

Abstract
Deficiency in diacylglycerol acyltransferase (DGAT1) is a rare cause of neonatal diarrhea, without a known mechanism or in vitro model.
Methods and Results:
A patient presenting at our institution at 7 weeks of life with failure to thrive and diarrhea was found by whole-exome sequencing to have a homozygous DGAT1 truncation mutation. Duodenal biopsies showed loss of DGAT1 and deficits in apical membrane transporters and junctional proteins in enterocytes. When placed on a very low-fat diet, the patient’s diarrhea resolved with normalization of brush border transporter localization in endoscopic biopsies. DGAT1 knockdown in Caco2-BBe cells modeled the deficits in apical trafficking, with loss of apical DPPIV and junctional occludin. Elevation in cellular lipid levels, including diacylglycerol (DAG) and phospholipid metabolites of DAG, was documented by lipid analysis in DGAT1 knockdown cells. Culture of the DGAT1 knockdown cells in lipid-depleted media led to re-establishment of occludin and return of apical DPPIV.
Discussion:
DGAT1 loss appears to elicit global changes in enterocyte polarized trafficking that could account for deficits in absorption seen in the patient. The in vitro modeling of this disease should allow for investigation of possible therapeutic targets.
Keywords: DGAT1, diarrhea, enterocyte, intestine, Rab11a, Caco2, occludin, CLDN4, JAM
Graphical Abstract Legend:
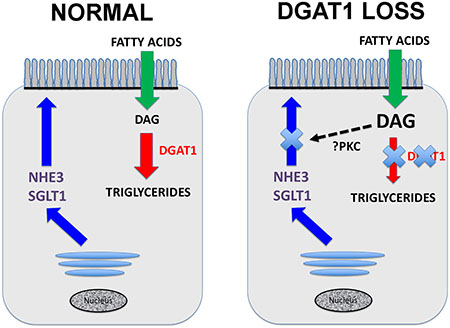
In the normal enterocyte DGAT1 catalyzes the conversion of diacylglycerol (DAG) to triglycerides. When DGAT1 is lost, then DAG accumulates in the cell, likely leading to activation of protein kinase C (PKC) isoforms which can inhibit the delivery of transporters such as NHE3 and SGLT1 to the apical brush border membranes.
Introduction
Congenital Diarrheal Disorders (CDD) represent heterogeneous enteropathies, typically presenting with unremitting diarrhea in the first few months of life 1–3. These diarrheal syndromes are predominately caused by inherited, autosomal recessive mutations that result in secretory or osmotic diarrhea, requiring significant dietary modification, total parental nutrition supplementation and, in rare cases for diseases such as MVID, intestinal transplantation. If unrecognized, patients can suffer from profound dehydration, multiorgan failure, and death 3. Recent work has categorized these diseases into four groups based on the cellular defect caused by a respective mutation: absorption and transport of nutrients and electrolytes, enterocyte differentiation and polarization, enteroendocrine cell differentiation, and modulation of the intestinal immune response 3. While the identification of the disease-associated genes aid diagnosis and disease management in patients, understanding the functionality of the mutations will allow for screening of new therapeutic targets.
The DGAT1 gene resides on human chromosome 8 and encodes diacyl CoA: diacylglycerol acyl transferase (DGAT), the enzyme responsible for catalyzing the final step in triglyceride synthesis, using diacylglycerol (DAG) and fatty acyl CoA 4. Mutations in this gene result in significant protein losing enteropathy, first described in two siblings born to non-consanguineous Ashkenazi Jewish parents 5. Both children were homozygous for a splice site mutation, resulting in the deletion of 25 amino acids and loss of DGAT1 function 5. These patients presented with diarrhea within the first week of life, hypoalbuminemia and hypertriglyceridemia. Two additional missense mutations were described in three children, all with less severe symptomatology. One child with a mutation in the membrane-bound O-acyl transferase domain of DGAT1 presented slightly later in life (2–4 months) without triglyceride abnormalities, while two twins of South Asian descent were found to have a p.L105P missense mutation resulting in partial loss of function, hypertriglyceridemia and a delay in symptom onset and severity 6,7. Most recently, 10 further patients with 5 different bi-allelic mutations have been reported with varying degrees of intestinal failure and variable onset of symptoms 8. These findings expand the clinical and molecular characteristics of DGAT1 deficiency and suggest variations in enzyme deficiency may drive a wider, unrecognized spectrum of congenital diarrhea phenotypes.
While DGAT1 deficiency falls into the second group of congenital diarrheal disease, i.e. defects in enterocyte differentiation and polarization, this classification is fairly arbitrary without an understanding of the mechanism by which DGAT1 deficiency drives diarrhea. DGAT1 knockout mice are noted to be lean, resistant to obesity and hypertriglyceridemia, making DGAT1 inhibition an attractive pharmacologic target for treatment of obesity 4,9. Yet when transitioned to human trials, the dose and tolerability of DGAT1 inhibitors was limited due to diarrhea, nausea and vomiting, interestingly, follows a similar profile to the symptoms experienced with infants with congenital DGAT1 mutations 10–12. The cause of diarrhea following DGAT1 loss remains unknown.
Our present studies have been directed by our experience in the presentation and diagnosis of a DGAT1 deficient patient. This child demonstrated protein losing enteropathy and diarrhea with documented deficiencies in apical transporter localization in the enterocyte brush border. Nevertheless, this child improved following initiation of a very low-fat diet with documented return of transporters to the apical brush border. Through in vitro modeling of this gene mutation using Caco2-BBe cells with DGAT1 knockdown, we have also evaluated alterations in trafficking and observed deficits in brush border trafficking similar to those seen in the child.
