

Mitochondrial utilization of the carbohydrate fuel for energy production is required for postnatal cerebellar development.
Abstract
While deregulation of mitochondrial metabolism and cytosolic glycolysis has been well recognized in tumor cells, the role of coordinated mitochondrial oxidation and cytosolic fermentation of pyruvate, a key metabolite derived from glucose, in physiological processes is not well understood. Here, we report that knockout of PTPMT1, a mitochondrial phosphoinositide phosphatase, completely blocked postnatal cerebellar development. Proliferation of granule cell progenitors, the most actively replicating cells in the developing cerebellum, was only moderately decreased, and proliferation of Purkinje cell progenitors did not seem to be affected in knockout mice. In contrast, generation of functional Bergmann glia from multipotent precursor cells (radial glia), which is essential for cerebellar corticogenesis, was totally disrupted. Moreover, despite a low turnover rate, neural stem cells were impaired in self-renewal in knockout mice. Mechanistically, loss of PTPMT1 decreased mitochondrial aerobic metabolism by limiting utilization of pyruvate, which resulted in bioenergetic stress in neural precursor/stem cells but not in progenitor or mature cells, leading to cell cycle arrest through activation of the AMPK-p19/p21 pathway. This study suggests that mitochondrial oxidation of the carbohydrate fuel is required for postnatal cerebellar development, and identifies a bioenergetic stress–induced cell cycle checkpoint in neural precursor/stem cells.
INTRODUCTION
Eukaryotic cells exhibit remarkable plasticity in metabolism during cellular processes. Cell metabolism and bioenergetics must be quickly organized according to the status of cellular activities. Perturbation of metabolic plasticity may be linked to dysregulated cell proliferation, differentiation, migration, and tumorigenesis. Mitochondria, the site of cellular respiration/oxidative phosphorylation, are key players of metabolic plasticity. These organelles provide a highly efficient route for eukaryotic cells to generate adenosine 5′-triphosphate (ATP) and produce building blocks for biosynthesis required for cell proliferation and differentiation. Pyruvate, a key metabolite of glucose (the major simple carbohydrate), is the master fuel for mitochondrial energy production and intermediate metabolism. This metabolite is at the central branch point of mitochondrial aerobic metabolism and cytosolic glycolysis. It can enter the mitochondria through mitochondrial pyruvate carriers (MPCs) for oxidation or be further reduced to lactate in the cytosol to complete the glycolytic pathway, which is an inefficient utilization of the carbon source for ATP production. Hence, the metabolic fate of pyruvate dictates the energy and intermediate metabolite output of the carbohydrate fuel, affecting metabolic plasticity and thus many aspects of cellular biochemistry, including fuel selection, oxygen consumption, redox status, ATP generation, and macromolecule biosynthesis (1). However, the molecular mechanisms regulating metabolic plasticity still remain poorly defined.
The brain has a high energy requirement, and glucose is the obligatory energy substrate of the adult and developing brain. Approximately 25% of the glucose and 20% of the oxygen consumed by the resting human body are dedicated to brain functions (2). However, not all glucose in the brain is oxidized in the mitochondria. Functional brain imaging studies have shown that in human adults and infants, 10 and 35%, respectively, of glucose entering the brain is metabolized through cytosolic glycolysis even in the presence of abundant oxygen (that is, aerobic glycolysis) (3, 4). Astrocytes generally exhibit much higher glycolytic flux than neurons (5, 6), and they have metabolic interactions with neurons through the astrocyte-neuron lactate shuttle (2). The nonoxidative glycolytic activity in the adult brain is restricted to specific regions, such as the dorsolateral prefrontal cortex, the superior and medial frontal gyrus, or the precuneus and posterior cingulate cortex, whereas in other regions, such as the cerebellum, it is virtually undetectable (7–9). However, given the diversity of cellular components in different regions of the brain and the complex interplay between mitochondrial metabolism and cytosolic glycolysis, it is still not well understood how these two important metabolic pathways are coordinated in various cell types during brain development and maintenance.
Highly dynamic cellular processes occur in brain development, especially cerebellar development. A complex pattern of neurogenesis and cell movements is involved in the formation of the cerebellar cortex and cerebellar nuclei. Cerebellar neurons are generated from two anatomically distinct germinal zones within the cerebellar anlage. The dorsomedial ventricular zone/neuroepithelium along the forth ventricle gives rise to Purkinje cells (PCs; the principal output neurons of the cerebellar cortex), GABAergic (γ-aminobutyric acid–containing) neurons of the cerebellar nuclei, cerebellar interneurons, as well as radial glia (RG), which are astroglia-like multipotent precursor cells (10, 11). The second germinal zone, the rhombic lip, generates the cerebellar granule cells (GCs), glutamatergic neurons of the cerebellar nuclei, and unipolar brush cells. Extensive cell interactions and migration are required to achieve the well-ordered cellular organization and laminal and foliated structures of the mature cerebellum, which develop primarily after birth. Formation of the PC layer (PCL) and migration of external granule layer (EGL) cells require radial scaffolds established by Bergmann glia (BG), astrocyte-like cells that are differentiated or transformed from multipotent precursor RG (10, 11). BG are first seen in the cerebellar cortex in the late embryonic period. By birth, the RG population has disappeared and replaced by BG, which are localized to the PCL and extend parallel processes to the pial surface. In addition, neurosphere-forming neural stem/progenitor cells (NSPCs) that have both self-renewal and differentiation capabilities are also present in the postnatal cerebellar parenchyma (12). They might be derived from RG and possibly contribute to the development and/or maintenance of the cerebellum. Previous studies have revealed cell signaling and transcriptional regulation of cell type specification, patterning, and homeostasis during cerebellar histogenesis (13–15). However, little is known about how cellular metabolism is involved and cooperatively facilitates development of this highly organized tissue structure.
PTPMT1, a PTEN (phosphatase and tensin homolog)–like phosphatase encoded by nuclear DNA, is localized to the mitochondria and is anchored at the mitochondrial inner membrane (16). It dephosphorylates phosphatidylinositol phosphates (PIPs), in particular, PI(3,5)P2 and PI5P (17, 18). Global knockout of PTPMT1 leads to developmental arrest and post-implantation lethality in mice (18, 19). Conditional knockout of PTPMT1 in the hematopoietic system causes hematopoietic failure and postnatal lethality (20). This phosphatase appears to facilitate mitochondrial metabolism by dephosphorylation of downstream PIP substrates (20). Accumulation of PIPs in PTPMT1 knockout mitochondria decreases mitochondrial metabolism but enhances cytosolic glycolysis (20), affecting cellular metabolic plasticity, although detailed mechanisms were unclear. In the present study, we use PTPMT1 knockout mice/cells as a model to determine the role of coordinated mitochondrial metabolism and glycolysis in brain development.
RESULTS
Knockout of PTPMT1 from neural precursor/stem cells blocked cerebellar development and compromised cerebral development
Our previous studies have shown that PTPMT1 plays a critical role in coordinating mitochondrial metabolism and cytosolic glycolysis (18, 20). To determine the role of balanced cellular metabolism in brain development, we generated neural cell–specific PTPMT1 knockout mice by crossing PTPMT1 conditional mice (20) with Nestin-Cre transgenic mice, which constitutively express Cre DNA recombinase in neural precursor cells beginning at embryonic day 10.5 (E10.5) (21). PTPMT1fl/fl/Nestin-Cre+ mice were born at a Mendelian ratio indistinguishable from their littermates. However, these mice subsequently displayed growth retardation and ataxia and invariably died before postnatal day 12 (P12) (Fig. 1A and fig. S1A). Histopathological examination of P8 brain tissues revealed a thinner cerebral cortex, a smaller hippocampus, and larger ventricles in these mice relative to control animals (fig. S1B). Detailed examination illustrated fewer neurons and increased astrocytes in the cerebral cortex and hippocampus in PTPMT1 knockout mice (fig. S1C). Most notably, however, these knockout mice had remarkably small cerebella (Fig. 1A). Compared to well-foliated and layered structures in control cerebella, foliation and lamination in the knockout cerebella were completely missing. This profound phenotype demonstrates a crucial role of PTPMT1 in cerebellar development.
Fig. 1. Depletion of PTPMT1 from neural precursor cells blocks postnatal cerebellar development.
