual independent mechanisms, demonstrating augmentation of channel activity through electrostatic interactions with both the pore and voltage-sensing domains of KV7.1.
Graphical Abstract
SUMMARY
Polyunsaturated fatty acid (PUFA) analogs represent a new class of potential anti-arrhythmic KV7.1 and KV7.1+KCNE1 channel activators. In this study, we describe dual independent activating effects of negatively charged PUFA analogs on KV7.1 and KV7.1+KCNE1 that are dependent on discrete channel motifs. PUFA analogs are critically dependent on K326 in S6 of KV7.1 to increase the maximum conductance and critically dependent on specific S4 arginines in KV7.1 to shift the voltage dependence of channel opening toward negative voltages. Our findings provide insights into how KV7.1+KCNE1 activators may interact electrostatically both with the pore domain and the voltage-sensing domain to augment channel activity. We believe that the molecular understanding of how PUFA analogs induce dual independent activating effects is an important step toward the development of effective anti-arrhythmic drugs that target KV7.1 channels.
INTRODUCTION
The slow delayed rectifier IKs channel generates one of the major repolarizing currents in the human heart (Nerbonne and Kass, 2005). Mutations in the genes encoding the IKs channel that attenuate channel function or trafficking cause long QT syndrome (Nerbonne and Kass, 2005), which increases the risk to suffer from ventricular fibrillation and sudden cardiac death (Nerbonne and Kass, 2005). Up to 30% of the patients with congenital long QT syndrome are not protected against potentially lethal arrhythmias by today’s main treatments (i.e., beta blockers and implantable defibrillator) (Goldenberg et al., 2006). An alternative treatment of long QT syndrome caused by reduced function of IKs would be to use compounds that augment IKs activity and thereby restore the ability of mutated IKs channels to generate repolarizing K+ currents, which in turn would restore a physiological QT interval. Unfortunately, there are currently no such compounds clinically available.
The IKs channel is formed by four KV7.1 alpha and one to four KCNE1 beta subunits (Murray et al., 2016; Nakajo et al., 2010; Plant et al., 2014) (Figure 1A). In this work, we will refer to the IKs channel as KV7.1+KCNE1. Transmembrane helices S1-S4 of KV7.1 form the voltage-sensing domains, which sense and respond to changes in the transmembrane voltage (Liin et al., 2015a). Positively charged arginines in S4 enable a stepwise outward movement of S4 in response to membrane depolarization (Zaydman et al., 2014). The movement of S4 leads to the opening of the ion-conducting pore domain, composed of segments S5 and S6 of all four KV7.1 subunits. KCNE1 has been proposed to be localized in the lipid-filled cleft between two voltage-sensing domains, close to S6 in the pore domain (Figure 1A) (Nakajo and Kubo, 2015). KCNE1 shifts the voltage dependence of opening to more positive voltages and slows the kinetics of channel opening (Barhanin et al., 1996; Sanguinetti et al., 1996). KCNE1 also alters the voltage sensor-to-gate coupling so that Kv7.1+KCNE1 channels only open when S4 is in the most activated state, whereas Kv7.1 channels open when S4 is in an intermediate activated state (Zaydman et al., 2014).
Figure 1. K326 Is Important for the N-AT Effect on Gmax.
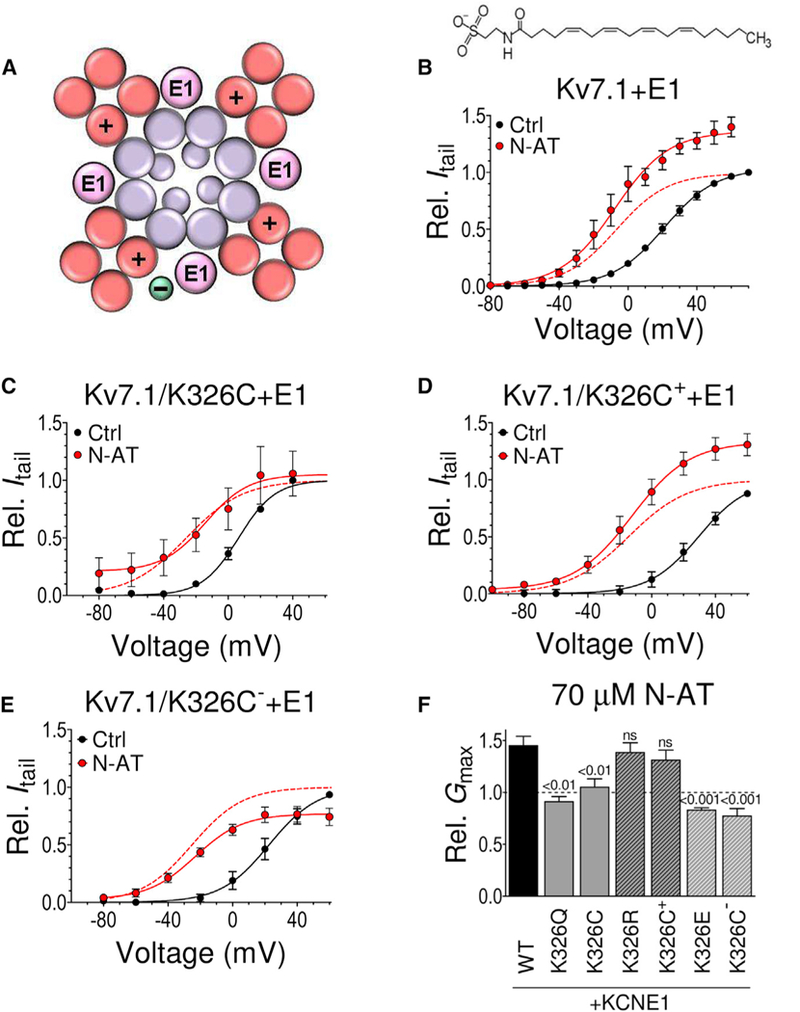
(A) Schematic top-down view of the Kv7.1+KCNE1 channel. S1-S4 helices (forming the voltage-sensing domains) in red, S5 and S6 helices and the central pore helices (forming the pore domain) in purple, and KCNE1 (E1) in pink. The positive sign denotes S4. The green circle denotes putative PUFA location.
(B) Effect of N-AT (70 μM, structure included) on WT Kv7.1+E1. n = 5. V50 (mV): control = +21.1 ± 1.1; N-AT = −7.5 ± 2.0. The dashed line is the N-AT curve normalized to 1 to facilitate comparison of V50.
(C) Retained effect of N-AT (70 μM) on V50 of Kv7.1/ K326C+E1, but lack of N-AT effect on Gmax. Same color coding as in (B). n = 5. V50 (mV): control = +6.4 ± 2.7; N-AT = −26.5 ± 3.0.
(D) Retained effect of N-AT (70 μM) on V50 and Gmax of MTSET+-modified Kv7.1/K326C+E1 (Kv7.1/K326C++E1). Same color coding as in (B). n = 4. V50 (mV): control = +29.4 ± 1.9; N-AT = − 14.5 ± 1.8.
(E) Retained effect of N-AT (70 μM) on V50 of MTSES−-modified Kv7.1/K326C+E1 (Kv7.1/K326C-+E1) but opposite N-AT effect on Gmax. Same color coding as in (B). n = 4. V50 (mV): control = +22.6 ± 2.0; N-AT = −25.1 ± 1.5.
(F) Summary of N-AT effect (70 μM) on Gmax of K326 mutants co-expressed with KCNE1. Statistics calculated using one-way ANOVA followed by Dunnett’s multiple-comparison test to compare the N-AT effect on each mutant to the N-AT effect on WT Kv7.1+E1. n = 6/5/6/5/4/4/4. Error bars denote SEM. For Kv7.1, we are unable to detect currents generated by K326 mutants and are therefore only able to study K326 mutants when co-expressed with KCNE1.
See also Figure S1.