Results
Clinical Findings and Genetics
Patient 1 was born at 39 weeks via spontaneous vaginal delivery following an uncomplicated pregnancy to a woman of Hispanic descent. The infant was referred to our facility at 7 weeks of life with failure to thrive and persistent, non-bloody, non-bilious emesis. At the time of admission, he weighed 3.11 kg (birth weight 3.26 kg) and was noted to have a multitude of electrolyte abnormalities, including hypophosphatemia, hypomagnesemia, and hypokalemia. He had profound Vitamin D deficiency (25-OH-Vitamin D Total <4ng/ml). He developed unremitting diarrhea, having 8–10 loose bowel movements a day, associated with significant protein loss (albumin nadir at 10 weeks of life was 1.0 g/dL).
On initial work-up, the patient was noted to have an elevated fecal fat neutral stain, although a 24-hour fecal fat level following this test was normal (1.2g). His other laboratory work-up, including genetic microarray, thyroid tests, fecal fat elastase, stool alpha1-antitrypsin, stool sodium, potassium, and chloride were all within normal limits. He underwent upper and lower endoscopy that grossly appeared normal (Figure 1A). Hematoxylin and eosin stains of biopsies were notable for blunting of the duodenal mucosa and patchy gastric metaplasia (data not shown).
Figure 1. Clinical presentation of patient with protein losing enteropathy.

A. Endoscopic visualization of the duodenum showed normal appearing mucosa. B. Height and weight growth charts for the patient over the first 2 years prior to diagnosis and then for months 24–60 after introduction of a low fat enteral diet.
Over his first three years of life, Patient 1 was readmitted to the hospital multiple times. His course was complicated by TPN dependence, multiple infectious complications and electrolyte abnormalities, hypogammaglobulinemia, venous thrombotic disease, Vitamin D deficiency, tetany and osteopenia.
Whole exome sequencing was performed for the patient at 3 years of age, revealing a homozygous mutation in DGAT1 (chromosome 8, 145541756 A → G) in the splice donor site 3′ of exon 8, altering the invariant GT to GC (Figure 2). This mutation was the same mutation in the two patients reported initially 5. The patient’s mother was heterozygous for the mutation (Figure 2).
Figure 2. Genotyping of patient and mother.

To confirm the diagnosis of DGAT1 mutation, Sanger sequencing was performed on the region of DNA amplified from a control human DNA sample and DNA isolated from blood from the patient and his mother. The Sanger sequencing showed that the mother was heterozygous for the mutation while the patient was homozygous.
Following identification of his homozygous DGAT1 mutation, based on the success reported in the original case report 5, the patient was switched to a very low-fat diet (Tolerex formula) resulting in the resolution of his diarrhea and discontinuation of his TPN requirement within 2 months. He showed dramatic improvement in his growth and height (Figure 1B), and at the time of this writing has not been admitted to the hospital in the 2 years since transitioning to the fat-restricted diet.
Loss of Apical Transporters and dysfunction of polarity.
Initial biopsies of the patient were reviewed to confirm the diagnosis of DGAT1 deficiency, with notable loss of immunostaining for DGAT1 in the patient’s duodenal tissue when compared to control (Figure 3A). Given previous work suggesting significant defects in intestinal apical transporters associated with congenital diarrheal diseases, we performed immunofluorescence staining of biopsy sections for brush border proteins and apical transporters to assess their presence in enterocytes and polarization in the setting of DGAT1 deficiency. Brush border microvilli in enterocytes appeared intact in the patient as exemplified by normal staining for Ezrin (Figure 3B). Nevertheless, we observed significant loss of apical transporters and enzymes in enterocytes at the tips of the villi, including loss of SGLT1, CD10, DPPIV (Figure 3B) and NHE3 (data not shown). Staining for MYO5B and Rab8a, known to be altered in microvillus inclusion disease 13, appeared reduced, but correctly localized to the subapical regions of enterocytes (Figure 3B). On transmission electron microscopy (Figure 4A–F), the brush border again appeared to be intact, although especially in the biopsy at 13 months of age, the microvilli themselves were often clustered together, forming a visual appearance of tasseling interspersed with areas of normal brush border (Figure 4D). In addition, in the biopsy at 13 months of age, subapical vesicular accumulations were observed in some groups of enterocytes. These vesicles often were located directly below the apical surface (Figure 4E,F).
Figure 3. Loss of transporters at villus tips in DGAT1 mutation patient duodenum.

A. Immunostaining for DGAT1 demonstrated a complete loss of DGAT1 in the patient compared to staining in a normal control duodenum. Bars = 10 μm. B. Immunostaining of patient duodenal mucosa showed normal distribution of ezrin at the apical brush border with a decrease of Myosin Vb (MYO5B) and Rab8a. A notable loss of DPPIV, SGLT1 and CD10 was observed in enterocytes at the villus tips in the patient duodenal enterocytes compared to control. Insets show higher magnification images of the immunostaining. C=Controls, P=Patients, Bars = 10 μm.
Figure 4. Alterations in intercellular junctions in enterocytes in the patient with DGAT1 mutation.

A-F. Transmission electron microscopic images of the patient duodenum. Images were taken in the upper villus or villus tips. At 3 months of age, the enterocytes showed a normal brush border structure in general (A-C). However, at 13 months of age, in some enterocytes areas of microvillus tasseling were observed (D). In addition, some cells showed unusual granules with heterogeneous contents below the apical membranes. Bars = 500 nm (C, D, F); 1 μm (E); 2 μm (A,B). G. Electron micrographs show regions with multiple junctional specializations in the enterocytes from the patient. H. Staining of sections of patient duodenum revealed mislocalization of claudin 4 (CLDN4), JAM and occludin to the entire lateral membrane. Insets show higher magnification images of the immunostaining. Bars = 10 μm.
A hallmark of DGAT1 diarrhea is the profound protein losing enteropathy of unknown etiology in these patients. This diagnosis was shared by our patient, who suffered from persistent hypoalbuminemia during his first few months of life. Using transmission electron microscopy, we focused our attention on junctional integrity of the patient’s tissue (Figure 4G). Multiple, expanded tight and adherens junctions were present in the lateral membranes in the duodenal enterocytes.