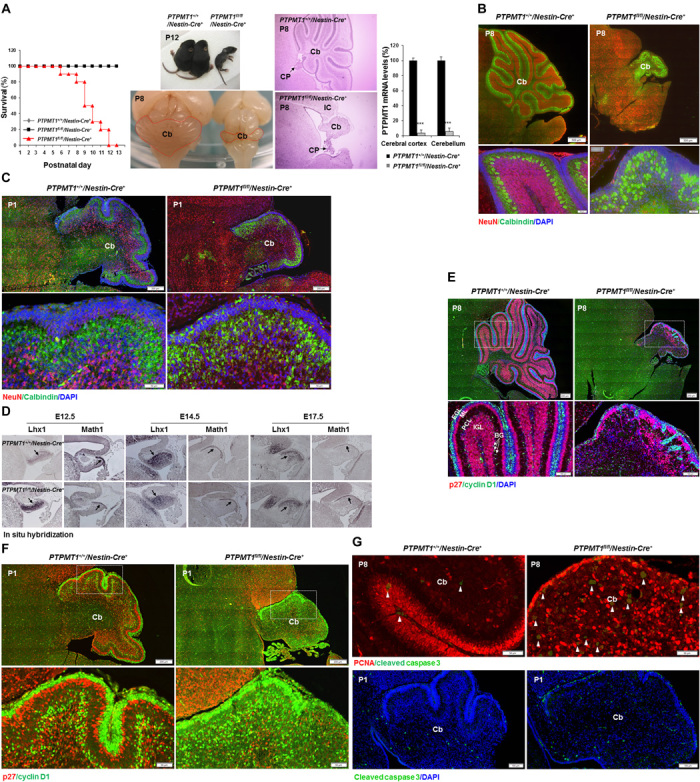
(A) Kaplan-Meier survival curves of PTPMT1fl/fl/Nestin-Cre+ (n = 18), PTPMT1+/+/Nestin-Cre+ (n = 20), and PTPMT1fl/fl/Nestin-Cre− (n = 18) mice. PTPMT1+/+/Nestin-Cre+ and PTPMT1fl/fl/Nestin-Cre+ mice and brains at P12 were photographed. Representative cerebella and cerebellar sections [hematoxylin and eosin (H&E) staining] of PTPMT1+/+/Nestin-Cre+ and PTPMT1fl/fl/Nestin-Cre+ mice at P8 are shown. Cb, cerebellum; IC, inferior colliculus; CP, choroid plexus. PTPMT1 mRNA levels in freshly isolated cerebra and cerebella with the indicated genotypes (n = 3) were determined by quantitative reverse transcription polymerase chain reaction (qRT-PCR). (B and C) Brain sections prepared from PTPMT1+/+/Nestin-Cre+ and PTPMT1fl/fl/Nestin-Cre+ mice at the indicated ages were processed for immunofluorescence staining with the indicated antibodies, followed by 4′,6′-diamidino-2-phenylindole (DAPI) counterstaining. (D) Cryosections of hindbrains with the indicated genotypes at E12.5, E14.5, and E17.5 were hybridized with digoxigenin (DIG)–labeled probes specific for mouse Lhx1 and Math1 mRNA. Arrows indicate Math1+ or Lhx1+ cells. (E to G) Brain sections prepared from PTPMT1+/+/Nestin-Cre+ and PTPMT1fl/fl/Nestin-Cre+ mice at the indicated ages were processed for immunofluorescence staining with the indicated antibodies, followed by DAPI counterstaining. EGL, external granule layer; PCL, Purkinje cell layer; IGL, internal granule layer; ML, molecular layer. Arrowheads in (G) indicate cleaved caspase 3+ apoptotic cells. Representative images from three mice per genotype are shown.
We examined cell populations in the aberrant cerebellum of PTPMT1 knockout mice at P8. GCs (NeuN+), the most abundant neurons in the cerebellum, were barely detected (Fig. 1B). The number of PCs (Calbindin+) did not decrease, but they were highly disorganized and featured a marked reduction in the number of dendrites relative to wild-type cells (Fig. 1B and fig. S2A). Examination of P1 cerebella revealed less severe defects in PTPMT1 knockout mice (Fig. 1C and fig. S2B)—GCs and PCs were readily detected, although foliation had not begun. Math1+ GC progenitors (GCPs) and Lhx1+ PC progenitors (PCPs) developed without noticeable defects in PTPMT1 knockout cerebellar primordium at E12.5, E14.5, and E17.5 (Fig. 1D). Collectively, these observations suggest that cerebellar development in PTPMT1 knockout mice was mainly blocked at the perinatal stage.
PTPMT1 ablation showed marginal effects on the proliferation of PCPs or GCPs
We examined proliferative and postmitotic cells in postnatal cerebella by immunostaining for cyclin D1 and p27, respectively. GCPs showed robust proliferation in the outmost half of the EGL in control P8 cerebella. In contrast, proliferating GCPs in PTPMT1 knockout mice at P8 greatly decreased (Fig. 1E). Similar results were obtained when using Ki67 or proliferating cell nuclear antigen (PCNA) as a marker to visualize replicating cells (fig. S2A). Active cell proliferation was observed at the EGL and in the parenchyma of P1 and E18.5 knockout cerebella (Fig. 1F and fig. S2, B to F). These proliferating cells were identified as GCPs based on colocalization of PCNA and the GCP marker Pax6 (fig. S2D). The density of Pax2+ interneurons was not changed in the knockout cerebella (fig. S2E), suggesting that interneuron development was not affected. p27+ postmitotic cells were present in the inner part of the EGL and the internal granule layer in control cerebella. These quiescent cells were also decreased and ectopically distributed in the knockout cerebella (Fig. 1, E and F). Immunostaining of cleaved caspase 3 or TUNEL (terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling) assays showed increased apoptosis in PTPMT1 knockout cerebella at P8 and P1 as compared to control tissues (Fig. 1G and fig. S2G). Apoptosis was also increased in knockout cerebra (see below). Because GCPs were proliferating in P1 knockout cerebella (fig. S2, B to E), the increased apoptosis and the markedly decreased number of GCs in P8 knockout cerebella (Fig. 1B) imply that nascent knockout GCs died without migrating and homing to the destination.
PC- and GC-specific PTPMT1 knockout mice exhibited only mild to moderate defects
To define the cell population that was responsible for the developmental arrest of the cerebellum in neural precursor/stem cell PTPMT1 knockout (PTPMT1fl/fl/Nestin-Cre+) mice, we generated PC-specific, GC-specific, and PC/GC double knockout mice by crossing PTPMT1fl/+ mice with PCP2 (L7)-Cre+ (22) and Atoh1 (Math1)-Cre+ (23) transgenic mice that express Cre specifically in PC and GC lineages, respectively. While GC-specific and PC/GC double PTPMT1 knockout mice displayed mild to moderate developmental defects in the cerebellum (the anterior lobules were abnormal but the foliation pattern was unaffected), structural changes in the PC-specific knockout cerebellum were minimal (Fig. 2, A to C), and the cerebellar phenotypes of PC/GC double PTPMT1 knockout mice were indistinguishable from those of GC single PTPMT1 knockout mice. In contrast to PTPMT1fl/fl/Nestin-Cre+ mice, which died between P9 and P12, none of these three lines of lineage-specific knockout mice died prematurely, although they manifested ataxia as evidenced by altered walking gaits (Fig. 2D). The fact that PC-specific, GC-specific, and PC/GC double PTPMT1 knockout mice had much milder developmental defects in the cerebellum than neural precursor/stem cell PTPMT1 knockout mice (PTPMT1fl/fl/Nestin-Cre+ mice) suggests that the profound cerebellar phenotypes of PTPMT1fl/fl/Nestin-Cre+ mice were not caused by defective GCPs, PCPs, or both.
Fig. 2. Deletion of PTPMT1 from PCPs and/or GCPs generates only mild to moderate effects on cerebellar development.

(A and B) PC-specific (PTPMT1fl/fl/PCP2-Cre+), GC-specific (PTPMT1fl/fl/Atoh1-Cre+), and PC/GC double knockout (PTPMT1fl/fl/PCP2-Cre+/Atoh1-Cre+) mice were generated. Cerebellar sections were prepared at 1 month of age for histopathological examination (H&E staining) (A) and immunofluorescence staining with the indicated antibodies (B). (C) PCs and GCs were isolated from PC-, GC-, and PC/GC double knockout mice at P4 (n = 3 per group). PTPMT1 mRNA levels in these cells were determined by qRT-PCR. (D) Footprint tests were performed to examine the gait of adult PC-specific, GC-specific, and PC/GC double knockout mice and control littermates. Dashed lines represent the direction of progression of walking. (E) PTPMT1 inducible knockout (PTPMT1fl/fl/CAG-Cre+-Esr) mice were generated and treated with tamoxifen by intraperitoneal injections (9 mg/40 g body weight, five doses over 10 days) at 6 weeks of age. Seven days after the last dose of tamoxifen administration, cerebellar sections were prepared for histopathological examination (H&E staining) and immunofluorescence staining with the indicated antibodies. (F) PTPMT1 mRNA levels in adult cerebral cortices and cerebella with the indicated genotypes (n = 4 per group) were determined by qRT-PCR. (G) Footprints of adult PTPMT1fl/fl/CAG-Cre+-Esr mice and PTPMT1+/+/CAG-Cre+-Esr littermates. Representative images from three mice per genotype are shown.
PTPMT1 depletion from adult cerebella resulted in only transient defects
To further determine the role of PTPMT1 in the cerebellum, we generated inducible PTPMT1 knockout mice (PTPMT1fl/fl/CAG-Cre+-Esr) by crossing PTPMT1fl/+ mice (20) with CAG-Cre+-Esr mice (24) and deleted PTPMT1 (>95%) from adult mice by tamoxifen administration (Fig. 2, E and F). Histopathological examination of the cerebellum in these animals revealed only mild histological changes, although increased apoptosis was detected (Fig. 2E). Depletion of PTPMT1 resulted in ataxia, tremor, and impaired motor coordination in the first several days following the gene deletion (Fig. 2G). These animals started to recover 10 days later and appeared normal thereafter, likely due to the regeneration from residual PTPMT1-undeleted cells, because PTPMT1 deletion efficiency in the cerebellum dropped to ~70% 50 days after tamoxifen administration. These data suggest that PTPMT1 plays a less important role in the maintenance than in the development of the cerebellum.
