

Abstract
Obesity and insulin resistance are associated with oxidative stress, which may be implicated in the progression of obesity-related diseases. The kinase JNK1 has emerged as a promising drug target for the treatment of obesity and type 2 diabetes. JNK1 is also a key mediator of the oxidative stress response, which can promote cell death or survival, depending on the magnitude and context of its activation. In this article, we describe a study in which the long-term effects of JNK1 inactivation on glucose homeostasis and oxidative stress in obese mice were investigated for the first time. Mice lacking JNK1 (JNK1−/−) were fed an obesogenic high-fat diet (HFD) for a long period. JNK1−/− mice fed an HFD for the long term had reduced expression of antioxidant genes in their skin, more skin oxidative damage, and increased epidermal thickness and inflammation compared with the effects in control wild-type mice. However, we also observed that the protection from obesity, adipose tissue inflammation, steatosis, and insulin resistance, conferred by JNK1 ablation, was sustained over a long period and was paralleled by decreased oxidative damage in fat and liver. We conclude that compounds targeting JNK1 activity in brain and adipose tissue, which do not accumulate in the skin, may be safer and most effective.—Becattini, B., Zani, F., Breasson, L., Sardi, C., D’Agostino, V. G., Choo, M.-K., Provenzani, A., Park, J. M., Solinas, G. JNK1 ablation in mice confers long-term metabolic protection from diet-induced obesity at the cost of moderate skin oxidative damage.
Keywords: type-2 diabetes, insulin resistance, metabolic inflammation, antioxidants, oxidative stress tolerance
It is now appreciated that the saturation of adipocyte lipid storage capacity, the consequent ectopic lipid deposition, metabolic inflammation, and insulin resistance are closely correlated events and hallmarks of obesity-associated diseases (1–6). JNK-1 and -2 emerged as a major link between obesity-driven lipotoxicity, metabolic inflammation, and insulin resistance (1, 2, 7, 8), with JNK1 being the most important isoform (8, 9). JNK1 is activated by saturated fatty acids, and mice lacking JNK1 have reduced adiposity, reduced metabolic inflammation, and improved insulin sensitivity (8, 10, 11). JNK1 was initially believed to promote insulin resistance by direct phosphorylation of the insulin receptor substrates (7, 8, 10, 12). However, current evidence indicates that part of the improved insulin sensitivity in mice lacking JNK1 is an indirect consequence of their leaner phenotype (11, 13), which depends on JNK1 activity in neurons (13–15). Moreover, JNK1 activity in myeloid cells plays an important role in the development of obesity-driven metabolic inflammation, which further promotes insulin resistance (11, 16, 17).
Although current studies consistently indicate that JNK1 is a promising drug target for the treatment of obesity-driven insulin resistance, the long-term efficacy of JNK1 ablation for the treatment of obesity-related diseases is unknown, and there are safety concerns. Obesity and diabetes are conditions associated with oxidative stress (18, 19), and JNK1 is a central mediator of the physiologic response to oxidative stress, with outcomes ranging from increased survival to cell death, depending on intensity, length, and context of its activation (20–22). JNK was shown to promote tolerance to oxidative stress and increase life span in Drosophila and in Caenorhabditis elegans, by a mechanism involving increased Forkhead box O (FoxO)-dependent antioxidant defense (23–26). JNK is a major signal transducer mediating FoxO activation in response to oxidative stress (20, 27), and controls the expression of the antioxidant proteins sestrin-2 (Sesn2) and heme oxygenase (HO)-1 via the transcription factor AP-1 (28–31). Mouse embryonic fibroblasts lacking JNK1 and -2 display increased sensitivity to hydrogen peroxide, indicating that JNK activity sustains oxidative stress tolerance in vertebrate cells (32). Finally, the well-documented action of sustained JNK activation in promoting apoptosis in response to oxidative stress may also play an important role in tissue homeostasis by eliminating irreversibly damaged cells, hence preventing the excessive inflammation that would result from a necrotic cell (20–22). However, the physiologic and pathophysiological relevance of JNK1-induced oxidative stress tolerance in vertebrates, in particular in mouse models of obesity and insulin resistance, is unknown. Indeed, whereas JNK1 inactivation is known to protect mice from diet-induced obesity, metabolic inflammation, and insulin resistance, it must be noted that current studies are limited to 20-wk exposure to a high-fat diet (HFD) (11). Hence, the long-term effects of JNK1 inactivation in obesity are unknown. To learn about the safety and efficacy of long-term JNK1 inactivation for the treatment of obesity-driven insulin resistance, we investigated metabolic homeostasis and oxidative stress in mice lacking JNK1 (JNK1−/−) fed an obesogenic HFD for a long period. Our results indicate that systemic inactivation of JNK1 leads to prolonged metabolic protection from diet-induced obesity and reduced oxidative damage in liver and adipose tissue, at the cost of increased predisposition to skin oxidative damage.
MATERIALS AND METHODS
In vivo studies
Mice were males on a pure C57BL6/J background, and JNK1−/− mice were bred as previously described (11). An HFD (60% of calories from fat) was purchased from Bio-Serv (diet F3282; Flemington, NJ, USA). The mice were kept at our standard facility in a 12 h light–dark cycle at room temperature (23°C). Two independent cohorts from different breeders were investigated. For the first cohort 15 wild-type (WT) mice and 15 JNK1−/− mice were weaned onto an HFD and maintained on it. For the second cohort, 9 WT mice and 12 JNK1−/− mice were also weaned onto the HFD and kept on it up to 44 wk of age (40 wk of HFD). Glucose tolerance (GTT) and insulin tolerance (ITT) tests were performed at various times (see Figs. 1 and 2) (33). Experimental procedures were authorized by the Cantonal Veterinary Committee (Service de la Sécurité Alimentaire et des Affaires Vétérinaires, Givisiez, Switzerland.).
