

Abstract
Acute myocardial infarction (AMI) and the heart failure (HF) that often follows are among the leading causes of death and disability worldwide. As such novel therapies are needed to reduce myocardial infarct (MI) size, and preserve left ventricular (LV) systolic function in order to reduce the propensity for HF following AMI. Mitochondria are dynamic organelles that can undergo morphological changes by two opposing processes, mitochondrial fusion and fission. Changes in mitochondrial morphology and turnover are a vital part of maintaining mitochondrial health, DNA stability, energy production, calcium homeostasis, cellular division, and differentiation, and disturbances in the balance of fusion and fission can predispose to mitochondrial dysfunction and cell death. Changes in mitochondrial morphology are governed by mitochondrial fusion proteins (Mfn1, Mfn2 and OPA1) and mitochondrial fission proteins (Drp1, hFis1, and Mff). Recent experimental data suggest that mitochondria undergo fission during acute ischemia/reperfusion injury (IRI), generating fragmented dysfunctional mitochondrial and predisposing to cell death. We and others have shown that genetic and pharmacological inhibition of the mitochondrial fission protein Drp1 can protect cardiomyocytes from acute IRI and reduce MI size. Novel components of the mitochondrial fission machinery, mitochondrial dynamics proteins of 49 kDa (MiD49) and mitochondrial dynamics proteins of 51 kDa (MiD51), have been recently described, which have been shown to mediating mitochondrial fission by targeting Drp1 to the mitochondrial surface. In this review article, we provide an overview of MiD49 and MiD51, and highlight their potential as novel therapeutic targets for treating cardiovascular diseases such as AMI, anthracycline cardiomyopathy, and pulmonary arterial hypertension.
Keywords: Mitochondrial dynamics, mitochondrial fission, mitochondrial fusion, Drp1, MiD49, MiD51, cardioprotection
Introduction
Acute myocardial infarction (AMI) and the heart failure (HF) that often follows are among the leading causes of death and disability worldwide. As such novel therapeutic targets need to be identified to reduce myocardial infarct (MI) size, and preserve left ventricular (LV) systolic function, in order to reduce the propensity for HF following AMI. Mitochondria are dynamic organelles that can undergo mitochondrial fusion and fission. Changes in mitochondrial morphology are essential for the maintenance of mitochondrial and cellular health and function. Fission is essential for mitochondrial transmission during cellular division, as mitochondria cannot be created de novo and are vital for many other cellular functions, including differentiation, mitochondrial transport, mitophagy, and apoptosis (Frazier et al., 2006; Twig et al., 2008; Chen and Chan, 2009). Drp1 is the key mediator of mitochondrial fission within cells (Smirnova et al., 2001; Yoon et al., 2001; Ingerman et al., 2005), and it forms higher order oligomers on the OMM, creating a ring-like structure around mitochondrial tubules to construct a division apparatus (Smirnova et al., 2001; Yoon et al., 2001). Drp1 is a cytosolic protein (Smirnova et al., 1998), regulated by post-translational modifications, that can promote or inhibit fission. Drp1 mediated fission cannot occur without endoplasmic reticulum (ER) pre-constriction of mitochondria, that occurs independently of Drp1 (Friedman et al., 2011). The reduced diameter of ER circumscribed mitochondria permits the formation of Drp1 and Dnm1 helices (Friedman et al., 2011). Drp1 assembly and GTPase activity also depend on the presence of actin, actin-binding proteins and myosin II polymerization at constriction sites (De Vos et al., 2005; Korobova et al., 2014; Ji et al., 2015).
The mammalian mitochondrial fission proteins: fission 1 protein (Fis1), mitochondrial fission factor (Mff), mitochondrial dynamics proteins of 49 kDa (MiD49) and mitochondrial dynamics proteins of 51 kDa (MiD51) allow Drp1 anchoring and oligomerization at the OMM constriction sites (Stojanovski et al., 2004; Otera et al., 2010; Palmer et al., 2011). Hydrolysis of GTP by Drp1 induces a conformational to cause constriction of the Drp1 helices, sufficient for mitochondrial scission (Otera et al., 2010; Mears et al., 2011)
In this review article, we provide an overview of the newly identified members of the mitochondrial fission machinery, MiD49 and MiD51, and highlight their potential as novel therapeutic targets for treating cardiovascular disease including AMI, anthracycline cardiomyopathy, and pulmonary arterial hypertension.
Discovery of the MiD49 and MiD51 proteins
MiD49 and MiD51 were originally identified as the Smith-Magenis syndrome chromosome region candidate gene 7 (SMCR7) and SMCR7-like (SMCR7L) proteins (Simpson et al., 2000), and were renamed mitochondrial dynamics proteins of 49 kDa and 51 kDa due to their mitochondrial activity and size (Palmer et al., 2011). Human MiD49 and MiD51 consist of 454aa and 463aa, respectively (Palmer et al., 2011; Zhao et al., 2011; Liu et al., 2013). Western blots of human cell lines have indicated variable expression levels in different cell types; however, many of the cell lines were derived from cancer cells (Liu et al., 2013). Realtime PCR of multiple tissue cDNA panels from human fetal and adult organs showed that MiD51 levels are significantly higher than MiD49, and β-actin, in fetal organs; but it is vastly reduced by adulthood. MiD49 mRNA levels become marginally higher than MiD51 in most adult cells, with the exception of expression in skeletal and heart cells, which is significantly higher in adult cells (Liu et al., 2013).
Despite mitochondrial dynamics being highly restricted in muscle cells, (Hom and Sheu, 2009), the expression of at least one MiD protein remains high in both stages of life (Liu et al., 2013), indicating the importance of their function. MiD protein expression was not as high in other cell types with high mitochondrial volume and energy demand, such as liver and kidney cells. These findings suggest that there may be functional differences between the two proteins. One distinct difference between the fetal and adult heart, which may be linked to the MiD proteins expression levels, is the metabolic switch which occurs soon after birth (Bartelds et al., 2000; Lopaschuk et al., 2010).