BG development was disrupted in PTPMT1 knockout cerebella
We next examined BG, which are derived from multipotent precursor cells RG and play a critical role in the correct patterning of cortical foliation and layering during cerebellar development (10, 11). In wild-type mice, BG, as visualized by immunostaining for glial fibrillary acidic protein (GFAP), brain lipid-binding protein (BLBP), and Nestin, were located to the PCL and extended long, radially oriented parallel processes to the basement membrane of the pial surface, forming glial scaffolds. In contrast, BLBP-labeled cells in PTPMT1 knockouts (PTPMT1fl/fl/Nestin-Cre+) did not have radial fibers. Nestin+/GFAP+/BLBP+ radial fibers were barely detected (Fig. 3, A and B, and fig. S3A). Moreover, the somata of BLBP+ cells were ectopically located in the molecular layer (Fig. 3, A and B). These aberrant BLBP+ cells might be either intermediate cells that had failed to mature into BG or stellate-like astrocytes that had prematurely developed from RG in the absence of PTPMT1. However, GFAP+ cells (Fig. 3A) were readily detected in the knockout cerebella, suggesting that parenchymal astrocytes could be formed in the knockouts.
Fig. 3. BG development is disrupted in neural precursor/stem cell PTPMT1 knockout mice.

(A to D) Cerebellar sections prepared from PTPMT1+/+/Nestin-Cre+ and PTPMT1fl/fl/Nestin-Cre+ mice at the indicated ages were processed for immunofluorescence staining with the indicated antibodies, followed by DAPI counterstaining. Representative images from three mice per group are shown. (E) Cerebellar cortical tissues dissected from PTPMT1+/+/Nestin-Cre+ and PTPMT1fl/fl/Nestin-Cre+ newborn pups (P0) and embryos at E17.5 were processed for microexplant culture. Seventy-two hours later, tissues and cells on the coverslips were processed for immunofluorescence staining for Nestin and GFAP, followed by DAPI counterstaining.
The loss of functional BG in PTPMT1 knockout cerebella happened mainly at the perinatal stage because smaller differences in BLBP+ cells between PTPMT1 knockout and control cerebella were noticed at P1 (Fig. 3C and fig. S3B). At E17.5, Nestin+ radial fibers (primarily from RG at this stage) were present without noticeable changes in PTPMT1-depleted cerebellar primordium, while nascent BLBP+Nestin+ cells were reduced (Fig. 3D). It is possible that loss of PTPMT1 prevented early precursor cells RG from undergoing differentiation into BG at the perinatal stage. Cerebellar microexplant culture with cerebellar cortical tissues dissected from P0 pups and E17.5 embryos showed that the capabilities of PTPMT1 knockout precursor cells to differentiate into progeny with GFAP+Nestin+ fibers were barely detectable (Fig. 3E).
To further define the developmental stage in which PTPMT1 depletion caused the block in cerebellar development, we generated inducible neural precursor/stem cell knockout mice (PTPMT1fl/fl/Nestin-Cre+-Esr1) by crossing PTPMT1fl/+ mice (20) with Nestin-Cre+-Esr1 mice (25). PTPMT1 was deleted from developing embryos by treating timed-pregnant female mice with tamoxifen. Tamoxifen treatment at E13.5 produced similar phenotypes in PTPMT1fl/fl/Nestin-Cre+-Esr1 pups that were born to those displayed by constitutive Nestin-Cre–mediated PTPMT1 knockout mice (PTPMT1fl/fl/Nestin-Cre+). All PTPMT1fl/fl/Nestin-Cre+-Esr1 pups died between P10 and P17. Cerebellar development in these knockout pups was blocked, and development of BG with radial fibers was completely disrupted (Fig. 4, A to C). Tamoxifen treatment at E18.5 (PTPMT1 was likely depleted after BG had developed) did not cause notable defects in cerebellar development (Fig. 4, D and E). These data verify that the role of PTPMT1 in cerebellar development is developmental stage specific and that defective BG development is responsible for the block of cerebellar development in PTPMT1 knockout (PTPMT1fl/fl/Nestin-Cre+) mice.
Fig. 4. Induced deletion of PTPMT1 from neural precursor cells at E13.5, but not E18.5, blocks cerebellar development.
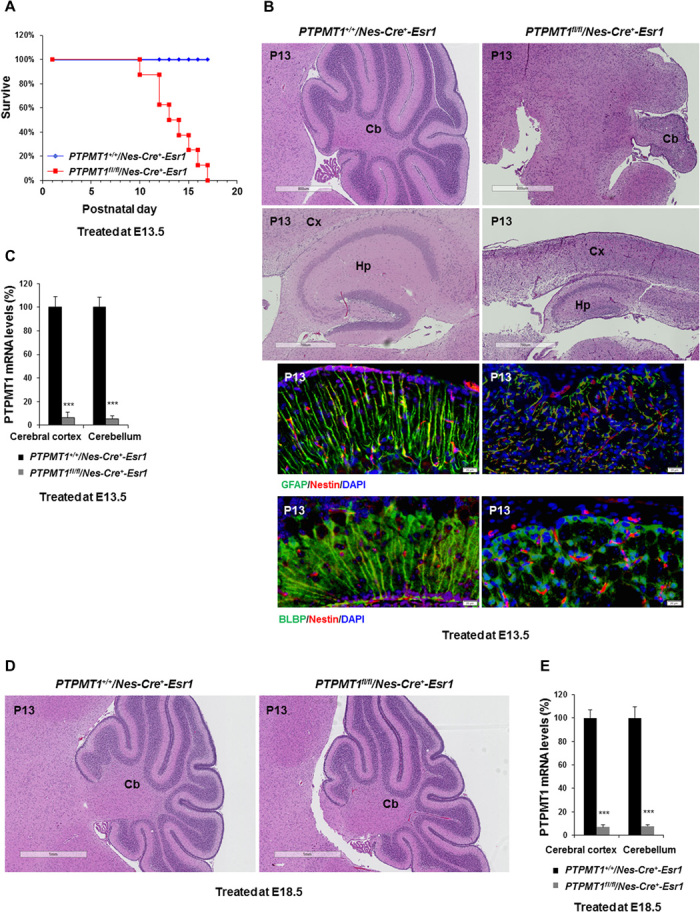
PTPMT1fl/+/Nestin-Cre+-Esr1 mice were generated and used to cross PTPMT1fl/+ mice. (A to C) Timed-pregnant female mice at E13.5 were administered tamoxifen by an intraperitoneal injection [2 mg dissolved in corn oil/ethanol (9:1) mixture]. After giving birth, lactating mothers were treated with tamoxifen again (83.5 mg/kg body weight, daily for 5 days). (A) Kaplan-Meier survival curves of PTPMT1fl/fl/Nestin-Cre+-Esr1 (n = 8) and PTPMT1+/+/ Nestin-Cre+-Esr1 (n = 24) pups. Representative cerebellar and cerebral sections (H&E staining) of PTPMT1fl/fl/Nestin-Cre+-Esr1 and PTPMT1+/+/ Nestin-Cre+-Esr1 pups at P13 are shown. Cx, cortex; Hp, hippocampus. Cerebellar sections were also processed for immunofluorescence staining with the indicated antibodies, followed by DAPI counterstaining. (B) Representative images. (C) PTPMT1 mRNA levels in freshly isolated cerebral cortices and cerebella with the indicated genotypes (n = 3 mice per group) were determined by qRT-PCR. (D and E) Timed-pregnant female mice at E18.5 were administered tamoxifen as above. Cerebellar sections (H&E staining) of PTPMT1fl/fl/Nestin-Cre+-Esr1 and PTPMT1+/+/ Nestin-Cre+-Esr1 pups at P13 are shown (D). PTPMT1 mRNA levels in freshly isolated cerebral cortices and cerebella with the indicated genotypes (n = 3 mice per group) were determined by qRT-PCR (E).
PTPMT1 depletion caused cell cycle arrest in NSPCs, leading to defective self-renewal and senescence
To determine the mechanisms by which PTPMT1 deficiency affects multipotent precursor cells, we examined NSPCs in the cerebellar parenchyma, which share certain traits with RG. Compared to controls, neurospheres derived from PTPMT1 knockout cerebellar NSPCs not only decreased in number but also were smaller in size (Fig. 5A) (PTPMT1 was nearly completely deleted in the knockout neurospheres). To further assess the self-renewing capability of NSPCs, we subjected primary neurospheres to the secondary neurosphere assay. Cells isolated from PTPMT1 knockout primary neurospheres produced fewer secondary neurospheres (Fig. 5A). Proliferation of PTPMT1 knockout neurosphere cells was essentially blocked (Fig. 5A), verifying the profound effect of PTPMT1 deficiency on the self-renewal of cerebellar NSPCs. The defect in the self-renewal of PTPMT1-depleted NSPCs was associated with increased cell death because apoptotic cells increased by nearly threefold in PTPMT1 knockout neurospheres (Fig. 5B). To further define the detrimental effects of PTPMT1 deficiency on NSPCs, we analyzed the cell cycle status of neurosphere cells. These analyses revealed a significant increase in the percentage of knockout cells in the G0 phase and a marked decrease in the percentages of G1 and S-G2-M cells (Fig. 5C), indicative of cell cycle arrest. Consistent with the cell cycle profile, p19 and p21, cyclin-dependent kinase inhibitors controlling the cell cycle progression, were robustly up-regulated in PTPMT1-depleted neurosphere cells (Fig. 5D). Moreover, p16, a senescence marker, was increased by more than threefold.