Figure 1.

JNK1−/− mice display sustained metabolic protection from an HFD, but also display signs of exacerbated skin aging. A) Survival curve of 15 WT mice and 15 JNK1−/− mice that were kept on an HFD from weaning to death. B) Growth curve of the mice. C) ITT of 48-wk-old WT mice and JNK1−/− mice on an HFD. D) Incidence of alopecia, overt alopecia, and hair depigmentation in 80-wk-old WT mice and JNK1−/− mice kept on an HFD.
Figure 2.
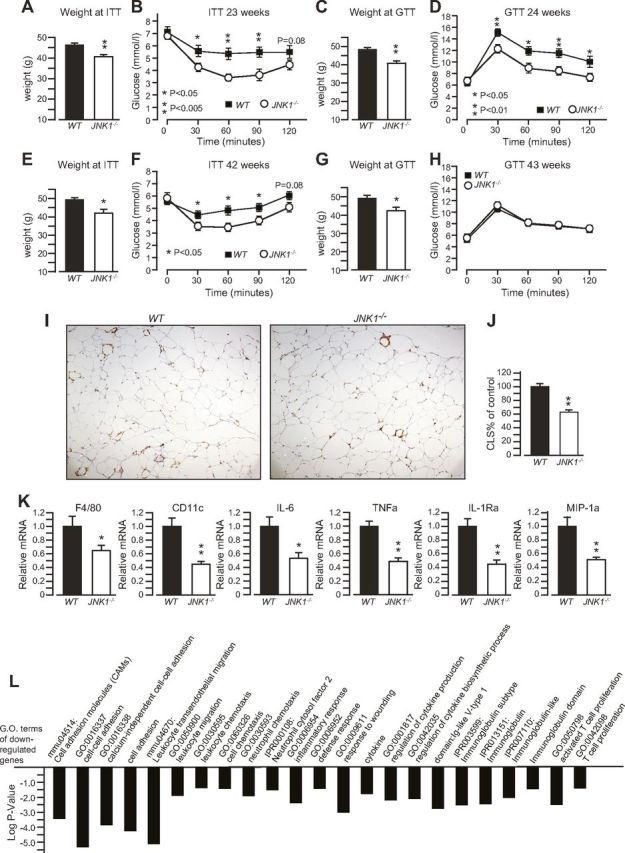
JNK1−/− mice display sustained protection against HFD-induced adipose tissue inflammation and insulin resistance. Body weight (A) and ITT results (B) of 23-wk-old WT mice and JNK1−/− mice kept on an HFD. Body weight (C) and GTT results (D) of 24-wk-old WT mice and JNK1−/− mice kept on an HFD. Body weight (E) and ITT results (F) of the same mice at the age of 42 wk. Body weight (G) and GTT results (H) of the same mice at the age of 43 wk. I) Mac2 staining of CLSs of adipose tissue sections from the mice at the age of 44 wk. J) Quantification of the number of CLSs from (I). K) Real-time qPCR analysis of the expression of proinflammatory genes in adipose tissue of the mice in (I). L) Significant terms from a gene ontology analysis of DNA microarray data, for genes that were down-regulated in adipose tissue of the JNK1−/− mice vs. their controls.
Molecular measurements
Total RNA was isolated from tissues by guanidinium–thiocyanate extraction.
For quantitative real-time PCR (qPCR), cDNA was prepared with a reverse transcription kit (Promega, Madison, WI, USA), and PCR was performed with a commercial SYBR green mix (Bio-Rad, Hercules, CA, USA) and specific primers (Supplemental Table S1). Data are expressed as relative RNA levels normalized on cyclophilin. For microarray analysis, total RNAs were hybridized in quadruplicate on a whole mouse genome 4x44K microarray (G4122F; AMADID 014868; Agilent Technologies, Santa Clara, CA, USA), scanned with the Agilent microarray scanner at 5 μm resolution, and analyzed numerically with the manufacturer’s software. All microarray data are available through the Gene Expression Omnibus database (National Institutes of Health, Bethesda, MD, USA; http://www.ncbi.nlm.nih.gov/geo/) under the accession number GSE73759. For immunoblot analysis, the antibodies were P-AKT Ser473, AKT, P-FoxO1 Ser356, P-FoxO1 Thr24, FoxO1, catalase, and superoxide dismutase (SOD)-2 (Cell Signaling Technology, Danvers, MA, USA); HO (Enzo Life Sciences, Farmingdale, NY, USA); and Sesn2 (Proteintec, Rosemont, IL, USA). Quantification of adipose tissue crown-like structures (CLSs) was performed as described elsewhere (33). Malondialdehyde (MDA) levels were measured with a commercial kit (Abcam, Cambridge, MA, USA). Macrophage immunostaining of liver sections was performed with F4/80 antibodies (AbD Serotec, Raleigh, NC, USA). Immunofluorescence analysis of FoxO proteins in skin sections was performed with anti-Fox03a (Cell Signaling Technology) or anti-Fox04 (Abcam), and images were acquired with the Apotome microscope (Zeiss, Thornwood, NY, USA) with a ×20 objective.