We recently described that analogs of polyunsaturated fatty acids (PUFAs) are IKs channel activators (Liin et al., 2015b, 2016). For example, the PUFA analog N-arachidonoyl taurine (N-AT) (Figure 1B) enhances the activity of the wild-type and mutated Kv7.1+KCNE1 channels and prevents arrhythmic beating in embryonic rat cardiomyocytes (Liin et al., 2015b, 2016). N-AT and other negatively charged PUFA analogs activate KV7.1 and KV7.1+KCNE1 channels by shifting the voltage of half-maximal activation (V50) toward more negative voltages (Liin et al., 2015b). We have proposed that PUFA analogs incorporate their lipid tails into the cell membrane in-between neighboring voltage-sensing domains of KV7.1, close to KCNE1 (Figure 1A, green circle) (Liin et al., 2015b, 2016). From that position, the negatively charged fatty acid head group interacts electrostatically with the positively charged S4 to facilitate the outward S4 movement and shift V50 toward negative voltages. In support of an electrostatic interaction between PUFA analogs and S4, we previously showed that mutating the first S4 arginine R1 to a glutamine removed the N-AT effect on V50 in KV7.1 (Liin et al., 2015b). Moreover, the positively charged PUFA analog arachidonoyl amine (AA+) right-shifted V50 of KV7.1 and KV7.1+KCNE1 (Liin et al., 2015b), as if the positively charged amine head electrostatically repels the outward S4 movement. The shift in V50 suggests that negatively charged PUFA compounds, such as N-AT, could rescue the function of arrhythmia-causing KV7.1+KCNE1 mutants that generate less K+ current because of defective voltage dependence or kinetics (Liin et al., 2016).
In our initial experiments testing PUFA analogs on KV7.1 and KV7.1+KCNE1, we noted that negatively charged PUFA analogs, such as N-AT, appeared to increase the K+ conductance of KV7.1 and KV7.1+KCNE1 at positive voltages (Liin et al., 2016), which we will refer to as a Gmax effect. As increased K+ conductance at those voltages cannot be explained by the shift in V50, there might be two activating effects of PUFA analogs on KV7.1 and KV7.1+KCNE1 channels. In this study, we determine the molecular mechanism underlying the dual activating effects of PUFA analogs on Kv7.1 and Kv7.1+KCNE1 channels. We propose that PUFA analogs such as N-AT increase the activity of Kv7.1 and Kv7.1+KCNE1 via two independent mechanisms, depending on discrete channel motifs. The molecular understanding of the dual activating effects of PUFA analogs on Kv7.1 and Kv7.1+KCNE1 will enable rational drug development of compounds that utilize these mechanisms.
RESULTS
K326 Is Important for the N-AT Effect on Gmax
K326, at the top of S6, is an interesting candidate for a residue important for the N-AT effect on Gmax by being a charged residue in the pore domain facing the lipid bilayer (Sun and MacKinnon, 2017; Vanoye et al., 2009). Whereas 70 μM N-AT increases Gmax of wild-type (WT) KV7.1+KCNE1 by 36% (Figures 1B and S1A [see Figures S1B and S1C for WT KV7.1 alone]), the charge-neutralizing K326Q and K326C mutations abolish the effect of N-AT on Gmax (Figures 1C, 1F, and S1D). Modifying the Kv7.1/K326C+KCNE1 mutant with the positively charged thiol reagent MTSET+ before testing the effect of N-AT or using the charge-preserving K326R mutation largely restores the WT-like increase in Gmax induced by N-AT (Figures 1D, 1F, and S1E). In contrast, modifying Kv7.1/K326C+KCNE1 with the negatively charged thiol reagent MTSES− or using the charge-reversing K326E mutation results in a 23% and 17% (p < 0.05, one-sample t test) N-AT-induced reduction in Gmax (Figures 1E,1F, and S1F). These data suggest that K326 at the top of transmembrane helix S6 is important for the N-AT effect on Gmax. The charge at position 326 determines the direction (increase versus decrease) of the effect on the Gmax, as if the interaction between N-AT and Kv7.1+KCNE1 that changes Gmax is electrostatic in nature.
Independent PUFA Analog Effects on Gmax and V50
Although mutations of K326 alter the N-AT effect on Gmax, we note that these mutations do not affect the ability of N-AT to shift V50: 70 μM of N-AT shifts V50 of all K326 mutants by about −30 mV, similar to WT Kv7.1+KCNE1 (Figures 1C-1E and S1G). This suggests that two independent mechanisms underlie the N-AT-induced increase in Gmax and left shift of V50. We test this idea by quantifying the effect of N-AT on Gmax in mutants with altered N-AT effect on V50. Mutating the first S4 arginine R1 (R228, Figure 2A) to a glutamine (R1Q) removes the V50 effect of N-AT on the KV7.1 channel (Liin et al., 2015b), but retains a Gmax effect of N-AT (Figures 2B and S2A) comparable to WT Kv7.1 (Figures S1B and S1C). Mutating R1 to a glutamine (R1Q) did neither alter the V50 effect nor the Gmax effect of N-AT on Kv7.1+KCNE1 (Figures 2C and S2B). Mutating the second S4 arginine R2 (i.e., R231, Figure 2A) to a glutamine (R2Q) removed the V50 effect but retained the Gmax effect of N-AT on Kv7.1+KCNE1 (Figures 2D and S2C). Thus, although mutation of specific S4 arginines abolish the V50 effect of N-AT on KV7.1 and KV7.1+KCNE1 (Figure 2E), N-AT still increases Gmax of these mutants (Figure 2F). Note that we use the KV7.1/R2Q/Q3R+KCNE1 mutant to test the importance of the second S4 arginine for PUFA analog effects, because the R2Q mutation needs to be combined with the Q3R (i.e., Q234R) mutation to retain voltage sensitivity (Panaghie and Abbott, 2007; Wu et al., 2010). KV7.1/R2Q/Q3R without KCNE1 co-expression is voltage insensitive (Wu et al., 2010), and we are therefore unable to test the N-AT effect on this mutant. The Q3R mutation alone does not alter the N-AT effect compared to WT (Figures 2E and 2F), suggesting that the abolished V50 effect on the KV7.1/R2Q/Q3R+KCNE1 mutant is caused by the R2Q mutation. Altogether, there is no correlation between the effect of N-AT on Gmax and V50 (Figure 2G).
Figure 2. Independent PUFA Analog Effects on Gmax and V50.
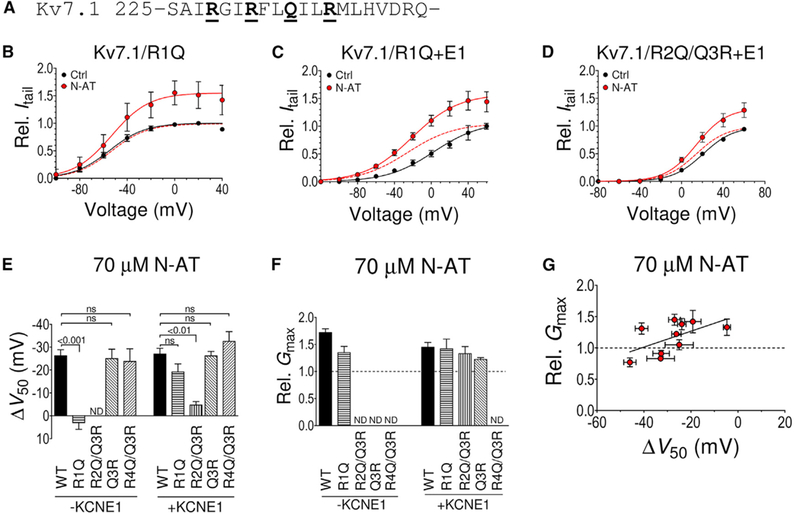
(A) Primary structure of human Kv7.1 transmembrane helix S4. The underlined bold residues denote S4 arginines R1-R4. Note that the residue at position R3 is uncharged in Kv7.1.
(B) Lack of effect of N-AT (70 μM) on V50 of Kv7.1/R1Q, but retained N-AT effect on Gmax. n = 6. V50 (mV): control = −55.1 ± 2.5; N-AT = −53.2 ± 3.9. The dashed line is the N-AT curve normalized to 1 to facilitate comparison of V50.
(C) Retained effect of N-AT (70 μM) on V50 and Gmax of Kv7.1/R1Q+E1. Same color coding as in (B). n = 6. V50 (mV): control = +2.7 ± 6.1; N-AT = −25.8 ± 5.0.
(D) Dramatically reduced effect of N-AT (70 μM) on V50 of Kv7.1/R2Q/Q3R+E1, but retained N-AT effect on Gmax. Same color coding as in (B). n = 7. V50 (mV): control = +20.6 ± 2.7; N-AT = +14.2 ± 2.5. Small error bars are covered by symbols.