Fig. 5. PTPMT1 depletion causes bioenergetic stress and cell cycle arrest in cerebellar NSPCs, leading to diminished self-renewal and senescence.

(A to C) Prominin+Lin− cells were sorted from individual PTPMT1fl/fl/Nestin-Cre+ and PTPMT1+/+/Nestin-Cre+ cerebella at P4 (n = 3 mice per group). These cells were assessed by the neurosphere assay. Neurospheres derived were counted after 7 days of culture. Representative neurospheres are shown on the left. Primary (1°) neurospheres were harvested and assayed by the neurosphere culture again. Secondary (2°) neurospheres derived were counted 7 days later. (A) Total cell numbers were determined at the indicated time points. Primary neurosphere cells were assayed by apoptosis analyses. (B) Annexin V+ apoptotic cells were quantified by fluorescence-activated cell sorting (FACS). (C) Cell cycle profiles of primary neurosphere cells were determined by Ki67 and Hoechst 33342 staining, followed by FACS analyses. (D) mRNA levels of the indicated cell cycle regulatory genes in primary neurosphere cells were determined by qRT-PCR (n = 6 mice per group). (E) Total DNA was extracted from neurospheres. Mitochondrial content was estimated by comparing mitochondrial DNA (mtDNA; cytochrome B) levels to genomic DNA levels by qPCR (n = 3 mice per group). NS, not significant. (F and G) Total cellular ATP and ROS levels in neurosphere cells were determined. (H) Prominin-1+Lin− cells were sorted from individual cerebella dissected from PTPMT1fl/fl/Nestin-Cre+ (n = 5) and PTPMT1+/+/Nestin-Cre+ (n = 3) pups at P4. Whole-cell lysates were prepared and examined by immunoblotting with the indicated antibodies. Representative results are shown. ACC, acetyl–coenzyme A (CoA) carboxylase. (I) Neurospheres generated from the cortices of PTPMT1fl/fl/Nestin-Cre+ and PTPMT1+/+/Nestin-Cre+ mice (n = 3 per group) were lysed and examined by immunoblotting with the indicated antibodies. Representative results are shown.
PTPMT1fl/fl/Nestin-Cre+ mice also displayed significant developmental defects in the cerebrum (fig. S1, B and C), and cell proliferative activities were decreased in the cortex, hippocampus, and subventricular zone where multipotent NSPCs and transient proliferating progenitors reside (fig. S4, A and B). Moreover, markedly increased apoptosis was detected in knockout cerebra (fig. S4C). To further verify the impact of PTPMT1 deletion on NSPCs, we also examined cerebral NSPCs. Similar defects to those of knockout cerebellar NSPCs were observed in the cerebral NSPCs of PTPMT1fl/fl/Nestin-Cre+ mice. The self-renewal of PTPMT1 knockout cerebral NSPCs was substantially decreased (fig. S5A). There was also a profound cell cycle arrest in PTPMT1-depleted neurosphere cells due to robust up-regulation of p19, p21, and p16, and apoptosis was markedly increased (fig. S5, B to D). In addition, neurosphere cells lacking PTPMT1 showed reduced neuronal but enhanced astrocyte differentiation (fig. S5E), suggesting that PTPMT1 is also important for balanced lineage specification of NSPCs.
The transcriptional factor p53 plays an important role in the cell cycle checkpoint and apoptosis; therefore, we asked whether the cell cycle arrest and diminished self-renewal of PTPMT1-depleted NSPCs could be rescued by deletion of p53. To this end, we generated PTPMT1fl/fl/Nestin-Cre+/p53−/− mice, in which both PTPMT1 and p53 were deleted in neural precursor/stem cells. These double mutants also died postnatally, as did PTPMT1 single knockouts (fig. S6A). Self-renewing capabilities of PTPMT1/p53 double knockout NSPCs were also greatly decreased (fig. S6B), similar to those of PTPMT1 single knockout cells. These data demonstrate that the cell cycle changes and apoptosis induced by PTPMT1 depletion were independent of p53.
PTPMT1 depletion induced bioenergetic stress
PTPMT1 is localized to the mitochondrial inner membrane where oxidative phosphorylation and ATP synthesis take place. We assessed the fundamental impact of PTPMT1 depletion on the mitochondria using neurosphere cells derived from the cerebella. The overall mitochondrial content in PTPMT1-ablated cells was comparable to that in wild-type cells (Fig. 5E). However, total cellular ATP levels in PTPMT1 knockout cells were decreased by ~50% (Fig. 5F). Likewise, levels of reactive oxygen species (ROS), the by-product of mitochondrial oxidative phosphorylation, were decreased by half in PTPMT1-depleted cells (Fig. 5G). [These ROS data also suggest that the increased apoptosis in PTPMT1 knockout cells (Fig. 5B) was not caused by oxidative damage]. We also examined cerebra-derived neurosphere cells. Essentially identical results were obtained (fig. S7, A to C). Adenosine 5′-monophosphate–activated kinase (AMPK), the master intracellular energy sensor (26), was highly activated in PTPMT1 knockout cerebellar stem cell–enriched prominin-1+Lin−(CD44−CD140α−PSA-NCAM−) cells (Fig. 5H) and cerebral neurosphere cells (Fig. 5I), as determined by phosphorylation of Thr172. Acetyl–coenzyme A (CoA) carboxylase, a target of AMPK and a negative regulator of fatty acid oxidation (27), was inhibited, as evidenced by a marked increase in the inhibitory phosphorylation of this enzyme (Fig. 5, H and I), implying increased fatty acid oxidation in PTPMT1 knockout cells. In agreement with previous findings that AMPK negatively regulates mammalian target of rapamycin (mTOR) (28, 29), the activity of the S6 ribosomal protein, a key downstream component of mTOR signaling, was greatly decreased (Fig. 5, H and I), suggesting a reduction in anabolic activity in PTPMT1-deleted cells.
Loss of PTPMT1 reprogrammed cellular metabolism by limiting mitochondrial utilization of pyruvate
We next determined mitochondrial aerobic metabolism in intact, viable neurosphere cells with full access to all metabolic substrates, by real-time measurement of oxygen consumption under basal conditions, in the presence of the mitochondrial inhibitor oligomycin, the mitochondrial uncoupling compound carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP), and the respiratory chain inhibitor rotenone. Compared to control cells, PTPMT1 knockout cells displayed much lower basal oxygen consumption rates (OCRs) and maximal oxidative capacity (Fig. 6A). Measurement of extracellular proton flux showed that PTPMT1-depleted cells had slightly increased basal extracellular acidification rates (ECARs; Fig. 6B). In response to the mitochondrial inhibition by oligomycin, ECARs were further elevated in control cells conceivably due to the diversion of pyruvate from oxidation in the mitochondria to fermentation in the cytosol. In contrast, this adaptive response to mitochondrial inhibition was dampened in PTPMT1-depleted cells, indicating that pyruvate was already largely reduced to lactate under basal conditions in these knockout cells.
Fig. 6. PTPMT1 depletion from the mitochondria reprograms cellular metabolism by limiting mitochondrial pyruvate utilization.
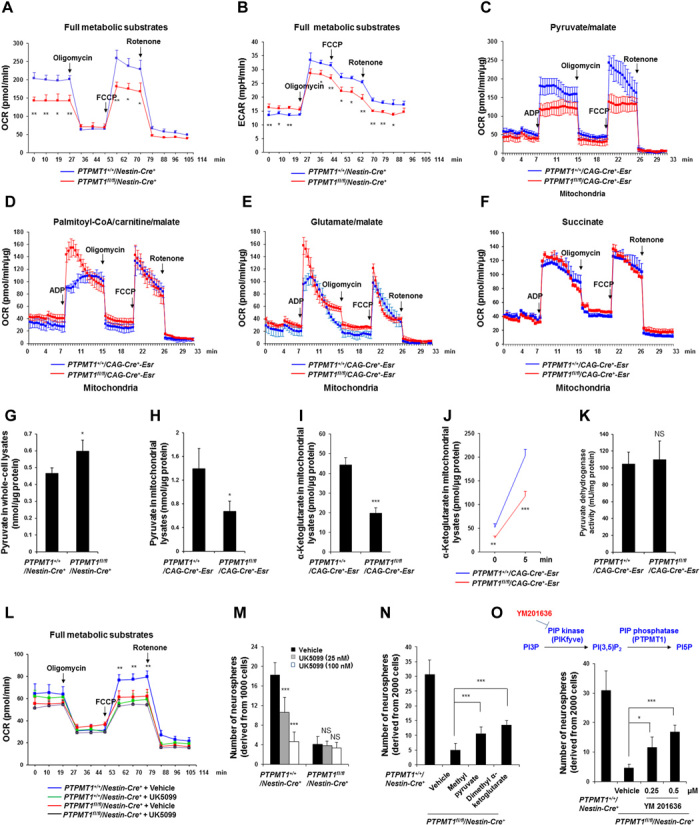
(A and B) Neurospheres derived from PTPMT1+/+/Nestin-Cre+ and PTPMT1fl/fl/Nestin-Cre+ mice (n = 3 mice per group) were dissociated into single cells. OCR (A) and ECAR (B) of these live cells in the medium containing all metabolic substrates were measured. Representative results are shown. (C to F) Mitochondria were isolated from adult brains dissected from PTPMT1fl/fl/CAG-Cre+-Esr mice and PTPMT1+/+/CAG-Cre+-Esr littermates (n = 3 mice per group) 10 days following tamoxifen administration. Oxygen consumption of the mitochondria was measured in the presence of pyruvate/malate (C), palmitoyl-CoA/carnitine/malate (D), glutamate/malate (E), or succinate (F), following the addition of ADP, oligomycin, FCCP, and rotenone. Representative results are shown. (G to I) Pyruvate and α-ketoglutarate levels in the lysates of the neurosphere cells derived from PTPMT1+/+/Nestin-Cre+ and PTPMT1fl/fl/Nestin-Cre+ mice (n = 3 mice per group) or in the lysates of the mitochondria isolated from the cerebella of adult PTPMT1fl/fl/CAG-Cre+-Esr mice and PTPMT1+/+/CAG-Cre+-Esr littermates (n = 3 mice per group) 10 days following tamoxifen administration were measured. (J and K) Mitochondria were isolated from the cerebella freshly dissected from adult PTPMT1fl/fl/CAG-Cre-Esr+ and PTPMT1+/+/CAG-Cre-Esr+ mice (n = 3 mice per group) as above. After being washed three times in mitochondrial assay solution (MAS) buffer, mitochondria were incubated with pyruvate/malate (5 mM) and ADP (4 mM) in a non-CO2 incubator. Five minutes later, mitochondria were collected, washed, and lysed. (J) α-Ketoglutarate levels in the mitochondrial lysates were measured. (K) PDH activities in the mitochondrial lysates were determined with a PDH assay kit following the manufacturer’s instructions. (L) Neurospheres derived from PTPMT1fl/fl/Nestin-Cre+ and PTPMT1+/+/Nestin-Cre+ mice (n = 3 mice per group) were dissociated into single cells. These cells were treated with the MPC inhibitor UK5099 (25 nM) or vehicle for 20 min. OCRs of the cells in the presence of all metabolic substrates were then measured as described above. Statistical analysis results between UK5099-treated and vehicle-treated PTPMT1+/+/Nestin-Cre+ cells are shown. (M to O) Cerebral cortices dissected from PTPMT1fl/fl/Nestin-Cre+ and PTPMT1+/+/Nestin-Cre+ mice (n = 3 per group) were assessed by the neurosphere assay in the presence of the MPC inhibitor UK5099 at the indicated concentrations (M), methyl pyruvate (20 mM), dimethyl α-ketoglutarate (6 mM) (N), or the PIKfyve inhibitor (YM201636) at the indicated concentrations (O). Neurospheres derived were counted after 7 days of culture.