MiD49 and MiD51 direct Drp1 to the mitochondrial surface
The MiD proteins were first described as mitochondrial fission proteins in 2011 (Palmer et al., 2011). The two proteins must be present at the OMM to induce mitochondrial fission, as the deletion of their transmembrane domain prevented OMM foci formation and Drp1 recruitment. The knockdown of either protein does not affect the expression of other mitochondrial dynamic proteins but significantly reduces mitochondrial Drp1 recruitment (Palmer et al., 2011).
In 2014, the structure of MiD51 was determined by two independent groups (Loson et al., 2014; Richter et al., 2014). The crystal structure of MiD49 was identified soon after (Loson et al., 2015). It was the identification of their crystal structures that helped to provide a better understanding of their function at the OMM. In 2011 Palmer et al., showed that both C-terminus GFP tagged MiD49 and MiD51 localized to the mitochondria (Simpson et al., 2000; Palmer et al., 2011). This was also observed by other groups, following V5 C-terminus epitope tagging of the MiD proteins (Zhao et al., 2011; Liu et al., 2013). Western blotting carried out after sodium carbonate extraction further supported the proteins’ localization at the mitochondria. Similar to other OMM proteins, such as Mfn2, Tom20 and Tim23, the MiD proteins were also sensitive to proteinase K treatment, thereby confirming that the MiD proteins are present at the OMM (Palmer et al., 2011).
Co-immunoprecipitation of GFP-tagged MiD proteins revealed their direct association with Drp1 (Palmer et al., 2011). MiD mutants lacking the N-terminal transmembrane domain (MiD49Δ1-49 and MiD51 Δ1-48), were shown to lose their ability to attach to the OMM. This resulted in their cytosolic interaction with Drp1, causing mitochondrial elongation (Palmer et al., 2011; Zhao et al., 2011; Liu et al., 2013; Palmer et al., 2013). The highly conserved residues within the transmembrane domain, amino acids 26-47 in MiD49 and 24-46 in MiD51 are essential for anchoring the proteins to the OMM, but are not required for Drp1 interaction (Palmer et al., 2011; Palmer et al., 2013).
Unlike Mff and hFis1 proteins, MiD49 and MiD51 have not been found to localize at peroxisomes. Interestingly, MiD overexpression causes a similar degree of peroxisome elongation as observed in Drp1 knockout cells. The increase in OMM MiD-Drp1 interaction reduced Drp1 availability for peroxisomes fragmentation (Palmer et al., 2013). Artificially expressing the MiD proteins on lysosomes redirected Drp1 recruitment to lysosomes. This outcome was very weakly observed with Mff, and Fis1 expression had no significant effect on Drp1 recruitment to lysosomes (Palmer et al., 2013). This evidence, as well as the absence of the MiD protein on peroxisomes, provides further evidence that MiD49 and MiD51 proteins can recruit Drp1 independent of Fis1 and Mff, and potentially with a higher affinity.
Foci formation of the MiD proteins was initially believed to be independent of Drp1, as foci formation could still occur following Drp1 knockdown (Palmer et al., 2011). This outcome may have been due to residual Drp1 interaction as mutant MiD proteins, incapable of Drp1 interaction, do not form foci and are evenly distributed across the OMM (Richter et al., 2014; Elgass et al., 2015). A closer look of these regions revealed that the MiD proteins align in a ring formation on the mitochondrial surface, similar to the establishment of Drp1 rings around mitochondrial tubules (Smirnova et al., 2001; Legesse-Miller et al., 2003; Palmer et al., 2011).
The inactive nucleotidyltransferase fold
MiD49 and MiD51 are compact globular proteins, comprising two α-helical regions held together by a central β-strand region (Loson et al., 2014; Richter et al., 2014; Loson et al., 2015). The membrane-proximal regions of the proteins are not as compact in structure. For this reason, deletion of the membrane-proximal region at the proteins’ N-terminal was carried out, to allow the production of high-resolution MiD49 and MiD51 crystal structures (Loson et al., 2014; Ma and Sun, 2014; Richter et al., 2014; Loson et al., 2015).
Human residues 23-48 are predicted to contain the transmembrane domain, and residues 50 to 123/8 are part of the membrane-proximal region (Ma and Sun, 2014; Richter et al., 2014). Expression of human MiD51 lacking the disordered region (MiD51Δ50-123), in wild-type and MiD51-null MEFs, had no negative effect on the proteins ability to successfully recruit Drp1 to the mitochondria (Richter et al., 2014). The functional purpose of this region may be to allow more flexibility during Drp1 oligomerization at the OMM. The deletion of residues 1-118 permitted the creation of high resolution crystallized human MiD51ΔN118 (Richter et al., 2014). Similarly, other groups also observed that the deletion of residues 1-128 allowed crystallization of human MiD51 at a resolution of 3.1 Å (Ma and Sun, 2014); and 1-133 residue deletion of mouse MiD51 (MiD51Δ1-133) permitted the identification of the native structure at a resolution of 2.2 Å (Loson et al., 2014).
Similarly, to crystallize MiD49, the membrane-proximal region which is also predicted to lack a secondary structure, was deleted. Due to further complications in creating a high-resolution crystal, MiD49 mutants were generated. Out of 12 mutants with alanine point substitution mutations, and lacking residues1-125, only one allowed crystallization (MiD49R218A). The MiD49R218A mouse mutant crystal structure was solved at a resolution of 2.4 Å, and retained its ability to recruit Drp1 (Loson et al., 2015).
The crystallized structure of both proteins revealed that these proteins belong to the nucleotide transferase (NTase) protein family, which normally bind nucleotide triphosphates, but lacked enzymatic activity. The NTase fold was found between the two alpha-helical regions connected by a central β-strand region (Loson et al., 2014; Richter et al., 2014; Loson et al., 2015). Both groups identified an additional electron density within this region of MiD51, suggesting the presence of a bound molecule. Surprisingly, fluorescence-based shift assay of MiD51 showed the highest level of stability with adenine diphosphate (ADP) binding, and the protein also weakly interacted with guanosine diphosphate (GDP). No significant structural change could be observed following MiD51-ADP or MiD51-GDP interaction (Loson et al., 2014; Richter et al., 2014). No ligand binding at the nucleotidetransferase cleft could be detected for MiD49, as its nucleotidetransferase clef is too small to allow successful nucleotide binding (Loson et al., 2015). Compared to MiD51, the only nucleotide binding residue conserved within the pocket is histidine (H193 in MiD49) (Loson et al., 2013).