Statistical analysis
Survival curves were analyzed by log-rank (Mantel-Cox) test and by Gehan-Breslow-Wilcoxon test. DNA microarray data were analyzed with the R software environment for statistical computing (http://www.r-project.org/) and the Bioconductor library of biostatistical packages (http://www.bioconductor.org/). Statistical significance was set at P < 0.05. The Database for Annotation, Visualization and Integrated Discovery (DAVID) resource was used for enrichment analysis of differentially expressed genes, with annotations from Gene Ontology, Kyoto Encyclopedia of Genes and Genomes (KEGG) (http://www.genome.jp/kegg/) and PFAM (http://www.pfam.xfam.org). A value of P < 0.05 denoted significant overrepresentation.
All other data are expressed as means ± sem; A value of P < 0.05 by Student’s t test indicates statistical significance.
RESULTS
JNK1−/− mice kept on an HFD display sustained protection from obesity and insulin resistance, but are predisposed to skin aging
Mice JNK1−/− and their WT controls were kept on an HFD after weaning, and we monitored life span, body weight, and glucose homeostasis. We did not find a statistically significant difference between the survival curves of WT and JNK1−/− mice, although we observed a nonsignificant tendency toward decreased age at 50% survival and increased maximum life span in JNK1−/− mice (Fig. 1A).
Body weight measurements showed that, while consuming an HFD, JNK1−/− mice were significantly leaner than control mice until the age of 65 wk, although by the age of 80 wk, body weight was similar between the genotypes (Fig. 1B). JNK1−/− mice at their plateau of body mass, at 50 wk of age, weighed ∼13 g less than their WT control. We performed GTTs and ITTs at different time points, and we observed that JNK1−/− mice displayed improved insulin tolerance vs. control mice, at least until 48 wk of age (Fig. 1C). GTT on 47-wk-old mice did not reveal any difference between groups (Supplemental Fig. S1). However, comparison of GTTs performed at different time points indicated that the absence of difference between groups at 47 wk of age was due to an improvement of glucose tolerance in control mice (Supplemental Fig. S1).
During our routine health checks of the mice we observed that, compared to WT controls, JNK1−/− mice displayed more signs of alopecia and hair depigmentation (Fig. 1D).
Overall, these data indicate that JNK1 ablation leads to a long-term protection from diet-induced obesity and insulin resistance, but also indicate that JNK1 activity may protect against skin aging.
JNK1−/− mice display sustained protection from obesity-induced adipose tissue inflammation and insulin resistance
To reproduce the observations described above and further learn about the role of JNK1 in prolonged HFD feeding, we investigated a second independent cohort of mice, which we euthanized at 44 wk of age, when differences in fur quality between genotypes were evident. We could reproduce the sustained leaner phenotype and long-term improved insulin tolerance of JNK1−/− mice, although by 43 wk of age, there was no difference in glucose tolerance between genotypes (Fig. 2A–H). The latter was explained by an improved glucose tolerance of the WT mice (Fig. 2D, H), which was consistent with that observed in the previous cohort (Supplemental Fig. S1). Mac-2 immunostaining of adipose tissue sections revealed that 44-wk-old JNK1−/− mice, kept on an HFD, displayed a decreased number of CLSs compared to their WT controls (Fig. 2I, J). Consistently, the expression of adipose tissue macrophage markers (F4/80 and CD11c) and of cytokines associated with classic M1 macrophage activation (IL-6, TNFα, IL-1Ra, and MIP-1α) was also decreased in white adipose tissue from JNK1−/− mice compared to WT mice (Fig. 2K). Genome-wide gene expression analysis revealed that the expression of several genes implicated in immunity was down-regulated in white adipose tissues from JNK1−/− mice, and gene ontology analysis of these down-regulated genes indicated reduced leukocyte recruitment, and milder immune responses to chronic obesity in adipose tissues from JNK1−/− mice compared to that in control mice (Fig. 2L). Altogether, our data indicate that JNK1 ablation in mice confers long-term protection from HFD-induced adipose tissue inflammation and insulin resistance.
JNK1−/− mice display sustained protection from HFD-induced hepatosteatosis but not from liver inflammation
Liver histology revealed that, relative to WT mice, JNK1−/− mice kept on HFD were protected from hepatic steatosis at least up to 44 wk of age and 40 wk of HFD feeding (Fig. 3A). However, immunostaining of liver sections with the macrophage marker F4/80 indicate that the number of liver macrophages was similar between WT and JNK1−/− mice (Fig. 3B, C). Consistently, qPCR analysis of liver samples indicated that F4/80 mRNA levels and expression of major proinflammatory cytokines were similar between JNK1−/− mice and WT control mice after long-term HFD feeding (Fig. 3D). Finally, genome-wide gene expression analysis of liver samples from these mice did not reveal significant differences in the expression of inflammatory genes between genotypes (data not shown). Overall, these results indicate that JNK1 ablation in mice confers sustained protection from the effects of chronic HFD feeding on hepatosteatosis, but not from liver inflammation.
Figure 3.