(E) Summary of N-AT effect (70 μM) on V50 of S4 arginine mutants with and without KCNE1 co-expression. Note that an arginine at the Q3 position is not able to rescue the N-AT effect on V50 in Kv7.1/R2Q. The Q3R and R4Q mutations do not affect the ability of N-AT to shift V50 of Kv7.1 without or with KCNE1 co-expressed.
(F) Summary of N-AT effect (70 μM) on Gmax of S4 arginine mutants with and without KCNE1 co-expression. ND, not determined. Effects on Gmax of R4Q/Q3R could not reliably be determined as control and N-AT recordings were performed on different oocytes (Experimental Procedures). Effects on Gmax ofQ3R without KCNE1 could not be reliably determined because of small currents. Statistics in (E) and (F) calculated using one-way ANOVA followed by Tukey’s multiple-comparison test to compare all columns (there was no statistical difference forany mutant compared to WT in F).n = 4/6/5/4/6/9/5/5/4 in (E) and 4/6/6/9/7/5 in (F).
(G) Graph illustrating lack of correlation between N-AT effects on V50 and Gmax. Included data: WT+E1, S4 mutants (R1Q+E1, R2Q/Q3R+E1, and Q3R+E1), and 326 mutants (K326C+E1, K326C++E1, K326C−+E1, K326Q+E1, K326R+E1, and K326E+E1). n as in Figures 1F, 2E, and 2F. The solid line represents linear regression, for which the slope is not significantly different from 0 (p = 0.18). Data are represented as mean ± SEM.
See also Figure S1
The positively charged PUFA analog AA+ (structure in Figure S2D), which was previously shown to shift V50 of KV7.1 and KV7.1+KCNE1 to more positive voltages (Liin et al., 2015b), reduces Gmax of KV7.1 and KV7.1+KCNE1 by 50%−70% (Figures S2D-S2G). Although the ability of AA+ to shift V50 is abolished or reduced by specific S4 arginine mutations (Figure S2F), the AA+ effect on Gmax is not (Figure S2G).
Thus, even though S4 mutants alter the ability of N-AT or AA+ to shift V50, N-AT and AA+ retain the ability to alter Gmax of these mutants, as if two independent mechanisms underlie the dual PUFA analog effect. We are unable to determine the impact of charge modification at position 326 for AA+ effects, as application of AA+ to KV7.1/K326 mutants quickly causes massive cell leakage. Despite this, the opposite effect of negatively (N-AT) and positively (AA+) charged PUFA analogs suggest that both the effect on Gmax and V50 are, at least in part, electrostatic. Different S4 arginines appear to be critical for the V50 effect in Kv7.1 and Kv7.1+KCNE1: R1Q removes the V50 effect of N-AT or AA+ on KV7.1, whereas R2Q has largest impact on the V50 effect on KV7.1 co-expressed with KCNE1. A plausible reason for the different critical S4 arginines in Kv7.1 and Kv7.1+KCNE1 will be proposed in Discussion.
Similar Concentration and pH Dependence for Gmax and V50 Effects
50% of the maximum N-AT-induced shift in V50 is caused by 31 ± 7 and 40 ± 13 μM N-AT for Kv7.1 and KV7.1+KCNE1, respectively (Figures 3A and 3B). The corresponding dose response for the N-AT effect on Gmax overlaps fairly well with the dose response for N-AT effects on V50 (Figures 3A and 3B). We previously found that the effect of the PUFA docosahexaenoic acid (DHA) on V50 of KV7.1 and KV7.1+KCNE1 depends on the extracellular pH (Liin et al., 2015b), as if the protonation state of the DHA head group determines the strength of the interaction with S4. The DHA effect on Gmax shows similar pH dependence as the DHA effect on V50 (Figures 3C and 3D). Moreover, KCNE1 equally alters the apparent pKa of the DHA effect on Gmax and V50 (Figures 3C and 3D).
Figure 3. Similar Concentration and pH Dependence for Effects on Gmax and V50.
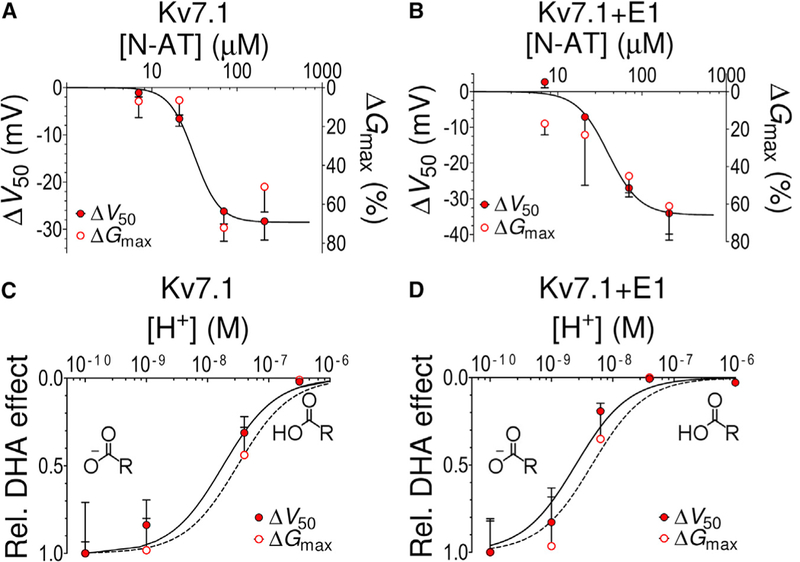
(A and B) Concentration-response curve for N-AT-induced shift of V50 (closed symbols) and increase in Gmax (open symbols) of (A) Kv7.1 and (B) Kv7.1+KCNE1. The continuous line is the best fit of ΔV50 data to Equation 2, with (A) c50 = 31.3 ± 6.8 μM, NH = 2.9, and n = 4 for each data point, and (B) c50 = 39.7 ± 13.2 μM, NH = 2.3, and n = 5 for each data point. c50 for the Gmax data could not be determined because of difficulties to reliably determine the Gmax effects at the lowest N-AT concentrations. The steep slope of the concentration-response curves (NH = 2.3-2.9) could partly be explained by cooperativity in the N-AT effect. Most of the steepness of the concentration-response curve is, however, most likely due to our inability to load more N-AT into the cell membrane at N-AT concentrations above 70 μM, either because of micelle formation by N-AT or because of electrostatic repulsion between membrane-incorporated N-AT molecules at high N-AT concentrations.
(C and D) Normalized pH dependence of DHA-induced (70 μM) shift of V50 (closed symbols) and increase in Gmax (open symbols) of (C) Kv7.1 and (D) Kv7.1+KCNE1. The continuous line is the best fit of ΔV50 data to Equation 2, with (C) c50 = 1.8 × 10−8 M (=pH 7.7, from Liin et al., 2015b) for ΔV50 and c50 = 3.1 × 10−8 M(=pH 7.5) for ΔGmax; NH = 1, and (D) c50 = 2.5 × 10−9 M(=pH 8.6, from Liin et al., 2015b) for ΔV50 and c50 = 4.4 × 10−9 M (=pH 8.4) for ΔGmax; NH = 1. The insets in (C) and (D) illustrate deprotonated and protonated DHA head groups. n = 3-13 for each data point. Error bars denote SEM.
See also Figure S3.
The similar pH dependence of DHA effects together with the similar apparent affinity of N-AT to induce a shift in V50 and increase Gmax is compatible with PUFA and PUFA analogs mediating the dual effects from a single binding site. Using the recent Kv7.1 structure (Sun and MacKinnon, 2017), one can envision a PUFA binding site in the lipid-filled cleft between two voltage-sensing domains, in which the PUFA molecule would be close enough to interact electrostatically with both arginines in S4 and K326 in the pore domain (Figure S3). However, we cannot rule out that there are two PUFA binding sites, one close to S4 and one close to K326, with similar affinities and pH dependences.