Interestingly MiD51-Drp1 recruitment still occurs in the absence of ADP binding (Loson et al., 2014; Richter et al., 2014). ADP binding is suggested to act as an essential cofactor to stabilise Drp1 spiral formation around mitochondria. Loson et al., found that MiD51-ADP binding can promote Drp1-Drp1 interaction, and its GTP hydrolysis activity, at basal levels (Loson et al., 2014). However, Richter et al., showed that Drp1 binding to MiD51 mutants that are unable to bind ADP or GDP, can still form rings around mitochondria and cause fission (Richter et al., 2014). If in fact ADP is an essential cofactor of MiD51, its activity may facilitate mitochondrial fission events when cellular ADP levels are high, and its low expression in the adult heart and skeletal muscles may be a protective evolutionary outcome to prevent mitochondrial fission during exercise or ischemic conditions. The weak binding of GDP at the nucleotidyltransferase fold of MiD51 may also be part of a stabilization or nucleotide sensing process, due to Drp1’s hydrolysis of GTP, to achieve the conformation change required to execute constriction, or aid Drp1 detachment (Mears et al., 2011; Richter et al., 2014; Kalia et al., 2018).
Binding of Drp1 to MiD proteins at the mitochondrial surface
X-ray crystallization of mouse and human MiD proteins, lacking their transmembrane and membrane-proximal regions, revealed the presence of a highly conserved surface loop on both proteins, essential for Drp1 binding and mitochondrial fission (shown in yellow in Figure 3) (Loson et al., 2014; Richter et al., 2014; Loson et al., 2015).
Figure 3: Mitochondrial ER interaction is essential for and Drp1 mediated fission.
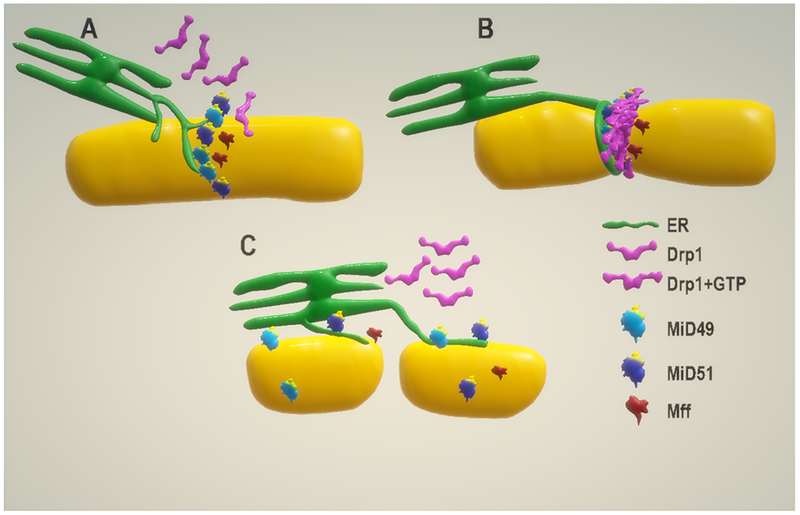
A) MiD foci interact with ER projections at the OMM. B) The ER circumscribes the mitochondrial tubule and constricts, to permit Drp1 oligomerization. GTP bound Drp1 molecules interact with the DRR of MiD proteins to form Drp1 rings. Mff molecules promote Drp1 GTPase activity, required for receptor dissociation, before to constriction. C) After Drp1 mediated fission, Drp1 rings disassemble and remain cytosolic until they are recruited to the OMM again.
MiD51 is capable of recruiting Drp1 puncta to the mitochondria even if it lacks the ability to bind to ADP or dimerize (Loson et al., 2014). The expression of GFP tagged MiD51 mutants lacking the defined loop at residues 238-242 (MiD51ΔPEYFP), although capable of still being present at the OMM of COS7 and MEF cells, were unable to recruit Drp1 to the mitochondria (Richter et al., 2014). The same was observed in 293T Fis1/Mff null cells, as Drp1 proteins remained cytosolic after the expression of two MiD51 mutants, one with a mutation prior to the loops (R234A R235A and N237A) and the other containing a mutation within the loop (E239A Y240A R243A). Similarly, it was shown that the disruption of the salt bridge (R235-D249) directly below the loop produced a similar outcome as the loop deficient mutants (Richter et al., 2014). The artificial expression of MiD51 at the lysosomal surface, established in a study by Palmer et al., showed that mutants lacking the four amino acid loop, could no longer redirect Drp1 to lysosomes (Palmer et al., 2013; Richter et al., 2014). The Drp1-binding motif is also present on the surface of MiD49, residues 230-234 (LEFHP) (Loson et al., 2015). Although MiD49 and MiD51 do not share the same amino acid sequence in this region, there is still a high sequence homology, and the loops are almost identical in their three-dimensional structure. The salt bridge that maintains the MiD49 loop’s structural integrity, an interaction between R227 and D241, is also present and essential for Drp1 recruitment. Co-immunoprecipitation carried out in 293T cells expressing mutants, lacking the salt bridge (MiD49R227A), led to a significant decline in Drp1 interaction. Live cell imaging in Fis1/Mff null cells, expressing the loop mutants also revealed a clear defect in recruiting cytosolic Drp1. These finding highlighted the importance of the loop sequence, and the stability of the structure, in its ability to successfully interact with Drp1.