We also examined mitochondrial metabolism of the entire cerebral cortex cell population, which is mainly composed of mature neural cells and intermediate progenitors. Mitochondrial oxygen consumption of PTPMT1 knockout cells was moderately decreased at the basal levels and maximal reserve capacities (fig. S8A). Homeostatic cellular ATP and ROS levels in the entire cortex cell population were not changed (fig. S8, B and C). Moreover, there was no significant change in the cell cycle status of the entire knockout cortex cell population (fig. S8D). These data indicate that mature neural cells and intermediate progenitors are more flexible than earlier precursor and stem cells in activating alternative energy production pathways in response to the same energy deficit caused by PTPMT1 depletion from the mitochondria.
To gain further insights into the acting mechanisms of PTPMT1 in mitochondrial metabolism, we isolated mitochondria from the cerebella of adult PTPMT1 knockout mice (PTPMT1fl/fl/CAG-Cre+-Esr) (to obtain sufficient amount of mitochondria) and measured mitochondrial respiratory responses to single, specific metabolic substrates. In the presence of pyruvate, the capacity of PTPMT1-depleted mitochondria to metabolize oxygen, which was triggered by ATP synthesis from adenosine 5′-diphosphate (ADP), was markedly decreased (Fig. 6C), suggesting that pyruvate utilization was impaired in PTPMT1-deficient mitochondria. Enhanced mitochondrial oxygen consumption was observed when fatty acids were supplied as the fuel (Fig. 6D). In addition, moderately increased respiratory responses were observed in PTPMT1-depleted mitochondria when glutamate, another important mitochondrial metabolic substrate, was provided (Fig. 6E). Notably, succinate, the substrate for complex II of the electron transport chain, resulted in similar OCRs in PTPMT1-depleted and control mitochondria (Fig. 6F), validating the intact functionality of the electron transport chain in the knockout mitochondria. These mechanistic data suggest that the fuel selection of PTPMT1-ablated mitochondria was altered.
We then measured pyruvate in neurosphere cells and in the mitochondria purified from knockout cerebella. Pyruvate levels in PTPMT1 knockout neurosphere cells increased (Fig. 6G), whereas its levels in the knockout mitochondria decreased by half (Fig. 6H). Levels of α-ketoglutarate, an important metabolite in the tricarboxylic acid cycle, were decreased by half in the knockout mitochondria (Fig. 6I). Mitochondrial acetyl-CoA levels were not significantly changed (data not shown), likely because acetyl-CoA could also be generated from free fatty acids and amino acids and that it could be transported outside of the mitochondria for lipid synthesis. Furthermore, we incubated freshly isolated mitochondria with pyruvate and determined pyruvate transportation efficiency by measuring acute production of α-ketoglutarate from extramitochondrial pyruvate. As shown in Fig. 6J, production of α-ketoglutarate in PTPMT1-depleted mitochondria was significantly decreased compared to that in control mitochondria. Because pyruvate transporter MPC1/2 were equally expressed in PTPMT1 knockout cells (Fig. 5, H and I), and the activity of pyruvate dehydrogenase (PDH), the enzyme that catalyzes the conversion of pyruvate into acetyl-CoA in the mitochondria, was not affected in the knockout mitochondria (Fig. 6K), the decrease in the mitochondrial pyruvate and α-ketoglutarate and the concomitant increase in the cytosolic pyruvate in PTPMT1 knockout cells suggest that pyruvate uptake/transportation of the knockout mitochondria was diminished.
To further verify that PTPMT1 depletion decreases mitochondrial metabolism by inhibiting pyruvate uptake, we treated wild-type and PTPMT1 knockout neurosphere cells with the MPC inhibitor UK5099 (30). Inhibition of mitochondrial uptake of pyruvate significantly decreased mitochondrial oxygen consumption in wild-type cells (Fig. 6L). This effect of UK5099 was minimal in PTPMT1 knockout cells, implying that pyruvate uptake/transportation in PTPMT1-ablated mitochondria was already decreased. Moreover, UK5099 inhibited neurosphere-forming capabilities in wild-type NSPCs in a dose-dependent manner, recapitulating the effect of PTPMT1 deficiency (Fig. 6M). However, this function of UK5099 was undetectable in PTPMT1 knockout cells. Furthermore, methyl pyruvate, which freely crosses the mitochondrial inner membrane and is cleaved by matrix esterase to generate intramitochondrial pyruvate, partially restored neurosphere-forming capabilities in PTPMT1 knockout NSPCs (Fig. 6N). Similar rescue effects were observed when PTPMT1-depleted cells were treated with dimethyl α-ketoglutarate (Fig. 6N), which generates α-ketoglutarate after entering the mitochondria. Collectively, these data provide additional support for the notion that loss of PTPMT1 reprogrammed cellular metabolism by limiting mitochondrial utilization/uptake of pyruvate.
Last, since PTPMT1 favors PIPs, especially PI(3,5)P2 and PI5P, as substrates (17, 18), we reasoned that if the mitochondrial metabolic alterations and thus the impaired function of PTPMT1 knockout neural precursor/stem cells were attributed to accumulation of PTPMT1 downstream substrates, then inhibition of PI(3,5)P2 (the major PIP substrate of PTPMT1) production would rescue these defects. To test this hypothesis, we treated PTPMT1 knockout NSPCs with YM201636, a selective inhibitor of type III PIP kinase PIKfyve (31) that is responsible for the generation of PI(3,5)P2. As shown in Fig. 6O, inhibition of PI(3,5)P2 production largely restored the neurosphere-forming capabilities of PTPMT1 knockout NSPCs. These rescue data reaffirm that the functional defects of PTPMT1 knockout neural precursor/stem cells are likely caused by excess PIPs in the mitochondria.
DISCUSSION
The understanding of the cellular and molecular mechanisms governing the development and maintenance of the central nervous system remains incomplete. Dysregulation of these mechanisms is associated with its developmental disorders, degenerative diseases, and cancer. Here, we demonstrate that the mitochondrial phosphatase PTPMT1 plays a critical role in the development of the central nervous system, especially the cerebellum. Deletion of PTPMT1 in neural precursor cells completely blocked cerebellar development at the perinatal stage, and cerebral development was also significantly compromised. Further mechanistic analyses showed that ablation of PTPMT1 decreased mitochondrial aerobic metabolism due to reduction in the utilization of the master metabolic substrate pyruvate (a key metabolite of glucose). This work reveals an essential role of PTPMT1 in coordinating mitochondrial metabolism and glycolysis in brain development and that mitochondrial oxidation of the carbohydrate fuel is required for neural precursor/stem cell function.
