

Abstract
Environmental contamination from legacy mine waste deposits is a persistent problem due to the long history of hard‐rock mining. Sulfide ore deposits can contain elevated levels of toxic metal(loid)s that, when mobilized by weathering upon O2 and H2O infusion, can result in groundwater contamination. Dry climate and lack of vegetative cover result in near‐surface pedogenic processes that produce fine‐particulate secondary minerals that can be translocated as geodusts leading to ingestion or inhalation exposure in nearby communities. In this study, in vitro bioassays were combined with synchrotron‐based X‐ray spectroscopy and diffraction to determine the potential risk for toxic element release from dust (PM10) samples into biofluid simulants. PM10 were isolated from across the oxidative reaction front in the top meter of tailings subjected to 50 years of weathering under semiarid climate and introduced to synthetic gastric and alveolar fluids. Aqueous concentrations were measured as a function of reaction time to determine release kinetics. X‐ray diffraction and absorption spectroscopy analyses were performed to assess associated changes in mineralogy and elemental speciation. In vitro bioaccessibility of arsenic and lead was highest in less‐weathered tailings samples (80–110 cm) and lowest in samples from the suboxic transition zone (40–52 cm). Conversely, zinc release to biofluids was greatest in the highly weathered near‐surface tailings. Results indicate that bioaccessibility of As and Pb was controlled by (i) the solubility of Fe2+‐bearing solids, (ii) the prevalence of soluble SO4 2−, and (iii) the presence of poorly crystalline Fe(III) oxide sorbents, whereas Zn bioaccessibility was controlled by the pH‐dependent solubility of the stable solid phase.
Keywords: in vitro bioassay, pharmacokinetic, XAS, arsenic (As), mine tailings, lead (Pb)
Key Points
Sulfide ore tailings undergo oxidative weathering that alters the speciation and lability of toxic metal(loid)s
Oxidative weathering of tailings decreases bioaccessibility in synthetic gastric fluid
The decrease in bioaccessibility with weathering can be attributed to an increase poorly crystalline ferric solids that are high‐affinity sorbents
1. Introduction
Tailings are the fine‐grained, uneconomical by‐products of sulfide ore processing (crushing, grinding, froth flotation, etc.) that, following deposition on a tailings pile, can pose health risks to proximal human populations (Lottermoser et al., 2011). Chalcophilic toxic metal(loid)s (e.g., As, Pb, Zn, Cd) are often associated with sulfide ore bodies (Misra, 2012) that contain valuable phases such as chalcopyrite (CuFeS2). The exposure of discarded waste material (tailings) to ambient weathering in the presence of dioxygen and water results in oxidative dissolution of the sulfide tailings material (Boulet & Larocque, 1998) concurrent with release of toxic metal(loid)s to aqueous solution or transfer to secondary solid phases. These weathering processes can result in health risks to surrounding populations when they transform contaminants to more labile forms that are then transported in air as fugitive dusts. Airborne particulate matter with an effective spherical diameter of less than 10 μm (PM10) can be inhaled or ingested. The exposure risk of PM10 can be evaluated in vivo and in vitro. However, in order to examine pharmacokinetic release from PM10 in bodily fluids, in vitro bioassays are employed. These offer highly reproducible, rapid, and inexpensive evaluation of metal(loid) bioaccessibility (Deshommes et al., 2012), which is defined as the fraction of total solid phase contaminant concentration released into a biofluid simulant in a given time frame. The bioaccessible fraction is, therefore, an estimate of the contaminant fraction available for interaction with a target organ (the actual in vivo value of which distinguishes the bioavailable fraction). The motivation of the current work was the hypothesis that weathering reactions, which control the diagenesis of mine tailings under climatic forcing, lead to changes in speciation (molecular form) of metal(loid) contaminants that, in turn, affect their bioaccessibility.
1.1. Weathering Reactions That Control Metal(loid) Speciation
Following the initial exposure of pyrite‐rich mine wastes to oxygenated water at the Earth's surface, oxidative dissolution of pyrite (FeS2) leads to the release of sulfate (SO4 2−), ferrous iron (Fe2+), and acid (H+) to interstitial pore waters (Hayes et al., 2014). Trace elements, including As, Pb, and Zn are released by oxidative dissolution of minor sulfide phases such as arsenopyrite (FeAsS), galena (PbS), and sphalerite (ZnS) (Basu & Schreiber, 2013; Rimstidt et al., 1994), potentially resulting in the formation of metastable secondary phases from supersaturated solutions (Blowes & Jambor, 1990). This collection of secondary phases may include metal sulfates such as melanterite [FeSO4·7H2O], copiapite [FeIIFe4 III(SO4)6(OH)2·22H2O], coquimbite [Fe2(SO4)3], gypsum (CaSO4·2H2O), anglesite (PbSO4), and goslarite (ZnSO4·7H2O) (Jambor et al., 2000). Further oxidation and acidification leads to the formation of tertiary Fe(II) and Fe(III) (hydr)oxides and (oxy)hydroxysulfates such as goethite [α‐FeOOH], ferrihydrite [5Fe2O3·9H2O], hematite [Fe2O3], schwertmannite [Fe8O8(OH)6(SO4)·10H2O], and jarosite [KFe3(SO4)2(OH)6] (Bigham et al., 1996; Bigham & Nordstrom, 2000). As the pH decreases due to further sulfide oxidation, ferric (hydr)oxides become increasingly unstable and (oxy)hydroxysulfates such as schwertmannite and jarosite become more prevalent (Bigham & Nordstrom, 2000). However, in semiarid environments, wet‐dry cycles and a lack of pore water through‐flux could facilitate the persistence of metastable minerals such as metal‐sulfate salts and Fe(III) (oxy)hydroxides that are typically solubilized under more humid and comparably acidic conditions (Meza‐Figueroa et al., 2009; Navarro et al., 2004). As tailings oxidation progresses, toxic element species (especially Pb2+ and AsO4 3−) become associated with these neo‐formed secondary phases by adsorption and coprecipitation (Fuller et al., 1993; Raven et al., 1998; Root et al., 2015; Savage et al., 2005; Waychunas et al., 1993).
As a result of its toxicity and association with health problems such as hyperkeratosis, diabetes, immunological diseases, bronchitis, and cardiac and renal diseases (Abernathy et al., 1999; Ahsan et al., 2006), arsenic is recognized as a potent carcinogen and major threat to human health and is therefore listed as number one on the Agency for Toxic Substances and Disease Registry (ATSDR) priority list of hazardous substances (ATSDR, 2016). In sulfide mine tailings, arsenic typically originates in sulfides such as arsenopyrite, As‐rich pyrite, orpiment (As2S3), and realgar (α‐As4S4) (Lengke et al., 2009; O'Day, 2006). Several extended X‐ray absorption fine structure (EXAFS) spectroscopy studies have found that arsenate (AsO4 3−) is the primary weathering product in the presence of sufficient oxygen, but that it can occur in multiple forms including the mineral scorodite (FeAsO4), poorly crystalline ferric arsenates, bidentate‐binuclear inner‐sphere adsorption complexes the surface of Fe(III) (oxyhydr)oxides, or coprecipitates with ferrihydrite or jarosite (Foster et al., 1998; Paktunc et al., 2004; Root et al., 2015; Sherman & Randal, 2003). Previous studies of As in mine waste have suggested that oxidative weathering increases bioavailability because AsO4 3− bearing phases are typically more soluble than the recalcitrant sulfide phases mentioned earlier (Craw & Bowell, 2014).
In contrast to arsenic, the primary detrimental effect of lead on human health is its neurotoxicity (Bressler & Goldstein, 1991), particularly in a pediatric or prenatal context (Lidsky & Schneider, 2003), placing it second on the ATSDR priority list of hazardous contaminants (ATSDR, 2016). In calcareous sulfide ore deposits, lead is typically associated with the sulfide mineral galena as well as metal carbonates (Ostergren et al., 1999). Oxidative weathering redistributes Pb2+ into secondary weathering products such as cerussite (PbCO3), anglesite (PbSO4), plumbojarosite [Pb0.5Fe3(SO4)2(OH)6], and the surfaces of amorphous metal oxide particles (particularly ferrihydrite) (Hayes et al., 2009; Morin et al., 1999; Ostergren et al., 1999; Root et al., 2015). In partially oxidized transition zones, lead mineralogy is dominated by Fe(III) oxide and sulfate phases that give way to plumbojarosite following oxidation and acidification (Hayes et al., 2012). However, in semiarid environments, there is evidence for persistence of metastable anglesite along with other metal sulfate salts (Hayes et al., 2012; Meza‐Figueroa et al., 2009). Similar to lead, Zn speciation undergoes a transformation from ZnS to ZnCO3 and ZnSO4·nH2O phases over the course of oxidative weathering (Hayes et al., 2011, 2009; Root et al., 2015).
1.2. Iron King Superfund Site as a Model Weathering System and Geodust Source
Arsenic, lead, and zinc are present at highly elevated solid phase concentrations (0.3 wt % ea) in the Iron King Mine Humboldt Smelter Superfund (IKMHSS) site—a legacy site north of Phoenix, AZ, United States that has been the subject of ongoing study (Csavina et al., 2012; Gil‐Loaiza et al., 2016; Honeker et al., 2016; Ramirez‐Andreotta et al., 2016; Stovern et al., 2016; Valentin‐Vargas et al., 2014). We previously reported on the mineral weathering trajectory and related speciation changes of major (Fe, S; Hayes et al., 2014) and trace (As, Pb, Zn; Root et al., 2015) elements in the IKMHSS bulk tailings by analyzing samples as a function of depth in the top 2 m of the tailings pile. The top 2 m were found to comprise the full sulfide tailings weathering continuum, including (i) a deep zone containing dominantly unweathered pyrite, (ii) a slightly shallower suboxic zone with diagenetic carbonates in addition to primary sulfides, (iii) a suboxic to partially oxidized transition zone dominated by Fe(III) oxyhydroxides, and (iv) an oxidized zone at the land surface composed primarily of Fe(III) oxyhydroxysulfates (Hayes et al., 2014). These mineralogical changes, apparent as a visible color change from gray at depth to orange at the surface (Figure S1 in the supporting information), are also associated with important transitions in As, Pb, and Zn molecular speciation. These elements were found to be associated with sulfide and carbonate phases in the anoxic zone, Fe (oxyhydr)oxide phases in the transition zone, and jarosites and efflorescent sulfate salts near the tailings surface (Root et al., 2015). Trace element release from tailings material into aqueous (including biological) fluids depends on the dissolution/precipitation and adsorption/desorption reactions associated with resident mineral phases.
Because of the semiarid climate of the IKHMSS site, wind dispersion of dust particles is the primary means by which residents of the nearby community of Dewey‐Humboldt, AZ are brought into contact with toxic metal(loid)s present in the tailings (Stovern et al., 2014). The fine texture of the deposited tailings and the lack of vegetative cover (due to the acidic pH and toxicity of the surface) generates a high risk of particle detachment and downwind transport into the nearby community, especially for the fine (clay and silt) particulate fraction (<40 μm), for which transport has been found to extend over regional scales (Csavina et al., 2012). Significant human health hazards result from inhalation or ingestion of contaminated particles (Morena et al., 2007) or later exposure to contaminated soil, water, or home garden crops following particle deposition (Ramirez‐Andreotta et al., 2013). When dust particles are ingested or inhaled, geochemical interactions between the particles and biofluids modulate health effects by controlling the rate of toxic substance release to solution (Kelley et al., 2002). Bioavailability studies have shown that actual exposure to toxic substances in ingested/inhaled materials may be only a fraction of the total contaminant load (Rodriguez et al., 1999; Ruby et al., 1996). The bioaccessibilities of toxic contaminants are controlled by their dissolution kinetics in gastric or lung fluid. The use of synthetic biofluids in in vitro experiments enables the estimation of time‐dependent changes in effective doses for subsequent correlation with toxicological response. Therefore, the objective of this study was to measure the bioaccessibility of As, Pb, and Zn in fine particulate matter as a function of tailings weathering extent, and hence depth across the weathering front. We hypothesized (i) that fractional bioaccessibility would be highest in the near surface of the tailings pile where secondary and tertiary phases are most prevalent, and lower at depth, and (ii) that bioaccessibility would correlate with speciation of the contaminants as measured by molecular spectroscopy.
2. Materials and Methods
2.1. Site Description
The IKHMSS, located near Dewey‐Humboldt, Arizona, United States, is a disused mine site that previously exploited a sulfide intrusive deposit located in a pre‐Cambrian metamorphosed andesitic tuff within the Bradshaw Mountains (Creasey, 1952). Before the mine halted operations in 1969, large amounts of metals [16,800 t Pb, 52,200 t Zn, 1,800 t Cu] were produced from 1906 to 1947, and small amounts of precious metals were extracted [105 t Ag, and 3390 kg Au] (Creasey, 1952), with the remaining wastes deposited as a tailings pile into a topographical depression located immediately east of the mine shaft (see Hayes et al., 2014; Root et al., 2015 for site details). A total of approximately 4 · 106 m3 of tailings were deposited at this site (Figure 1).
Figure 1.

IKMHSS tailings pile. (a) Aerial view of tailings site and adjacent town of Dewey‐Humboldt, AZ to NE (from Google Earth™), (b) side view of mine tailings pile from NE, (c) view of sample pit excavation and sampling.