To evaluate further the integrity of the tight junctions and the ability of enterocytes to establish these junctions in the setting of apparent altered polarity, we immunostained for Claudin 4 (CLDN4), JAM, and occludin. While these proteins localized to the apical and apicolateral border of enterocytes in control neonatal tissue, they were grossly redistributed in the DGAT1 mutant patient. In the case of JAM1 and cccludin, there was a decrease in immunostaining in the apical junctional area with redistribution to the basal lateral membrane as well as cytoplasmic relocalization of these junctional markers (Figure 4H). Basolateral markers, including E-cadherin and p120, remained unchanged (data not shown).
Return of apical transporters following treatment with a very low-fat diet.
Six months following the patient’s transition to a low fat enteral formula, and subsequent improvement in diarrhea, weight and growth, a repeat endoscopy was performed. Duodenal tissue samples were again evaluated for apical brush border proteins. Remarkably, the apical transporters NHE3 and SGLT1were now present on the enterocyte brush border in the tips of villi (Figure 5), suggesting that placement on a very low-fat diet had ameliorated the apical trafficking deficit. Despite the patient’s clinical improvement and apparent resolution of defects in polarization, the distribution of JAM and occludin was still aberrant, although the distribution of claudin 4 appeared to have substantially normalized (Figure 5).
Figure 5. Return of apical transporters to the brush border after 6 months of very low-fat diet.

Staining of duodenal biopsies taken 6 months after initiation of the very low-fat diet reveal normal brush border localization of DPPIV, SGLT1, CD10, and NHE3 throughout the villi. Claudin 4 localization was similar to the normal control. However, the expression of occludin and JAM were still diminished and localized along the lateral membranes. Bar = 10 μm.
In vitro knockdown of DGAT1 expression in Caco2-BBe cells models apical trafficking dysfunction.
To examine the effects of the loss of DGAT1 on the establishment of apical polarity, we stably knocked down DGAT1 expression in Caco2-BBe cells, a subclone from the Caco2 parental cell line 14, using lentiviral shRNA. The Caco2-BBe cell line was selected specifically for the extensive microvilli that develop when the cells are allowed to polarize. DGAT1 protein expression was reduced by greater than 80% compared to the control cells lines (Supplemental Figure 1A). Previous work has documented elevated levels of lipids, specifically triglycerides, in limited screens performed in patients with DGAT1 mutations 8. To evaluate the ability of DGAT1 knockdown (DGAT1-KD) Caco2-BBe cells to model this metabolic lipid malfunction, we performed a lipid analysis on both control Caco2-BBe cells and DGAT1-KD cells (Supplemental Figure 1B and Table 1). When compared to controls, DGAT1-KD cells demonstrated an over two-fold increases in phosphatidylglycerol and lyso-cardiolipin, with decreases in their precursor phosphatidic acid. These cells also had notable increases in diacylglycerol (36.76 vs 21.61 nmol/mg).
To evaluate the ability of DGAT1-KD cells to form an effective lumen and polarize, we then grew both Caco2-BBe control cells and DGAT1-KD Caco2-BBe cells in matrigel, and stained for DPPIV and occludin (Figure 6). In control cells, we noted a normal appearing central lumen with apical distribution of DPPIV outlining this luminal structure and F-actin highlighting the brush border. In DGAT-KD cells, DDPIV levels in the apical brush bordered were reduced, with the presence of DPPIV staining subapically (Figure 6). While the majority of cysts formed with the control cell line exhibited the single central lumen (88%), only a subset (28%) of the DGAT1-KD cysts exhibited a single lumen morphology. Additionally, the lumens of the DGAT1-KD cysts were not as smooth as the lumens of the control cyst. The lumen in the majority of the cysts formed by the DGAT1-KD cell line (72%) formed scrambled lumens, with an apical surface marked by DPPIV and F-actin. However, instead of forming a smooth internal hollow ball, the apical surface transitioned throughout the center of the cyst. When single cysts were selected for closer evaluation, DPPIV appeared to collect around multiple cystic structures within the cyst, unable to present a clear, polarized lumen when compared to control cells (Figure 6). When we stained the cysts for occludin (Figure 6), the controls exhibited the typical apical lateral junction staining pattern, while the DGAT1 knockdown cysts had a decrease at the apical lateral junctions with occludin now found at the basal lateral junction (arrows in Figure 6B), similar to the pattern observed in the patient with the DGAT1 mutation.
Figure 6. Knockdown of DGAT1 in Caco2-BBe cells grown in a 3D matrix alters the newly formed apical membrane.

The Caco2-BBe control (top row) and the DGAT1-KD (rows 2 and 3) cell lines were grown for 7 days as 3D cultures in Matrigel. (A) The cysts were stained for DDPIV (green in merge), DGAT1 (red in merge), F-actin (cyan in merge) and DAPI (blue in merge). The majority (88%) of the controls exhibited a smooth round central lumen with apical DPPIV and F-actin, while only a small subset of the DGAT1 knockdown cysts exhibited a central lumen (28%) with the majority exhibited a scrambled lumen (72%). (B) The cysts were stained for DDPIV (green in merge), DGAT1 (red in merge), Occludin (cyan in merge) and F-actin (blue in merge). Arrowheads indicate DPPIV, occludin and F-actin at the basal lateral junction. Bar=10 μM
Exposure to extremely low fat media ameliorates the phenotype in DGAT1-KD cells.
We also evaluated the behavior of DGAT1-KD on permeable Transwell filters. The DGAT1-KD and Scrambled control Caco-2BBe cell lines were grown for 7 days post-confluence. Figure 7A demonstrates that while Scrambled Control cells polarized with strong expression of DPPIV at the apical brush border, the DGAT1-KD cells showed markedly reduced apical staining for DPPIV. Compared to the Scrambled Control cells the DGAT1-KD cells also showed decreases in occludin at the tight junctions (Figure 7 A). We also evaluated whether there was an increase in leakiness with knockdown of DGAT1 by analyzing polarized monolayers in Ussing Chambers. Compared with Scrambled controls, the DGAT1-KD cells showed no change in short circuit current and demonstrated a significant increase in transmembrane resistance (Supplemental Figure 1C,D). These studies indicate that loss of DGAT1 in the Caco-2BBe cells did not lead to an opening of tight junctions.