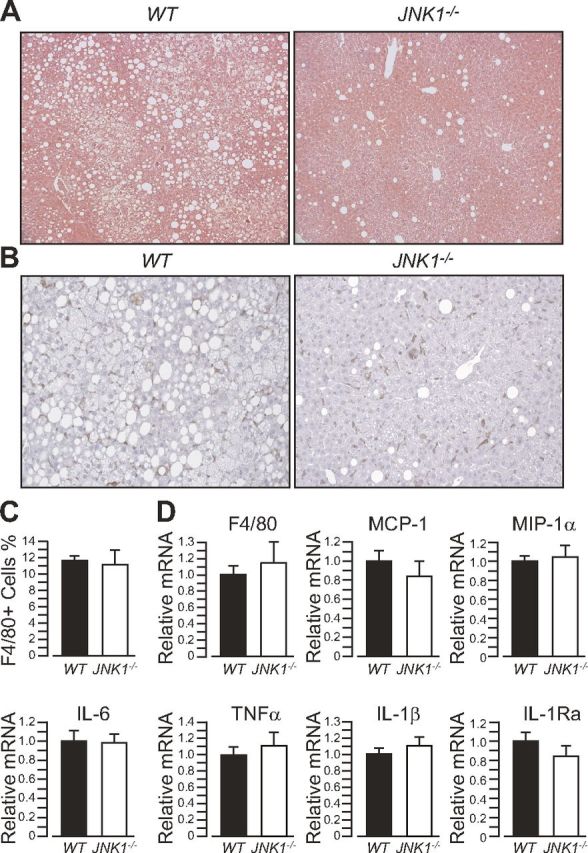
JNK1−/− mice display sustained protection against HFD-induced steatosis, but not against liver inflammation. A) Hematoxylin and eosin staining of representative liver sections of 44-wk-old WT mice and JNK1−/− mice kept on an HFD. B) F4/80 immunostaining of liver sections from the same mice. C) Quantification of the number of F4/80+ cells from the liver sections in (B). D) Real-time qPCR analysis of the expression of proinflammatory genes in mouse liver.
JNK1−/− mice chronically fed an HFD are predisposed to skin damage and inflammation
JNK1−/− mice and WT controls kept on HFD were closely monitored for signs of alopecia and hair depigmentation. Consistent with the data from the previous cohort (Fig. 1), we observed that JNK1−/− mice develop alopecia and hair depigmentation before their WT controls (Fig. 4A, B). By the age of 44 wk, both JNK1−/− mice and WT controls displayed at least some minor sign of alopecia (compared to young mice); however, it was more pronounced in JNK1−/− mice. Furthermore, we did not detect hair depigmentation in WT mice up to 44 wk of age, whereas 75% of JNK1−/− mice showed some depigmentation, which was quite variable within the group, ranging from minor depigmentation to overt (Fig. 4A, B). Histologic analysis of skin samples showed that epidermal thickness was significantly increased in JNK1−/− mice, which also displayed signs of inflammation (Fig. 4C, D). qPCR analysis revealed an induction of the expression of genes associated with M1 macrophage activation (IL-6, -1β, and -1Ra) and decreased expression of genes associated with alternative M2 macrophage activation (MGL-1, MRC-2, and FIZZ1) (Fig. 4E). These data indicate that skin macrophages of JNK1−/− mice are polarized toward an M1 proinflammatory phenotype, which is consistent with the skin histology data, and in marked contrast with what was observed in adipose tissue. To further learn about the pathways altered in the skin of JNK1−/− mice, we performed genome-wide gene expression analysis of skin samples and analyzed the data by gene ontology analysis. The results revealed that several genes related to keratinization, epidermis development, hair cycle, pigmentation, and inflammation were up-regulated in skin samples from JNK1−/− mice compared with gene expression in samples from WT controls (Fig. 4F). This pattern of gene expression is consistent with the idea of increased tissue damage, inflammation, and repair in the skin of JNK1−/− mice.
Figure 4.

JNK1−/− mice fed an HFD for a prolonged period are predisposed to skin damage and inflammation. A) Representative photographs of 44-wk-old WT and JNK1−/− mice kept on an HFD. B) Incidence of hair depigmentation, alopecia (any sign), and mild-to-overt alopecia in the same mice. C) Hematoxylin and eosin staining of representative skin sections of the mice. D) Average epidermal thickness of 44-wk-old WT and JNK1−/− mice kept on an HFD. E) Real-time qPCR analysis of the expression of M1 cytokines (top) and M2 genes (bottom) in skin samples of WT and JNK1−/− mice. F) Significant terms from a gene ontology analysis of DNA microarray data for genes with expression induced in the skin of JNK1−/− mice compared to WT controls.
Taken together, our data indicate that JNK1−/− mice kept on an HFD for a prolonged period are predisposed to skin damage and inflammation.
JNK1 ablation has opposite effects on fat and skin oxidative damage in mice fed an HFD for a prolonged period
To evaluate the effects of JNK1 ablation on oxidative stress damage in mice kept on an HFD, we measured the levels of the lipid peroxidation product MDA in different tissues from the WT mice and JNK1−/− mice described above.
MDA levels were similar between genotypes in kidney and heart, were slightly but significantly decreased in liver, and were markedly decreased in white adipose tissue from JNK1−/− mice compared with levels in WT controls (Fig. 5A). By contrast, MDA levels were significantly elevated in skin samples from JNK1−/− mice compared to WT controls indicating increased oxidative damage in this tissue. To further learn about the mechanisms of increased oxidative damage in the skin of JNK1−/− mice, we measured mRNA levels of HO-1, a potent antioxidant and anti-inflammatory enzyme with expression that has been shown to be elevated in keratinocyte cell cultures by oxidative stress inducers and by lipid peroxidation products in a JNK1-dependent manner (34, 35). HO-1 mRNA levels were similar between genotypes in kidney, heart, and liver, but were markedly reduced in white adipose tissue of JNK1−/− mice compared to WT controls (Fig. 5B). This pattern of gene expression parallels the MDA levels in these tissues and is consistent with the concept of HO-1 as an oxidative stress and lipid-peroxidation–responsive gene. However, we also observed that HO-1 mRNA levels were significantly lower in the skin of JNK1−/− mice vs. WT controls, despite increased MDA levels in the former. This observation is consistent with keratinocyte cell culture studies indicating a major role for JNK1 in the induction of HO-1 gene expression in response to oxidative stress and lipid peroxidation products. We then used real-time qPCR to measure the expression of several genes implicated in oxidative stress tolerance in skin samples from WT mice and JNK1−/− mice.