Since tailings deposition ended in 1969, the tailings surface has been subjected to nearly a half century of oxidative weathering of the original material, initially composed primarily of sulfide minerals including pyrite, arsenopyrite, sphalerite (ZnS), and galena. Metal(loid) contaminants such as As, Pb, and Zn that were already present at concentrations above remediation limits, were released from larger particulate sulfide forms during this oxidative weathering process and incorporated into colloidal precipitates that today pose a significant risk to the nearby community due to the potential for wind erosion and the spread of contaminated dust into populated areas (Csavina et al., 2012).
The tailings profile is subjected to periodic water erosion events that remove the oxidized surficial tailings and reveal the deeper zone of sulfidic particulates to weathering influences, as well as potential wind and water transport. Hence, particulate matter across the full extent of the weathering gradient is potentially susceptible to airborne transport off‐site. For this reason, bioaccessibility studies were conducted on the full reaction front from oxidized to sulfidic tailings material following the isolation of fine particulate matter (PM10), as discussed in detail below. In addition, this approach enables the results of this study to be used to help predict the bioaccessibility of toxic metal(loid)s at other semiarid sites where the extent of oxidative weathering may be different.
2.2. Sample Collection and PM10 Isolation
A tailings profile was collected by excavating a fresh pit within the top 2 m, thereby exposing the redox gradient from surface oxidized tailings to the reduced sulfide stable regions at depth (Figure S1). Samples were designated A–G from the surface to 1.1 m below the surface. Boundaries of the genetic horizons were determined based on color, texture, and the presence or absence of other morphological features such as concretions and cementation. Approximately 20 kg of material were composited for each depth increment, homogenized, sieved to 2 mm at the site, and then stored at 4°C. Samples were air dried, with C–G depths being dried under anoxic conditions in a glove box to avoid oxidation (H2:N2 = 2:98, Coy, MI), prior to separation of the PM10 dust fraction.
The respirable fraction of each dried sample was isolated using a laboratory dust fractionator (Figure S2), composed of a dust generation chamber, a Teflon®‐coated fractionation module (Cyclone PM10 Model URG‐2000‐30EA, URG Corp., NC,), and a collection chamber. Details regarding the theory and functioning of the dust fractionator are discussed elsewhere (Gonzales et al., 2014). This device separated the transported dust into two size fractions; a coarse fraction (diameter > 10 μm) and a fine fraction (diameter < 10 μm, i.e., PM10). The obtained fine fraction (PM10) was employed in the experiments described below.
2.3. In Vitro Bioaccessibility Assays
To investigate the in vitro bioaccessibility of tailings fine particulate matter (PM10), two synthetic biofluids—designed to mimic aqueous geochemical conditions in the human gastrointestinal and respiratory systems—were used. The synthetic gastric fluid (SGF) was composed of a 0.4 M glycine solution with the pH adjusted to 1.80 using reagent‐grade concentrated HCl, adapted from a standard operating procedure developed by the Solubility/Bioavailability Research Consortium (Kelley et al., 2002). Lung fluid simulant (Boisa et al., 2014) was synthesized as a modified Gamble's solution amended to include organic salts and amino acids, with the pH adjusted to 7.4 using 1 M HCl (see Table S1 for precise composition) (Menka et al., 2014; Scholze & Conradt, 1987; Takaya et al., 2006). When agitated continuously under physiological conditions, these synthetic biofluids mimic the conditions present at the particle‐fluid interface following tailings ingestion or inhalation. Synthetic biofluids were stored at 4°C until use.
To mimic the physiological conditions of the stomach, all gastric IVBAs were performed at 37°C maintained by a shaker incubator (Bioexpress Genemate Mini Incubator Shaker, UT) and under anoxic conditions within an anoxic chamber (Coy, MI). A measured mass of 10.0 mg of PM10 tailings from each layer were added to 1.8 mL polypropylene microcentrifuge tubes and reacted with 1.00 g of synthetic gastric fluid in triplicate. The reaction vessels were then covered with aluminum foil to avoid chemical changes due to photocatalysis (Cornell & Schwertmann, 2003) and agitated in an end‐over‐end rotator within the anoxic chamber for periods between 10 s and 4 hr (each time step was run in triplicate). The reactions were then terminated and solids separated by centrifugation at 5,000 g (Flexifuge, Argos) for 1 min and 0.5 mL of supernatant were aspirated and diluted in ~4.5 mL reagent‐grade 0.01 M HCl (pH 2.0, same as synthetic gastric fluid). This 5 mL sample was then filtered immediately using an acid‐washed Pall Acrodisk 0.2 μm syringe filter and diluted in 2% w/w HNO3 for acid preservation prior to elemental analysis by inductively coupled plasma mass spectrometry (ICP‐MS, Agilent 7700, Santa Clara, CA) and sulfate analysis by ion chromatography (IC, Dionex, LC20 Chromatograph with ED40 Electrochemical Detector). The remaining supernatant was then aspirated and discarded and the pellet was washed in ultrapure deionized water, and recentrifuged for 1 min at 5,000 g. The supernatant solution was aspirated and discarded, and solid samples were frozen and stored at −125°C prior to solid phase analysis.
The procedure for the lung IVBAs was similar to that for the gastric IVBAs, except that lung fluid simulants were employed and reactions were conducted under oxic conditions. After the batch reactions were terminated, the supernatant solution was diluted in ultrapure deionized water without acidification and filtered. Further dilutions were acid preserved with 2 w/w% HNO3 prior to chemical analysis using ICPMS and IC. The bioaccessible fraction (BAc) is defined as the experimentally determined element of interest concentration in a synthetic biofluid divided by the total concentration, determined by lithium metaborate fusion with acid digestion and analysis by ICPMS. Details of ICP‐MS precision are in the SI. The initial metal(loid) release rates, which correspond to dissolution rates under conditions far from equilibrium, were determined from the first time point (farthest from equilibrium) for triplicate reactors; d[X]/dt = (moles X released in 10 s)/10 s.
2.4. Synchrotron‐Based PM10 Characterization Before and After Reaction
Concentrations of Fe, As, Pb, and Zn in the PM10 solids were determined using microwave‐assisted aqua regia digestion by EPA Method 3050B, and total elemental analysis by ICP‐MS. X‐ray diffraction data were collected on beam line (BL) 11–3 at the Stanford Synchrotron Radiation Lightsource (SSRL) operating at ~12,735 eV (λ = 0.976 Å). The instrument was operated in transmission mode with a focused spot size of 150 μm, and the scans were collected using a MARCCD detector calibrated to a LaB6 standard. Approximately 0.01 g of unreacted or reacted PM10 samples were packed between two layers of clear tape (Scotch TapeTM). The samples were rastered by 1 mm in x and y directions during six 15 s exposures. Laue patterns were averaged and integrated to 2‐D diffractograms using the Python‐coded beamline software Wxdiff (http://openhub.net/p/wxdiff). The background from sample support tape was subtracted with no further background removal to include contributions from X‐ray amorphous phases. The patterns were converted from 0.976 Å to the conventional Cu Kα 1.54 Å for pattern identification using the X‐Pert HighScore Plus software (PANalytical).
Speciation of As, S, and Fe in reacted and unreacted PM10 samples was determined using synchrotron X‐ray absorption spectroscopy (XAS) on BLs 4–3 (Fe, S) and 11–2 (As) at SSRL. A double‐crystal monochromator (Si [220] for As; Si [111] for Fe, S) detuned 40% to suppress higher‐order harmonics was used with either a 100‐element Ge detector (BL 11–2 for As) or a passivated implanted planar silicon (PIPS) detector (BL 4–3 for Fe and S). Vertical slits of 2 mm and horizontal slits adjusted to between 2 and 10 mm were used to maximize the fluorescence signal without saturating the detector. Energy calibration was achieved for S by designating the first peak of Na2S2O3 as 2,472.04 eV, for Fe by designating the maximum of the first derivative of an Fe foil as 7,112 eV, and for As by designating the As foil first derivative peak as 11,867 eV. Both Fe and As measurements were collected at <77 K with a liquid N2 cryostat used to keep the operating temperature well below the Debye temperatures for Fe (470 K) and As (282 K) (Kittel, 2005) to minimize spectral contributions from lattice vibrations and beam‐induced chemical changes to the sample. At least three scans of all reacted and unreacted tailings samples were collected in fluorescence mode. Collection of As and Fe data was achieved with Z‐1 filters with Soller slits used to minimize the contributions of elastic and Compton scattering.
Spectral data quality assessment and averaging was performed using SIXPack v1.2 (Webb, 2006). Background subtraction and normalization to the postedge oscillations of averaged spectra were done by fitting a polynomial spline function to the linear preedge parameters (Fe, As: −150 eV to −30 eV) and a postedge quadratic function extending to the end of the scanned region using the Athena program v0.9.24 (Ravel & Newville, 2005).
Linear combination fitting (LCF) of the Fe and As EXAFS and S XANES spectra were processed for quantitative analysis using Athena software (Ravel & Newville, 2005). Fits to the Fe EXAFS (k 3‐weighted, k = 2–10 Å) were limited to four or less components and As EXAFS (k 3‐weighted, k = 2–10 Å) to three or fewer components from a reference library of over 20 references unless other components were indicated by secondary analysis such as X‐ray Diffraction (XRD) or Fourier Transform‐Infrared Spectroscopy (Hayes et al., 2014; Root et al., 2015). The reference spectra were obtained from SSRL BLs 4–3 and 11–2 under similar conditions. All LCF components were constrained to be nonnegative, but the sums of the components were not forced to unity. The LCF software returns a linear combination of reference standards as well as fit statistics comparing the linear combination to the actual input spectrum, and the best fits are determined by comparing the R and χ2 values of each linear combination as well as the feasibility of the existence of the best fit references in each sample.
2.5. Geochemical Modeling
Aqueous geochemical data including known background electrolyte concentrations, measured total elemental concentrations, pH, and SO4 2− were used to calculate saturation indices of selected minerals at each measured time point in the reaction using the SpecE8 program in Geochemist's Workbench V10.0 (Bethke, 1994, 2008). Thermodynamic data were obtained from a built‐in database (thermo.dat.v8.R7) amended to include internally consistent thermodynamic constants for schwertmannite (Bigham et al., 1996) and a theoretical plumbojarosite endmember (Forray et al., 2010). Due to the very low solubility of Fe(III) oxide precipitates at circumneutral pH, all aqueous Fe in lung fluid was assumed to be Fe2+ (aq). For the gastric IVBA experiments, which were conducted under acidic (pH 1.8 conditions), aqueous Fe redox state was measured on selected samples after 2 hr using the ferrozine method (Stookey, 1970). The ferrozine determined ferrous/ferric ratio was used to infer the distribution of Fe between Fe(II) and Fe(III) species as a function of sample depth. On this basis, 100% of the Fe released from samples A–C, 80% released from samples D and E, and 50% released from samples F and G was designated as Fe3+ (aq). All thermodynamic data used for geochemical modeling are listed in Table 1.
Table 1.
Mineral Species Used for Geochemical Modeling
| Mineral name | Congruent dissolution reaction | Log K a | Refb | |
|---|---|---|---|---|
| Pb‐Jarosite |
|
−25.5 | 1 | |
| K‐Jarosite |
|
−11.0 | 2 | |
| H‐Jarosite |
|
−5.39 | 2 | |
| Na‐Jarosite |
|
−5.32 | 2 | |
| Schwertmannite |
|
18.0 | 3 | |
| Ferrihydrite | Fe(OH)3 + 3H+ ⇄ Fe3+ + 3H2O | 3.2 | 2 | |
| Goethite | FeOOH + 3H+ ⇄ Fe3+ + 2H2O | 0.49 | 2 | |
| Hematite | Fe2O3 + 6H+ ⇄ 2Fe3+ + 3H2O | −1.42 | 2 | |
| Siderite |
|
−10.59 | 2 | |
| Smithsonite |
|
−10.9 | 2 | |
| Cerussite |
|
−13.2 | 2 | |
| Fe(OH)2 (am) | Fe(OH)2 (am) + 2H+ ⇄ Fe2+ + 2H2O | 13.49 | 2 |
log K = equilibrium constant at 25°C, IVBA conditions were at 37°C, which necessitated modeling with second constant (K 40°C) calculated with the Van't Hoff equation; , where ΔH° = standard enthalpy change, R = gas constant.
3. Results
3.1. Characterization of PM10 Tailings
3.1.1. Elemental Analysis
The Fe, As, Pb, and Zn concentrations obtained by EPA 3050B are summarized in Table 2. The Fe profile exhibits two concentration maxima (B, D), corresponding to accumulations of jarosite (5–20 cm) and ferrihydrite (30–40 cm), respectively. The As concentration reaches a maximum in the redox transition zone between 30 and 80 cm. Concentrations of Pb are at a maximum in the surface tailings (A–C), enriched to >10 g/kg at 5–20 cm (B), and relatively depleted at 40–52 cm (E). A Zn enrichment is noted at 30–40 cm relative to shallower zones, but Zn concentrations are much higher (~25 times) in deep tailings where the pH is above 4.
Table 2.