Figure 7. DPPIV and Occludin losses in the DGAT1 knockdown cell line are reversed in lipid-free media.

(A) Scrambled control and DGAT1 knockdown Caco-2BBe cell lines were grown on Transwells for 7 days in normal sera then one set of DGAT1-KD cells were washed and refed with extremely low fat media for 48 hours. Cells were immunostained for DGAT1 (green), DPPIV (red) and occludin (blue). Occludin returned to tight junctions completely while DPPIV at the apical membrane partially recovered. (B) Quantitation of the return of DPPIV and occludin in the Caco-2BBe DGAT1 knockdown cell line incubated with extremely low fat media. A minimum of 12 fields from 3 separate experiments were quantified for each cell condition. Bar=10 μM Student t-test *= p0.0001, **=p0.0005.
We next sought to evaluate whether re-expression of DGAT1 in Caco-2BBe with DGAT1 knockdown could ameliorate the phenotype of loss of DPPIV from the apical membrane and occludin from the tight junctions. The Caco-2BBe DGAT1-KD cells were transiently infected with lentivirus coding for GFP-DGAT1 (Supplemental Figure 2). Re-expression of GFP-DGAT1 caused return of DPPIV to the apical membrane and re-establishment of occludin at tight junctions (Supplemental Figure 2).
As noted above, when the patient was placed on an extremely low fat diet, diarrhea subsided and apical transporters returned to the brush border in enterocytes at the villus tips. To determine if a similar recovery would be observed in our Caco-2BBe cell model, the DGAT1-KD Caco-2BBe cell line was grown for 7 days post-confluence, then switched to media containing charcoal-stripped fetal bovine sera for another 48 hours (extremely low fat media). A full recovery of occludin was observed with a partial recovery of apical DPPIV when the cells were grown in extremely low fat media (Figure 7B,C), indicating that the apical membrane trafficking defect could also be ameliorated in our cell model system as was observed in the patient.
Discussion
Congenital diarrheal diseases are predominantly associated with genetic mutations in trafficking pathways that establish apical brush border transporters and maintain junctional polarity. Diarrhea results from an inability to absorb luminal fluid as well as leakiness within intercellular junctions. These infants require dietary modification and TPN and have a high rate of morbidity and mortality. Deficiency in DGAT1 is a recently recognized, serious cause of neonatal diarrhea. Since its description in 2012, three genetic mutations have been identified resulting in variable phenotypes of this disease, and suggesting that, like many cases of congenital diarrhea diagnoses, there remains much to learn regarding the presentation, prevalence and etiology of DGAT1 mutation-driven diarrhea 5–7. Despite the growing recognition of congenital diarrheal pathologies, and the increasing diversity of diagnosis resulting in significant diarrheal disease, the establishment of in vitro models to study the physiology behind these and other diseases remains elusive in the literature.
This study describes the presentation, work up, diagnosis and treatment of one such congenital diarrhea case. Whole exome sequencing of the patient with enteropathy confirmed a loss of function mutation in DGAT1 identical to the mutation originally described in two Ashkenazi Jewish siblings 5. The delayed onset of symptoms despite identical mutations described by Haas suggests that, within a single loss of function mutation, variations in the penetrance of the phenotype are present. The carboxyl terminal truncation mutation resulting from the loss of a splice donor site leads to destabilization of the protein and subsequently its degradation, as demonstrated by a complete loss of DGAT1 protein staining in the patient. Duodenal tissue from this patient demonstrated loss of apical transporters and redistribution of junctional proteins. However, following treatment with a very low fat diet, there was resolution of the patient’s diarrhea, dramatic improvement in both weight and height growth charts, and a concurrent return of apical transporters when examined by immunofluorescence, although junctional proteins, occludin and JAM, remained mislocalized.
Our patient was transitioned to the very low-fat enteral formula, Tolerex®, which contains 2% of calories from fat. Previous reports describe successful treatment with anywhere from 4–10% calories from fat, yet given this rare disease and limited literature, much of the dietary intervention is based on experience with a handful of diagnosed patients 5,6. The processing of fat is far more complex, and while DGAT1 loss is known to affect triglyceride levels and has been described as creating hypertriglyceridemia, this was not observed in either our patient, or in the patients with p.L105P mis-sense mutation 6. To model DGAT1 loss in vitro, we successfully knocked down DGAT1 in Caco2-BBe cells. This model demonstrated similar dysfunction in apical trafficking as well as alterations in junctional proteins. Loss of DGAT1 appears to affect a variety of lipid pathways in vitro as observed in the DGAT1 knockdown Caco2-BBe cells. Of note, in the DGAT1 knockdown cells, we observed elevations in diacylglycerol consistent with the blockade of processing into triglycerides. In addition, we observed elevations in downstream metabolites including phosphatidylserine and cardiolipin, suggesting that elevation in diacylglycerol may alter phospholipid pools within cells. It seems likely that these changes in phospholipids and elevations in diacylglycerol may alter intracellular signaling. In particular, elevations in diacylglycerol and phosphatidylserine might be expected to impact activation of protein kinase C isoforms. Previous studies have noted that activation of protein kinase C isoforms can inhibit the delivery of transporters to the apical membrane 15. Thus, in our patient, the recovery of apical trafficking to the brush border of enterocytes at the villus tips following very low-fat enteral feeding may accrue from reduced levels of intracellular diacylglycerol and normalization of intracellular phospholipid pools. We also observed recovery of DPPIV and occludin in the Caco-2BBe DGAT1-KD cell line when the cells were transferred to extremely low fat media, indicating that the Caco-2BBe DGAT1-KD cells can be used a model system for the disease. These data further suggest that alterations in cellular lipid levels may impact cell proliferation, junction establishment, polarization and differentiation in enterocytes. Future studies are required to evaluate the role of protein kinase C isoforms in DGAT1 deficiency. Moreover, the in vitro model described here will allow further study of this pathway, with the goal of developing possible pharmacologic targets to prevent diarrhea seen in children with DGAT1 mutations.