An Increase in the Open Probability Could Explain the Increase in Gmax
N-AT could increase Gmax by increasing the single-channel conductance or by increasing the open probability Po by stabilizing the open conductive conformation of the filter and reducing the flickering of Kv7.1 channels (Chakrapani et al., 2011; Gibor et al., 2007; Seebohm et al., 2003). To test whether the N-AT-induced increase in Gmax is due to changes in the single-channel conductance or the open probability Po, we measure the effect of N-AT on Gmax of the Kv7.1 mutants S209F and I204F, which have reported open probabilities Po of 0.4 and 0.04, respectively, compared to 0.2 for WT Kv7.1+KCNE1 (Eldstrom et al.,2015). If N-AT increases Gmax by increasing the single-channel conductance, we expect the same increase for these two mutants compared to WT Kv7.1+KCNE1, because they all have the same reported single-channel conductance (Eldstrom et al., 2015). However, if N-AT increases Gmax by stabilizing the open conducting filter and thereby increases Po, we expect smaller Gmax effects on the “high Po” Kv7.1/S209F+KCNE1 mutant than on WT Kv7.1+KCNE1, because the mutant already has an increased Po in control solution and there is therefore no room for much more increase in Po by PUFAs. In contrast, we expect larger Gmax effects on the “low Po” Kv7.1/I204F+KCNE1 mutant than on WT Kv7.1+KCNE1. Whereas 70 μM N-AT shifts V50 of Kv7.1/S209F+KCNE1, Kv7.1/I204F+KCNE1, and WT Kv7.1+KCNE1 all by about −30 mV, the effect of 70 μM N-AT on Gmax is clearly altered: The Gmax effect is smaller for Kv7.1/S209F+KCNE1 (Figure 4A: GmaxPUFA/Gmax = 1.2 ± 0.1, n = 7) and larger for Kv7.1/I204F+KCNE1 (Figure 4B: GmaxPUFA/Gmax =1.8 ± 0.2, n = 7) than for WT Kv7.1+KCNE1 (GmaxPUFA/ Gmax = 1.4). The effect of N-AT on Gmax of Kv7.1/S209F+KCNE1 and Kv7.1/I204F+KCNE1 matches well the predicted effect on these mutants with different baseline Po (Figure 4C).
Figure 4. An Increase in the Open Probability Could Explain the Increase in Gmax.
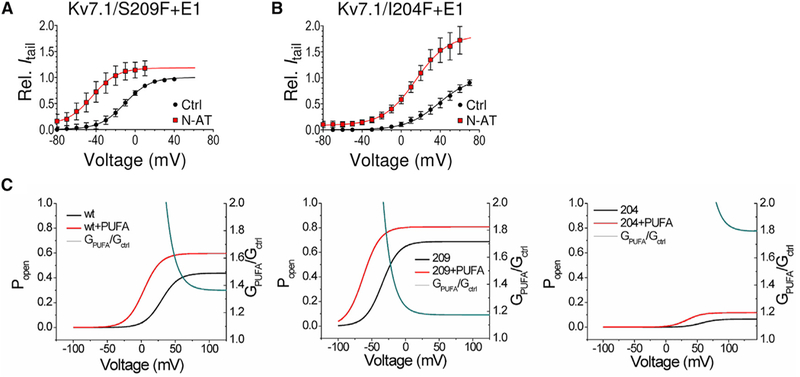
(A and B) Ability of N-AT (70 μM) to increase Gmax and shift V50 of KV7.1/S209F+KCNE1 (A, high open-probability mutant) and KV7.1/I204F+KCNE1 (B, low openprobability-mutant). The maximum conductance for N-AT is normalized to the maximum conductance in control solution for each cell. n = 5-7.
(A) V50 (mV): control = −10.6 ± 5.8; N-AT = −44.0 ± 5.7.
(B) V50 (mV): control = +38.4 ± 7.5; N-AT = +14.1 ± 3.9. Data are represented as mean ± SEM.
(C) Simulated effect of N-AT on WT KV7.1+KCNE1 (left), KV7.1/S209F+KCNE1 (middle, high open-probability mutant), and KV7.1/I204F+KCNE1 (right, low openprobability mutant). The black lines represent open probability (Popen) as a function of voltage in control and red in the presence of N-AT. The blue lines show the simulated effect of N-AT on the conductance at indicated voltages (GPUFA/GCtrl). A macroscopic three-state model (resting, activated, and open states) was used, with a voltage-sensing step between the resting and activated states and a gate-opening step between the activated and open states (see Experimental Procedures). The effect of N-AT was modeled as a shift of the voltage dependence of ΔV50 = −30 mV for the first step and an increase in the equilibrium constant by a factor of LPUFA = 1.9 for the second step. Due to the modal behavior of single KV7.1+KCNE1 channels, we had to calculate the predicted N-AT effect on each of the published single-channel traces for WT and the S209F mutation (Eldstrom et al., 2015) and then adjust the Popen in the macroscopic three-state model to Popen = 0.4 for WT and Popen = 0.66 for S209F mutant (and we call these effective Popen), to get the effective Popen in the macroscopic three-state model to display the expected Gmax increase as calculated from the single-channel data (see Experimental Procedures for details).
Molecular Modeling Reveals Structural Rearrangements at the Selectivity Filter by Pulling on K326
The experimental data are consistent with that N-AT electrostatically attracts K326 away from the pore and thereby increases Gmax by increasing Po. To get further insight into what underlies this increase in Gmax, we use molecular-dynamics simulations of the cryo-electron microscopy (EM) Kv7.1 model embedded in a lipid bilayer. We mimic the electrostatic N-AT effect on K326 by pulling each K326 residue away from the pore toward the lipid bilayer (Figure 5A; see Experimental Procedures for details). The pull force selected is based on a previous work on a similar system that evaluated which force that would allow gradual rearrangement of side-chain interactions rather than lead to deformation of the channel (Conti et al., 2016). In three control simulations without pull force applied, we find that K326 interacts primarily with the phospholipid POPC (1,2-palmitoyl-oleoyl-sn-glycero-3-phosphocholine), in the lipid-filled space between neighboring voltage-sensing domains, and D301, in the outer vestibule, through hydrogen bonds and salt bridges (Figures 5B and S4A-S4C). In three pull simulations of 300 ns each, the salt bridge between K326 and D301 breaks completely, as K326 interacts almost exclusively with POPC (Figures 5C, S4B, and S4C). During the last 150 ns ofthe pull simulations, K326 forms a hydrogen bond with POPC >30% of the time. The intimate interaction between K326 and the negatively charged POPC in the pull simulations supports the hypothesis that negatively charged N-AT may interact directly with K326.
Figure 5. Molecular Modeling Reveals Structural Rearrangements at the Selectivity Filter by Pulling on K326.
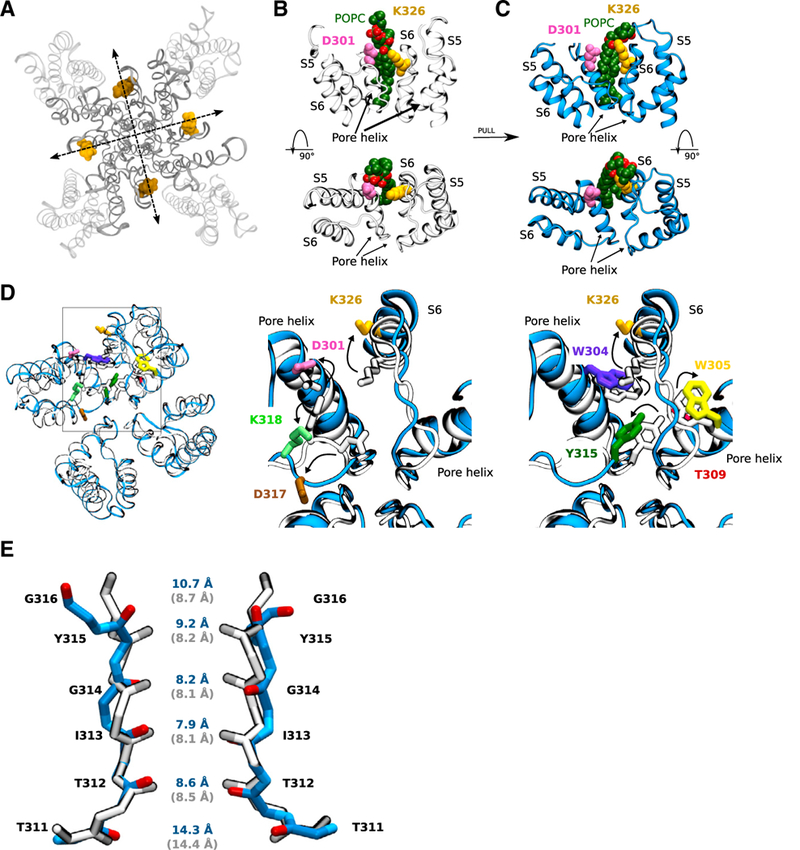
(A) Top view of the Kv7.1 channel depicting the pull direction for each pair of K326 residues located on opposite channel subunits.