Chemical and Mineralogical Composition of the PM10 Fractionsa
| Depthb (cm) | pHc | Sd | Fed | ASd | Pbd | Znd | Major componentse | Minor componentsf | |
|---|---|---|---|---|---|---|---|---|---|
| A | 0–5 | 2.27 | 2310 | 3370 | 40.4 | 29.1 | 31.0 | qz, jrs, fh | gyp, chl, alb |
| B | 5–20 | 2.31 | 3330 | 4690 | 30.0 | 54.4 | 26.8 | qz, jrs | gyp, chl, alb, fh |
| C | 20–30 | 2.36 | 1850 | 2940 | 51.7 | 38.7 | 44.9 | qz, gyp, jrs, schw | chl, alb, fh |
| D | 30–40 | 2.10 | 1890 | 5480 | 77.0 | 26.0 | 184 | qz, gyp, jrs, fh | chl, alb |
| E | 40–52 | 2.92 | 2100 | 2700 | 114 | 16.9 | 104 | qz, pyr, fh, gyp | chl, alb, jrs |
| F | 52–80 | 4.90 | 1560 | 2380 | 85.4 | 24.2 | 763 | qz, pyr, chl | gyp, fh, ank, alb |
| G | 80–110 | 5.84 | 1690 | 2780 | 66.5 | 27.5 | 581 | qz, pyr, chl | fh, ank, alb |
Location of PM10 collection shown in Figure 1.
Sampling interval below the surface.
The pH measured on supernatant solution from 2:1 mass ratio of DI water to PM10.
Elemental concentration on air dry mass basis (mmol/kg).
Major components comprise ≥10% of the total PM10 composition based on XRD and XAS results.
Minor components <10% identified using XRD and XAS results. qz = quartz; jrs = jarosite; fh = ferrihydrite; schw = schwertmannite; gyp = gypsum; chl = chlorite; ank = ankerite; alb = albite; pyr = pyrite.
3.1.2. Mineralogy
The major and minor components of PM10 mineral assemblage from layers A–G are summarized in Table 2. The PM10 of the IKMHSS tailings profile exhibited the same mineral constituents observed in the bulk fraction (Root et al., 2015). However, the PM10 fractions were more heavily enriched in secondary precipitates (ferrihydrite, jarosite, etc.) relative to quartz and primary sulfides. They also showed higher concentration of associated metal(loids), with corresponding enrichment factors for PM10 relative to bulk tailings of 1.6 (Fe), 1.5 (As), 2.5 (Pb), and 1.2 (Zn), consistent with prior reports for other tailings (Kim et al., 2011). The gradient in composition of the horizons reflected a metal sulfide reaction front, with the mineralogy of the initially deposited material composed of pyrite, quartz, feldspar, chlorite, and carbonates (horizons F, G). The redox transition zone (horizons D, E) was dominated by a mix of pyrite, quartz, ferrihydrite, carbonates, and gypsum. The upper layers (A, B, and C) were dominated by gypsum, quartz, ferrihydrite, and jarosite. Schwertmannite, a ferric hydroxysulfate, was most prevalent at 20–30 cm depth but also detectable down to 52 cm.
3.1.3. Fe and As Speciation
The Fe speciation across the tailings profile from Fe Kα EXAFS (based on full spectra and linear combination fit [LCF] results shown in Figure S3) reveal a sharp near‐surface pyrite weathering sequence (Figure 2) consistent with the mineralogy determined by XRD (Table 2). Most of the Fe in the unweathered tailings was present in pyrite, with minor amounts of Fe‐rich chlorite and carbonate (ankerite). Shallower depths, subjected to moderate oxidative weathering (D, E), show ferrihydrite as the dominant Fe phase in the transition zone. Conversely, in layer C, the Fe mineralogy was dominated by a mixture of jarosite and schwertmannite. At the surface, where tailings are most acidic due to sulfide oxidation, the Fe phases are a mixture of jarosite and ferrihydrite.
Figure 2.
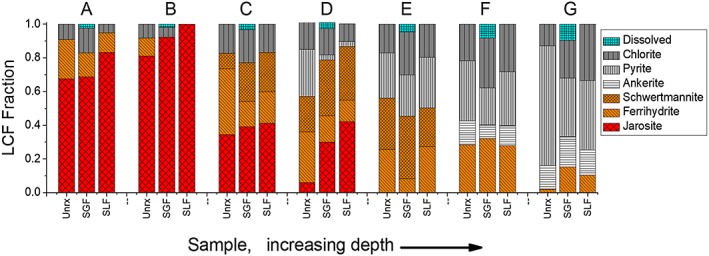
Solid phase iron speciation changes with depth for unreacted (Unrx), synthetic gastric fluid extracted (SGF), and synthetic lung fluid extracted (SLF) PM10 tailings at depths A–G (depths in Table 2). Fractional components of iron phases were determined by linear combination fits of the Fe Kα k 3‐EXAFS (extended X‐ray absorption fine structure). EXAFS and fits to reference spectra shown in Figure S3, fit summary in Table S3. The fraction that was dissolved, that is, the bioaccessible fraction, was calculated from the mass of Fe removed relative to the unreacted tailings. Sample D SLF was fit over a truncated range of 2–9 due to diminished data quality at high k (Å−1).
Prior work has shown that the As in unweathered bulk IKMHSS tailings is present as arsenopyrite, but with increased weathering, it is subjected to oxidation and redistribution to secondary Fe(III) bearing phases (Root et al., 2015). The prevalence of various As solid phase species in the isolated PM10 particles was determined on the basis of LCFs to As Kα EXAFS data (Figure S4). Quantitative summaries of these LCF results are presented in Figure 3. In sample G (90–110 cm), despite the predominance of primary minerals and small amounts of detectable ferrihydrite and jarosite in the bulk tailings, most of the As present at this depth in the PM10 fraction was incorporated into the ferrihydrite and jarosite secondary phases. Hence, the As speciation of PM10 differed significantly from that of the bulk tailings, which at 1 m depth was dominated by arsenopyrite (Root et al., 2015). Upon further oxidative weathering (i.e., with approach from depth to the surface) the As speciation is increasingly fit to arsenate adsorbed to ferrihydrite and jarosite (Figure 3). Variation of Pb and Zn speciation with depth in the bulk IKMHSS tailings (summarized in section 1.1) is discussed in detail in Root et al. (2015).
Figure 3.
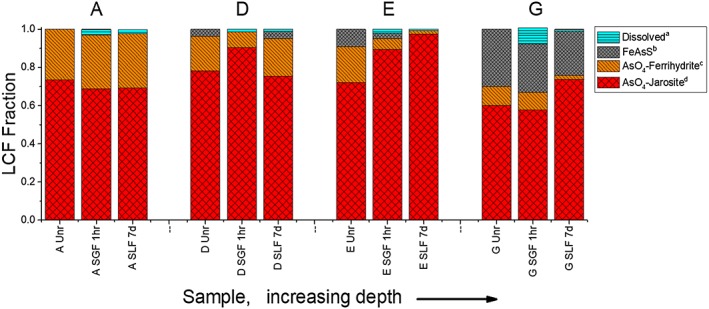
Solid phase arsenic speciation changes with depth for unreacted (Unrx), synthetic gastric fluid extracted (SGF), and synthetic lung fluid extracted (SLF) tailings. Fractional components of arsenic species were determined by linear combination fits of the As Kα k 3‐EXAFS (extended X‐ray absorption fine structure) of untreated (Unrx) and gastric (SGF) and lung (SLF) fluid reacted PM10 samples from depths A (0–5 cm), D (30–40 cm), E (40–52 cm), and G (80–110 cm). Fit components are detailed in Table S4, EXAFS, and fits to reference spectra shown in Figure S4. aThe dissolved fraction, operationally defined as bioaccessible, was calculated from the mass of As removed relative to the unreacted tailings; bFeAsS, arsenopyrite Ontario, Canada (Root et al., 2015); cAsO4‐Ferrihydrite = synthetic ferrihydrite with adsorbed arsenate (Gao et al., 2013); dsynthetic AsO4‐bearing jarosite (Root et al., 2015).
3.2. Kinetic Release in SGF
Kinetics of Fe, As, Pb, Zn, and SO4 2− release from PM10 upon reaction with acidic, anoxic SGF showed a strong dependence on sample location along the weathering profile (Figures 4, 5, 6), with trends that were element specific. All samples showed a similar trend in As release (Figure 4), with a rapid initial release followed by a slow approach to steady state between 15 min and 1 hr after initiation of the reaction. The gastric bioaccessibility (i.e., fractional release) for As at 1 hr ranged from 0.014 for sample D to 0.087 for sample G (Figure 4). In general, the deeper sulfidic tailings layers (F, G) showed the highest gastric As bioaccessibility, followed by the surface layers (A, B), with the redox transition zone (C–E) showing the lowest values. All initial release rates and equilibrium values for the SGF tests are recorded in Table 3, while Figure 6a relates gastric bioaccessibility to depth. Although samples C–E showed the highest initial release rates (0.11–0.12 mmolAs · kg−1 · s−1), the steady state gastric bioaccessibility values were lower for these samples than all of the others tested.
Figure 4.
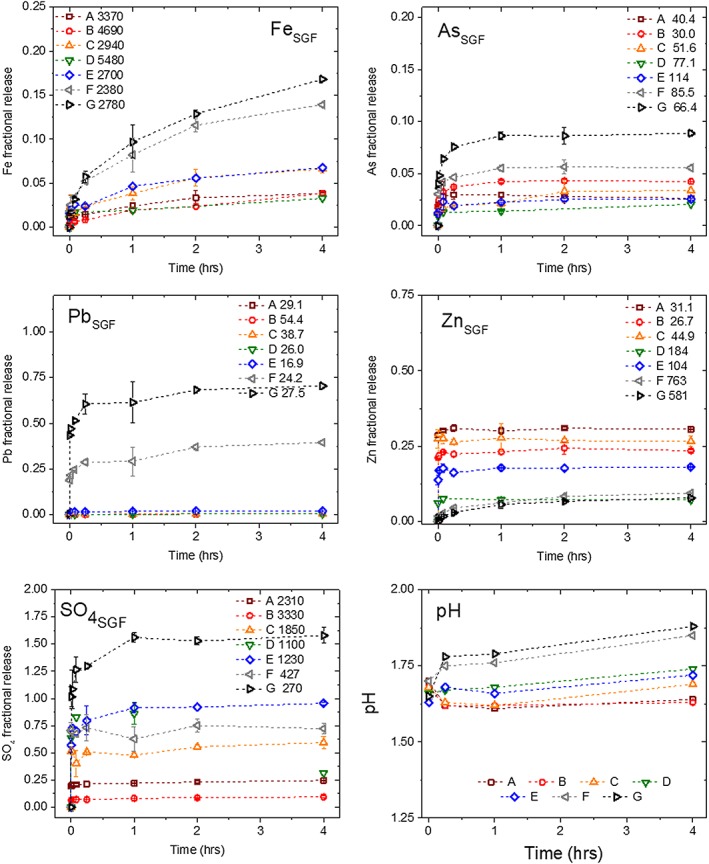
Fractional release of Fe, As, Pb, Zn, and SO4 and from PM10 tailings to solution during synthetic gastric fluid (SGF) IVBA with samples A–G, depths given in Table 2. Inset initial total concentrations of [Fe, As, Pb, Zn, SO4] are given in mmol/kg sample, plotted values are the fractional release relative to total PM10 concentration (Table 2). The inset total sulfate concentration is the fraction of the total S attributed to sulfate containing phases determined by S XANES and included all of the nonpyrite phases (Table S2). The pH panel shows the solution pH during 4 h IVBA reaction time. In all panels maroon squares show depth A(SGF), red circles show depth B(SGF), orange triangles show depth C(SGF), green inverted triangles show depth D(SGF), blue diamonds show depth E(SGF), gray left pointing triangles show depth F(SGF), and black right‐ pointing triangles show depth G(SGF). Please refer to electronic version for correct colors. Errors shown are standard deviations of triplicate measurements, error bars not visible are smaller than plot symbol.
Figure 5.