A recent study by Kalia et al., has produced valuable, evidence to further our understanding of the mechanism of interaction between Drp1 and the MiD proteins. The authors’ in vitro findings provided further evidence to support the importance of the MiD binding loop, within the dynamin recruitment region (DRR), for successful Drp1 binding (shown in yellow in Figure 3) (Kalia et al., 2018). The surprising discovery made from cryo-electron microscopy of Drp1 and MiD49/ MiD51 was that each MiD DRR interacts with four Drp1 molecules, at four possible surfaces within each Drp1 chain (receptor interfaces 1-4), to form cofilaments (Kalia et al., 2018). DRR interaction with interfaces 1 and 2 are only possible following Drp1 nucleotide biding which is otherwise inaccessible (Figure 3) (Kalia et al., 2018). The presence of stalk loops at interfaces 3 and 4 (LINs and L2 loop, respectively) are also essential components for successful receptor interaction. The authors suggest that the inability to interact with MiD49 or MiD51 may explain the impaired Drp1 activity observed in conditions linked to the LINs loop mutation (Chang et al., 2010; Sheffer et al., 2016; Vanstone et al., 2016; Kalia et al., 2018). GTP hydrolysis by Drp1 results in MiD49 or MiD51 dissociation before constriction of the Drp1 rings. MiD association was found to be structurally incompatible with the Drp1 rings during constriction, further indicating that receptor dissociation is required prior to constriction (Kalia et al., 2018).
MiD protein dimerization
The homodimer formation of MiD51 was first identified in 2011 before the protein’s structure was determined. Wild-type MiD51, as well as transmembrane deficient mutants, were able to form homodimers, but the deletion of residues 49-195 prevented dimerization (Zhao et al., 2011). Dimerization of MiD51 is not essential for Drp1 recruitment, but it has been shown to be necessary to cause fission. Cells expressing mutants, incapable of forming dimers undergo mitochondrial fission to a lesser degree after CCCP or antimycin A treatment (Loson et al., 2014). MiD49 did not crystallize as a dimer, as the protein lacks the residues used by MiD51 to dimerize. The authors suggest that there may be weak dimerization present, which could not be detected in the truncated protein (Loson et al., 2015). These findings indicate that the MiD proteins could have alternative mechanisms of Drp1 recruitment. A recent study has identified that the MiD proteins form cofilaments with Drp1 molecules by binding to four Drp1 proteins, at four receptor interface sites (Kalia et al., 2018). As these studies were predominantly carried out using mutants or performed in vitro, it is still not clear if MiD dimer formation occurs in cells and if this is essential prior to the formation of higher-order structures with Drp1.
Interaction of MiD proteins with the endoplasmic reticulum during fission
Mitochondrial ER constriction is an essential step for mitochondrial fission. Small projections from the ER wrap around mitochondrial tubules, reducing their diameter by roughly 30%, to permit the formation of Drp1 helices (Friedman et al., 2011; Elgass et al., 2015). Recently, the simultaneous presence of MiD49/MiD51 foci and their interaction with the ER has been identified at ER-mitochondria constriction sites (Figure 3) (Elgass et al., 2015). Interestingly less than 40% of ER-mitochondria in contact with the MiD foci were located at constriction sites (Richter et al., 2014; Elgass et al., 2015). This suggests that the MiD proteins may have pleiotropic roles, such as ER tethering to facilitate inter-organelle signaling. The single knockout of MiD49 or MiD51 did not affect ER-mitochondria constriction sites, but the interaction was significantly reduced following the knockdown of both proteins (Elgass et al., 2015). These findings further support the importance of the role of MiD proteins in the mitochondrial fission machinery and the need to knockdown of both proteins to inhibit ER interaction and mitochondrial fission (Palmer et al., 2013). Interestingly, Mff deletion did not affect ER-mitochondria contact and constriction, but their presence at these sites is believed to have a regulatory effect on the MiD proteins (Friedman et al., 2011; Elgass et al., 2015; Osellame et al., 2016).
MiD49 and MiD51 activity and regulation during physiological, stress and disease conditions
CCCP mitochondrial uncoupling induces Drp1 mediated mitochondrial fission. Alterations to the fission machinery can prevent this response, reduce mitochondrial fission and protect the cells against apoptosis (Frank et al., 2001; Palmer et al., 2011; Loson et al., 2013). Osellame et al., investigated the level of protection against apoptotic signaling by targeting the fission proteins. The single knockout of OMM fission proteins was protective; however, the cells deficient of Drp1, MiD49/MiD51/Mff or MiD49/MiD51/Mff/Fis1 were most protected against CCCP treatment (Osellame et al., 2016). The additional knockout of Fis1 did not make a significant difference to mitochondrial connectivity or cellular protection, with no significant difference in the level of cytochrome c release between, MiD49/MiD51/Mff and MiD49/MiD51/Mff/Fis1 knockout cells (Osellame et al., 2016). In this study, MiD49 and MiD51 were shown to have a prominent role in mitochondrial fission and intrinsic apoptotic signaling sensitivity. As well as modulating mitochondrial morphology, this outcome may be linked to their involvement in regulating the cristae structure. MiD49/MiD51 knockout cells are significantly more resistant against cristae remodeling and cytochrome c release (Otera et al., 2016).
The membrane-associated ring-CH-type finger 5 (MARCH5) regulates MiD49 activity and leads to its breakdown during stress conditions, subsequently reducing mitochondrial fragmentation. The knockdown of MARCH5 restores MiD49’s ability to induce mitochondrial fragmentation during stress (Xu et al., 2016). This mechanism may be part of a natural protective pathway against mitochondrial fragmentation, to protect the cell during stress conditions such as IRI. Drp1 and Mff negatively regulate this activity to reduce ubiquitination of MiD49 (Cherok et al., 2017).
Mff is also capable of regulating MiD49 and MiD51 activity during mitochondrial fission (Elgass et al., 2015; Osellame et al., 2016). The MiD proteins have been identified to have an inhibitory effect on Drp1 GTPase activity (Osellame et al., 2016), which may aid MiD-Drp1 interaction and provide sufficient time for Drp1 polymerization around mitochondrial tubules. Reversely, Mff molecules promote Drp1 GTPase activity (Osellame et al., 2016), an essential step for Drp1 OMM receptor detachment before the constriction of Drp1 helices (Francy et al., 2015; Kalia et al., 2018). For this reason, the presence of Mff at mitochondrial constriction sites may be an important regulatory component for executing mitochondrial fission (Figure 3). MiD51 was found to exert a significantly stronger inhibitory effect than Mff’s stimulatory effect on Drp1 GTPase activity, which may be the cause of the fused mitochondrial morphology observed in cells overexpressing the MiD proteins (Palmer et al., 2011; Zhao et al., 2011; Liu et al., 2013; Loson et al., 2013; Palmer et al., 2013; Osellame et al., 2016).