Figure 5.

Loss of JNK1 in mice fed an HFD for a prolonged period protects liver and adipose tissue, but predisposes skin to oxidative damage. A) MDA levels in different tissues from 44-wk-old WT and JNK1−/− kept on an HFD. B) Real-time qPCR analysis of the expression of HO-1 in the tissues. C) qPCR analysis of the expression of genes implicated in oxidative stress tolerance in the skin of the same mice. D) Immunoblot analysis of catalase, SOD2, HO-1, and Sesn-2 protein levels in the skin samples in (C). E) Quantification of the immunoblots panel D. WAT, white adipose tissue.
The results show that mRNA levels of Sesn-2 and catalase were also significantly decreased in skin samples from JNK1−/− mice (Fig. 5C). These differences may explain the increased oxidative damage in the skin of JNK1−/− mice, despite the systemic metabolic protection and decreased oxidative damage in fat and liver. Catalase, in particular, has been shown to play a major role in skin and hair tolerance to oxidative stress (36, 37). Immunoblot analysis indicated that HO-1 and Sesn-2 protein levels were only marginally reduced in JNK1−/− mice, and the differences were not statistically significant. However, catalase and SOD2 protein levels were markedly reduced in skin samples from JNK1−/− mice. mRNA levels of catalase, Sesn-2, and SOD-2 were not reduced in adipose tissue, liver, heart, and kidney samples from JNK1−/− mice vs. their controls (Supplemental Fig. S2). Because catalase and SOD2 expression are controlled by FoxO transcription factors, we evaluated the activation of FoxOs proteins and of their regulator AKT in the skin of the mice. Our data showed that AKT activation and FoxO1 phosphorylation by AKT were similar in the skin of WT and JNK1−/− mice (Supplemental Fig. S3A, B). Furthermore, immunostaining of skin sections with FoxO3a- and FoxO4-specific antibodies revealed an almost complete nuclear localization of FoxO3a and FoxO4 in the skins of both genotypes (Supplemental Fig. S3C).
Altogether, our results indicate that systemic JNK1 ablation leads to decreased oxidative damage in liver and white adipose tissue and, by contrast, increased skin oxidative damage and reduced expression antioxidant genes specifically in the skin.
DISCUSSION
Several studies have demonstrated that JNK1 ablation protects mice from diet-induced obesity and insulin resistance (8–11, 16, 17). However, these medium-term studies do not exceed 20 wk of dietary treatment, and thus the long-term effects of JNK1 inhibition in obesity are largely unknown. In this study, we investigated for the first time the late effects of JNK1 inactivation in aging mice exposed for a prolonged period to an obesogenic HFD. Our JNK1−/− mice kept on an HFD were mildly predisposed to skin oxidative damage and inflammation. However, the metabolic protection from the effects of dietary fat caused by JNK1 ablation was largely sustained over least 40 wk of an HFD, and this improved metabolism was paralleled by reduced oxidative damage in liver and adipose tissue. The protection from oxidative damage observed in liver and fat of JNK1−/− mice can thus be explained by decreased adiposity and reduced adipose tissue inflammation observed in the mice, which is believed to have major local and systemic effects (17). In line with this interpretation, MDA levels were significantly but modestly reduced in liver and markedly reduced in white adipose tissue of JNK1−/− mice vs. their WT controls. Furthermore, we observed that, although the JNK1−/− mice displayed long-term protection from fatty liver, hepatic inflammation in them was similar to that in WT controls after 40 wk of an HFD, indicating limited protection in this tissue and a more specific role for JNK1 in the control of adipose tissue inflammation. It is possible that the lack of long-term protection from liver inflammation observed in the JNK1−/− mice is the result of a compensatory action from JNK2. Indeed, targeted deletion of JNK1 in liver has been reported to cause hepatic microsteatosis, whereas compound deletion of JNK1 and -2 was protective (38, 39). This observation suggests that simultaneous targeting of JNK1 and -2 may be a more effective antidiabetogenic strategy than targeting JNK1 in isolation. However, that mouse embryonic fibroblasts lacking JNK1 and -2 are more sensitive to hydrogen peroxide is a possible concern (32). Indeed, our observation that JNK1−/− mice fed an HFD over a long period displayed increased skin oxidative damage and reduced antioxidant gene expression implies a role for JNK1 in promoting oxidative stress tolerance in this tissue.