One of the interesting findings in this study is that different cell populations in the cerebellum have remarkably distinct reliance on optimal mitochondrial metabolism for cellular functions. Cerebellar development involves several cell populations, and cellular activities in the developing cerebellum are extremely dynamic. Cell type specification of precursor cells, rapid cell proliferation, and cell migration/patterning occur during cerebellar development, which are thought to require tremendous energy supply and robust biosynthesis. Glucose is the most important metabolic substrate for the brain (2), and previous studies have shown that astrocytes generally exhibit much higher glycolytic flux than neurons (5, 6). The glucose transport capacity and glycolytic rate of BG (specialized astroglial cells) are several-fold higher than those of PCs (32), although PCs are estimated to spend more energy than BG. Unexpectedly, we found that proliferation of GCPs, the most actively replicating neurons in the developing cerebellum, was only moderately reduced and that PCP proliferation did not seem to be affected by PTPMT1 depletion, which impaired mitochondrial metabolism of glucose but enhanced cytosolic glycolysis (Fig. 6, A and C). PC-specific, GC-specific, and PC/GC double PTPMT1 knockout mice displayed only mild-to-moderate developmental defects in the cerebellum, and depletion of PTPMT1 from perinatal and adult cerebella produced only minimal transient pathological effects (Figs. 2, A to G, and 4D). In contrast, multipotent precursor cells (RG) were much more severely affected in PTPMT1fl/fl/Nestin-Cre+ mice. RG differentiation into BG was essentially blocked in microexplant culture (Fig. 3E), and correctly localized BG with radial fibers were undetectable in PTPMT1 knockout postnatal cerebella (Fig. 3, A to D). Moreover, self-renewal of PTPMT1-depleted NSPCs was severely impaired, and they underwent pronounced senescence (Fig. 5A and fig. S5A). The differential responses of multipotent precursor/stem cells and PCs/GCs to the same metabolic changes caused by loss of PTPMT1 suggest that neural precursor/stem cells rely more on efficient mitochondrial metabolism of glucose than on progenitors and mature cells.
PTPMT1 knockout PCPs, GCPs, and mature neural cells tolerated well the reprogrammed cellular metabolism caused by PTPMT1 deficiency. This was partly because reduction of pyruvate oxidation in the mitochondria and accumulation of pyruvate in the cytosol in PTPMT1 knockout cells might be actually beneficial for the biosynthesis required for these progenitor cell proliferation, and compensatorily increased utilization of alternative fuels (fatty acids and glutamate) in PTPMT1-depleted mitochondria (Fig. 6, D and E) might be sufficient for energy production. In addition, progenitors and mature cells appear to be more flexible than immature precursor/stem cells in activating alternative energy production pathways in response to the energy stress. Steady-state cellular ATP levels were well maintained in the whole-cortex cell population in PTPMT1 knockout mice (fig. S8B), whereas ATP levels in knockout NSPCs were decreased by half (Fig. 5F and fig. S7B).
Another interesting finding in this report is that mitochondrial metabolism in PTPMT1-depleted NSPCs was only moderately perturbed, but cellular responses (cell cycle arrest and senescence) were rather profound. This phenomenon is reminiscent of conditions involving nuclear DNA damage or mitotic spindle assembly checkpoints, in which a single double-strand DNA break or misalignment of a single chromosome causes complete cell cycle arrest. Because the energy stress sensor AMPK was highly activated in PTPMT1 knockout cells (Fig. 5, H and I), it is likely that the bioenergetic stress signal elicited by PTPMT1 ablation was sensed and propagated by a cascade of events, including hyperactivation of AMPK and robust up-regulation of cyclin-dependent kinase inhibitors p19/p21/p16, leading to the amplified cellular consequences (Fig. 5, A to D, and fig. S5, A to D). The observation that multipotent RG and NSPCs were much more sensitive and responsive than PCPs, GCPs, and mature neural cells to the same metabolic alterations suggests that the bioenergetic/metabolic stress–activated cell cycle checkpoint is specific for early precursor and stem cells, but not intermediate progenitors or mature cells.
Mechanistically, loss of PTPMT1 appears to decrease mitochondrial aerobic metabolism and cause bioenergetic/metabolic stress by limiting utilization/uptake of pyruvate. This notion is supported by the fact that ATP synthesis–driven oxygen consumption in PTPMT1-depleted mitochondria markedly decreased when pyruvate was provided as an exclusive fuel source, whereas oxygen consumption of these same mitochondria increased when fatty acids or glutamate was supplied as the substrate (Fig. 6, C to E). Moreover, levels of pyruvate and α-ketoglutarate in the mitochondria decreased by half (Fig. 6, H and I), while pyruvate in the cytosol increased in PTPMT1 knockout cells (Fig. 6G). Acute production of α-ketoglutarate from extramitochondrial pyruvate in PTPMT1-depleted mitochondria was significantly lower than that in control mitochondria (Fig. 6J). In addition, the observations that PTPMT1 deficiency and MPC inhibition produced similar effects on mitochondrial metabolism (Fig. 6L) and that methyl pyruvate and dimethyl α-ketoglutarate partially rescued PTPMT1 knockout NSPC self-renewal (Fig. 6N) also support that mitochondrial utilization/uptake of pyruvate was diminished in PTPMT1-depleted cells.
Nevertheless, it remains to be determined how PTPMT1 deficiency reduces mitochondrial uptake of pyruvate. PTPMT1 is localized to the inner membrane of the mitochondria where pyruvate transporters also reside. This phosphatase primarily dephosphorylates PIPs (17, 18). Our previous studies have shown that PIPs directly promote fatty acid–induced activation of uncoupling protein 2 (UCP2) (20), a C4 metabolite transporter whose elevated activity inhibits mitochondrial metabolism but enhances cytosolic glycolysis (33–35). It is possible that PTPMT1 facilitates mitochondrial metabolism by inhibiting UCP2. Accordingly, depletion of PTPMT1 leads to buildup of downstream PIP substrates in the mitochondria, which, in turn, enhance UCP2 function and thereby critically alter cellular metabolic activities. Inhibition of PI(3,5)P2 (the major PIP substrate of PTPMT1) production by a selective inhibitor of the upstream type III PIP kinase PIKfyve largely rescued the NSPC defects caused by PTPMT1 deficiency (Fig. 6O). Another possibility is that accumulated PIPs might directly inhibit MPCs. Further studies are required to investigate how hyperactivation of UCP2 inhibits mitochondrial uptake/transportation of pyruvate and whether PIPs might physically and functionally interact with MPCs.
MATERIALS AND METHODS
Mice
PTPMT1fl/+ mice were generated in our previous study (20). Nestin-Cre+ (21), Nestin-Cre+-Esr1 (25), PCP2-Cre+ (22), Atoh1-Cre+ (23), CAG-Cre+-Esr (24), and p53+/− (36) mice were purchased from the Jackson Laboratory. Mice of the same age, sex, and genotype were mixed and then randomly grouped for subsequent analyses (investigators were not blinded). All mice were kept under specific pathogen–free conditions in Division of Animal Resources at Emory University. All animal procedures complied with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee.
Immunohistochemistry and immunofluorescence staining
Paraffin-embedded tissue sections were prepared at a thickness of 5 μm. Sections were dewaxed, rehydrated, and heated in antigen unmasking buffer [10 mM citric acid and 0.05% Tween 20 (pH 6.0)], followed by incubation in 3% H2O2 in methanol for 10 min to abolish endogenous peroxidases. After blocking sections with 10% normal goat serum (NGS) in tris-buffered saline (TBS) for 30 min, primary antibodies were applied for 16 hours at 4°C. Sections were then rinsed in 1% NGS in TBS, followed by incubation in 10% NGS for 10 min. For fluorescent signals of immunoreactivity, fluorescein isothiocyanate (FITC)– or phycoerythrin-conjugated secondary antibodies were used and counterstained with DAPI. Sections were mounted with Fluoromount-G.
In situ hybridization
Mouse brains were fixed with 4% paraformaldehyde (PFA), embedded in optimal cutting temperature (OCT) compound, and cryosectioned at a thickness of 10 μm. Sections were postfixed in 4% PFA/phosphate-buffered saline (PBS), acetylated, and incubated for 1 hour at room temperature in prehybridization buffer [50% formamide, 5× SSC, 1× Denhardt’s solution, yeast transfer RNA (tRNA; 250 μg/ml), and herring sperm DNA (500 μg/ml)]. Sections were hybridized overnight at 65°C in hybridization buffer [50% deionized formamide, 1× Denhardt’s solution, 300 mM NaCl, 20 mM tris-HCl (pH 8.0), 5 mM EDTA, 10 mM Na2HPO4 (pH 7.4), 10% dextran sulfate, and yeast tRNA (0.5 mg/ml)] containing DIG–uridine 5′-triphosphate–labeled probes for Lhx1 and Math1. Probes were synthesized using a DIG labeling kit (Roche, Indianapolis, IN) according to the manufacturer’s protocol. After hybridization, sections were incubated overnight at 4°C with an antibody against DIG that was conjugated with alkaline phosphatase (Roche, Indianapolis, IN). Bound probe was visualized by incubating slides in bromochloroindolyl phosphate–nitro blue tetrazolium (BCIP-NBT) at 4°C for several hours to several days in the dark. Coverslips were mounted with glycerol or mounting media.
Isolation of PCs and GCPs
Cerebella dissected from mice at P4 were minced and incubated in PBS containing 0.25% trypsin and 0.1% deoxyribonuclease at 37°C for 20 min. Trypsin was inactivated by addition of Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS). Cells were collected, resuspended in PBS, and then fractionated by two-layer Percoll gradient (35% Percoll and 65% Percoll in PBS) centrifugation. Astroglial cells were collected from the top of the gradient. PCs were collected from the upper interface of the gradient. GCPs were harvested from the 35%/65% interface of the Percoll gradient. These cells were washed in DMEM containing 10% FBS for subsequent experiments.