(B) In the WT system (no pull force applied), K326 interacts with a nearby POPC lipid, shown in green, and D301 on the neighboring subunit through salt bridge formation.
(C) The effects of the pull force (the arrow indicates that the pull force has been applied in-between B and C) on K326 is visible through an increase in the number of hydrogen bonds formed with the nearby POPC lipid, and the consequent breaking of salt bridge with D301. The upper panel shows a side view, whereas the lower panel displays a top-down view.
(D) Structure of two neighboring subunits in the pull system (blue) is compared to the control system (gray) by superimposing the structures. Residues involved in causing key structural rearrangements in and behind the selectivity filter induced by pulling K326 away from the pore are displayed in various colors for the pull system.
(E) Comparison of the selectivity filter of the pull (blue) and the control (gray) structures. The average backbone carbon distances between each pair of residues located in the selectivity filter ofthe final 150 ns ofthe molecular dynamics (MD) simulations is displayed in blue for the pull systems and in gray for the control systems.
See also Figures S4–S7 and Video S1.
In the pull simulations, we observe structural rearrangement of key residues nearby to K326. We observe altered interactions within the network of charged residues D301, D317, and K318 behind the selectivity filter and aromatic residues W304, W305, and Y315 in and behind the selectivity filter. In the pull simulations, K318 in the outer vestibule interacts more frequently with D317 (especially through salt bridges, Figures S4D and S4E) at the expense ofthe interaction with D301 in the pore helix (Figures S4F and S4G). Simultaneously, Y315 in the selectivity filter interacts less frequently with W305 and T309 in the pore helix (Figures S5A-S5C), whereas W304 in the pore helix tends to move away from the selectivity filter (Figure S5D). Key structural rearrangements induced by pulling K326 away from the pore are summarized in Figure 5D and Video S1. Several ofthe identified amino acids have been previously described to be important for the stability of the selectivity filter and for K+ conductance in K+ channels. For instance, W304 and D317 correspond to W67 and D80 in KcsA, which are part ofthe so-called inactivation triad important for slow inactivation of KcsA (Cordero-Morales et al., 2011). D301 corresponds to R64 in KcsA, which also is critical for slow inactivation of KcsA (Poveda et al., 2014).
In our pull simulations, we observe a slight decrease in the electrostatic potential at the external and internal entrance of the selectivity filter, with a maximum decrease in the electrostatic potential of 33.8 ± 10.5 mV at the internal entrance (z = 12.5 Å; Figures S6A-S6C). We also measure the backbone carbon distance between each pair of residue located in the selectivity filter. The pairwise distances for each system across the three replicates show that the distance between opposing Y315 and G316 residues typically increases in the last 150 ns of the pull simulations, whereas the distance between opposing T311, T312, I313, and G314 remains largely unchanged (Figures 5E and S6D). As a consequence, the distance between backbone carbon atoms for Y315 and G316 increases from 8.19 ± 0.02 and 8.67 ± 0.03 Å, respectively, in the control simulations to 9.19 ± 0.05 and 10.70 ± 0.05 Å, respectively, by the end of the pull simulations. In addition, by comparing the density of K+ at coordination sites S1-S4 in our control system and pull system, we find that the density map is slightly shifted and more smeared in the pull simulations, as if the ions have a slightly different coordination pattern (Figure S7A).
N-AT Reduces the Relative Rubidium Permeability
To test whether the rearrangements in and near the selectivity filter observed in the molecular dynamics simulations alter the relative permeability of different ions, we assess the ratio of rubidium to potassium ion permeability in Kv7.1 and Kv7.1+KCNE1. As previously described, Kv7.1 has a high rubidium-to-potassium permeability ratio compared to Kv7.1+KCNE1 (Figures 6A-6C) (Pusch et al., 2000; Zaydman et al., 2014). We find that N-AT (35 μM) significantly decreases the rubidium-to-potassium permeability ratio in both Kv7.1 and Kv7.1+KCNE1 (Figures 6A-6C). These data are in agreement with the idea that N-AT induces structural rearrangements in and near the selectivity filter, promoting potassium over rubidium permeability.
Figure 6. N-AT Reduces the Relative Rubidium Permeability.
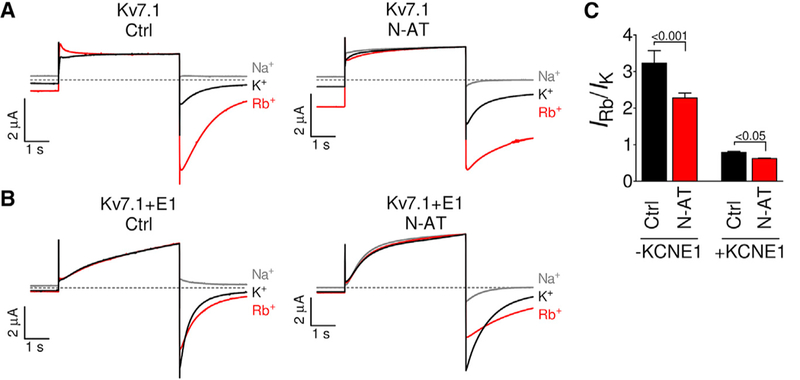
(A and B) Left panels: representative currents from Kv7.1 (A) and Kv7.1+KCNE1 (B) in external control solutions containing 100 mM Na+, K+, or Rb+.
(A and B) Right panels: representative currents from Kv7.1 (A) and Kv7.1+KCNE1 (B) in external N-AT-supplemented (35 μM) solutions containing 100 mM Na+, K+, or Rb+. Currents were normalized to the maximum outward currents.
(C) Summary of the rubidium-to-potassium permeability ratio in control solution and in the presence of N-AT. Statistics calculated using one-way ANOVA followed by Bonferroni’s multiple-comparison test. n = 6/6/6/9. Data are represented as mean ± SEM.
DISCUSSION
We here show that PUFA analogs, such as N-AT, have two independent effects on Kv7.1 and Kv7.1+KCNE1 that modulate the activity of these channels. One effect is on the pore domain and changes the maximum conductance, whereas the second effect is on the voltage-sensing domain and shifts the voltage dependence of opening. We show that the PUFA-induced effect on Gmax is due to an electrostatic interaction between the head group of the PUFA and the charged K326 residue in the pore domain, which induces structural rearrangements at the selectivity filter. We also show that the PUFA-induced shifts in V50 are primarily caused by electrostatic interactions between the head group of the PUFA analog and specific arginines in S4 (R1 in Kv7.1 and R2 in Kv7.1+KCNE1).
We propose the following model for the two independent effects of PUFA analogs on Kv7.1 and Kv7.1+KCNE1: A previous study suggested a gating model in which Kv7.1 opens from an intermediate S4 conformation, whereas Kv7.1+KCNE1 opens from a fully activated S4 conformation (Zaydman et al., 2014) (Figure 7). Our data could be explained using this model if we assume that the head group of the PUFA analog is close to R1 (R228) in the intermediate S4 conformation, but close to R2 (R231) in the fully activated S4 conformation (Figure 7). We therefore propose that the V50 effect of PUFA analogs in Kv7.1 is primarily caused by electrostatic interactions between the head group of the PUFA analog and R1 to shift the voltage dependence of the first horizontal transition in our model (Figure 7A). Because Kv7.1 opens after this first S4 transition at negative voltages, a PUFA analog-induced shift in this first S4 transition would also shift the voltage dependence of Kv7.1 opening (V50). However, any PUFA analog-induced shift in the second S4 transition at more positive voltages would not significantly affect the voltage dependence of Kv7.1 opening (V50), because Kv7.1 opens already after the first S4 transition at more negative voltages. This model would explain why neutralization of R1 abolishes the PUFA analog effect on V50 in Kv7.1.
Figure 7. Schematic Models of PUFA Analog Effects on Kv7.1 and Kv7.1+KCNE1.