A limitation of our study is that only male mice were investigated; thus, the impact of female steroids on the observed phenotype remains unresolved. Another major limitation of our study is that the use of systemic JNK1 inactivation does not allow the identification of the specific cell type implicated in the increased skin oxidative damage observed in JNK1−/− mice. Therefore, our data do not exclude a non–cell-autonomous action of JNK1 in promoting oxidative stress tolerance. Nonetheless, a role for JNK in the control of antioxidant gene expression in cultured skin cells has been reported (34, 35, 40, 41), and that the skin was the only tissue of JNK1−/− mice displaying increased oxidative damage is consistent with the idea that JNK1 activity within a specific skin cell type may protect against local oxidative damage. Indeed, because JNK1 deletion improves metabolic homeostasis and reduces adiposity, the expected systemic outcome of JNK1 deletion would be reduced oxidative stress, as observed in liver and adipose tissue. We found that mice lacking JNK1 and having prolonged exposure to an HFD displayed, specifically in skin cells, reduced mRNA and protein levels of catalase, an enzyme playing a major role in skin tolerance to oxidative stress, whose reduced expression correlates strongly with hair depigmentation (36, 37). Furthermore, we observed a marked reduction of SOD2 protein levels and decreased mRNA levels of the antioxidant genes Sesn-2 and HO-1 in the skin of JNK1 mice. Because catalase and SOD2 expression is controlled by FoxO transcription factors, it is possible that the effects of JNK1 activity on skin catalase levels are mediated by JNK1s effect on FoxOs proteins (27). However, other mechanisms may also be involved, as we did not observe signs of reduced FoxO1 activation and of FoxO3a and -O4 nuclear localization in skin samples from JNK1−/− mice. Furthermore, SOD2, HO-1, and Sesn-2 have been reported to be directly regulated by the JNK-AP1 pathway in a cell-autonomous fashion (28–31, 40). Hence, the increased oxidative damage in the skin of JNK1−/− mice could be explained by a local effect on antioxidant gene expression. We observed some discrepancies between mRNAs and protein levels of the antioxidant genes with down-regulated expression in the skin of JNK1−/− mice. Sesn2 and HO-1 mRNA levels were significantly reduced in the skin of JNK1−/− mice, whereas we observed only a marginal, statistically nonsignificant, reduction in SOD2 mRNA levels. By contrast, SOD2 protein content was markedly reduced in the skin of JNK1−/− mice, whereas HO-1 and Sesn2 protein levels were only marginally reduced, and these differences did not reach statistical significance. Only catalase was consistently reduced at the mRNA and protein levels, although the observed differences in protein levels were larger than for the differences in mRNA content. The reasons for these apparent discrepancies are not clear and may suggest posttranscriptional regulation mechanisms. However, it should be considered that these genes are rhythmically expressed in a circadian fashion (42), and the observed differences in mRNA and protein content may be caused by a lag time between the circadian changes in mRNA and protein levels consequent to the relative abundance and stability of these proteins.
Overall, our data show that the effects of JNK1 ablation on oxidative stress tolerance in obese mice are specific for the skin and are less dramatic than those reported in the Drosophila and Caenorhabditis elegans models (23–26). Furthermore, we show that protection against obesity, adipose tissue inflammation, steatosis, and insulin resistance is largely sustained over a long period in JNK1−/− and is paralleled by reduced oxidative damage in white adipose tissue and liver. Our results suggest that prolonged use of drugs targeting JNK may predispose patients to skin oxidative damage; however, this possible side effect can be easily detected and may be largely preventable by minimizing skin exposure to oxidative stress sources such as irritants and UV radiation. Indeed, JNK is a UV-responsive kinase (43) that is activated in human skin by UV light (44).
We conclude that JNK1 should be regarded as an effective and relatively safe target for long-term treatment of obesity and insulin resistance and that compounds that target JNK1 in brain and adipose tissue and do not accumulate in the skin could be the more effective and safer drug candidates.
Supplementary Material
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
ACKNOWLEDGMENTS
This work was supported by grants from the Swiss National Science Foundation, the Swedish Diabetes Foundation, the Swedish Research Council, and a startup package from the University of Gothenburg (to G.S.). Author contributions: B. Becattini and F. Zani performed most of the experiments, analyzed data, and contributed to the discussion; L. Breasson and C. Sardi performed experiments; V. G. D’Agostino and A. Provenzani performed the DNA microarray analysis; M.-K. Choo and J. M. Park performed the skin histopathological analysis and discussed the skin data; G. Solinas conceived the project, the experimental design, and wrote the manuscript; B. Becattini, F. Zani, A. Provenzani, M.-K. Choo and J. M. Park, significantly contributed to the writing of the manuscript with their comments; and all authors approved the paper. The authors declare no conflicts of interest.