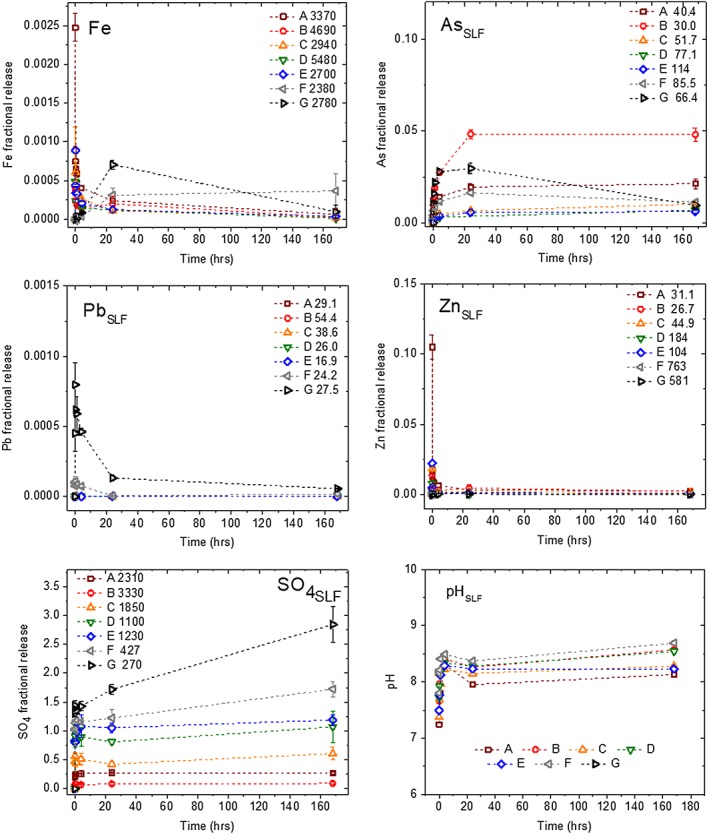
Fractional release of Fe, As, Pb, Zn, and SO4 from PM10 tailings to solution during synthetic lung fluid (SLF) IVBA with samples A–G for 7 days, depths given in Table 2. Initial total values of [Fe, As, Pb, Zn, and SO4] are mmol/kg. Plotted values are the fractional release relative to the initial total concentration, Pb was not detected in A–E. The plotted sulfate concentration is the fraction of the total S attributed to sulfate containing phases determined by S XANES, and included all of the nonpyrite phases (Table S2). The solution pH during 7 d IVBA reaction time is shown. Maroon squares show depth A(SLF), red circles show depth B(SLF), orange triangles show depth C(SLF), green inverted triangles show depth D(SLF), blue diamonds show depth E(SLF), gray left pointing triangles show depth F(SLF), and black right pointing triangles show depth G(SLF). Please refer to electronic version for correct colors. Errors shown are standard deviations of triplicate measurements, error bars not visible are smaller than plot symbol.
Figure 6.
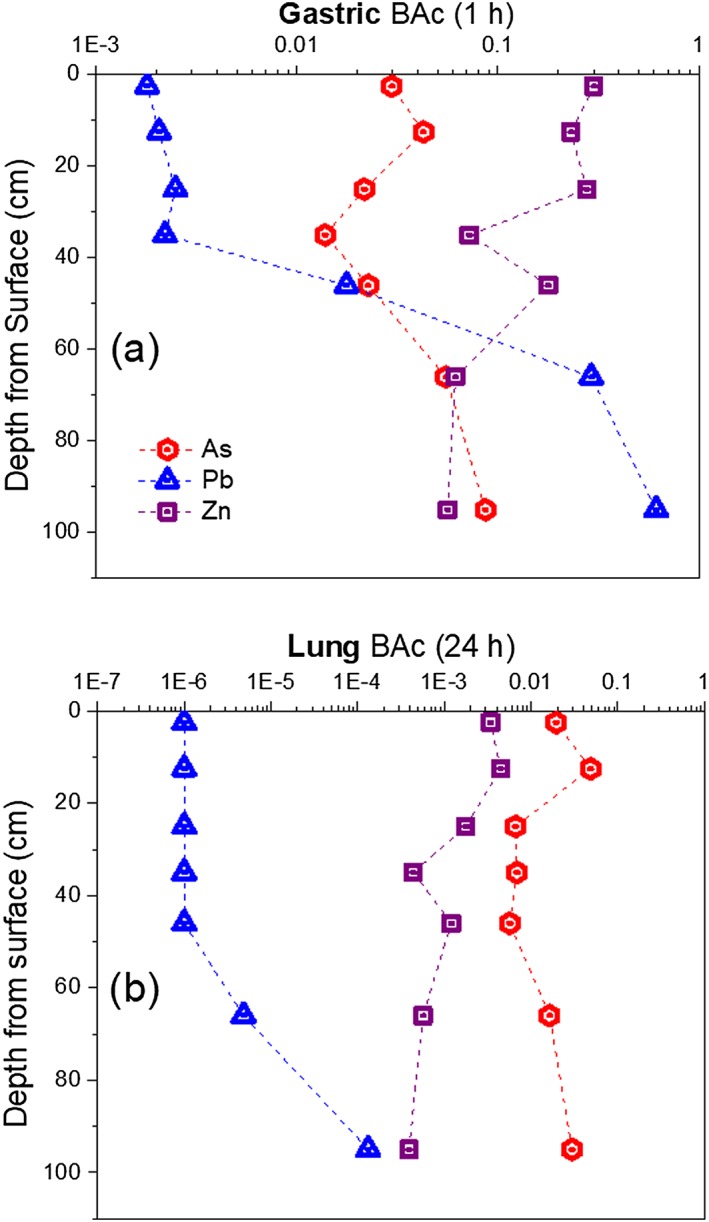
Bioaccessibility expressed as mole fraction released of As, Pb, and Zn from PM10 tailings to solution as a function of depth for (a) SGF IVBA interaction with samples A–G (panels A‐G, respectively) for 1 h; and (b) SLF for 24 h. All values of [As, Pb, Fe, Zn] are expressed in terms of mole fractional released to IVBA.
Table 3.
Initial Release Rates (After 10 s) and Percent Bioaccessibilities of As, Pb, Fe, and Zn in Gastric Fluid After 1 Hr
| Sample | Depth (cm) | Initial As release ratea (mmol · s−1 · kg−1 solid) | %IVBAAs1hr b | Initial Pb release rate (mmol · s−1 · kg−1 solid) | %IVBAPb1hr | Initial Fe release rate (mmol · s−1 · kg−1 solid) | %IVBAFe1hr | Initial Zn release rate (mmol · s−1 · kg−1 solid) | %IVBAZn 1hr |
|---|---|---|---|---|---|---|---|---|---|
| A | 0–5 | 0.074 | 2.96% | 0.000650 | 0.18% | 3.81 | 2.42% | 0.889 | 30.1% |
| B | 5–20 | 0.056 | 4.25% | 0.00145 | 0.21% | 1.92 | 1.94% | 0.567 | 23.1% |
| C | 20–30 | 0.063 | 2.16% | 0.00854 | 0.25% | 4.19 | 3.85% | 1.23 | 27.7% |
| D | 30–40 | 0.074 | 1.38% | 0.00244 | 0.22% | 5.83 | 1.92% | 1.12 | 7.21% |
| E | 40–52 | 0.136 | 2.26% | 0.0124 | 1.76% | 4.32 | 4.67% | 1.44 | 17.8% |
| F | 52–80 | 0.260 | 5.52% | 0.455 | 29.2% | 4.36 | 8.23% | 1.24 | 6.14% |
| G | 80–110 | 0.265 | 8.65% | 0.458 | 61.4% | 3.48 | 9.70% | 0.586 | 5.61% |
Initial release is operationally defined as the mmol/kg released from mine tailings after 10 s reaction by IVBA.
Percent IVBA after 1 hr reaction.
In contrast to the case for As, Pb release kinetics varied significantly across the profile. Rates of Pb release for samples A through E were less than 0.01 mmolPb · kg−1 · s−1 initially (Table 3) and decreased steadily until reaching apparent equilibrium (Figure 4). Conversely, samples F and G showed high initial release rates in the first 10 s, when most of the Pb released over the course of the experiment occurred, and rates slowed dramatically as concentration increased gradually before reaching apparent equilibrium. The total gastric bioaccessibility for Pb after 1 hr increased greatly with depth across the reaction front, with order of magnitude increases in BAc between samples D and E, and again between samples E and F (Table 3 and Figure 6a).
A depth‐dependent bimodal release of Zn in SGF fluid was observed with rapid release kinetics for all depths from 0 to 52 cm (A–E) and a slower release observed for 52–110 cm (Figures 4 and 6a). Nearly 100% of the released Zn was observed at the first sampling time point (10 s) from 0 to 52 cm depth, representing 20–30% of the total solid phase concentration, whereas subsequent time steps showed no additional release (Table 3). At depths F and G the release of Zn was not instantaneous, the fractional release was much lower (5–6%), and the release curve achieved a maximum at 2 hr (Table 3 and Figure 4).
Iron was released into gastric fluid at a rate considerably higher than that observed for Pb (Figure 4), consistent with the high total Fe concentration in the fine fraction across the tailings profile. After an initial release of Fe into solution that varied in magnitude from 0.058 mmolFe · kg−1 · s−1 for sample B to 0.616 mmolFe · kg−1 · s−1 for sample F (Table 3), the Fe concentration in solution did not appear to reach equilibrium over the course of the SGF treatment. The BAc for Fe after 1 hr of reaction time varied by fivefold, from 0.02 for sample D to 0.1 for sample G (Table 3).
Sulfate release into gastric fluid also showed depth dependence with highest values for transition zone samples (C–E). All of the samples showed a large initial release of SO4 2− in the first 10 s of reaction, but the magnitude of this release was highest in samples C–E, where XRD indicated the largest proportion of sulfate salts, for example, gypsum (Figure 4). Following this initial release, sulfate concentrations in the extraction fluid continued to increase for all of the samples, with apparent equilibrium being reached between 15 min and 2 hr, depending on the sample (Figure 4).
The dissolution/precipitation reactions occurring during reaction of tailings material with gastric fluid consume or release H+ and OH− ions, therefore pH measurements at each time point in the reaction were recorded (Figure 4, pH). After an initial decrease in pH following tailings addition to gastric fluid, which was more pronounced in the acidic surface tailings, the pH increased progressively until the reaction was terminated after 4 hr. This observed increase in pH was greater for the sulfidic tailings than the oxidized tailings closer to the surface.
Saturation indices (SI) of SGF solutions with respect to selected Fe(III) (oxyhydr)oxide and oxyhydroxysulfate species evolved over the course of the reaction (Figure S5). As Fe was released from oxidized surface samples, solutions approached saturation with respect to ferrihydrite asymptotically. Although solutions were supersaturated relative to goethite and hematite, no precipitation of these phases was observed, as discussed below. Solutions remained below saturation for H3O‐jarosite while SI was >10 for plumbojarosite (Forray et al., 2010) in all samples. Solutions were undersaturated with respect to schwertmannite (SI < −8) in all samples (Bigham et al., 1996).
3.3. Reaction Kinetics in Synthetic Lung Fluid
For the circumneutral, oxic synthetic lung fluid (SLF) experiments, arsenic release patterns were similar across samples A through E; for all of these samples, rapid initial releases during the first 4 hr were followed by a more gradual increase in concentration toward apparent equilibrium between 4 and 24 hr. Whereas As concentrations in those samples stabilized after 24 hr, the As concentrations in samples F and G showed large decreases of 30% (F) and 70% (G) after 24 hr, indicating that an As removal mechanism became active at longer times for these deeper samples (Figure 5).
Samples A–E showed no detectable releases of Pb into lung fluid because concentrations were below the ICP‐MS detection limit (method detection limit ~10 μg/kg). Conversely, reaction of samples F and G with lung fluid resulted in an initial Pb release of up to 0.0012 mmolPb · kg−1 · s−1 before reaching equilibrium values at low Pb concentrations, near the limit of detection (Figure 5).
The release of Zn in SLF fluid was greatest at shallow depths (0–5 cm) and short exposure times, with a release of over 10% of the total Zn at the first time point (10 s) (Figure 5). Subsequent aqueous concentrations decreased to near the detection limit for the remaining time points, and was <0.5% at all depths by 24 hr (Table 4).
Table 4.
Initial Release Rates and Percent Bioaccessibilities for As, Pb, Fe, and Zn in Lung Fluid
| Sample | Depth (cm) | Initial As release ratea (mmol · s−1 · kg−1 solid) | %IVBAAs24h b | Initial Pb release ratea (mmol · s−1 · kg−1 solid) | %IVBAPb24h b | Initial Fe release ratea (mmol · s−1 · kg−1 solid) | %IVBAFe24h b | Initial Zn release ratea (mmol · s−1 · kg−1 solid) | %IVBAZn24h b |
|---|---|---|---|---|---|---|---|---|---|
| A | 0–5 | 0.034 | 1.95% | <0.00001 c | <0.0002% c | 0.833 | 0.024% | 0.326 | 0.342% |
| B | 5–20 | 0.011 | 4.83% | <0.00001 c | <0.0001% c | 0.183 | 0.020% | 0.054 | 0.445% |
| C | 20–30 | 0.0093 | 0.660% | <0.00001 c | <0.0001% c | 0.266 | 0.014% | 0.040 | 0.177% |
| D | 30–40 | 0.011 | 0.687% | <0.00001 c | <0.0002% c | 0.555 | 0.009% | 0.027 | 0.044% |
| E | 40–52 | 0.015 | 0.567% | <0.00001 c | <0.0003% c | 0.240 | 0.013% | 0.019 | 0.120% |
| F | 52–80 | 0.021 | 1.63% | 0.00018 | 0.00048% | n.d. | 0.031% | 0.011 | 0.057% |
| G | 80–110 | 0.032 | 2.96% | 0.0012 | 0.0132% | n.d. | 0.071% | 0.007 | 0.039% |
Initial release is operationally defined as the mmol/kg released from mine tailings after 10 s reaction by IVBA.
Percent IVBA after 24 hr reaction time.
In italics, n.d. = not detected, the result was below the limit of detection, where LOD = method detection limit (MDL) × dilution factor (~ ×100), ICP‐MS [Fe]MDL = 0.03 μmol/L, [Zn]MDL = 0.013 μmol/L, [As]MDL = 0.002 μmol/L, [Pb]MDL = 0.0001 μmol/L.