Recent understanding of several congenital diarrheal diseases has been aided by development of genetic mouse models that recapitulate the major pathologies in these diseases 16,17. Unfortunately, present mouse models of DGAT1 loss do not recapitulate the pathology and diarrhea observed in human patients. While mice express both DGAT1 and DGAT2, their enteric system predominately uses DGAT2 for lipid processing, with DGAT2 null mice born significantly smaller than their wild type counterparts, surviving for only a few hours, and with significant reductions in triglycerides, free fatty acids, and glucose 4,18. In contrast, DGAT1 null mice do not replicate diarrhea seen clinically in children and appear phenotypically otherwise healthy. They have notable decreases in adipose tissue and show resistance to high-fat diets and the development of insulin resistance, along with increased activity and energy expenditure 11,19. This has led to the development of DGAT1 inhibitors as a possible treatment for obesity and hypertriglyceridemia, with results dose limited by the development of diarrhea 10,20. The inability to model this disease in vivo makes an in vitro human cell model system an attractive option. In our present study, we have demonstrated the ability to identify a gene mutation associated with a congenital diarrheal disease and then efficiently develop an in vitro model to investigate the physiology and mechanisms underlying the patient’s disease. This approach will be advantageous in identifying and targeting therapeutics in undiagnosed congenital diarrheal diseases.
Methods
Whole exome sequencing
100 μl blood was used to purify DNA on the MagNA Pure Compact System (Roche). The isolated DNA was quantified using the Qubit dsDNA BR (broad range) (Thermo Fischer Scientific). Additionally, the integrity of the extracted genomic DNA was evaluated on 0.8% agarose gel. For exome library preparation, barcoded libraries were constructed using the NEBNext DNA Library prep kit (New England Biologicals). Exome capture was performed on 1000 ng of the barcoded libraries using the SeqCap EZ Human Exome Kit v3.0 (Roche) as per manufacturer’s instruction. The final libraries were sequenced on Illumina Hiseq2500 v4 (Illumina) as paired end 100bp. Fastqs were aligned to the human reference genome GRCh37 using BWA version 0.6.2. Bams were then cleaned and realigned using GATK2 Best Practices. Variant calling was performed and the VCF file was uploaded into Fabric Enterprise (formerly Omicia, Fabric Genomics, Oakland, CA) for variant analysis.
Sanger sequencing
For Sanger sequencing confirmation of the mutation, genomic DNA purified from blood as above from the patient, his mother and a normal control were used to amplify and sequence the region around the putative mutation. Briefly, 1 μl of purified DNA was utilized in a PCR reaction with Accuprime Pfx Mix (Agilent) using specific sense (ctcccactctgctgtgctcgtagcctc) and anti-sense (gaggaagtagtagagatctggaatgggaatg) primers with 42 cycles of amplification (95C 30s, 60 C 30s, 68C 40s). The resulting fragments were resolved on a 1% agarose gel, isolated (Qiagen) and submitted for sequencing with both the sense and anti-sense primers.
Cell lines, real-time PCR analysis, lentivirus-mediated DGAT1-KD.
Human colonic epithelial Caco2-BBe cell lines were grown in DMEM. All cell media was supplemented with L-glutamine (Cellgro), nonessential amino acids (Cellgro), penicillin/streptomycin (Cellgro), 10% fetal bovine serum (Hyclone). Cell culture dishes were purchased from Costar. For growth in extremely low fat media, charcoal treated serum (Omega Scientific, Inc FB-04: fetal bovine serum, charcoal-dextran treated) was substituted for the Hyclone fetal bovine serum, the cells were washed twice in PBS, then incubated for 48 hours in the extremely low fat media.
A lentiviral shRNA vector targeting DGAT1 (Sigma Aldrich TRCN0000036151) and a control shRNA (Open Biosystems, RHS4346) were used for transduction of Caco2-BBe cells. The Caco-2BBe cells were transduced with lentiviral media produced in HEK cells and selected using puromycin (Cellgro).
For rescue of the Caco-2BBe DGAT1-KD cell line, DGAT1 was amplified from mouse intestine and cloned as an EGFP-chimera into the lentiviral inducible expression vector pIND20 21. The GFP-DGAT1 lentiviral media produced by the HEK cells was concentrated with Lenti-X concentrator (Takara 631231), resuspended in PBS to a titer of 107 virus particles per 1 ml and stored in 100 μl aliquots at −80°C. Caco-2BBe scrambled KD and DGAT1-KD cell lines were plated on Transwells and grown for 7 days post confluence (see below). To enhance lentiviral infection of the Caco-2BBe cell lines, cells were washed with PBS, then switched to low calcium media to depolarize the cells and 50 μl of the concentrated lentivirus was added to the top compartment of the Transwells. The cells were incubated with the virus in low calcium conditions overnight and the next morning the media was replaced with normal media supplemented with 1 μg/ml of Doxycycline to induce transcription of GFP-DGAT1 and incubated overnight. The next day the cells were fixed and stained as below.
Human tissue preparation
Samples of duodenum from the patient with DGAT1 deficiency were obtained via endoscopic biopsies. All tissue samples were obtained through protocols approved by the Vanderbilt University Medical Center IRB. All biopsy samples were washed in PBS and fixed in 10% formalin. Samples were stored in 70% ethanol, embedded in paraffin, and sectioned for immunostaining.
Transmission Electron Microscopy.
Specimens were processed for transmission electron microscopy (TEM) and imaged in the Cell Imaging Shared Resource-Research Electron Microscopy facility at Vanderbilt University.