Glossary
- CLS
crown-like structure
- GTT
glucose tolerance
- HFD
high-fat diet
- HO
heme oxygenase
- ITT
insulin tolerance
- MDA
malondialdehyde
- qPCR
quantitative PCR
- Sesn
sestrin
- SOD
superoxide dismutase
- WT
wild-type
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Solinas G. (2012) Molecular pathways linking metabolic inflammation and thermogenesis. Obes. Rev. (Suppl 2), 69–82 [DOI] [PubMed] [Google Scholar]
- 2.Solinas G., Karin M. (2010) JNK1 and IKKbeta: molecular links between obesity and metabolic dysfunction. FASEB J. , 2596–2611 [DOI] [PubMed] [Google Scholar]
- 3.Virtue S., Vidal-Puig A. (2008) It’s not how fat you are, it’s what you do with it that counts. PLoS Biol. , e237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Unger R. H., Clark G. O., Scherer P. E., Orci L. (2010) Lipid homeostasis, lipotoxicity and the metabolic syndrome. Biochim. Biophys. Acta , 209–214 [DOI] [PubMed] [Google Scholar]
- 5.Hotamisligil G. S. (2006) Inflammation and metabolic disorders. Nature , 860–867 [DOI] [PubMed] [Google Scholar]
- 6.Donath M. Y. (2014) Targeting inflammation in the treatment of type 2 diabetes: time to start. Nat. Rev. Drug Discov. , 465–476 [DOI] [PubMed] [Google Scholar]
- 7.Aguirre V., Uchida T., Yenush L., Davis R., White M. F. (2000) The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307). J. Biol. Chem. , 9047–9054 [DOI] [PubMed] [Google Scholar]
- 8.Hirosumi J., Tuncman G., Chang L., Görgün C. Z., Uysal K. T., Maeda K., Karin M., Hotamisligil G. S. (2002) A central role for JNK in obesity and insulin resistance. Nature , 333–336 [DOI] [PubMed] [Google Scholar]
- 9.Tuncman G., Hirosumi J., Solinas G., Chang L., Karin M., Hotamisligil G. S. (2006) Functional in vivo interactions between JNK1 and JNK2 isoforms in obesity and insulin resistance. Proc. Natl. Acad. Sci. USA , 10741–10746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Solinas G., Naugler W., Galimi F., Lee M. S., Karin M. (2006) Saturated fatty acids inhibit induction of insulin gene transcription by JNK-mediated phosphorylation of insulin-receptor substrates. Proc. Natl. Acad. Sci. USA , 16454–16459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Solinas G., Vilcu C., Neels J. G., Bandyopadhyay G. K., Luo J. L., Naugler W., Grivennikov S., Wynshaw-Boris A., Scadeng M., Olefsky J. M., Karin M. (2007) JNK1 in hematopoietically derived cells contributes to diet-induced inflammation and insulin resistance without affecting obesity. Cell Metab. , 386–397 [DOI] [PubMed] [Google Scholar]
- 12.Aguirre V., Werner E. D., Giraud J., Lee Y. H., Shoelson S. E., White M. F. (2002) Phosphorylation of Ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action. J. Biol. Chem. , 1531–1537 [DOI] [PubMed] [Google Scholar]
- 13.Sabio G., Cavanagh-Kyros J., Barrett T., Jung D. Y., Ko H. J., Ong H., Morel C., Mora A., Reilly J., Kim J. K., Davis R. J. (2010) Role of the hypothalamic-pituitary-thyroid axis in metabolic regulation by JNK1. Genes Dev. , 256–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsaousidou E., Paeger L., Belgardt B. F., Pal M., Wunderlich C. M., Brönneke H., Collienne U., Hampel B., Wunderlich F. T., Schmidt-Supprian M., Kloppenburg P., Brüning J. C. (2014) Distinct roles for JNK and IKK activation in Agouti-related peptide Neurons in the development of obesity and insulin resistance. Cell Reports , 1495–1506 [DOI] [PubMed] [Google Scholar]
- 15.Belgardt B. F., Mauer J., Wunderlich F. T., Ernst M. B., Pal M., Spohn G., Brönneke H. S., Brodesser S., Hampel B., Schauss A. C., Brüning J. C. (2010) Hypothalamic and pituitary c-Jun N-terminal kinase 1 signaling coordinately regulates glucose metabolism. Proc. Natl. Acad. Sci. USA , 6028–6033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han M. S., Jung D. Y., Morel C., Lakhani S. A., Kim J. K., Flavell R. A., Davis R. J. (2013) JNK expression by macrophages promotes obesity-induced insulin resistance and inflammation. Science , 218–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perry R. J., Camporez J. P., Kursawe R., Titchenell P. M., Zhang D., Perry C. J., Jurczak M. J., Abudukadier A., Han M. S., Zhang X. M., Ruan H. B., Yang X., Caprio S., Kaech S. M., Sul H. S., Birnbaum M. J., Davis R. J., Cline G. W., Petersen K. F., Shulman G. I. (2015) Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell , 745–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Furukawa S., Fujita T., Shimabukuro M., Iwaki M., Yamada Y., Nakajima Y., Nakayama O., Makishima M., Matsuda M., Shimomura I. (2004) Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Invest. , 1752–1761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Urakawa H., Katsuki A., Sumida Y., Gabazza E. C., Murashima S., Morioka K., Maruyama N., Kitagawa N., Tanaka T., Hori Y., Nakatani K., Yano Y., Adachi Y. (2003) Oxidative stress is associated with adiposity and insulin resistance in men. J. Clin. Endocrinol. Metab. , 4673–4676 [DOI] [PubMed] [Google Scholar]
- 20.Shaukat Z., Liu D., Hussain R., Khan M., Gregory S. L. (2016) The role of JNK signalling in responses to oxidative DNA damage. Curr. Drug Targets 17, 154–163 [DOI] [PubMed] [Google Scholar]
- 21.Wagner E. F., Nebreda A. R. (2009) Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer , 537–549 [DOI] [PubMed] [Google Scholar]