Iron release from samples A–E to lung fluid displayed kinetic behavior similar to that of Pb, with an initial release of 0.183 (B) to 0.833 (A) mmolFe · kg−1 · s−1 followed by a decrease to equilibrium concentrations after approximately 4–24 hr (Figure 5). Samples F and G released Fe to lung fluid at a low initial rate, but the aqueous Fe concentrations accumulated slowly over time, eventually reaching apparent equilibrium at a slightly higher level than that for the shallower depth samples. Solution phase Fe concentrations for sample G then decreased by about 85% between 24 hr and 7 days reaction time. Initial release rates and bioaccessibilities of Fe in lung fluid at 24 hr are given in Table 4.
While sulfate is a reagent used to prepare synthetic lung fluid, releases of sulfate were apparent from concentrations significant in excess of the amount added initially (560 μM). Sulfate releases from all samples to lung fluid were large initially; within the first 4 hr of over 80% of the total dissolved over the course of the reaction (Figure 5). Afterward, sulfate release from samples A, B, and D reached apparent equilibrium after about 24 hr, while release from samples C–F continued to increase until the end of the reaction.
All reactions showed an initial small decrease in pH immediately after the samples were contacted with SLF, followed by a steady increase in pH as the reaction progressed before termination after 7 days. This increase in pH was highest for the deeper sulfidic tailings samples F and G, with pH of each reaction increasing to above 8.7 at the end of these experiments. It is noteworthy that this increase in pH was concurrent with decreases in Fe and As solution concentrations.
SI were highly dependent on the chosen redox state of Fe in the SLF solutions. Assigning all aqueous Fe to Fe(III) resulted in solution phase supersaturation with respect to ferrihydrite, goethite, hematite, and K‐jarosite early in the reaction, with the saturation index decreasing as Fe precipitated out of solution (Figure S6). The SI for ferrihydrite remained greater than 5 in all samples. Assigning all aqueous Fe to Fe(II) resulted in supersaturation of cerussite (PbCO3), smithsonite (ZnCO3), and siderite (FeCO3) in all reactions, especially within the first hour of reaction time, and supersaturation of solution with respect to Fe(II) hydrolysis products was observed for sample G (Figure S6).
The IVBA values for As, Pb, and Zn in SGF at 1 hr and SLF at 24 hr, which are the times most commonly used in standard metrics of BAc for these biofluid simulants, are summarized to show depth‐dependent trends in Figure 6. These data indicate inverse trends in BAc values for Pb and Zn. For both gastric and lung simulants, Pb BAc values were highest for the sulfide‐rich PM10 resident at depth and lowest for the oxidized material near the surface, whereas Zn showed higher BAc values for the oxidized surficial materials than the primary sulfides at depth. The depth‐dependent trends for As are more complex but also consistent across biofluid types. For As, the oxidized surface tailings (A, B) and the reduced layers (F, G) showed the highest BAc values, with the layers comprising the redox transition zone (C–E) showing the lowest values (Table 4 and Figure 5).
3.4. Changes in IVBA‐Reacted PM10 Samples
The PM10 samples were subjected to solid phase characterizations using synchrotron‐based XAS and XRD techniques both before and after reaction with SGF and SLF solutions. Iron K‐edge EXAFS data are reported prereaction and postreaction for all sample depths (Figures 2 and S3) and As K‐edge EXAFS data are reported for samples A, D, E, and G (Figures 3 and S4). Results from prereaction and postreaction XRD analyses are presented in Figures S7 and S8 for samples A, E, and G to represent the surface, redox transition, and stable sulfide zones, respectively.
3.4.1. Solid Phase Transformations of Surface Samples in SGF
Initial mineralogical composition of the surface samples (A–C in Figure S1a) included quartz, jarosite, gypsum, ferrihydrite, and chlorite. Rapid sulfate release data are consistent with dissolution of kinetically labile sulfates including gypsum and other efflorescent sulfate salts (Figure S7). XRD peaks assigned to jarosite in sample A decreased following the first time step followed by alternating periods of jarosite dissolution and precipitation (Figure S7). There was minimal change to the quartz diffraction peaks, which provided a baseline for variation in other diffraction peaks (Figure S7). Iron XAS data showed an increase in jarosite‐like coordination following reaction with SGF with a corresponding decrease in ferrihydrite (Figures 2 and S3). The most dramatic change was observed for depth B, where ferrihydrite‐like Fe coordination was eliminated from Fe XAS detection after 1 hr of reaction time. In contrast, arsenic XAS showed little or no shift in the prevalence of AsO4 in jarosite‐like versus ferrihydrite‐like coordination in the surface PM10 (Figures 3 and S4).
3.4.2. Redox Transition‐Zone PM10 Alteration in SGF
Prior to treatment with SGF, the suboxic transition zone samples (D and E in Figure S1a) were composed of a mix of pyrite, ferrihydrite, schwertmannite, quartz, gypsum, and a minor amount of jarosite. The sulfate salt fraction (represented by gypsum) of these samples dissolved immediately upon contact with SGF (Figure S7). Ferrihydrite loss coupled with jarosite and schwertmannite precipitation was indicated in XRD and Fe K‐edge EXAFS data for postreacted sample D, whereas in sample E, ferrihydrite loss resulted in schwertmannite precipitation, but no indication of jarosite formation, according to Fe EXAFS and S XANES data (Figure 2 and Tables S2 and S3). The behavior of As in the transition zone samples varied by depth; sample D showed redistribution of As from jarosite‐like to ferrihydrite‐like coordination, while sample E showed As transfer into jarosite‐like coordination (Figure 3). The As EXAFS were best fit to arsenopyrite and two As(V) phases (jarosite and ferrihydrite). As such, the As(V) redistribution between ferrihydrite and jarosite at depth D may not be resolved in the As EXAFS spectrum.
3.4.3. Deep Subsurface PM10 Alteration in SGF
The deeper samples F and G were composed primarily of pyrite, quartz, carbonates, chlorite, and smaller amounts of ferrihydrite and gypsum (Table 2). As with other PM10 samples, sulfate salts were dissolved immediately. This was followed by dissolution of carbonates, pyrite, and ferrihydrite present in both samples. Carbonate dissolution occurred at a slower rate than ferrihydrite and pyrite, and Fe EXAFS suggested a relative increase in ankerite after 1 hr, although this increase was within the 0.05 standard error of an EXAFS LCF fit (Figure 2) and the ankerite XRD peaks did not increase in intensity (Figure S7). Neither jarosite nor schwertmannite precipitation was observed for these deeper samples, but an increase in ferrihydrite at the expense of pyrite was observed (Figure 2). Arsenic K‐edge EXAFS for sample G showed loss of arsenopyrite accompanied the largest As release to solution among the treatments (Figure 3).
3.4.4. Surface PM10 Alteration in SLF
The efflorescent sulfate salts in the surface samples dissolved immediately upon introduction into synthetic lung fluid. Iron EXAFS data showed that the ferrihydrite component decreased and the jarosite component increased in all surface samples over the course of the experiments (Figure 2). Because S XANES showed that the schwertmannite component was removed over the course of the lung fluid reaction (Table S2), the entire poorly crystalline Fe component in the Fe XAS fits was assigned to ferrihydrite. Despite the decrease in the ferrihydrite component and increase in jarosite component as measured by Fe EXAFS (Figure 2), the As K‐edge EXAFS data showed again the opposing trend of As redistribution from jarosite to ferrihydrite in sample A (Figure 3).
3.4.5. Redox Transition Zone PM10 Alteration in SLF
Sulfur XANES data for samples D and E indicated the dissolution of sulfate salts during SLF reaction and a large increase in jarosite‐like sulfur (Table S2). Iron EXAFS data for these samples showed a decrease in pyrite (greatest decrease for the deepest sample G), a relative increase in chlorite, and a modest increase in ferrihydrite during reaction (Figure 2 and Tables S2 and S3). The relative quality of the Fe EXAFS for sample D diminished at higher k(Å−1) relative to other samples; therefore, the LC fit range was reduced to k = 2 to 9 compared to k = 2 to 12 for other fits. Iron XAS LC fits for sample D indicated that SLF reaction resulted in a loss of ferrihydrite and an accumulation of jarosite and schwertmannite, whereas reaction of sample E resulted in little change in Fe speciation (Figure 2). No statistically significant changes were observed in sample D As speciation, but As XAS spectra of sample E showed redistribution of AsO4 3− from ferrihydrite to jarosite (Figure 3).
3.4.6. Deep Subsurface PM10 Alteration in SLF
Prior to treatment with synthetic lung fluid, samples F and G were dominated by pyrite, quartz, ankerite, and chlorite, with substantive ferrihydrite observed in sample F. The quartz, chlorite, and ankerite XRD peaks did not decrease in size over the course of the reaction, but a decrease in pyrite was observed in both XRD (e.g., Figure S8) and XAS (Figure 2) data sets, which showed a decrease in the pyrite component, a modest increase in the ferrihydrite component (particularly in sample G), and a relative increase in chlorite. Arsenic EXAFS LC fits indicated a small decrease in arsenopyrite and ferrihydrite‐like coordination, along with an accretion of jarosite‐like coordination (Figure 3).
4. Discussion
Results of this study were partially contrary to our initial prediction that toxic metal(loid)s in secondary and tertiary weathering products (prevalent in the oxic near surface of the tailings) would show greater gastric and lung fluid bioaccessibility than parent sulfide minerals (most prevalent at depth). While this was indeed observed for Zn, the inverse trend was observed for Pb, and As showed a more complex pattern of BAc with depth (Figure 6). The molecular mechanisms underlying this observation, however, can be derived from the detailed characterization of geochemical changes occurring both within the weathering profile and during the IVBA experiments themselves.
As noted, the behavior of Zn was consistent with our initial hypothesis; BAc was greatest in the weathered near‐surface tailings. Gastric fluid bioaccessibility was inversely correlated to the depth of the PM10 sample collection (Figure 6). As weathering extent increased with approach from depth to the surface, secondary Zn weathering products (e.g., goslarite) were observed to accumulate (also, see Root et al., 2015) and such phases evidently result in greater solubilization in gastric or lung biofluid than is observed for the parent sulfides (sphalerite). Interestingly, Zn released to lung fluid under circumneutral conditions, which includes 32.2 mmol/L NaCO3, was rapidly repartitioned to the solid phase (Figure 5). This was attributed to the precipitation of smithsonite (ZnCO3) at the higher pH of SLF, for which solutions were supersaturated at early time steps (Figure S6b). The mechanisms limiting bioaccessibility of As and Pb in gastric and lung fluid simulants are, therefore, the focus of the latter portion of this section, following initial discussion of the relevant geochemical controls.
4.1. Mineralogical and Morphological Characterization of the Tailings PM10
4.1.1. Unweathered Tailings
The composition of the fine particulate matter in each layer was a result of (1) fine particles inherited from unweathered parent material, (2) secondary precipitates forming in situ as a result of oxidative weathering, and (3) illuviation and immobilization of particles and solutes from upper layers of the tailings profile. In the absence of oxidative weathering, the PM10 fraction of the tailings is dominated by pyrite, quartz, chlorite, and carbonate fragments generated by anthropogenically enhanced physical weathering (Table 2). Although Fe in these samples is concentrated primarily in sulfide, carbonate, and chlorite phases, small amounts of secondary precipitates present even in these deeper samples indicate that incipient weathering processes have contributed secondary precipitates such as As‐bearing Fe(III) oxyhydroxides and oxyhydroxysulfate salts in the fine fractions that are below detection using the same methods applied to the bulk tailings (Hayes et al., 2014).
4.1.2. Partially Oxidized Tailings
Tailings PM10 in the redox transition zone (D and E) were composed of a mixture of primary minerals and secondary mineral products, with secondary precipitates becoming increasingly dominant toward the surface (Tables S2 and S3). Pyrite, ankerite, and chlorite are depleted relative to the unweathered tailings samples, and ferrihydrite, the initial secondary phase produced by pyrite oxidation, is the dominant secondary phase in samples D and E (Figure 2). Cementation that was observed in the partially oxidized layers in situ was most likely a result of precipitation of ferrihydrite. Total elemental concentrations in the partially oxidized samples (Table 2) show enrichment of Fe and As in the fine fraction compared to the surface and unweathered tailings samples. Iron enrichment reaches 31% in sample D (compared to 16% in the layer above), indicating either concentration of Fe(III) secondary precipitates in the fine fraction or precipitation of Fe leached from the surface (Table 2). Hayes et al. (2014) previously observed Fe enrichment in the bulk fraction of partially oxidized layers from a similar excavation at the IKMHSS tailings site, suggesting that Fe(III) leaching is the primary process contributing to Fe enrichment in these samples. Arsenic enrichment is particularly pronounced in sample E, most likely due to the dominance of ferrihydrite in this layer. Arsenic is known to exhibit a strong affinity for ferrihydrite (Waychunas et al., 1993), and ferrihydrite partitioning into the PM10 fraction is probably a major factor contributing to As enrichment in the partially oxidized PM10 samples. This has an effect on the IVBA of As in this layer, as As‐loaded ferrihydrite is known to dissolve slowly in dilute HCl (Paige et al., 1997).