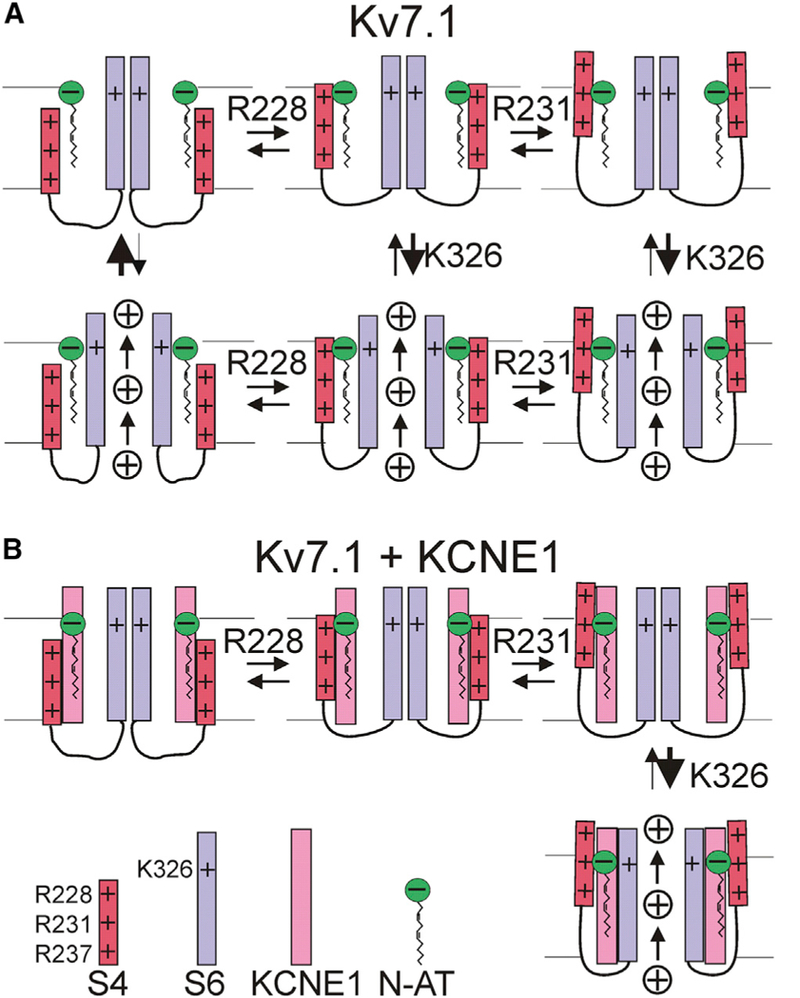
(A) Effects of negatively charged PUFA analog on S4 movement (horizontal transitions) and pore opening (vertical transition) in Kv7.1. The Kv7.1 model is based on a published model (Zaydman et al., 2014), which describes that S4 moves in two transitions from a resting position at negative voltage, to an intermediate position around 0 mV and to a fully activated position at positive voltages. The first S4 transition has a V50 around −30 mV and the second around +20 mV. In this model, Kv7.1 can open from the intermediate S4 conformation, and the second S4 transition does not significantly increase the open probability. The residue number near each transition denotes the proposed residue mainly responsible for the effects induced by charged PUFA analogs on that specific transition. In our model, the PUFA analog electrostatically interacts with R228 (R1) in S4 to promote an intermediate S4 conformation and R231 (R2) to promote a fully activated S4 conformation. Because Kv7.1 opens from the intermediate S4 position, the effect of PUFA on the first S4 transition will have the largest effect on the V50 of channel opening for Kv7.1. In addition, the PUFA analog electrostatically interacts with K326 in S6 to increase K+ conductance by increasing the open probability (and potentially increasing the K+ ion concentration or single-channel conductance).
(B) Same as in (A) but for Kv7.1+KCNE1. The Kv7.1+KCNE1 model is based on a published model (Zaydman et al., 2014), which describes that S4 in Kv7.1+KCNE1 also undergoes two S4 transitions (with the first S4 transition has a V50 around −80 mV and the second around +20 mV), but that Kv7.1+KCNE1 only can open from a fully activated S4 conformation (Barro-Soria et al., 2014). Because Kv7.1+KCNE1 opens only from the fully activated S4 position, the effect of PUFA on the second S4 transition will have the largest effect on the V50 of channel opening for Kv7.1+KCNE1. In addition, the PUFA analog electrostatically interacts with K326 in S6 to increase K+ conductance.
In contrast, in our model, the V50 effect on Kv7.1+KCNE1 is primarily caused by electrostatic interactions between the head group of the PUFA analog and R2 that shift the voltage dependence of the second horizontal transition (Figure 7B). Because Kv7.1+KCNE1 opens after this second S4 transition at positive voltages, a PUFA-induced shift in this second S4 transition would also shift the voltage dependence of Kv7.1+KCNE1 opening (V50). However, any PUFA-induced shift in the first S4 transition at more negative voltages would have less of an effect on the voltage dependence of Kv7.1+KCNE1 opening (V50), because Kv7.1+KCNE1 only opens after the second S4 transition at more positive voltages. This model would explain why neutralization of R2 abolishes the PUFA analog effect on V50 in Kv7.1+KCNE1, whereas neutralization of R1 only reduces the effect on Kv7.1+KCNE1. The inability of other S4 arginines to compensate for these neutralizations suggests that PUFA analogs shift the voltage dependence of channel opening by interacting specifically with certain S4 arginines during each step of voltage activation of these channels. A previous study by Ottos- son et al. (2014) found that R1 is not critical for the activating effect of naturally occurring PUFA on the Drosophila Shaker Kv channel. As the Shaker channel only opens from a fully activated S4 conformation (Bezanillaetal., 1994), similarto Kv7.1+KCNE1, our model raises the possibility that PUFA might interact with R2, rather than R1, also in Shaker.
In our model, the effect of PUFA analogs on Gmax is due to an electrostatic interaction between the head group of the PUFA and K326 that increases Gmax (Figure 7, vertical transitions). We anticipate that the interaction between the negatively charged head group and K326 is electrostatic in the form of a salt bridge, similar to the POPC-K326 interaction observed in the molecular dynamics simulation. However, we expect the interaction with a PUFA analog to be of higher affinity and longer lasting in comparison to a POPC lipid. Direct interactions between negatively charged phospholipid head groups and the corresponding lysine in KcsA (i.e., K89) have been proposed to promote inactivation of the selectivity filter by promoting formation of the W67-E71-D80 inactivation triad (Poveda et al., 2014). In Kv7.1, we instead propose that the interaction between the PUFA analog and K326 induces structural rearrangements in and near the selectivity filter that increase the open probability Po and thereby increase Gmax. We cannot rule out that the PUFA analogs also have some minor effects on the single-channel conductance. The ability of K+ channels to efficiently conduct ions depends on a number of features of the selectivity filter. Although the molecular details are not fully understood—especially not for channels with small single-channel conductance and fast-frequency flickering such as Kv7.1 (Cui, 2016)—the electrostatic potential near the selectivity filter (Carvacho et al., 2008) and the diameter (Gibor et al., 2007) and ion occupancy profile (Chakrapani et al., 2011; Demo and Yellen, 1992; Zhou and MacKinnon, 2003) of the selectivity filter have been proposed to be important for K+ conductance. We therefore focus on these features and propose three possible mechanisms for how N-AT increases Gmax: electrostatic potential, selectivity filter diameter, and ion occupancy profile.
As has been previously suggested for charged residues near the selectivity filter of the BK channel (Carvacho et al., 2008), pulling K326 away from the ion-permeation pathway may increase K+ conductance by making the electrostatic potential at the entrances of the selectivity filter more negative (see Figures S6A-S6C). Thus, a more negative electrostatic potential induced by pulling K326 away from the pore may increase Gmax by providing an electrostatic potential that increases the local K+ concentration and K+ occupancy at the external end of the selectivity filter. K+ occupancy at the external end of the selectivity filter has previously been shown to stabilize the open state of Kv channels by preventing C-type inactivation (Lopez-Barneo et al., 1993). However, Kv7.1+KCNE1 is not known to undergo C-type inactivation and is not sensitive to changes in extracellular K+ concentrations (Larsen et al., 2011).
Increased maximal distance between Y315 of opposing subunits together with flexibility in this upper part of the selectivity filter was previously proposed to promote K+ conductance in Kv7.1 by preventing slow inactivation (Gibor et al., 2007). Thus, pulling K326 away from the pore may increase Gmax by widening the upper part of the selectivity filter (see Figures 5E and S6D), which could stabilize the open state.