- 22.Luo J. L., Kamata H., Karin M. (2005) IKK/NF-kappaB signaling: balancing life and death: a new approach to cancer therapy. J. Clin. Invest. , 2625–2632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oh S. W., Mukhopadhyay A., Svrzikapa N., Jiang F., Davis R. J., Tissenbaum H. A. (2005) JNK regulates lifespan in Caenorhabditis elegans by modulating nuclear translocation of forkhead transcription factor/DAF-16. Proc. Natl. Acad. Sci. USA , 4494–4499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang M. C., Bohmann D., Jasper H. (2005) JNK extends life span and limits growth by antagonizing cellular and organism-wide responses to insulin signaling. Cell , 115–125 [DOI] [PubMed] [Google Scholar]
- 25.Wang M. C., Bohmann D., Jasper H. (2003) JNK signaling confers tolerance to oxidative stress and extends lifespan in Drosophila. Dev. Cell , 811–816 [DOI] [PubMed] [Google Scholar]
- 26.Uno M., Honjoh S., Matsuda M., Hoshikawa H., Kishimoto S., Yamamoto T., Ebisuya M., Yamamoto T., Matsumoto K., Nishida E. (2013) A fasting-responsive signaling pathway that extends life span in C. elegans. Cell Reports , 79–91 [DOI] [PubMed] [Google Scholar]
- 27.Eijkelenboom A., Burgering B. M. (2013) FOXOs: signalling integrators for homeostasis maintenance. Nat. Rev. Mol. Cell Biol. , 83–97 [DOI] [PubMed] [Google Scholar]
- 28.Lee J. H., Budanov A. V., Karin M. (2013) Sestrins orchestrate cellular metabolism to attenuate aging. Cell Metab. , 792–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang X. Y., Wu X. Q., Deng R., Sun T., Feng G. K., Zhu X. F. (2013) Upregulation of sestrin 2 expression via JNK pathway activation contributes to autophagy induction in cancer cells. Cell. Signal. , 150–158 [DOI] [PubMed] [Google Scholar]
- 30.Yi L., Li F., Yong Y., Jianting D., Liting Z., Xuansheng H., Fei L., Jiewen L. (2014) Upregulation of sestrin-2 expression protects against endothelial toxicity of angiotensin II. Cell Biol. Toxicol. , 147–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kietzmann T., Samoylenko A., Immenschuh S. (2003) Transcriptional regulation of heme oxygenase-1 gene expression by MAP kinases of the JNK and p38 pathways in primary cultures of rat hepatocytes. J. Biol. Chem. , 17927–17936 [DOI] [PubMed] [Google Scholar]
- 32.Ventura J. J., Cogswell P., Flavell R. A., Baldwin A. S. Jr., Davis R. J. (2004) JNK potentiates TNF-stimulated necrosis by increasing the production of cytotoxic reactive oxygen species. Genes Dev. , 2905–2915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Becattini B., Marone R., Zani F., Arsenijevic D., Seydoux J., Montani J. P., Dulloo A. G., Thorens B., Preitner F., Wymann M. P., Solinas G. (2011) PI3Kγ within a nonhematopoietic cell type negatively regulates diet-induced thermogenesis and promotes obesity and insulin resistance. Proc. Natl. Acad. Sci. USA , E854–E863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ray P. D., Huang B. W., Tsuji Y. (2015) Coordinated regulation of Nrf2 and histone H3 serine 10 phosphorylation in arsenite-activated transcription of the human heme oxygenase-1 gene. Biochim. Biophys. Acta , 1277–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng R., Heck D. E., Mishin V., Black A. T., Shakarjian M. P., Kong A. N., Laskin D. L., Laskin J. D. (2014) Modulation of keratinocyte expression of antioxidants by 4-hydroxynonenal, a lipid peroxidation end product. Toxicol. Appl. Pharmacol. , 113–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kauser S., Westgate G. E., Green M. R., Tobin D. J. (2011) Human hair follicle and epidermal melanocytes exhibit striking differences in their aging profile which involves catalase. J. Invest. Dermatol. , 979–982 [DOI] [PubMed] [Google Scholar]
- 37.Maresca V., Flori E., Briganti S., Mastrofrancesco A., Fabbri C., Mileo A. M., Paggi M. G., Picardo M. (2008) Correlation between melanogenic and catalase activity in in vitro human melanocytes: a synergic strategy against oxidative stress. Pigment Cell Melanoma Res. , 200–205 [DOI] [PubMed] [Google Scholar]
- 38.Vernia S., Cavanagh-Kyros J., Barrett T., Tournier C., Davis R. J. (2016) Fibroblast growth factor 21 mediates glycemic regulation by hepatic JNK. Cell Rep. , 2273–2280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sabio G., Cavanagh-Kyros J., Ko H. J., Jung D. Y., Gray S., Jun J. Y., Barrett T., Mora A., Kim J. K., Davis R. J. (2009) Prevention of steatosis by hepatic JNK1. Cell Metab. , 491–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Son Y. O., Hitron J. A., Cheng S., Budhraja A., Zhang Z., Lan Guo N., Lee J. C., Shi X. (2011) The dual roles of c-Jun NH2-terminal kinase signaling in Cr(VI)-induced apoptosis in JB6 cells. Toxicol. Sci. , 335–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zheng R., Heck D. E., Black A. T., Gow A., Laskin D. L., Laskin J. D. (2014) Regulation of keratinocyte expression of stress proteins and antioxidants by the electrophilic nitrofatty acids 9- and 10-nitrooleic acid. Free Radic. Biol. Med. , 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Patel S. A., Velingkaar N. S., Kondratov R. V. (2014) Transcriptional control of antioxidant defense by the circadian clock. Antioxid. Redox Signal. , 2997–3006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hibi M., Lin A., Smeal T., Minden A., Karin M. (1993) Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. , 2135–2148 [DOI] [PubMed] [Google Scholar]
- 44.Fisher G. J., Talwar H. S., Lin J., Lin P., McPhillips F., Wang Z., Li X., Wan Y., Kang S., Voorhees J. J. (1998) Retinoic acid inhibits induction of c-Jun protein by ultraviolet radiation that occurs subsequent to activation of mitogen-activated protein kinase pathways in human skin in vivo. J. Clin. Invest. , 1432–1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.