Isolation of NSPCs from the cerebellum
Cerebella were dissected in ice-cold PBS from P4 pups. After meninges were removed, cerebella were minced and incubated in PBS containing 0.25% trypsin and 0.1% deoxyribonuclease at 37°C for 20 min. Trypsin was inactivated by incubation with DMEM containing 10% FBS and penicillin (100 U/ml)–streptomycin (100 μg/ml) (PS). The mixture was passed through a cell strainer to remove debris and centrifuged at 3000 rpm for 5 min. The stem cell–enriched cell population was purified by sorting cells that expressed prominin-1 (CD133) and lacked the lineage markers CD44 (astrocyte progenitor marker), CD140α (oligodendrocyte progenitor marker), and PSA-NCAM (neuronal progenitor marker). Prominin-1+Lin− cells were cultured in B27/neurobasal medium supplemented with basic fibroblast growth factor (bFGF; 20 ng/ml), epidermal growth factor (EGF; 20 ng/ml), 2 mM l-glutamine, and PS.
The neurosphere assay
The neurosphere assay was performed to determine self-renewal capacity in NSPCs. NSPC-containing cerebral cortices dissected from E14.5 embryos were dissociated into single cells by trypsinization and mechanical dissociation. These cells or prominin-1+Lin− cells freshly sorted from P4 cerebella were suspended and maintained in B27/neurobasal medium supplemented with bFGF (20 ng/ml), EGF (20 ng/ml), 1 mM l-glutamine, and PS in 96-well plates at a density of 1000 cells/200 μl per well. After 7 days of incubation, neurospheres formed in the culture were counted under an optical microscope, and a minimum of 10 wells were counted. For the secondary neurosphere assay, primary neurospheres were collected and dissociated into a single-cell suspension and reseeded as above. Secondary neurospheres derived were enumerated 7 days later.
Apoptosis and cell cycle analyses
Neurospheres derived from cerebral cortices were harvested and dissociated into single cells. These cells or prominin-1+Lin− cells freshly isolated from cerebella at P4 were stained with annexin V–FITC and 7-amino-actinomycin D (7-AAD) using Annexin V–FITC Apoptosis Detection Kit I (BD Biosciences). Percentages of the annexin V–positive (early apoptotic cells) and annexin V/7-AAD double-positive cells (late apoptotic cells) were quantified by FACS. Neurosphere cells or prominin-1+Lin− cells freshly isolated from cerebella at P4 were fixed at 4°C overnight and then stained with FITC-labeled anti-Ki67 antibody followed by Hoechst 33342 (20 μg/ml) staining. Percentages of the cells in G0, G1, and S-G2-M phases were determined by FACS.
Neuronal and astroglial differentiation of NSPCs
Neurospheres (enriched with cerebral NSPCs) derived from E14.5 cerebral cortices were dissociated into single cells. These cells were seeded at 20,000 cells/ml onto coverslips precoated with poly-d-lysine (0.1 mg/ml) in serum-free B27/neurobasal medium with no growth factors. Cells were incubated in this adhesion culture for 5 days to allow neuronal differentiation. FBS (2%) was added on day 5 to enhance neuronal differentiation and astroglial differentiation. Differentiated cells were examined by immunofluorescence staining 2 days later (day 7).
Cerebellar microexplant culture
Cerebellar microexplants were cultured as previously described (37). P0 or E17.5 cerebella were stripped of meninges and deep cerebellar nuclei, and the remaining cortical tissues were minced and suspended in PBS. Explants were placed, pial surface uppermost, on the surface of millicell cell culture inserts (pore size, 0.4 μm; MilliporeSigma) on sterile medium (B27/neurobasal medium supplemented with 1 mM l-glutamine and PS) in culture dishes for 3 days. Tissues were fixed in 4% PFA in PBS for 1 day and then processed for immunofluorescence staining.
Measurement of cellular respiration, extracellular acidification, ATP, and ROS levels
Measurement of respiration of living cells with full access to all metabolic substrates was performed using a Seahorse XF24 analyzer (20). Respiration (OCR) was measured under basal conditions in the presence of the mitochondrial inhibitor oligomycin (1.2 μM), the mitochondrial uncoupling compound FCCP (5 μM), and the respiratory chain inhibitor rotenone (1 μM). Glycolytic activities were measured simultaneously using the same instrument based on the ECAR. Total cellular ATP levels were assessed using an ATP bioluminescent somatic cell assay kit (Sigma-Aldrich, St. Louis, MO), following the instructions provided by the manufacturer. To measure cellular ROS levels, cells were loaded with 2′-7′-dichlorofluorescein diacetate (5 μM) at 37°C for 15 min. ROS (H2O2) levels were then determined by FACS.
Measurement of respiration in isolated mitochondria
Mitochondria were freshly isolated from brains. Respiration of mitochondria was measured using a Seahorse XF24 analyzer as previously described (38). Mitochondria (10 μg of protein) were plated in each well of an XF24 plate in 50 μl of 1× MAS (pH 7.4; 70 mM sucrose, 220 mM mannitol, 5 mM KH2PO4, 5 mM MgCl2, 2 mM Hepes, 1 mM EGTA, and 0.2% fatty acid–free bovine serum albumin). XF24 plates were centrifuged at 4°C for 20 min at 2000g, and then 450 μl of MAS containing 5 mM pyruvate/5 mM malate, 5 mM glutamate/5 mM malate, 40 μM palmitoyl-CoA/40 μM carnitine/5 mM malate, or 10 mM succinate was added to each well. Plates were incubated at 37°C in a non-CO2 incubator for 8 to 10 min. OCRs were measured under basal conditions in the presence of ADP (4 mM), oligomycin (1.5 μM), FCCP (4 μM), and rotenone (1 μM), as described above.
Mitochondrial PDH activity assay, pyruvate, acetyl-CoA, and α-ketoglutarate measurement
Mitochondria were freshly isolated from brain tissues and washed two times with MAS. Mitochondrial pellets or neurosphere cells derived from cortices were lysed in buffer [50 mM tris-HCl (pH 7.4), 1% NP-40, 0.25% Na-deoxycholate, 150 mM NaCl, 1 mM EDTA, 1 mM NaF, 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride, and aprotinin/leupeptin (10 μg/ml)]. Lysates were subjected to the PDH assay, pyruvate, acetyl-CoA, and α-ketoglutarate measurement using PDH activity colorimetric assay, pyruvate assay, acetyl-CoA assay, and α-ketoglutarate assay kits purchased from BioVision, following the manufacturers’ instructions.
Footprint test
The footprint test was used to compare the gait of PTPMT1 knockout mice with that of control mice. To obtain footprints, the hind- and forefeet of the mice were coated with red and blue nontoxic ink, respectively. The animals were then allowed to walk on a fresh sheet of white paper on the floor of the runway to generate footprints. The footprint patterns were documented.
Statistical analysis
Data are presented as means ± SD of all mice analyzed in multiple independent experiments (that is, biological replicates; cells, mitochondria, etc. analyzed were not pooled but isolated from single individual mouse). Statistical significance was determined using an unpaired two-tailed Student’s t test. *P < 0.05, **P < 0.01, ***P < 0.001.
Supplementary Material
Acknowledgments
We are grateful to M. A. Smith and S. Siedlak (Case Western Reserve University) and T. Caspary and A. Long (Emory University) for their insightful advice and expert technical assistance. Funding: This work was supported by NIH grants DK092722 and HL130995 (to C.-K.Q.). Author contributions: H.Z., W.-M.Y., J.S., and D.H. conducted the research and summarized the data. S.K., J.Y.L., Y.S., A.M.K., and J.C. provided critical reagents, discussed the work, and edited the manuscript. C.-K.Q. designed the experiments and directed the entire study. H.Z. and C.-K.Q. wrote the manuscript with input from the other authors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/10/eaat2681/DC1
Fig. S1. PTPMT1 depletion from neural precursor cells compromises cerebral development.
Fig. S2. Active proliferation of GCPs and increased apoptosis in postnatal PTPMT1 knockout cerebella.
Fig. S3. Correctly localized BG with radial fibers are missing in PTPMT1 knockout cerebella.
Fig. S4. Decreased cell proliferation and increased apoptosis in PTPMT1 knockout cerebra.
Fig. S5. Loss of PTPMT1 causes cell cycle arrest in cerebral NSPCs, leading to defective self-renewal and senescence.
Fig. S6. Deletion of p53 does not rescue PTPMT1 knockout mice or NSPC self-renewal.
Fig. S7. Loss of PTPMT1 causes bioenergetic stress in cerebral NSPCs.
Fig. S8. The entire cerebral cortex cell population is less affected by PTPMT1 ablation.