The selectivity filter of K+ channels typically alternates between open conductive and closed non-conductive conformations during fast flickering and slow inactivation. Although the detailed structural determinants of the conductive and non-conductive filter are not completely understood, it is clear that mutations or diverse ions that alter the relative ion occupancy at coordination sites S1-S4 may stabilize or destabilize the open conductive filter (Chakrapani etal., 2011; Demo and Yellen, 1992; Pusch et al., 2000; Seebohm et al., 2003; Zhou and MacKinnon, 2003). Thus, pulling K326 away from the pore may increase Gmax by inducing subtle alterations in the ion occupancy profile at coordination sites S1-S4 (see Figure S7A), which could stabilize the open state (Chakrapani et al., 2011; Seebohm et al., 2003).
Thus, a decreased electrostatic potential, a widening of the selectivity filter, and a higher mobility of potassium ions might all contribute to the higher Po in Kv7.1 by stabilizing the open state but also contribute to the effect on Gmax by mechanisms independent of Po (e.g., increasing the local K+ concentration and thereby increasing the single-channel conductance). N-AT-induced facilitation of K+ conductance is in accordance with our experiments that show that N-AT reduces the rubidium-to-potassium permeability ratio of both Kv7.1 and Kv7.1+KCNE1. However, N-AT increases the Gmax even in symmetrical high-K+ solutions (Figure S7B), suggesting that the changes in Gmax are not due to changes in K+/Na+ selectivity.
The Gmax and V50 effects by PUFA analogs on Kv7.1 and Kv7.1+KCNE1 channels have similar N-AT concentration dependence and pH dependence, as if a single PUFA binding site in a Kv7.1 subunit and a single PUFA analog affects both the pore domain (to alter Gmax) and the voltage-sensing domain (to shift V50) (Figure S3). However, we cannot rule out that there are two PUFA binding sites with similar affinities and pH dependences in Kv7.1 channels. In Kv1.4 channel, it was recently proposed that naturally occurring PUFAs interact with two distinct PUFA binding sites in the voltage-sensing domain and the pore domain to shift the voltage dependence and induce inactivation, respectively (Farag et al., 2016). To conclude, we anticipate that the detailed molecular understanding of the dual independent activating effects of PUFA analogs on Kv7.1 and Kv7.1+KCNE1 will aid in future drug development of compounds that utilize these mechanisms.
EXPERIMENTAL PROCEDURES
Additional information and resources are included in Supplemental Experimental Procedures.
Two-Electrode Voltage-Clamp Electrophysiology
Experiments were approved by The Linkoping Animal Ethics Committee at Linkoping University. Mutationswere introduced into Kv7.1 using site-directed mutagenesis (QuikChange; Stratagene, CA, USA). cRNA was prepared from linearized DNA using the T7 mMessage mMachine transcription kit (Ambion, TX, USA). 50 nL of cRNA (~50 ng of Kv7.1 for Kv7.1-only expression, 25 ng of Kv7.1 plus 8 ng of KCNE1 for co-expression) were injected into each Xenopus laevis oocyte. Currents were measured using the two-electrode voltage-clamp technique (CA-1B amplifier; Dagan, MN, USA), sampled at 1-3.3 kHz, and filtered at 500 Hz. The control solution contained the following (in mM): 88 NaCl, 1 KCl, 15 HEPES, 0.4 CaCl2, and 0.8 MgCl2 (pH adjusted to using NaOH). Activation curves were generally elicited by stepping from a holding voltage of −80 mV to test voltages between −90 and +70 mV (3- to 5-s durations and 10-mV increments), followed by a tail voltage of −30 mV. N-AT (CAS 119959-65-8; Cayman Chemical, MI, USA) was stored, diluted, and applied to the oocyte chamber as previously described (Liin et al., 2015b). Arachidonoyl amine was synthesized as previously described (Borjesson et al., 2010). 1 mM MTSET+ ([2-(trimethylammonium)ethyl] methanethiosulfonate, bromide; CAS 91774-25-3) or 10 mM MTSES− (sodium[2-sulfonatoethyl]methanethiosulfonate; CAS 184644-83-5; Toronto
Research Chemicals) were applied continuously for 7-10 min to the bath solution using a gravity-driven perfusion system.
Electrophysiological Analysis
To quantify effects on the G(V), tail currents (measured shortly after initiation of tail voltage) were plotted against the pre-pulse (test) voltage and fitted to the following:
| (Equation 1) |
where V50 is the midpoint and s is the slope factor (shared slope for control and N-AT curves within the same cell). In figures showing Itail versus voltage, the control curves are normalized between 0 and the fitted Gmax. N-AT or AA+ curves are normalized to the control curve (symbols) (more details are in Supplemental Experimental Procedures). PUFA analog-induced alterations in the maximum conductance, relative Gmax, was calculated as the ratio of the fitted Gmax in the presence and absence of PUFA analog.
To quantify the concentration dependence and pH dependence of the PUFA analog-induced ΔV50 (or ΔGmax), the following equation was used:
| (Equation 2) |
where ΔVmax is the maximal shift, C50 is the concentration causing 50% of the maximal shift, c is the concentration of PUFA analog (or H+), and Nh is the Hill slope. The data in Figures 3A and 3B has been scaled to allow comparison. The data in Figures 3C and 3D have been normalized between 0 and 1.
The rubidium-to-potassium permeability ratio was assessed as previously described (Zaydman et al., 2014), by first stepping the voltage to +60 mV for 5 s followed by −60 mV for 3 s. The following solutions were used (in mM): 100 Na+ (96 NaCl, 4 KCl, 5 HEPES, 1.8 CaCl2, and 1 MgCl2; pH adjusted to using NaOH), 100 K+ (100 KCl, 5 HEPES, 1.8 CaCl2, and 1 MgCl2; pH adjusted to 7.4 using KOH), or 100 Rb+ (96 RbCl, 4 KCl, 5 HEPES, CaCl2, and 1 MgCl2; pH adjusted to 7.4 using RbOH).
Kinetic Model Simulations
Simulations of the N-AT effect on the conductance (Gpufa/Gcw) were done using a kinetic model in Berkeley Madonna (Berkeley, CA). A macroscopic three-state model (resting, activated, and open states) was used, with a voltage-sensing step between the resting and activated states and a gate-opening step between the activated and open states. The N-AT effect was modeled as a shift of the voltage dependence of ΔV50 = −30 mV for the first step and an increase in the equilibrium constant by a factor of LPUFA =1.9 for the second step (more details are in Supplemental Experimental Procedures).
Molecular Simulations
The cryo-EM structure of Kv7.1 (Sun and MacKinnon, 2017) was embedded in a POPC bilayer with explicit solvent using the CHAMM36 force field for both the protein and the lipids (Best et al., 2012). Electrostatic interactions were calculated with the particle mesh Ewald algorithm at every step (Essmann et al., 1995). In order to simulate the electrostatic effect of N-AT, each K326 residue was pulled away from the pore by applying steered molecular dynamics (more details are in Supplemental Experimental Procedures).
Statistics
Average values are expressed as mean ± SEM. p < 0.05 is considered as statistically significant. Statistics were calculated using one-way ANOVA followed by Tukey’s multiple-comparison test to compare all columns, Bonferroni’s multiple-comparison test to compare pair of columns, or Dunnett’s multiple-comparison test to compare the N-AT effect on each mutant to the N-AT effect on WT. One-sample t test was used to compare the alteration in Gmax to a hypothetical value of 1. Statistical tests are denoted in each figure legend.
Supplementary Material
Highlights.
Polyunsaturated fatty acid (PUFA) analogs are new potential anti-arrhythmic drugs
PUFA analogs interact both with the KV7.1 channel pore and voltage sensors
The two interactions are independent and electrostatic in both cases
This could lead to development of anti-arrhythmic drugs that target KV7.1 hannels
ACKNOWLEDGMENTS
We thank Drs. F. Elinder and E. Lindahl forvaluable comments. This work was supported by the National Institutes of Health (R01GM109762; R01HL131461), American Heart Association (14GRNT20380041), the Swedish Society for Medical Research, the Swedish Research Council (2017-02040), Magnus Bergvall’s Foundation, and the Lions Foundation. S.Y. was supported by the Center for Systems Neurobiology at Linkoping University. Computational resources were provided by the Swedish National Infrastructure for Computing (SNIC 2016/1-543; 2017/1-290).
Footnotes
Data Availability
The data of this study are available upon request.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and one video and can be found with this article online at https://doi.Org/10.1016/j.celrep.2018.08.031.