Embedding:
Samples were fixed in 2.5% glutaraldehyde in 0.1M cacodylate buffer, pH 7.4 at room temperature (RT) 1 hour then transferred to 4°C, overnight. The samples were washed in 0.1M cacodylate buffer, then incubated 1 hour in 1% osmium tetraoxide at RT then washed with 0.1M cacodylate buffer. Subsequently, the samples were dehydrated through a graded ethanol series and then 3 exchanges of 100% ethanol. Next, the samples were incubated for 5-minutes in 100% ethanol and propylene oxide (PO) followed by 2 exchanges of pure PO. Samples were then infiltrated with Epon 812 resin and PO. Next day, the samples went through resin to PO exchange then incubated with pure epoxy resin overnight then incubated in pure epoxy resin and then polymerize at 60°C.
Sectioning and Imaging
500–1000nm thick sections were cut for ultra-structure identification. Then 70–80nm ultra-thin sections were cut of the region of interest and post-stained with Uranyl acetate and Reynold’s lead citrate. Samples were subsequently imaged on the Philips/FEI Tecnai T12 electron microscope at various magnifications.
Western blot
The Caco2-BBe DGAT1-KD and Caco2-BBe control cell lines were plated on 10cm dishes for 15 days, washed with PBS, and lysed in RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) for 10 minutes at 4°C. The lysates were sonicated for 25 seconds and centrifuged at 16,000 g for 10 minutes. For all samples, protein concentrations were measured by DirectDetect (EMD Millipore, Billerica, MA, USA) and 80 μg of protein were loaded onto a NuPage 4–12% Bis-Tris gel (Invitrogen). The proteins were transferred onto Odyssey nitrocellulose membrane (LI-COR, Lincoln, NE, USA). Blots were air-dried for one hour at room temperature, then blocked for one hour in Odyssey TBS Blocking Buffer (LI-COR) and probed with Rabbit anti-DGAT1 and Rabbit anti-VDAC for 18 hours in 0.01% Tween-20/Odyssey TBS Blocking Buffer (TBS-BB/0.01%T) at 4oC. Blots were washed in TBS-BB/0.01%T followed by a one hour incubation with Donkey anti-Rabbit IgG labeled with 800 nm fluorescent dye (LI-COR cat# 926–32213) and diluted as the primary antibody. Blots were washed 3 times in TBS-BB/0.01%T and fluorescence was detected using an Odyssey Fc (LI-COR). The resulting JPEGs were opened in ImageJ and the area under the peak was calculated and the relative density was normalized. The densities were then adjusted to the control (VDAC) and statistical significance was determined by an unpaired student t-test.
Caco2-BBe growth, staining and rescue on Transwells.
For growth of the Caco2-BBe and the Caco2-BBe DGAT1 knockdown cell lines on Transwells, 200 μl of cell stock (150,000 cells per 1 ml of media) were plated onto 12 mm Transwells (CoStar 3460). The cells were grown for 7 days post-confluence then washed twice for 5 minutes with PBS, fixed with 4% paraformaldehyde for 20 minutes, washed again then blocked and extracted for 30 minutes with 10% normal donkey serum (NDS, Jackson ImmunoResearch), 0.3% Triton X-100, PBS. The Transwells were washed with 0.05% Tween-20 PBS (PBS-T) then incubated overnight at 4oC with primary antibodies diluted in 1% NDS in PBS-T (see List of Antibodies in Table 1). The next day the Transwells were washed 3 times for 15 minutes in PBS-T, incubated for 1 hour with secondary antibodies, washed 3 times for 15 minutes with PBS-T and once for 15 minutes with PBS, the Transwell filters were separated from the plastic supports and mounted on glass slides with Prolong Gold antifade (Invitrogen P36934). The monolayers were imaged either on a Zeiss 710 Meta Inverted confocal microscope with a 63×/1.40 LD Plan-Apochromat oil immersion lens or a Olympus 60×/1.45 Plan-Apochromat oil immersion lens and the images were processed utilizing Imaris software (Bitplane). For quantification of fluorescence intensity, the 3D stacks of the image slices were first converted to 2D with Imaris software, the 2D images were then measured for mean intensity using ImageJ software (NIH). Both of the confocal microscopes and the Imaris workstation were located in the Vanderbilt University Cell Imaging Shared Resource.
Table 1:
Antibodies used (W:western blot; IF: immunofluorescence)
| Target | Company and Catalog number | Species | Dilution |
|---|---|---|---|
| Claudin 4 | Invitrogen 329400 | Ms | 1:100 |
| CD10 | Abcam ab951 | Ms | 1:100 |
| DGAT1 | Santa Cruz sc-32861 | Rb | IF-1:100 W-1:1000 |
| DPPIV | R&D AF1180 | Gt | 1:200 |
| E-cadherin | Abcam ab11512 | Rt | 1:200 |
| Ezrin | DSHB CPTC-1-S | Ms | 1:50 |
| JAM1 | Abcam ab52647 | Rb | 1:100 |
| Myosin Vb | Goldenring Lab (ref 12) | Ch | 1:400 |
| NHE3 | Novus NBP1–82574 | Rb | 1:200 |
| Occludin | Invitrogen 73–1500 | Ms | 1:200 |
| P120 | BD biosciences 610133 | Ms | 1:200 |
| Phallodin | Invitrogen A22287 | 1:200 | |
| Rab8a | Goldenring Lab (ref 12) | Rb | 1:400 |
| SGLT1 | Abcam ab14655 | Rb | 1:200 |
| VDACC | Abcam ab15895 | Rb | W-1:1000 |
Üssing chambers
DGAT1KD or scrambled control Caco-2BBe cells were grown on the Snapwells (Costar, Cat. # 3801) and inserted into the sliders with an aperture = 1.13 cm2 (P2302; Physiologic Instruments, San Diego, CA). Luminal and basolateral baths were filled with 4 ml Krebs-Ringer solution (117 mM NaCl, 4.7 mM KCl, 1.2 mM MgCl2, 2.5 mM CaCl2, 1.2 mM NaH2PO4, 25 mM NaHCO3, 5 mM glucose), maintained at 37°C using a water-recirculating heating system. The solution was bubbled with a gas mixture of 95% O2-5% CO2 to maintain the pH at 7.4.