REFERENCES AND NOTES
- 1.Vander Heiden M. G., Cantley L. C., Thompson C. B., Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 324, 1029–1033 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Magistretti P. J., Allaman I., A cellular perspective on brain energy metabolism and functional imaging. Neuron 86, 883–901 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Raichle M. E., Posner J. B., Plum F., Cerebral blood flow during and after hyperventilation. Arch. Neurol. 23, 394–403 (1970). [DOI] [PubMed] [Google Scholar]
- 4.Settergren G., Lindblad B. S., Persson B., Cerebral blood flow and exchange of oxygen, glucose, ketone bodies, lactate, pyruvate and amino acids in infants. Acta Paediatr. Scand. 65, 343–353 (1976). [DOI] [PubMed] [Google Scholar]
- 5.Itoh Y., Esaki T., Shimoji K., Cook M., Law M. J., Kaufman E., Sokoloff L., Dichloroacetate effects on glucose and lactate oxidation by neurons and astroglia in vitro and on glucose utilization by brain in vivo. Proc. Natl. Acad. Sci. U.S.A. 100, 4879–4884 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bolaños J. P., Almeida A., Moncada S., Glycolysis: A bioenergetic or a survival pathway? Trends Biochem. Sci. 35, 145–149 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Vaishnavi S. N., Vlassenko A. G., Rundle M. M., Snyder A. Z., Mintun M. A., Raichle M. E., Regional aerobic glycolysis in the human brain. Proc. Natl. Acad. Sci. U.S.A. 107, 17757–17762 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goyal M. S., Hawrylycz M., Miller J. A., Snyder A. Z., Raichle M. E., Aerobic glycolysis in the human brain is associated with development and neotenous gene expression. Cell Metab. 19, 49–57 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bauernfeind A. L., Barks S. K., Duka T., Grossman L. I., Hof P. R., Sherwood C. C., Aerobic glycolysis in the primate brain: Reconsidering the implications for growth and maintenance. Brain Struct. Funct. 219, 1149–1167 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Leto K., Arancillo M., Becker E. B., Buffo A., Chiang C., Ding B., Dobyns W. B., Dusart I., Haldipur P., Hatten M. E., Hoshino M., Joyner A. L., Kano M., Kilpatrick D. L., Koibuchi N., Marino S., Martinez S., Millen K. J., Millner T. O., Miyata T., Parmigiani E., Schilling K., Sekerková G., Sillitoe R. V., Sotelo C., Uesaka N., Wefers A., Wingate R. J., Hawkes R., Consensus paper: Cerebellar development. Cerebellum 15, 789–828 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buffo A., Rossi F., Origin, lineage and function of cerebellar glia. Prog. Neurobiol. 109, 42–63 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Lee A., Kessler J. D., Read T. A., Kaiser C., Corbeil D., Huttner W. B., Johnson J. E., Wechsler-Reya R. J., Origin, lineage and function of cerebella glia. Nat. Neurosci. 8, 723–729 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang V. Y., Zoghbi H. Y., Genetic regulation of cerebellar development. Nat. Rev. Neurosci. 2, 484–491 (2001). [DOI] [PubMed] [Google Scholar]
- 14.Roussel M. F., Hatten M. E., Cerebellum development and medulloblastoma. Curr. Top. Dev. Biol. 94, 235–282 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Millen K. J., Gleeson J. G., Cerebellar development and disease. Curr. Opin. Neurobiol. 18, 12–19 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pagliarini D. J., Wiley S. E., Kimple M. E., Dixon J. R., Kelly P., Worby C. A., Casey P. J., Dixon J. E., Involvement of a mitochondrial phosphatase in the regulation of ATP production and insulin secretion in pancreatic beta cells. Mol. Cell 19, 197–207 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Pagliarini D. J., Worby C. A., Dixon J. E., A PTEN-like phosphatase with a novel substrate specificity. J. Biol. Chem. 279, 38590–38596 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Shen J., Liu X., Yu W. M., Liu J., Nibbelink M. G., Guo C., Finkel T., Qu C. K., A critical role of mitochondrial phosphatase Ptpmt1 in embryogenesis reveals a mitochondrial metabolic stress-induced differentiation checkpoint in embryonic stem cells. Mol. Cell Biol. 31, 4902–4916 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang J., Guan Z., Murphy A. N., Wiley S. E., Perkins G. A., Worby C. A., Engel J. L., Heacock P., Nguyen O. K., Wang J. H., Raetz C. R., Dowhan W., Dixon J. E., Mitochondrial phosphatase PTPMT1 is essential for cardiolipin biosynthesis. Cell Metab. 13, 690–700 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu W. M., Liu X., Shen J., Jovanovic O., Pohl E. E., Gerson S. L., Finkel T., Broxmeyer H. E., Qu C. K., Metabolic regulation by the mitochondrial phosphatase PTPMT1 is required for hematopoietic stem cell differentiation. Cell Stem Cell 12, 62–74 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tronche F., Kellendonk C., Kretz O., Gass P., Anlag K., Orban P. C., Bock R., Klein R., Schütz G., Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat. Genet. 23, 99–103 (1999). [DOI] [PubMed] [Google Scholar]
- 22.Barski J. J., Dethleffsen K., Meyer M., Cre recombinase expression in cerebellar Purkinje cells. Genesis 28, 93–98 (2000). [PubMed] [Google Scholar]
- 23.Matei V., Pauley S., Kaing S., Rowitch D., Beisel K. W., Morris K., Feng F., Jones K., Lee J., Fritzsch B., Smaller inner ear sensory epithelia in Neurog 1 null mice are related to earlier hair cell cycle exit. Dev. Dyn. 234, 633–650 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hayashi S., McMahon A. P., Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: A tool for temporally regulated gene activation/inactivation in the mouse. Dev. Biol. 244, 305–318 (2002). [DOI] [PubMed] [Google Scholar]
- 25.Burns K. A., Ayoub A. E., Breunig J. J., Adhami F., Weng W. L., Colbert M. C., Rakic P., Kuan C. Y., Nestin-CreER mice reveal DNA synthesis by nonapoptotic neurons following cerebral ischemia hypoxia. Cereb. Cortex 17, 2585–2592 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Hardie D. G., AMP-activated protein kinase: An energy sensor that regulates all aspects of cell function. Genes Dev. 25, 1895–1908 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Viollet B., Guigas B., Leclerc J., Hébrard S., Lantier L., Mounier R., Andreelli F., Foretz M., AMP-activated protein kinase in the regulation of hepatic energy metabolism: From physiology to therapeutic perspectives. Acta Physiol. 196, 81–98 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hardie D. G., AMPK: Positive and negative regulation, and its role in whole-body energy homeostasis. Curr. Opin. Cell Biol. 33, 1–7 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Liang J., Mills G. B., AMPK: A contextual oncogene or tumor suppressor? Cancer Res. 73, 2929–2935 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Halestrap A. P., The mitochondrial pyruvate carrier. Kinetics and specificity for substrates and inhibitors. Biochem. J. 148, 85–96 (1975). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jefferies H. B., Cooke F. T., Jat P., Boucheron C., Koizumi T., Hayakawa M., Kaizawa H., Ohishi T., Workman P., Waterfield M. D., Parker P. J., A selective PIKfyve inhibitor blocks PtdIns(3,5)P2 production and disrupts endomembrane transport and retroviral budding. EMBO Rep. 9, 164–170 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barros L. F., Courjaret R., Jakoby P., Loaiza A., Lohr C., Deitmer J. W., Preferential transport and metabolism of glucose in Bergmann glia over Purkinje cells: A multiphoton study of cerebellar slices. Glia 57, 962–970 (2009). [DOI] [PubMed] [Google Scholar]
- 33.Bouillaud F., UCP2, not a physiologically relevant uncoupler but a glucose sparing switch impacting ROS production and glucose sensing. Biochim. Biophys. Acta 1787, 377–383 (2009). [DOI] [PubMed] [Google Scholar]
- 34.Diano S., Horvath T. L., Mitochondrial uncoupling protein 2 (UCP2) in glucose and lipid metabolism. Trends Mol. Med. 18, 52–58 (2012). [DOI] [PubMed] [Google Scholar]
- 35.Vozza A., Parisi G., De Leonardis F., Lasorsa F. M., Castegna A., Amorese D., Marmo R., Calcagnile V. M., Palmieri L., Ricquier D., Paradies E., Scarcia P., Palmieri F., Bouillaud F., Fiermonte G., UCP2 transports C4 metabolites out of mitochondria, regulating glucose and glutamine oxidation. Proc. Natl. Acad. Sci. U.S.A. 111, 960–965 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jacks T., Remington L., Williams B. O., Schmitt E. M., Halachmi S., Bronson R. T., Weinberg R. A., Tumor spectrum analysis in p53-mutant mice. Curr. Biol. 4, 1–7 (1994). [DOI] [PubMed] [Google Scholar]
- 37.Fischer G., Künemund V., Schachner M., Neurite outgrowth patterns in cerebellar microexplant cultures are affected by antibodies to the cell surface glycoprotein L1. J. Neurosci. 6, 605–612 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rogers G. W., Brand M. D., Petrosyan S., Ashok D., Elorza A. A., Ferrick D. A., Murphy A. N., High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria. PLOS ONE 6, e21746 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/10/eaat2681/DC1
Fig. S1. PTPMT1 depletion from neural precursor cells compromises cerebral development.
Fig. S2. Active proliferation of GCPs and increased apoptosis in postnatal PTPMT1 knockout cerebella.
Fig. S3. Correctly localized BG with radial fibers are missing in PTPMT1 knockout cerebella.
Fig. S4. Decreased cell proliferation and increased apoptosis in PTPMT1 knockout cerebra.
Fig. S5. Loss of PTPMT1 causes cell cycle arrest in cerebral NSPCs, leading to defective self-renewal and senescence.
Fig. S6. Deletion of p53 does not rescue PTPMT1 knockout mice or NSPC self-renewal.
Fig. S7. Loss of PTPMT1 causes bioenergetic stress in cerebral NSPCs.
Fig. S8. The entire cerebral cortex cell population is less affected by PTPMT1 ablation.