Mitochondrial dynamics are essential for maintaining mitochondrial health and function. Defects of the mitochondrial dynamics machinery lead to cellular dysfunction and tissue pathologies (Bach et al., 2003; Frazier et al., 2006; Chan, 2012; Mishra and Chan, 2014). Recently the homozygous nonsense mutation of the MiD49 gene has been identified in an affect individual to cause progressive muscle weakness and exercise intolerance. Isolated patient fibroblasts expressed a highly elongated mitochondrial network, higher mtDNA, abnormal cristae structure and a higher expression of ETC complexes, than healthy control cells (Bartsakoulia et al., 2018). These findings were the first to implicate the role of MiD proteins in humans.
MiD49 and MiD51 as potential novel targets for preventing anthracycline cardiomyopathy and pulmonary artery hypertension
Researchers have identified that MiD expression is significantly increased in pulmonary arterioles of patients with pulmonary arterial hypertension (Chen et al., 2018). Contrary to previous MiD studies, pulmonary artery smooth muscle cells overexpressing of the proteins expressed fragmented mitochondrial networks. siRNA knockdown of the MiD49 and MiD51 proteins, without altering Mff or Fis1 expression, promoted mitochondrial elongation and significantly decreased pulmonary artery smooth muscle cell proliferation (Chen et al., 2018).
MiD49 and MiD51 as potential novel targets for preventing anthracycline cardiomyopathy
The role of MiD49 has a therapeutic target for preventing anthracycline cardiomyopathy has been recently explored. The use of Doxorubicin (DOX) as a chemotherapeutic agent for treating a wide variety of human cancers has been limited by its cardiotoxic effects, the underlying mechanisms of which remain unclear. Zhou et al (Zhou et al., 2017) found that Foxo3a was downregulated in the cardiomyocyte and mouse heart in response to DOX therapy, and Foxo3a attenuated DOX-induced mitochondrial fission and apoptosis in cardiomyocytes. Cardiac-specific Foxo3a transgenic mice showed reduced mitochondrial fission, apoptosis and Dox cardiotoxicity, and Foxo3a was found to directly inhibit transcription of MiD49. Knockdown of MiD49 was demonstrated to reduce DOX-induced mitochondrial fission and apoptosis in cardiomyocytes. Also, knockdown of MiD49 was found to protect the heart against DOX-induced cardiotoxicity. These interesting findings suggest that Dox-induced cardiotoxicity is induced by attenuation of Fox3a and MiD49-mediated mitochondrial fission, providing a novel therapeutic target for preventing anthracycline cardiomyopathy.
MiD49 and MiD51 as potential novel targets for cardioprotection
Fusion and fission proteins are highly expressed in adult cardiomyocytes, despite the compact arrangement of mitochondria. Inhibiting Drp1-mediated fission upon reperfusion is cardioprotective and linked to a reduction in mitochondrial permeability transition pore opening (Ong et al., 2010; Wang et al., 2011; Din et al., 2013). These cardioprotective effects are likely only to be protective against acute ischemia/reperfusion injury (IRI) if achieved acutely, as mitochondrial fusion and fission are an essential part of maintaining mitochondrial health (Chan, 2012). Permanent inhibition of Drp1 activity has been shown to be detrimental in cardiomyocytes, causing accumulation of dysfunctional mitochondria, cardiac remodeling, cardiac failure and premature death in mice (Kageyama et al., 2014; Ikeda et al., 2015; Song et al., 2015). Drp1 mutation in humans leads to a significant increase in mitochondrial and peroxisome fragmentation, causing severe developmental conditions and premature death (Waterham et al., 2007).
Our group has investigated the role of MiD49 and MiD51 as targets of cardioprotection. In accordance to previous studies, MiD49 and MiD51 knockdown caused a significant increase in mitochondrial elongation within cardiac cell lines (Palmer et al., 2011; Loson et al., 2013; Palmer et al., 2013). Their knockdown resulted in a significant reduction in mitochondrial fragmentation during simulated ischemia and a faster recovery of mitochondrial morphology upon simulated reperfusion. Interestingly, MiD deficient cells were less likely to undergo mitochondrial calcium overload upon reperfusion, which is one of the contributing factors to MPTP opening (Yellon and Hausenloy, 2007). This outcome may be due to a reduction of the MiD proteins’ ER/SR interaction (Elgass et al., 2015), which has been shown to protect cardiomyocytes against IRI, by reducing calcium overload upon reperfusion (Hall et al., 2016).
Dual knockout of MiD49 and MiD51 is embryonically lethal; therefore, in vivo investigations were carried out using whole body MiD49 knockout transgenic mice. MiD49 expression is higher in the adult heart, and for this reason, we believed that using a MiD49 knockout mouse colony was a better animal model for our initial studies (Liu et al., 2013). The absence of any overt myocardial pathologies as a result of MiD49 knockout, allowed the investigation of cardiac ischemia-reperfusion susceptibility by LAD coronary artery occlusion, compared to wild-type littermates, without the influence of confounding factors. There was no significant difference in mitochondrial size, cardiac phenotype or MI size following in vivo LAD occlusion between MiD49 knockout and WT mice. From our in vitro and in vivo studies, we believe that both MiD49 and MiD51 must be targeted to cause a significant change in mitochondrial morphology. We believe that these proteins should only be inhibited acutely to prevent adverse effects on cardiac structure and function, as a result of chronic manipulation of the dynamic machinery (Papanicolaou et al., 2011; Papanicolaou et al., 2012; Ikeda et al., 2015; Hall et al., 2016).