The short-circuit current across Snap-wells was determined by a voltage clamp (Physiologic Instruments) at zero potential difference with compensation for solution resistance. Short-circuit current (Isc) and transepithelial resistance (Rt, Ω●cm2) were continuously recorded by the DataQ system (Physiologic Instruments). Positive values of Isc indicate luminal-to-serosal current flow, e.g., anion secretion or cation absorption. After a 60 min stabilization period, the basal Isc and Rt were determined.
Experiments were repeated 4 times with individually cultured sets of cells and data were expressed as mean ± SEM. Significance was determined by t-test.
Caco2-BBe cyst growth and staining.
For growth of the Caco2-BBe and the Caco2-BBe DGAT1 knockdown cell lines as cysts, 100 μl of cell stock (150,000 cells per 1 ml of media) was mixed with 75 μl of Matigel (Corning #356234) per well of an 8-chamber Lab-Tek coverglass slide (Thermo #155411). The cysts were grown for 7 days then washed 4× 15 minutes with PBS, fixed with 4% paraformaldehyde for 40 minutes, washed again then blocked and extracted for 2 hours with 10% normal donkey serum (NDS, Jackson ImmunoResearch), 0.3% Triton X-100, PBS. The cysts were washed with 0.05% Tween-20 PBS (PBS-T) then incubated overnight at 4oC with primary antibodies diluted in 1% NDS in PBS-T (see List of Antibodies in Table 1). The next day the cysts were washed 6 times for 15 minutes in PBS-T, incubated for 4 hours with secondary antibodies and phalloidin, washed 6 times for 15 minutes with PBS-T and twice for 15 minutes with PBS, then overlayed with Prolong Gold with DAPI (Invitrogen P36931). The cysts were imaged on a Zeiss 710 Meta Inverted confocal microscope with a 40×/1.10 LD C-Apochromat water immersion lens and the images were processed utilizing Imaris software (Bitplane). Both the microscope and workstation were located in the Vanderbilt University Cell Imaging Shared Resource.
Histology.
Normal human duodenum sections and those from the patient with the DGAT1 mutation were deparaffinized and were submitted to antigen retrieval in a pressure cooker using the target retrieval solution (Dako North America Inc.). Serum-free protein block (Dako North America Inc.) was used for blocking the tisssue, and immunofluorescence staining was performed. In short, tissue sections were incubated overnight at 4°C with primary antibodies diluted in Antibody Diluent with Background Reducing Components (Dako North America Inc.). Appropriate secondary antibodies were conjugated for immunofluorescence with Alexa Fluor 488, Alexa Fluor 568, Alexa Fluor 647, Cy3, or Cy5 (1-hour incubation at room temperature). Primary antibodies are listed in Table 1. Tissue sections were washed 3 times in PBS and mounted with ProLong Gold plus DAPI (Invitrogen). All imaging was performed using the Olympus FV-1000 or Zeiss Axiophot microscope equipped with an Axiovision digital imaging system (Zeiss, Jena GmBH, Germany).
Lipid Analysis.
Three hundred μL of ice-cold PBS (10× diluted) was added to an Eppendorf tube which contained either the Caco-2BBe control or DGAT1 knockdown cells. Individual cell suspensions were homogenized for one minute using a disposable soft tissue homogenizer. An aliquot of 25 μL was pipetted to determine the protein content (BCA protein assay kit, Thermo Scientific, Rockford, IL). The rest of homogenate was accurately transferred into a disposable glass culture test tube, and a mixture of lipid internal standards was added prior to lipid extraction for quantification of all reported lipid species. Lipid extraction was performed by using a modified Bligh and Dyer procedure as described previously 22. Each lipid extract was resuspended into a volume of 500 μL of chloroform/methanol (1:1, v/v) per mg of protein and flushed with nitrogen, capped, and stored at −20 °C for lipid analysis. Part of the lipids extract was taken out to be derivatized with DMG (dimethylglycine) for DAG analysis as described 23.
For ESI direct infusion analysis, lipid extracts or DAG derivatives were further diluted to a final concentration of ~500 fmol/μL, and the mass spectrometric analysis was performed on a QqQ mass spectrometer (Thermo TSQ VANTAGE, San Jose, CA) equipped with an automated nanospray device (TriVersa NanoMate, Advion Bioscience Ltd., Ithaca, NY) as described 24. Identification and quantification of lipid species from mass spectrometric analysis were conducted as previously described 25.
Supplementary Material
Synopsis:
Mutational loss of DGAT1 leads to congenital diarrhea due to a loss of apical trafficking of key ion transporters (NHE3 and SGLT1) to the enterocyte brush border and deficits in intercellular junctions. Treatment with a very low-fat diet reverse these deficits. Knockdown of DGAT1 in Caco2-BBe cells recapitulates apical trafficking defects and alterations in junctional proteins. Diacylglycerol acyltransferase, DGAT; diacylglycerol, DAG; protein kinase, PKC.
Acknowledgements
This work was supported by the National Institute of Health (NIH) grants R01 DK48370 and R01 DK70856 to JRG and a gift from the Christine Volpe Fund and a Pilot Translational Research Award to SA and JRG funded by the Vanderbilt Digestive Disease Center (P30 DK058404) and the Vanderbilt Institute for Clinical and Translational Research (UL1 TR002243). CS and VGW were supported by T32 HD060554 and T32 DK007673. CP-Z was supported by T32 CA106183. This work was supported by core resources of the Vanderbilt Digestive Disease Center, (P30 DK058404) the Vanderbilt-Ingram Cancer Center (P30 CA68485), and imaging supported by both the Vanderbilt Combined Imaging Shared Resource (CA68485, DK20593, DK58404, DK59637, EY08126) and the Vanderbilt Digital Histology Shared Resource, supported by VA Shared Instrumentation Grant (1IS1BX003097).