4.1.3. Surface Tailings
The PM10 of samples A–C are representative of late stage oxidative weathering; no residual sulfides were detected (Table 2 and Figure 2). As a result, this portion of the tailings profile is highly acidic and sulfate enriched, promoting the transformation of ferrihydrite into Fe(III) oxyhydroxysulfate phases. Hydronium‐jarosite and schwertmannite are prevalent minerals in these samples, as is predictable from thermodynamic considerations given the high acidity, high sulfate conditions at the tailings surface. The persistence of ferrihydrite is attributed to its metastability under the semiarid conditions at the site (Hayes et al., 2014). Low water content at the tailings surface leads to hypersaline conditions in the pore water, diminishing solvation of ferrihydrite and enhancing the persistence of efflorescent salts such as gypsum, which were also observed in the surface samples. Gypsum was especially common in sample C (Table 2), likely the result of sulfate leaching from above.
4.2. Degree of Weathering Affects Gastric Bioaccessibility
Gastric BAc of elements in tailings PM10 is determined by the chemical and mineralogical compositions of the solid and aqueous phases under anoxic conditions. Elements are released into solution by dissolution and desorption processes taking place at solid‐solution interfaces, and removed from solution by coprecipitation and adsorption. Therefore, the kinetics of contaminant release to solution are determined by mineralogical changes in the solid phase over time and supersaturation of the biofluid with respect to potential secondary solids. The factors found to control these mineralogical changes and, by extension, toxic element concentrations in solution, included oxidation state and activity of Fe and S, sulfate site activity, and the presence of nucleation sites for precipitation of new surfaces, as discussed in turn below.
4.2.1. Fe Oxidation State
The low solubility of Fe(III) bearing secondary solids limited BAc of As in the oxidized zone of the tailings materials. Fe(III) (oxy)hydroxides and oxyhydroxysulfates, which are major by‐products of sulfide mineral oxidation, have been found to be important sinks for trace elements, especially As (Foster et al., 1998; Paktunc et al., 2004; Root et al., 2015; Sherman & Randal, 2003) and Pb (Hayes et al., 2009; Morin et al., 1999; Ostergren et al., 1999; Root et al., 2015). These phases require high Fe3+ activity and supersaturation for nucleation and crystal growth. The deep, unweathered tailings samples (F and G) have Fe mineral assemblages dominated by ferrous sulfides and carbonates (Table 2). Secondary ferrous minerals are more soluble than ferric counterparts, and the Fe2+ released by proton‐promoted dissolution of these minerals in the absence of dioxygen is unable to drive the formation of ferric As‐ and Pb‐bearing precipitates such as ferrihydrite and jarosite. Metal carbonates are also highly soluble under acidic conditions and exhibit rapid dissolution, while metal sulfides were found to be surprisingly soluble under acidic, low‐O2 conditions. Acid volatilization of sulfides under these conditions may have occurred, but was not confirmed by H2S release.
4.2.2. Adsorbent Surfaces
The suboxic transition samples were consistently found to have the lowest As bioaccessible fraction of all samples tested in this study (Figure 5). This is attributed to higher levels of short‐range‐order (SRO) Fe(III) oxides and oxyhydroxysulfates, such as ferrihydrite and schwertmannite, observed in these samples. Ferrihydrite and schwertmannite have a high capacity for adsorption of AsO4 3− (Burton et al., 2009; Raven et al., 1998) and a more limited capacity for Pb2+ sorption (Webster et al., 1998) under acidic conditions. The prevalence of high‐surface area SRO metal oxide phases in the transition zone samples implies that adsorption to these surfaces may lower the BAc of oxyanion species (especially As). Supporting this contention is the fact that the bioaccessible As fraction was inversely related to the poorly crystalline ferric mineral content (ferrihydrite plus schwertmannite) present in each sample prior to reaction (Figure 7). The one exception was noted with the lung fluid sample B, where no ferrihydrite was detected after reaction (Figure 2). Prior work has shown that ferrihydrite with coprecipitated and/or adsorbed AsO4 3−, as observed in samples D and E, has a slower dissolution rate in dilute HCl than pure ferrihydrite (Paige et al., 1997). Because AsO4 3− forms bidentate binuclear complexes on ferrihydrite surfaces (Waychunas et al., 1993), it links and protects Fe(III) centers against proton‐ and ligand‐promoted dissolution (Paige et al., 1997). Incorporation of arsenate and other trace element species has also been shown to retard the dissolution of schwertmannite (Regenspurg & Peiffer, 2005).
Figure 7.
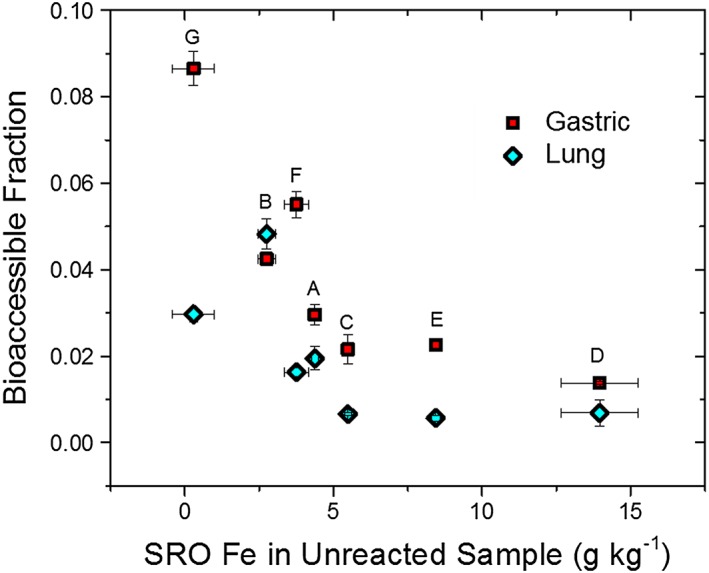
Relationship of As bioaccessible fraction in lung and gastric fluid to the mass fraction of Fe in short‐range‐ordered (SRO) Fe(III) oxyhydroxides (SRO Fe = Total Fe × SRO Fe EXAFS fit, composed of ferrihydrite and schwertmannite contributions to the LCF). Standard errors calculated from measured experimental error. NIST SRM 2709a [As]T = 8610 (reported range 6400–10000, NIST Special Publication 260–172, 2010).
4.2.3. Sulfate Salts
An accumulation of evaporite salts, particularly gypsum, was observed in the transition‐zone PM10 samples because of sulfate and metal cations leaching from the surface horizons or capillary wicking from deeper tailings, and this sulfate played a key role in limiting bioaccessibility (Table 2). When these samples were introduced into gastric fluid, they dissolved immediately and released SO4 2− to solution, that, along with Fe3+, promoted precipitation of jarosite and potentially other alunite‐type minerals (such as beudantite [PbFe3(OH)6SO4AsO4] and segnitite [PbFe3(AsO4)2(OH,H2O)6]; Dutrizac & Jambor, 2000). Arsenate and lead incorporation into these newly precipitated minerals further decreased the fractional bioaccessibility in samples with high levels of sulfate salts compared to samples F and G, whose very low levels of sulfate salts result from the fact that the sulfide parent minerals had not yet undergone oxidation.
Lead bioaccessibility had a stronger dependence on jarosite precipitation than did As bioaccessibility, most likely due to low solubility of Pb‐bearing jarosite (Forray et al., 2010; Kashkay et al., 1975). Based on prior studies of Pb molecular speciation in the IKMHSS tailings (Root et al., 2015), we contend that the low Pb BAc is likely due to the recalcitrance of plumbojarosite to dissolution during the reaction.
The near‐surface PM10 samples (0–20 cm) were depleted by an order of magnitude in Zn relative to deeper PM10 tailings (52–110 cm; Table 2). However, the fractional release of Zn was greatest in the highly weathered near‐surface tailings (Figure 6). The dominant Zn species in the near‐surface tailings was efflorescent salt goslarite (Root et al., 2015), which is highly soluble at low pH (Hayes et al., 2009), and accounts for the in vitro release of Zn from the IKHMSS PM10 fractions. The bioaccessibility of Zn is likely controlled by the solubility of zinc‐containing sulfate salts.
4.2.4. Nucleation and Growth of Jarosite Crystals
Although the presence of aqueous sulfate as a thermodynamic driver of jarosite precipitation was an important factor controlling Pb and As BAc in this study, there were kinetic limitations to jarosite precipitation that affected bioaccessibility. Jarosite is a long‐range‐ordered, crystalline mineral with a high surface energy (Morse & Casey, 1988; Yang et al., 2015), and there are, therefore, kinetic limitations to its homogenous nucleation even under acidic, high‐sulfate conditions when jarosite formation is thermodynamically favorable (Beckmann, 2013). In the absence of existing crystal template surfaces that facilitate heterogeneous nucleation and crystal growth, amorphous precipitates such as schwertmannite with lower surface energies are initially favored, and then these undergo rate‐limited transformation to more crystalline jarosite by an Ostwald ripening process (Morse & Casey, 1988). In samples where jarosite was present as a templating surface (A–E), it showed a relative accumulation, precipitation likely occurring via growth of existing crystals. However, in sample F, where jarosite was not detectable in the Fe EXAFS LC fits (Figure 2), schwertmannite formation was indicated by S‐XANES (Table S2), even though the solution was supersaturated relative to a Pb‐bearing (plumbo‐)jarosite. Because schwertmannite is a less effective sink for As and Pb than ferrihydrite or jarosite (Acero et al., 2006; Carlson et al., 2002), we observed increased BAc here relative to the locations in the profile where jarosite (as a template for crystal growth) was present.
4.3. Factors Controlling Lung Bioaccessibility
In vitro bioaccessibilities for all measured elemental species were generally lower in lung fluid than gastric fluid, consistent with the circumneutral pH of the former (Figure 6). Solubility product constants for most Fe(III) oxides are on the order of 10−4 (Cornell & Schwertmann, 2003), and solubilities under circumneutral conditions are exceedingly low. Other metal oxides and metal carbonates also exhibit low solubility under these conditions. As a result, the mechanism of metal cation release from the tailings samples is most likely hydration of soluble salts such as sulfates followed by hydrolysis and precipitation of amorphous metal hydroxides and carbonates. The most prevalent of these was ferrihydrite, which has a high adsorption affinity for As. Because the synthetic lung fluid is oxygenated, oxidative dissolution of pyrite was also found to play a role in the release of metal(loids) in vitro.
4.3.1. Metal Cation Hydrolysis and Precipitation
Dissolution of ferric solids followed by rapid reprecipitation of Fe3+ (aq) in the form of Fe(III) oxyhydroxides (particularly ferrihydrite) and oxyhydroxysulfates in the SLF‐reacted samples from layers A–E is consistent with the fact that such ferric solids are sparingly soluble under oxic, circumneutral conditions. The higher initial rate of Fe and SO4 2− release from samples A, C, D, and E is consistent with a substantial portion of the Fe in these samples being in the form of soluble sulfate salts (Figure 4). This larger Fe release results in a substantially lower BAc for As during lung fluid reaction, as AsO4 3− is readily adsorbed or incorporated into the structure of neo‐formed ferrihydrite (Waychunas et al., 1993) and jarosite (Savage et al., 2005).
Iron release from samples F and G showed the opposite trend: Fe was released gradually to substantially higher concentrations, and it did not precipitate from solution for the first 24 hr (Figure 4). This suggests that most of the Fe released from these samples was in the form of Fe2+ resulting from oxidative dissolution of pyrite, as solutions comprising Fe(III) at the measured concentrations would be highly supersaturated with respect to ferrihydrite and other Fe(III) oxides.
4.3.2. Redox Processes
Because the synthetic lung fluid used in these experiments was oxygenated, oxidative processes had a substantive effect on element release, especially for sulfide‐rich samples. When exposed to O2(g) and water, metal sulfide particle surfaces undergo oxidative dissolution [equation (1)], acidifying the solution and releasing Fe2+, SO4 2−, and any sulfide‐associated As and Pb:
| (1) |
Under the oxic, circumneutral conditions that exist in synthetic lung fluid, aqueous Fe2+ is unstable and rapidly oxidized to Fe3+ (aq). This results in solution phase supersaturation with respect to ferrihydrite [equation (2)]:
| (2) |
This ferrihydrite precipitation is consistent with Fe K‐edge XAS results for samples F and G, and it accompanied concurrent decreases in aqueous Fe and As concentrations (Figures 2 and 4).
4.3.3. Effect of Ligand Complexation on Mineral Dissolution and Precipitation
Desorption and ligand exchange likely contributed to the release of toxic species (especially AsO4 3−) into solution. Inorganic and organic ligands in synthetic lung fluid (i.e., PO4 3−, tartrate and citrate) have high affinities for the ferrihydrite surface and can compete with AsO4 3− for binding sites (Grafe et al., 2002; Jain & Loeppert, 2000), whereby ligand exchange results in the release of AsO4 3− to solution.