Conclusions
MiD49 and MiD51 are mammalian Drp1 binding proteins, located at the outer mitochondrial membrane, and serve as important components of the mitochondrial fission machinery. As fission proteins, their deletion promotes mitochondrial elongation. The level of interplay between MiD49 and MiD51 still remains unclear. The two proteins are very similar in structure, both capable of ER interaction and bind Drp1 at the same region; however, some studies have shown that targeting just one of the proteins is sufficient to induce changes in mitochondrial morphology. The two proteins expression varies with age and tissue type, suggesting that there may be functional differences between the two proteins. MiD51’s ability to bind GTP or ADP at its nucleotidetransferase cleft suggests that the protein’s activity may be interconnected with cellular metabolism. Further investigation is required to identify if MiD51 is more sensitive to metabolic changes, such as anaerobic respiration or ischemia, due to its ability to bind nucleotides. We speculate that this may be the reason that MiD51 expression is low in high energy demand tissues such as the heart and skeletal muscles. Recent studies have revealed that changes in the MiD proteins’ expression can result in various human diseases, and targeting these proteins can aid restore a balance in mitochondrial dynamics. These findings further highlight the importance of the MiD proteins’ as components of the cellular fusion and fission machinery and may serve as potential therapeutic targets for treating cardiovascular diseases such as AMI, anthracycline cardiomyopathy, and pulmonary arterial hypertension.
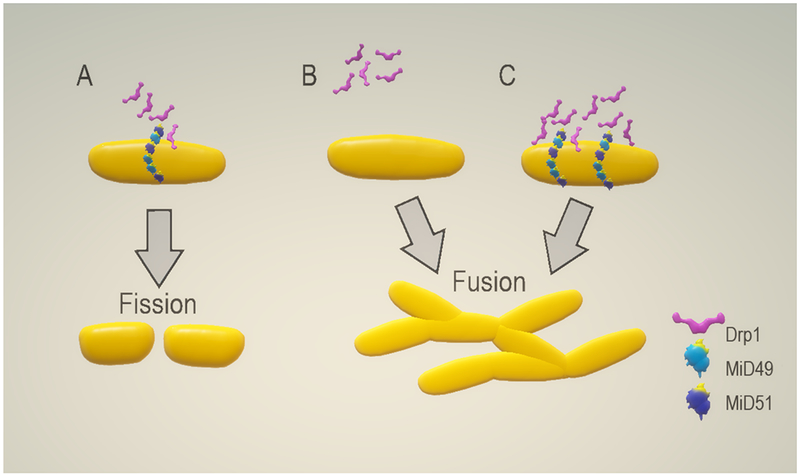
Figure 1: Fusion and Fission are essential for cellular health and function.
Mitochondria are dynamic organelles that can change their morphology by two highly regulated, opposing processes, known as mitochondrial fission and fusion. These mechanisms are essential for various cellular processes as well as maintaining a healthy mitochondrial network. A) Mitochondrial fusion helps to restore mitochondrial membrane potential to improve ATP production and gain stability by the mixing of matrix contents such as DNA and calcium. B) Fission allows easier movement of mitochondrial tubules, as well as removal of damaged mitochondria, that can contaminate the rest of the network.
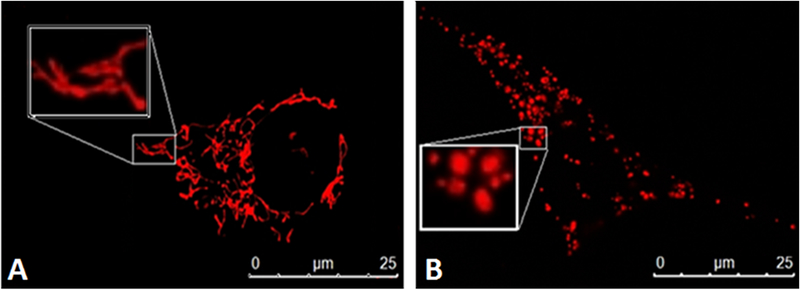
Figure 2: MiD depletion or overexpression promotes mitochondrial elongation.
MiD49 and MiD51 are important components of the mitochondria fission machinery A) The MiD proteins are OMM receptors of Drp1 required for mitochondrial fission. The deletion or the overexpression MiD49 and/or MiD51 induces mitochondrial elongation and the formation of mitochondrial networks (B and C, respectively).
Acknowledgments
Funding
This project was funded by NIH grant R01HL81863 to WAB. PS was funded by the British Heart Foundation (grant number FS/13/41/30368). SBO is funded by the Singapore Ministry of Health’s National Medical Research Council under its Open Fund – Young Individual Research Grant (OF-YIRG) - OFYIRG16may031 as well as a Khoo Postdoctoral Fellowship Award (Duke-NUS-KPFA/2016/ 0010) from the Estate of Tan Sri Khoo Teck Puat, Singapore. DJH was supported by the British Heart Foundation (FS/10/039/28270), the National Institute for Health Research University College London Hospitals Biomedical Research Centre, Duke-National University Singapore Medical School, Singapore Ministry of Health’s National Medical Research Council under its Clinician Scientist-Senior Investigator scheme (NMRC/CSA-SI/0011/2017) and Collaborative Centre Grant scheme (NMRC/CGAug16C006), and the Singapore Ministry of Education Academic Research Fund Tier 2 (MOE2016-T2-2-021). This article is based upon work from COST Action EU-CARDIOPROTECTION CA16225 supported by COST (European Cooperation in Science and Technology).
Footnotes
Conflicts of interest
The authors declare that they have no conflicts of interest.