DECLARATION OF INTERESTS
A patent application (#62/032,739) based on these results has been submitted by the University of Miami with S.I.L. and H.P.L. identified as inventors.
REFERENCES
- Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, and Romey G (1996). KyLQT1 and lsK(minK) proteins associate to form the Iks cardiac potassium current. Nature 384, 78–80. [DOI] [PubMed] [Google Scholar]
- Barro-Soria R, Rebolledo S, Liin SI, Perez ME, Sampson KJ, Kass RS, and Larsson HP (2014). KCNE1 divides the voltage sensor movement in KCNQ1/KCNE1 channels into two steps. Nat. Commun. 5, 3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best RB, Zhu X, Shim J, Lopes PEM, Mittal J, Feig M, and Mackerell AD Jr. (2012). Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone 4, C and side-chain C1 and c2 dihedral angles. J. Chem. Theory Comput 8, 3257–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezanilla F, Perozo E, and Stefani E (1994). Gating of Shaker K+ channels: II The components of gating currents and a model of channel activation. Biophys. J. 66,1011–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borjesson SI, Parkkari T, Hammarstrom S, and Elinder F (2010). Electrostatic tuning of cellular excitability. Biophys. J. 98, 396–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvacho I, Gonzalez W, Torres YP, Brauchi S, Alvarez O, Gonzalez- Nilo FD, and Latorre R (2008). Intrinsic electrostatic potential in the BK channel pore: role in determining single channel conductance and block. J. Gen. Physiol. 737, 147–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrapani S, Cordero-Morales JF, Jogini V, Pan AC, Cortes DM, Roux B, and Perozo E (2011). On the structural basis of modal gating behavior in K+ channels. Nat. Struct. Mol. Biol. 18, 67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti L, Renhorn J, Gabrielsson A, Turesson F, Liin SI, Lindahl E, and Elinder F (2016). Reciprocal voltage sensor-to-pore coupling leads to potassium channel C-type inactivation. Sci. Rep. 6, 27562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero-Morales JF, Jogini V, Chakrapani S, and Perozo E (2011). A multipoint hydrogen-bond network underlying KcsA C-type inactivation. Biophys. J. 100, 2387–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J (2016). Voltage-dependent gating: novel insights from KCNQ1 channels. Biophys. J. 110, 14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demo SD, and Yellen G (1992). Ion effects on gating of the Ca2+-activated K+ channel correlate with occupancy of the pore. Biophys. J. 61, 639–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldstrom J, Wang Z, Werry D, Wong N, and Fedida D (2015). Microscopic mechanisms for long QT syndrome type 1 revealed by single-channel analysis of Iks with S3 domain mutations in KCNQ1. Heart Rhythm 12, 386–394. [DOI] [PubMed] [Google Scholar]
- Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, and Pedersen LG (1995). A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593. [Google Scholar]
- Farag NE, Jeong D, Claydon T, Warwicker J, and Boyett MR (2016). Polyunsaturated fatty acids inhibit Kv1.4 by interacting with positively charged extracellular pore residues. Am. J. Physiol. Cell Physiol. 311, C255–C268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibor G, Yakubovich D, Rosenhouse-Dantsker A, Peretz A, Schottelndre- ier H, Seebohm G, Dascal N, Logothetis DE, Paas Y, and Attali B (2007). An inactivation gate in the selectivity filter of KCNQ1 potassium channels. Biophys. J. 93, 4159–4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldenberg I, Moss AJ, Zareba W, McNitt S, Robinson JL, Qi M, Tow- bin JA, Ackerman MJ, and Murphy L (2006). Clinical course and risk stratification of patients affected with the Jervell and Lange-Nielsen syndrome. J. Cardiovasc. Electrophysiol. 17, 1161–1168. [DOI] [PubMed] [Google Scholar]
- Larsen AP, Steffensen AB, Grunnet M, and Olesen SP (2011). Extracellular potassium inhibits Kv7.1 potassium channels by stabilizing an inactivated state. Biophys. J. 101, 818–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liin SI, Barro-Soria R, and Larsson HP (2015a). The KCNQ1 channel- remarkable flexibility in gating allows for functional versatility. J. Physiol. 593,2605–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liin SI, Silvera Ejneby M, Barro-Soria R, Skarsfeldt MA, Larsson JE, Starck Harlin F, Parkkari T, Bentzen BH, Schmitt N, Larsson HP, and Elinder F (2015b). Polyunsaturated fatty acid analogs act antiarrhythmically on the cardiac IKs channel. Proc. Natl. Acad. Sci. USA 112, 5714–5719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liin SI, Larsson JE, Barro-Soria R, Bentzen BH, and Larsson HP (2016). Fatty acid analogue N-arachidonoyl taurine restores function of Iks channels with diverse long QT mutations. eLife 5, e20272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Barneo J, Hoshi T, Heinemann SH, and Aldrich RW (1993). Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors Channels 1, 61–71. [PubMed] [Google Scholar]
- Murray C.l., Westhoff M, Eldstrom J, Thompson E, Emes R, and Fedida D (2016). Unnatural amino acid photo-crosslinking of the IKs channel complex demonstrates a KCNE1:KCNQ1 stoichiometry of up to 4:4. eLife 5, e11815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajo K, and Kubo Y (2015). KCNQ1 channel modulation by KCNE proteins via the voltage-sensing domain. J. Physiol. 593, 2617–2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajo K, Ulbrich MH, Kubo Y, and Isacoff EY (2010). Stoichiometry of the KCNQ1-KCNE1 ion channel complex. Proc. Natl. Acad. Sci. USA 107, 18862–18867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerbonne JM, and Kass RS (2005). Molecular physiology of cardiac repolarization. Physiol. Rev. 85, 1205–1253. [DOI] [PubMed] [Google Scholar]
- Ottosson NE, Liin S.l., and Elinder F (2014). Drug-induced ion channel opening tuned by the voltage sensor charge profile. J. Gen. Physiol. 143, 173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panaghie G, and Abbott GW (2007). The role of S4 charges in voltage- dependent and voltage-independent KCNQ1 potassium channel complexes. J. Gen. Physiol. 129, 121–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant LD, Xiong D, Dai H, and Goldstein SA (2014). Individual IKs channels at the surface of mammalian cells contain two KCNE1 accessory sub-units. Proc. Natl. Acad. Sci. USA 111, E1438–E1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poveda JA, Giudici AM, Renart ML, Molina ML, Montoya E, Fernan- dez-Carvajal A, Fernandez-Ballester G, Encinar JA, and Gonzalez-Ros JM (2014). Lipid modulation of ion channels through specific binding sites. Biochim. Biophys. Acta 1838, 1560–1567. [DOI] [PubMed] [Google Scholar]
- Pusch M, Bertorello L, and Conti F (2000). Gating and flickery block differentially affected by rubidium in homomeric KCNQ1 and heteromeric KCNQ1/ KCNE1 potassium channels. Biophys. J. 78, 211–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, and Keating MT (1996). CoassemblyofKyLQT1 and minK(IsK) proteins to form cardiac Iks potassium channel. Nature 384, 80–83. [DOI] [PubMed] [Google Scholar]
- Seebohm G, Sanguinetti MC, and Pusch M (2003). Tight coupling of rubidium conductanceand inactivation in human KCNQ1 potassium channels. J. Physiol. 552, 369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, and MacKinnon R (2017). Cryo-EM structure ofaKCNQ1/CaM complex reveals insights into congenital long QT syndrome. Cell 169, 1042–1050.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanoye CG, Welch RC, Daniels MA, Manderfield LJ, Tapper AR, Sanders CR, and George AL Jr. (2009). Distinct subdomains of the KCNQ1 S6 segment determine channel modulation by different KCNE sub-units. J. Gen. Physiol. 134, 207–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Pan H, Delaloye K, and Cui J (2010). KCNE1 remodels the voltage sensor of Kv7.1 to modulate channel function. Biophys. J. 99, 3599–3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaydman MA, Kasimova MA, McFarland K, Beller Z, Hou P, Kinser HE, Liang H, Zhang G, Shi J, Tarek M, and Cui J (2014). Domain-domain interactions determine the gating, permeation, pharmacology, and subunit modulation of the IKs ion channel. eLife 3, e03606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, and MacKinnon R (2003).Theoccupancyofions in the K+ selectivity filter: charge balance and coupling of ion binding to a protein conformational change underlie high conduction rates. J. Mol. Biol. 333, 965–975. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.