During the reaction of surficial samples A–D with lung fluid, Fe XAS fits consistently indicated substantive redistribution of Fe from ferrihydrite and schwertmannite to jarosite (Figure 2). Although solutions were supersaturated relative to jarosite, this observation was initially surprising because this transformation requires dissolution of ferrihydrite prior to jarosite precipitation, and the solubility of ferrihydrite at neutral pH is low. However, it is known that metal complexing anions, including Cl− (Cornell & Schwertmann, 2003) and citrate (Schwertmann, 1991), which were present in the biofluid simulants, can accelerate and enhance ferrihydrite dissolution by complex formation with Fe(III). In addition, jarosite exhibits low solubility under circumneutral to acidic conditions, and the initial presence of jarosite as a template for surface nucleation likely increased the rate of jarosite precipitation and growth in the lung fluid reactions.
4.3.4. Schwertmannite transformation
Schwertmannite, a short‐range order Fe(III) oxyhydroxysulfate, is an important component of samples in the redox transition zone (C–E), and possibly, other samples due to its stability under acidic conditions when SO4 2− is present. Prior studies have shown that exposing schwertmannite to circumneutral aqueous conditions (similar to the synthetic lung fluid; Davidson et al., 2008; Schroth & Parnell, 2005), promotes its transformation to goethite through ferrihydrite as a metastable intermediate. This transformation occurs due to elimination of SO4 2− from the schwertmannite structure, a process indicated in this experiment by changes in Fe and S aqueous phase concentrations and solid phase speciation (Tables S2 and S3). Schroth and Parnell (2005) and Acero et al. (2006) observed that elements such as As and Pb were released from schwertmannite during its transformation to goethite, but it is unknown if this release occurred during its initial transformation to the ferrihydrite intermediate.
5. Conclusions
This experiment investigated the kinetics of As, Pb, and Zn release into synthetic gastric and lung biofluids from tailings samples collected across a gossan oxidative weathering profile formed over ~50 years of in situ weathering. Contrary to our initial predictions, we observed that oxidative weathering decreased the bioaccessibility of As and Pb by adsorption and coprecipitation with ferric iron, including the formation of secondary precipitates such as jarosite capable of sequestering As and Pb. Conversely, Zn bioaccessibility did increase with weathering extent, as its release was found to be governed by oxygen and pH‐dependent mineral stability. By revealing molecular mechanisms controlling contaminant bioaccessibility, the results of these experiments should inform measures to limit contaminant bioaccessibility at IKMHSS and similar tailings sites.
Supporting information
Supporting Information S1
Acknowledgments
This research was supported by NIEHS Superfund Research Program grant 2 P42 ES04940. We thank Steven Schuchardt, president of North American Industries, for providing access to the IKMHSS site and help with irrigation and weather station and Stanford Synchrotron Radiation Laboratory, operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences for providing resources for mineral analysis. Special thanks to Scott White for extensive work in establishing and maintaining the field site and supervising all sampling efforts. The reader is referred to the supporting information for the data used to generate figures, graphs, and plots in the manuscript. The views of authors do not necessarily represent those of the NIEHS, NIH.
Thomas, A. N. , Root, R. A. , Lantz, R. C. , Sáez, A. E. , & Chorover, J. (2018). Oxidative weathering decreases bioaccessibility of toxic metal(loid)s in PM10 emissions from sulfide mine tailings. GeoHealth, 2, 118–138. 10.1002/2017GH000118
References
- Abernathy, C. O. , Liu, Y. P. , Longfellow, D. , Aposhian, H. V. , Beck, B. , Fowler, B. , et al. (1999). Arsenic: Health effects, mechanisms of actions, and research issues. Environmental Health Perspectives, 107(7), 593–597. 10.1289/ehp.99107593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acero, P. , Ayora, C. , Torrento, C. , & Nieto, J. M. (2006). The behavior of trace elements during schwertmannite precipitation and subsequent transformation into goethite and jarosite. Geochimica et Cosmochimica Acta, 70(16), 4130–4139. 10.1016/j.gca.2006.06.1367 [DOI] [Google Scholar]
- Ahsan, H. , Chen, Y. , Parvez, F. , Argos, M. , Hussain, A. I. , Momotaj, H. , et al. (2006). Health effects of arsenic longitudinal study (HEALS): Description of a multidisciplinary epidemiologic investigation. Journal of Exposure Science & Environmental Epidemiology, 16(2), 191–205. 10.1038/sj.jea.7500449 [DOI] [PubMed] [Google Scholar]
- ATSDR (2016). Agency for Toxic Substances and Disease Registry's Priority List of Hazardous Substances. 2016. Retrieved from http://www.atsdr.cdc.gov/SPL/index.html
- Basu, A. , & Schreiber, M. E. (2013). Arsenic release from arsenopyrite weathering: Insights from sequential extraction and microscopic studies. Journal of Hazardous Materials, 262, 896–904. 10.1016/j.jhazmat.2012.12.027 [DOI] [PubMed] [Google Scholar]
- Beckmann, W. (2013). Crystallization: Basic concepts and industrial applications. Weinheim, Germany: Wiley‐VCH; 10.1002/9783527650323 [DOI] [Google Scholar]
- Bethke, C. M. (1994). The Geochemist's Workbench (Version 2.0): A user's guide to Rxn, Act2,Tact, React, and Gtplot, edited, p. 213, Hydrogeology Program, University Ill.
- Bethke, C. M. (2008). Geochemical and biogeochemical reaction modeling. Cambridge, UK: Cambridge University Press. [Google Scholar]
- Bigham, J. M. , & Nordstrom, D. K. (2000). Iron and aluminum hydroxysulfates from acid sulfate waters. Reviews in Mineralogy and Geochemistry, 40(1), 351–403. 10.2138/rmg.2000.40.7 [DOI] [Google Scholar]
- Bigham, J. M. , Schwertmann, U. , Traina, S. J. , Winland, R. L. , & Wolf, M. (1996). Schwertmannite and the chemical modeling of iron in acid sulfate waters. Geochimica et Cosmochimica Acta, 60(12), 2111–2121. 10.1016/0016-7037(96)00091-9 [DOI] [Google Scholar]
- Blowes, D. W. , & Jambor, J. L. (1990). The pore‐water geochemistry and the mineralogy of the vadose zone of sulfide tailings, Waite Amulet, Quebec, Canada. Applied Geochemistry, 5(3), 327–346. 10.1016/0883-2927(90)90008-S [DOI] [Google Scholar]
- Boisa, N. , Elom, N. , Dean, J. R. , Deary, M. E. , Bird, G. , & Entwistle, J. E. (2014). Development and application of an inhalation bioaccessibility method (IBM) for lead in the PM10 size fraction of soil. Environment International, 70, 132–142. 10.1016/j.envint.2014.05.021 [DOI] [PubMed] [Google Scholar]
- Boulet, M. P. , & Larocque, A. C. L. (1998). A comparative mineralogical and geochemical study of sulfide mine tailings at two sites in New Mexico, USA. Environmental Geology, 33(2‐3), 130–142. 10.1007/s002540050233 [DOI] [Google Scholar]
- Bressler, J. P. , & Goldstein, G. W. (1991). Mechanisms of lead neurotoxicity. Biochemical Pharmacology, 41(4), 479–484. 10.1016/0006-2952(91)90617-E [DOI] [PubMed] [Google Scholar]
- Burton, E. D. , Bush, R. T. , Johnston, S. G. , Watling, K. M. , Hocking, R. K. , Sullivan, L. A. , & Parker, G. K. (2009). Sorption of As (V) and As (III) to schwertmannite. Environmental Science and Technology, 43(24), 9202–9207. 10.1021/es902461x [DOI] [PubMed] [Google Scholar]
- Carlson, L. , Bigham, J. M. , Schwertmann, U. , Kyek, A. , & Wagner, F. (2002). Scavenging of As from acid mine drainage by schertmannite and ferrihydrite: A comparison with synthetic analogues. Environmental Science and Technology, 36(8), 1712–1719. 10.1021/es0110271 [DOI] [PubMed] [Google Scholar]
- Cornell, R. M. , & Schwertmann, U. (2003). The iron oxides. Weinheim: John Wiley; 10.1002/3527602097 [DOI] [Google Scholar]
- Craw, D. , & Bowell, R. J. (2014). The characterization of arsenic in mine waste. Reviews in Mineralogy and Geochemistry, 79(1), 473–505. 10.2138/rmg.2014.79.10 [DOI] [Google Scholar]
- Creasey, S. C. (1952). Geology of Iron King mine, Yavapai County, Arizona. Economic Geology, 47(1), 24–56. 10.2113/gsecongeo.47.1.24 [DOI] [Google Scholar]
- Csavina, J. , Field, J. , Taylor, M. P. , Gao, S. , Landazuri, A. , Betterton, E. A. , & Saez, E. A. (2012). A review on the importance of metals and metalloids in atmospheric dust and aerosol from mining operations. Science of the Total Environment, 433, 58–73. 10.1016/j.scitotenv.2012.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson, L. E. , Shaw, S. , & Benning, L. G. (2008). The kinetics and mechanisms of schwertmannite transformation to goethite and hematite under alkaline conditions. American Mineralogist, 93(8–9), 1326–1337. 10.2138/am.2008.2761 [DOI] [Google Scholar]
- Deshommes, E. , Tardif, R. , Edwards, M. , Sauvé, S. , & Prévost, M. (2012). Experimental determination of the oral bioavailability and bioaccessibility of lead particles. Chemistry Central Journal, 6, 138 10.1186/1752-153X-6-138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutrizac, J. E. , & Jambor, J. L. (2000). Jarosites and their application in hydrometallurgy. Reviews in Mineralogy and Geochemistry, 40(1), 405–452. 10.2138/rmg.2000.40.8 [DOI] [Google Scholar]
- Forray, F. L. , Smith, A. M. L. , Drouet, C. , Navrotsky, A. , Wright, K. , Hudson‐Edwards, K. A. , & Dubbin, W. E. (2010). Synthesis, characterization and thermochemistry of a Pb‐jarosite. Geochimica et Cosmochimica Acta, 74(1), 215–224. 10.1016/j.gca.2009.09.033 [DOI] [Google Scholar]
- Forray, F. L. , Smith, A. M. L. , Navrotsky, A. , Wright, K. , Hudson‐Edwards, K. A. , & Dubbin, W. E. (2014). Synthesis, characterization and thermochemistry of synthetic Pb–As, Pb–Cu and Pb–Zn jarosites. Geochimica et Cosmochimica Acta, 127 107–119. [Google Scholar]
- Foster, A. L. , Brown, G. E. Jr. , Tingle, T. N. , & Parks, G. A. (1998). Quantitative arsenic speciation in mine tailings using X‐ray absorption spectroscopy. American Mineralogist, 83(5‐6), 553–568. 10.2138/am-1998-5-616 [DOI] [Google Scholar]
- Fuller, C. C. , Davis, J. A. , & Waychunas, G. A. (1993). Surface chemistry of ferrihydrite: Part 2. Kinetics of arsenate adsorption and coprecipitation. Geochimica et Cosmochimica Acta, 57(10), 2271–2282. 10.1016/0016-7037(93)90568-H [DOI] [Google Scholar]
- Gao, X. , Root, R. A. , Farrell, J. , Ela, W. , & Chorover, J. (2013). Effect of silicic acid on arsenate and arsenite retention mechanisms on 6‐L ferrihydrite: A spectroscopic and batch adsorption approach. Applied Geochemistry, 38, 110–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil‐Loaiza, J. , White, S. A. , Root, R. A. , Solis‐Dominguez, F. A. , Hammond, C. M. , Chorover, J. , & Maier, R. M. (2016). Phytostabilization of mine tailings using compost‐assisted direct planting: Translating greenhouse results to the field. The Science of the Total Environment, 565, 451–461. 10.1016/j.scitotenv.2016.04.168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzales, P. , Felix, O. , Alexander, C. , Lutz, E. , Ela, W. , & Eduardo Sáez, A. (2014). Laboratory dust generation and size‐dependent characterization of metal and metalloid‐contaminated mine tailings deposits. Journal of Hazardous Materials, 280, 619–626. 10.1016/j.jhazmat.2014.09.002 [DOI] [PubMed] [Google Scholar]
- Grafe, M. , Eick, M. J. , Grossl, P. R. , & Saunders, A. M. (2002). Adsorption of arsenate and arsenite on ferrihydrite in the presence and absence of dissolved organic carbon. Journal of Environmental Quality, 31(4), 1115–1123. 10.2134/jeq2002.1115 [DOI] [PubMed] [Google Scholar]