References
- Bach D, Pich S, Soriano FX, Vega N, Baumgartner B, Oriola J, Daugaard JR, Lloberas J, Camps M, Zierath JR, Rabasa-Lhoret R, Wallberg-Henriksson H, Laville M, Palacin M, Vidal H, Rivera F, Brand M, Zorzano A (2003) Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. The Journal of biological chemistry 278:17190–17197. [DOI] [PubMed] [Google Scholar]
- Bartelds B, Knoester H, Smid GB, Takens J, Visser GH, Penninga L, van der Leij FR, Beaufort-Krol GC, Zijlstra WG, Heymans HS, Kuipers JR (2000) Perinatal changes in myocardial metabolism in lambs. Circulation 102:926–931. [DOI] [PubMed] [Google Scholar]
- Bartsakoulia M, Pyle A, Troncosco D, Vial J, Paz-Fiblas MV, Duff J, Griffin H, Boczonadi V, Lochmuller H, Kleinle S, Chinnery PF, Grunert S, Kirschner J, Eisner V, Horvath R (2018) A novel mechanism causing imbalance of mitochondrial fusion and fission in human myopathies. Human molecular genetics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DC (2012) Fusion and fission: interlinked processes critical for mitochondrial health. Annual review of genetics 46:265–287. [DOI] [PubMed] [Google Scholar]
- Chang CR, Manlandro CM, Arnoult D, Stadler J, Posey AE, Hill RB, Blackstone C (2010) A lethal de novo mutation in the middle domain of the dynamin-related GTPase Drp1 impairs higher order assembly and mitochondrial division. The Journal of biological chemistry 285:32494–32503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Chan DC (2009) Mitochondrial dynamics--fusion, fission, movement, and mitophagy--in neurodegenerative diseases. Human molecular genetics 18:R169–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K-H, Dasgupta A, Lin J, Potus F, Bonnet S, Iremonger J, Fu J, Mewburn J, Wu D, Dunham-Snary K, Theilmann AL, Jing Z-C, Hindmarch C, Ormiston ML, Lawrie A, Archer SL (2018) Epigenetic Dysregulation of the Drp1 Binding Partners MiD49 and MiD51 Increases Mitotic Mitochondrial Fission and Promotes Pulmonary Arterial Hypertension: Mechanistic and Therapeutic Implications. Circulation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherok E, Xu S, Li S, Das S, Meltzer WA, Zalzman M, Wang C, Karbowski M (2017) Novel regulatory roles of Mff and Drp1 in E3 ubiquitin ligase MARCH5–dependent degradation of MiD49 and Mcl1 and control of mitochondrial dynamics. Molecular Biology of the Cell 28:396–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos KJ, Allan VJ, Grierson AJ, Sheetz MP (2005) Mitochondrial function and actin regulate dynamin-related protein 1-dependent mitochondrial fission. Current biology : CB 15:678–683. [DOI] [PubMed] [Google Scholar]
- Din S, Mason M, Volkers M, Johnson B, Cottage CT, Wang Z, Joyo AY, Quijada P, Erhardt P, Magnuson NS, Konstandin MH, Sussman MA (2013) Pim-1 preserves mitochondrial morphology by inhibiting dynamin-related protein 1 translocation. Proceedings of the National Academy of Sciences of the United States of America 110:5969–5974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgass KD, Smith EA, LeGros MA, Larabell CA, Ryan MT (2015) Analysis of ER-mitochondria contacts using correlative fluorescence microscopy and soft X-ray tomography of mammalian cells. Journal of cell science 128:2795–2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francy CA, Alvarez FJ, Zhou L, Ramachandran R, Mears JA (2015) The mechanoenzymatic core of dynamin-related protein 1 comprises the minimal machinery required for membrane constriction. The Journal of biological chemistry 290:11692–11703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ (2001) The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Developmental cell 1:515–525. [DOI] [PubMed] [Google Scholar]
- Frazier AE, Kiu C, Stojanovski D, Hoogenraad NJ, Ryan MT (2006) Mitochondrial morphology and distribution in mammalian cells. Biological chemistry 387:1551–1558. [DOI] [PubMed] [Google Scholar]
- Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK (2011) ER tubules mark sites of mitochondrial division. Science (New York, NY) 334:358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall AR, Burke N, Dongworth RK, Kalkhoran SB, Dyson A, Vicencio JM, Dorn GW II, Yellon DM, Hausenloy DJ (2016) Hearts deficient in both Mfn1 and Mfn2 are protected against acute myocardial infarction. Cell death & disease 7:e2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hom J, Sheu SS (2009) Morphological dynamics of mitochondria--a special emphasis on cardiac muscle cells. Journal of molecular and cellular cardiology 46:811–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, Nomura M, Mihara K, Egashira K, Ohishi M, Abdellatif M, Sadoshima J (2015) Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circulation research 116:264–278. [DOI] [PubMed] [Google Scholar]
- Ingerman E, Perkins EM, Marino M, Mears JA, McCaffery JM, Hinshaw JE, Nunnari J (2005) Dnm1 forms spirals that are structurally tailored to fit mitochondria. The Journal of Cell Biology 170:1021–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji WK, Hatch AL, Merrill RA, Strack S, Higgs HN (2015) Actin filaments target the oligomeric maturation of the dynamin GTPase Drp1 to mitochondrial fission sites. eLife 4:e11553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kageyama Y, Hoshijima M, Seo K, Bedja D, Sysa-Shah P, Andrabi SA, Chen W, Hoke A, Dawson VL, Dawson TM, Gabrielson K, Kass DA, Iijima M, Sesaki H (2014) Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. The EMBO Journal 33:2798–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia R, Wang RY, Yusuf A, Thomas PV, Agard DA, Shaw JM, Frost A (2018) Structural basis of mitochondrial receptor binding and constriction by DRP1. Nature 558:401–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korobova F, Gauvin TJ, Higgs HN (2014) A role for myosin II in mammalian mitochondrial fission. Current biology : CB 24:409–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legesse-Miller A, Massol RH, Kirchhausen T (2003) Constriction and Dnm1p Recruitment Are Distinct Processes in Mitochondrial Fission. Molecular Biology of the Cell 14:1953–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Yu R, Jin SB, Han L, Lendahl U, Zhao J, Nister M (2013) The mitochondrial elongation factors MIEF1 and MIEF2 exert partially distinct functions in mitochondrial dynamics. Experimental cell research 319:2893–2904. [DOI] [PubMed] [Google Scholar]
- Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC (2010) Myocardial fatty acid metabolism in health and disease. Physiological reviews 90:207–258. [DOI] [PubMed] [Google Scholar]