References
- 1.Terrin G, Tomaiuolo R, Passariello A, et al. Congenital diarrheal disorders: an updated diagnostic approach. International journal of molecular sciences. 2012;13(4):4168–4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Passariello A, Terrin G, Baldassarre ME, De Curtis M, Paludetto R, Berni Canani R. Diarrhea in neonatal intensive care unit. World J Gastroenterol. 2010;16(21):2664–2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berni Canani R, Terrin G, Cardillo G, Tomaiuolo R, Castaldo G. Congenital diarrheal disorders: improved understanding of gene defects is leading to advances in intestinal physiology and clinical management. J Pediatr Gastroenterol Nutr. 2010;50(4):360–366. [DOI] [PubMed] [Google Scholar]
- 4.Yen CL, Stone SJ, Koliwad S, Harris C, Farese RV Jr. Thematic review series: glycerolipids. DGAT enzymes and triacylglycerol biosynthesis. Journal of lipid research. 2008;49(11):2283–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haas JT, Winter HS, Lim E, et al. DGAT1 mutation is linked to a congenital diarrheal disorder. J Clin Invest. 2012;122(12):4680–4684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gluchowski NL, Chitraju C, Picoraro JA, et al. Identification and characterization of a novel DGAT1 missense mutation associated with congenital diarrhea. Journal of lipid research. 2017;58(6):1230–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stephen J, Vilboux T, Haberman Y, et al. Congenital protein losing enteropathy: an inborn error of lipid metabolism due to DGAT1 mutations. Eur J Hum Genet. 2016;24(9):1268–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Rijn JM, Ardy RC, Kuloglu Z, et al. Intestinal Failure and Aberrant Lipid Metabolism in Patients With DGAT1 Deficiency. Gastroenterology. 2018;155(1):130–143e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen HC, Farese RV Jr. Inhibition of triglyceride synthesis as a treatment strategy for obesity: lessons from DGAT1-deficient mice. Arterioscler Thromb Vasc Biol. 2005;25(3):482–486. [DOI] [PubMed] [Google Scholar]
- 10.Maciejewski BS, LaPerle JL, Chen D, et al. Pharmacological inhibition to examine the role of DGAT1 in dietary lipid absorption in rodents and humans. Am J Physiol Gastrointest Liver Physiol. 2013;304(11):G958–969. [DOI] [PubMed] [Google Scholar]
- 11.Smith SJ, Cases S, Jensen DR, et al. Obesity resistance and multiple mechanisms of triglyceride synthesis in mice lacking Dgat. Nat Genet. 2000;25(1):87–90. [DOI] [PubMed] [Google Scholar]
- 12.Nakajima K, Chatelain R, Clairmont KB, et al. Discovery of an Orally Bioavailable Benzimidazole Diacylglycerol Acyltransferase 1 (DGAT1) Inhibitor That Suppresses Body Weight Gain in Diet-Induced Obese Dogs and Postprandial Triglycerides in Humans. Journal of medicinal chemistry. 2017;60(11):4657–4664. [DOI] [PubMed] [Google Scholar]
- 13.Knowles BC, Roland JT, Krishnan M, et al. Myosin Vb uncoupling from RAB8A and RAB11A elicits microvillus inclusion disease. J Clin Invest. 2014;124(7):2947–2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peterson MD, Mooseker MS. Characterization of the enterocyte-like brush border cytoskeleton of the C2BBe clones of the human intestinal cell line, Caco-2. J Cell Sci. 1992;102 (Pt 3):581–600. [DOI] [PubMed] [Google Scholar]
- 15.Cha B, Chen T, Sarker R, et al. Lysophosphatidic acid stimulation of NHE3 exocytosis in polarized epithelial cells occurs with release from NHERF2 via ERK-PLC-PKCdelta signaling. Am J Physiol Cell Physiol. 2014;307(1):C55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weis VG, Knowles BC, Choi E, et al. Loss of MYO5B in mice recapitulates Microvillus Inclusion Disease and reveals an apical trafficking pathway distinct to neonatal duodenum. Cell Mol Gastroenterol Hepatol. 2016;2(2):131–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schneeberger K, Vogel GF, Teunissen H, et al. An inducible mouse model for microvillus inclusion disease reveals a role for myosin Vb in apical and basolateral trafficking. Proc Natl Acad Sci U S A. 2015;112:12408–12413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stone SJ, Myers HM, Watkins SM, et al. Lipopenia and skin barrier abnormalities in DGAT2-deficient mice. J Biol Chem. 2004;279(12):11767–11776. [DOI] [PubMed] [Google Scholar]
- 19.Chen HC, Smith SJ, Ladha Z, et al. Increased insulin and leptin sensitivity in mice lacking acyl CoA:diacylglycerol acyltransferase 1. J Clin Invest. 2002;109(8):1049–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Denison H, Nilsson C, Lofgren L, et al. Diacylglycerol acyltransferase 1 inhibition with AZD7687 alters lipid handling and hormone secretion in the gut with intolerable side effects: a randomized clinical trial. Diabetes Obes Metab. 2014;16(4):334–343. [DOI] [PubMed] [Google Scholar]
- 21.Meerbrey KL, Hu G, Kessler JD, et al. The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo. Proc Natl Acad Sci U S A. 2011;108(9):3665–3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han X Lipidomics: Comprehensive Mass Spectrometry of Lipids. Hoboken, NJ: Wiley; 2016. [Google Scholar]
- 23.Wang M, Hayakawa J, Yang K, Han X. Characterization and quantification of diacylglycerol species in biological extracts after one-step derivatization: a shotgun lipidomics approach. Anal Chem. 2014;86(4):2146–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han X, Yang K, Gross RW. Microfluidics-based electrospray ionization enhances the intrasource separation of lipid classes and extends identification of individual molecular species through multi-dimensional mass spectrometry: development of an automated high-throughput platform for shotgun lipidomics. Rapid Commun Mass Spectrom. 2008;22(13):2115–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang K, Cheng H, Gross RW, Han X. Automated lipid identification and quantification by multidimensional mass spectrometry-based shotgun lipidomics. Anal Chem. 2009;81(11):4356–4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.