- Hayes, S. M. , O'Day, P. A. , Webb, S. M. , Maier, R. M. , & Chorover, J. (2011). Changes in zinc speciation with mine tailings acidification in a semiarid weathering environment. Environmental Science & Technology, 45(17), 7166–7172. 10.1021/es201006b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes, S. M. , Root, R. A. , Perdrial, N. , Maier, R. M. , & Chorover, J. (2014). Surficial weathering of iron sulfide mine tailings under semi‐arid climate. Geochimica et Cosmochimica Acta, 141, 240–257. 10.1016/j.gca.2014.05.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes, S. M. , Webb, S. M. , Bargar, J. R. , O'Day, P. A. , Maier, R. M. , & Chorover, J. (2012). Geochemical weathering increases lead bioaccessibility in semi‐arid mine tailings. Environmental Science & Technology, 46(11), 5834–5841. 10.1021/es300603s [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes, S. M. , White, S. A. , Thompson, T. L. , Maier, R. M. , & Chorover, J. (2009). Changes in lead and zinc lability during weathering‐induced acidification of desert mine tailings: Coupling chemical and micro‐scale analyses. Applied Geochemistry, 24(12), 2234–2245. 10.1016/j.apgeochem.2009.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honeker, L. K. , Root, R. A. , Chorover, J. , & Maier, R. M. (2016). Resolving colocalization of bacteria and metal(loid)s on plant root surfaces by combining fluorescence in situ hybridization (FISH) with multiple‐energy micro‐focused X‐ray fluorescence (ME muXRF). Journal of Microbiological Methods, 131, 23–33. 10.1016/j.mimet.2016.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain, A. , & Loeppert, R. H. (2000). Effect of competing anions on the adsorption of arsenate and arsenite by ferrihydrite. Journal of Environmental Quality, 29(5), 1422–1430. 10.2134/jeq2000.00472425002900050008x [DOI] [Google Scholar]
- Jambor, J. L. , Nordstrom, D. K. , & Alpers, C. N. (2000). Metal‐sulfate salts from sulfide mineral oxidation. Reviews in Mineralogy and Geochemistry, 40(1), 303–350. 10.2138/rmg.2000.40.6 [DOI] [Google Scholar]
- Kashkay, C. M. , Borovskaya, Y. B. , & Babazade, M. A. (1975). Determination of deltaGf 298 of synthetic jarosite and its sulfate analogues. Geokhimiya, 5, 778–784. [Google Scholar]
- Kelley, M. E. , Grauning, S. E. , Schoof, R. A. , & Ruby, M. V. (2002). Assessing oral bioavailability of metals in soil. Columbus, OH: Battelle Press. [Google Scholar]
- Kim, C. S. , Wilson, K. M. , & Rytuba, J. J. (2011). Particle‐size dependence on metal(loid) distributions in mine wastes: Implications for water contamination and human exposure. Applied Geochemistry, 26(4), 484–495. 10.1016/j.apgeochem.2011.01.007 [DOI] [Google Scholar]
- Kittel, C. (2005). Introduction to solid state physics (8th ed.). Hoboken, NJ: John Wiley. [Google Scholar]
- Lengke, M. F. , Sanpawanitchakit, C. , & Tempel, R. N. (2009). The oxidation and dissolution of arsenic‐bearing sulfides. The Canadian Mineralogist, 47(3), 593–613. 10.3749/canmin.47.3.593 [DOI] [Google Scholar]
- Lidsky, T. I. , & Schneider, J. S. (2003). Lead neurotoxicity in children: Basic mechanisms and clinical correlates. Brain, 126(1), 5–19. 10.1093/brain/awg014 [DOI] [PubMed] [Google Scholar]
- Lottermoser, B. G. , Schnug, E. , & Haneklaus, S. (2011). Cola soft drinks for evaluating the bioaccessibility of uranium in contaminated mine soils. Science of the Total Environment, 409(18), 3512–3519. 10.1016/j.scitotenv.2011.05.043 [DOI] [PubMed] [Google Scholar]
- Menka, N. , Root, R. , & Chorover, J. (2014). Bioaccessibility, release kinetics, and molecular speciation of arsenic and lead in geo‐dusts from the Iron King Mine Federal Superfund site in Humboldt, Arizona. Reviews on Environmental Health, 29(1–2), 23–27. 10.1515/reveh-2014-0009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meza‐Figueroa, D. , Maier, R. M. , de la O‐Villanueva, M. , Gomez‐Alvarez, A. , Moreno‐Zazueta, A. , Rivera, J. , et al. (2009). The impact of unconfined mine tailings in residential areas from a mining town in a semi‐arid environment: Nacozari, Sonora, Mexico. Chemosphere, 77(1), 140–147. 10.1016/j.chemosphere.2009.04.068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra, K. G. (2012). Introduction to geochemistry: Principles and applications. Hoboken, NJ: Wiley‐Blackwell. [Google Scholar]
- Morena, T. , Oldroyd, A. , McDonald, I. , & Gibbons, W. (2007). Preferential fractionation of trace metals‐metalloids into PM10 resuspended from contaminated gold mine tailings and Rodalquilar, Spain. Water, Air, and Soil Pollution, 179(1‐4), 93–105. 10.1007/s11270-006-9216-9 [DOI] [Google Scholar]
- Morin, G. , Ostergren, J. , Juillot, F. , Ildefonse, P. , Calas, G. , & Brown, G. E. J. (1999). XAFS determination of the chemical form of lead in smelter‐contaminated soils and mine tailings: Importance of sorption processes. American Mineralogist, 84(3), 420–434. 10.2138/am-1999-0327 [DOI] [Google Scholar]
- Morse, J. W. , & Casey, W. H. (1988). Ostwald processes and mineral paragenesis in sediments. American Journal of Science, 288(6), 537–560. 10.2475/ajs.288.6.537 [DOI] [Google Scholar]
- Navarro, A. , Collado, D. , Carbonell, M. , & Sanchez, J. A. (2004). Impact of mining activities on soils in a semi‐arid environment: Sierra Almagrera district, SE Spain. Environmental Geochemistry and Health, 26(3‐4), 383–393. 10.1007/s10653-005-5361-0 [DOI] [PubMed] [Google Scholar]
- O'Day, P. A. (2006). Chemistry and mineralogy of arsenic. Elements, 2(2), 77–83. 10.2113/gselements.2.2.77 [DOI] [Google Scholar]
- Ostergren, J. , Brown, G. , & Parks, G. (1999). Quantitative speciation of lead in selected mine tailings from Leadville, CO. Environmental Science & Technology, 33(10), 1627–1636. 10.1021/es980660s [DOI] [Google Scholar]
- Paige, C. R. , Snodgrass, W. J. , Nicholson, R. V. , & Scharer, J. M. (1997). An arsenate effect on ferrihydrite dissolution kinetics under acidic oxic conditions. Water Research, 31(9), 2370–2382. 10.1016/S0043-1354(97)00107-3 [DOI] [Google Scholar]
- Paktunc, D. , Foster, A. , Heald, S. , & Laflamme, G. (2004). Speciation and characterization of arsenic in gold ores and cyanidation tailings using X‐ray absorption spectroscopy. Geochimica et Cosmochimica Acta, 68(5), 969–983. 10.1016/j.gca.2003.07.013 [DOI] [Google Scholar]
- Ramirez‐Andreotta, M. D. , Brusseau, M. L. , Beamer, P. , & Maier, R. M. (2013). Home gardening near a mining site in an arsenic‐endemic region of Arizona: Assessing arsenic exposure dose and risk via ingestion of home garden vegetables, soils, and water. Science of the Total Environment, 454‐455, 373–382. 10.1016/j.scitotenv.2013.02.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez‐Andreotta, M. D. , Lothrop, N. , Wilkinson, S. T. , Root, R. A. , Artiola, J. F. , Klimecki, W. , & Loh, M. (2016). Analyzing patterns of community interest at a legacy mining waste site to assess and inform environmental health literacy efforts. Journal of Environmental Studies and Sciences, 6(3), 543–555. 10.1007/s13412-015-0297-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravel, B. , & Newville, M. (2005). ATHENA, ARTEMIS, HEPHAESTUS: Data analysis for X‐ray absorption spectroscopy using IFEFFIT. Journal of Synchrotron Radiation, 12(4), 537–541. 10.1107/S0909049505012719 [DOI] [PubMed] [Google Scholar]
- Raven, K. P. , Jain, A. , & Loeppert, R. H. (1998). Arsenite and arsenate adsorption on ferrihydrite: Kinetics, equilibrium, and adsorption envelopes. Environmental Science and Technology, 32(3), 344–349. 10.1021/es970421p [DOI] [Google Scholar]
- Regenspurg, S. , & Peiffer, S. (2005). Arsenate and chromate incorporation in schwertmannite. Applied Geochemistry, 20(6), 1226–1239. 10.1016/j.apgeochem.2004.12.002 [DOI] [Google Scholar]
- Rimstidt, D. R. , Chermak, J. A. , & Gagen, P. M. (1994). Rates of reaction of galena, sphalerite, chalcopyrite, and arsenopyrite with Fe (III) in acidic solutions In Alpers C. N. & Blowes D. W. (Eds.), Environmental Geochemistry of Sulfide Oxidation (pp. 2–13). Washington, DC: American Chemical Society. [Google Scholar]
- Rodriguez, R. R. , Basta, N. T. , Casteel, S. W. , & Pace, L. W. (1999). An in vitro gastrointestinal method to estimate bioavailable arsenic in contaminated soils and solid media. Environmental Science and Technology, 33(4), 642–649. 10.1021/es980631h [DOI] [Google Scholar]
- Root, R. A. , Hayes, S. M. , Hammond, C. M. , Maier, R. M. , & Chorover, J. (2015). Toxic metal(loid) speciation during weathering of iron sulfide mine tailings under semi‐arid climate. Applied Geochemistry, 62, 131–149. 10.1016/j.apgeochem.2015.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruby, M. V. , Davis, A. , Schoof, R. , Eberle, S. , & Sellstone, C. M. (1996). Estimation of lead and arsenic bioavailability using a physiologically based extraction test. Environmental Science and Technology, 30(2), 422–430. 10.1021/es950057z [DOI] [Google Scholar]
- Savage, K. S. , Bird, D. K. , & O'Day, P. A. (2005). Arsenic speciation in synthetic jarosite. Chemical Geology, 215(1‐4), 473–498. 10.1016/j.chemgeo.2004.06.046 [DOI] [Google Scholar]
- Scholze, H. , & Conradt, R. (1987). An in vitro study of the chemical durability of siliceous fibers. The Annals of Occupational Hygiene, 31, 683–692. [Google Scholar]
- Schroth, A. W. , & Parnell Jr, R. A. (2005). Trace metal retention through the schwertmannite to goethite transformation as observed in a field setting, Alta Mine, MT. Applied Geochemistry, 20(5), 907–917. 10.1016/j.apgeochem.2004.09.020 [DOI] [Google Scholar]
- Schwertmann, U. (1991). Solubility and dissolution of iron oxides In Chen Y., & Hadar Y. (Eds.), Iron nutrition and interactions in plants (pp. 3–27). Dordrecht, Netherlands: Kluwer Academic Publishers; 10.1007/978-94-011-3294-7_1 [DOI] [Google Scholar]
- Sherman, D. , & Randal, S. R. (2003). Surface complexation of arsenic(V) to iron(III) (hydr)oxides: Structural mechanism from ab initio molecular geometries and EXAFS spectroscopy. Geochimica et Cosmochimica Acta, 67(22), 4223–4230. 10.1016/S0016-7037(03)00237-0 [DOI] [Google Scholar]
- Stookey, L. L. (1970). Ferrozine— A new spectrophotometric reagent for iron. Analytical Chemistry, 42(7), 779–781. [Google Scholar]
- Stovern, M. , Felix, O. , Csavina, J. , Rine, K. P. , Russell, M. R. , Jones, M. R. , et al. (2014). Simulation of windblown dust transport from a mine tailings impoundment using a computational fluid dynamics model. Aeolian Research, 14, 75–83. 10.1016/j.aeolia.2014.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stovern, M. , Guzman, H. , Rine, K. P. , Felix, O. , King, M. , Ela, W. P. , et al. (2016). Windblown dust deposition forecasting and spread of contamination around mine tailings. Atmosphere, 7(2), 16 10.3390/atmos7020016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaya, M. , Shinohara, Y. , Serita, F. , Ono‐Ogasawara, M. , Otaki, N. , Toya, T. , et al. (2006). Dissolution of functional materials and rare earth oxides into pseudo alveolar fluid. Industrial Health, 44(4), 639–644. 10.2486/indhealth.44.639 [DOI] [PubMed] [Google Scholar]
- Valentin‐Vargas, A. , Root, R. A. , Neilson, J. W. , Chorover, J. , & Maier, R. M. (2014). Environmental factors influencing the structural dynamics of soil microbial communities during assisted phytostabilization of acid‐generating mine tailings: a mesocosm experiment. The Science of the Total Environment, 500‐501, 314–324. 10.1016/j.scitotenv.2014.08.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waychunas, G. A. , Rea, B. A. , Fuller, C. C. , & Davis, J. A. (1993). Surface chemistry of ferrihydrite: Part 1. EXAFS studies of the geometry of coprecipitated and adsorbed arsenate. Geochimica et Cosmochimica Acta, 57(10), 2251–2269. 10.1016/0016-7037(93)90567-G [DOI] [Google Scholar]
- Webb, S. M. (2006). SixPACK: A graphical user interface for XAS analysis using IFEFFIT. Physica Scripta, T115, 1011–1014. [Google Scholar]
- Webster, J. G. , Swedlund, P. J. , & Webster, K. S. (1998). Trace metal adsorption onto an acid mine drainage iron (III) oxyhydroxysulfate. Environmental Science and Technology, 32(10), 1361–1368. 10.1021/es9704390 [DOI] [Google Scholar]
- Yang, X. , Zhu, M. , Kang, F. , Cao, S. , Chen, R. , Liu, H. , & Wei, Y. (2015). Formation mechanism of a series of trigonal antiprismatic jarosite‐type compounds. Journal of Crystal Growth, 429, 49–55. 10.1016/j.jcrysgro.2015.07.037 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information S1