- Loson OC, Song Z, Chen H, Chan DC (2013) Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Molecular Biology of the Cell 24:659–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loson OC, Meng S, Ngo H, Liu R, Kaiser JT, Chan DC (2015) Crystal structure and functional analysis of MiD49, a receptor for the mitochondrial fission protein Drp1. Protein science : a publication of the Protein Society 24:386–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loson OC, Liu R, Rome ME, Meng S, Kaiser JT, Shan SO, Chan DC (2014) The mitochondrial fission receptor MiD51 requires ADP as a cofactor. Structure (London, England : 1993) 22:367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Sun F (2014) Expression, purification, crystallization and preliminary crystallographic study of the cytoplasmic domain of the mitochondrial dynamics protein MiD51. Acta crystallographica Section F, Structural biology communications 70:596–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mears JA, Lackner LL, Fang S, Ingerman E, Nunnari J, Hinshaw JE (2011) Conformational changes in Dnm1 support a contractile mechanism for mitochondrial fission. Nature structural & molecular biology 18:20–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra P, Chan DC (2014) Mitochondrial dynamics and inheritance during cell division, development and disease. Nature reviews Molecular cell biology 15:634–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ (2010) Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 121:2012–2022. [DOI] [PubMed] [Google Scholar]
- Osellame LD, Singh AP, Stroud DA, Palmer CS, Stojanovski D, Ramachandran R, Ryan MT (2016) Cooperative and independent roles of the Drp1 adaptors Mff, MiD49 and MiD51 in mitochondrial fission. Journal of cell science 129:2170–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otera H, Miyata N, Kuge O, Mihara K (2016) Drp1-dependent mitochondrial fission via MiD49/51 is essential for apoptotic cristae remodeling. The Journal of Cell Biology 212:531–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, Mihara K (2010) Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. The Journal of Cell Biology 191:1141–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer CS, Osellame LD, Laine D, Koutsopoulos OS, Frazier AE, Ryan MT (2011) MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO reports 12:565–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer CS, Elgass KD, Parton RG, Osellame LD, Stojanovski D, Ryan MT (2013) Adaptor proteins MiD49 and MiD51 can act independently of Mff and Fis1 in Drp1 recruitment and are specific for mitochondrial fission. The Journal of biological chemistry 288:27584–27593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papanicolaou KN, Ngoh GA, Dabkowski ER, O’Connell KA, Ribeiro RF Jr., Stanley WC, Walsh K (2012) Cardiomyocyte deletion of mitofusin-1 leads to mitochondrial fragmentation and improves tolerance to ROS-induced mitochondrial dysfunction and cell death. American journal of physiology Heart and circulatory physiology 302:H167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papanicolaou KN, Khairallah RJ, Ngoh GA, Chikando A, Luptak I, O’Shea KM, Riley DD, Lugus JJ, Colucci WS, Lederer WJ, Stanley WC, Walsh K (2011) Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Molecular and cellular biology 31:1309–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter V, Palmer CS, Osellame LD, Singh AP, Elgass K, Stroud DA, Sesaki H, Kvansakul M, Ryan MT (2014) Structural and functional analysis of MiD51, a dynamin receptor required for mitochondrial fission. The Journal of Cell Biology 204:477–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffer R, Douiev L, Edvardson S, Shaag A, Tamimi K, Soiferman D, Meiner V, Saada A (2016) Postnatal microcephaly and pain insensitivity due to a de novo heterozygous DNM1L mutation causing impaired mitochondrial fission and function. American journal of medical genetics Part A 170:1603–1607. [DOI] [PubMed] [Google Scholar]
- Simpson JC, Wellenreuther R, Poustka A, Pepperkok R, Wiemann S (2000) Systematic subcellular localization of novel proteins identified by large-scale cDNA sequencing. EMBO reports 1:287–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova E, Shurland DL, Ryazantsev SN, van der Bliek AM (1998) A human dynamin-related protein controls the distribution of mitochondria. The Journal of Cell Biology 143:351–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova E, Griparic L, Shurland DL, van der Bliek AM (2001) Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Molecular Biology of the Cell 12:2245–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M, Mihara K, Chen Y, Scorrano L, Dorn GW (2015) Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell metabolism 21:273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojanovski D, Koutsopoulos OS, Okamoto K, Ryan MT (2004) Levels of human Fis1 at the mitochondrial outer membrane regulate mitochondrial morphology. Journal of cell science 117:1201–1210. [DOI] [PubMed] [Google Scholar]
- Twig G, Elorza A, Molina AJA, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS (2008) Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. The EMBO Journal 27:433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanstone JR, Smith AM, McBride S, Naas T, Holcik M, Antoun G, Harper ME, Michaud J, Sell E, Chakraborty P, Tetreault M, Majewski J, Baird S, Boycott KM, Dyment DA, MacKenzie A, Lines MA (2016) DNM1L-related mitochondrial fission defect presenting as refractory epilepsy. European journal of human genetics : EJHG 24:1084–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JX, Jiao JQ, Li Q, Long B, Wang K, Liu JP, Li YR, Li PF (2011) miR-499 regulates mitochondrial dynamics by targeting calcineurin and dynamin-related protein-1. Nature medicine 17:71–78. [DOI] [PubMed] [Google Scholar]
- Waterham HR, Koster J, van Roermund CWT, Mooyer PAW, Wanders RJA, Leonard JV (2007) A Lethal Defect of Mitochondrial and Peroxisomal Fission. New England Journal of Medicine 356:1736–1741. [DOI] [PubMed] [Google Scholar]
- Xu S, Cherok E, Das S, Li S, Roelofs BA, Ge SX, Polster BM, Boyman L, Lederer WJ, Wang C, Karbowski M (2016) Mitochondrial E3 ubiquitin ligase MARCH5 controls mitochondrial fission and cell sensitivity to stress-induced apoptosis through regulation of MiD49 protein. Molecular Biology of the Cell 27:349–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellon DM, Hausenloy DJ (2007) Myocardial reperfusion injury. The New England journal of medicine 357:1121–1135. [DOI] [PubMed] [Google Scholar]
- Yoon Y, Pitts KR, McNiven MA (2001) Mammalian dynamin-like protein DLP1 tubulates membranes. Molecular Biology of the Cell 12:2894–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Liu T, Jin S, Wang X, Qu M, Uhlen P, Tomilin N, Shupliakov O, Lendahl U, Nister M (2011) Human MIEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission. The EMBO Journal 30:2762–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Li R, Liu C, Sun T, Htet Aung LH, Chen C, Gao J, Zhao Y, Wang K (2017) Foxo3a inhibits mitochondrial fission and protects against doxorubicin-induced cardiotoxicity by suppressing MIEF2. Free radical biology & medicine 104:360–370. [DOI] [PubMed] [Google Scholar]