

Abstract
Advanced prostate cancer initially responds to androgen deprivation therapy (ADT), but the disease inevitably recurs as castration-resistant prostate cancer (CRPC). Although CRPC initially responds to abiraterone and enzalutamide, the disease invariably becomes non-responsive to these agents. Novel approaches are required to circumvent resistance pathways and extend survival, but the mechanisms underlying resistance remain poorly defined. Our group previously showed the histone lysine-N-methyltransferase EZH2 to be overexpressed in prostate cancer and quantitatively associated with progression and poor prognosis. In this study, we screened a library of epigenetic inhibitors for their ability to render CRPC cells sensitive to enzalutamide and found that EZH2 inhibitors specifically potentiated enzalutamide-mediated inhibition of proliferation. Moreover, we identified antisense oligonucleotides (ASO) as a novel drug strategy to ablate EZH2 and AR expression, which may have advantageous properties in certain settings. RNA-seq, ChIP-seq, and ATAC-seq demonstrated that EZH2 inhibition altered the AR cistrome to significantly upregulate AR signaling, suggesting an enhanced dependence of CRPC cells on this pathway following inhibition of EZH2. Combination treatment with ASO targeting EZH2 and AR transcripts inhibited prostate cancer cell growth in vitro and in vivo better than single agents. In sum, this study identifies EZH2 as a critical epigenetic regulator of ADT resistance and defines ASO-based co-targeting of EZH2 and AR as a promising strategy for treatment of CRPC.
INTRODUCTION
Prostate cancer remains the most commonly diagnosed cancer and the second leading cause of cancer-related deaths in American men, with most mortalities resulting from advanced metastatic tumors (1). Since its development and progression depend on signals through the androgen receptor (AR), physical and pharmacological castration became therapeutic mainstays for advanced disease (2). While initially responsive to androgen deprivation therapy (ADT), these tumors almost invariably recur in an incurable androgen-independent form, called castration-resistant prostate cancer (CRPC). The pathophysiological underpinnings of CRPC are many, but frequently involve the continued activation of AR despite low levels of androgens (2). To address this phenomenon, contemporary antiandrogen therapy, typified by the second-generation antagonist enzalutamide, seeks to directly bind and target AR, blocking its intracellular activity. Though clinical outcomes are superior to conventional castration, enzalutamide-treated CRPC tumors still uniformly recur (3,4).
The role of epigenetic modifications in CRPC has become an important area of inquiry. For example, we and others have shown that inhibition of BET bromodomain chromatin reader proteins attenuates growth of AR-wild-type and AR-mutant CRPC (5,6). Our group has also shown that the histone-lysine N-methyltransferase EZH2 is overexpressed and associated with poor prognosis in prostate cancer (7). While EZH2 has been described as a transcriptional repressor that methylates histone H3 lysine 27 (H3K27Me3) to mediate epigenetic silencing of multiple tumor suppressors, studies also suggest that EZH2 possesses additional activities in prostate cancer (8). Recently, EZH2 has been found to be highly expressed in neuroendocrine prostate cancer (NEPC); progression to NEPC includes loss of lineage-specific AR-mediated gene expression and gain-of-function in EZH2 (9,10). Another study in a NEPC model showed that EZH2 inhibition could reactivate AR signaling and sensitivity to drugs (11), while an additional showed that NEPC may represent a late-stage with loss of plasticity, and AR expression/signaling was not amenable to EZH2 targeting (12). Whether inhibition of EZH2 can impact AR signaling during resistance to current AR targeting therapies (e.g., enzalutamide) outside of NEPC has not been defined.
In this study, we identified EZH2 as a key regulator of sensitivity to AR-targeted therapies in CRPC adenocarcinoma models through an unbiased epigenetic inhibitor screen. EZH2 inhibition had profound effects on the AR cistrome and signaling, suggesting an enhanced dependence on this pathway. Furthermore, we tested antisense oligonucleotides (ASOs) targeting EZH2 and AR as an alternative strategy to target oncogenic factors, which may be beneficial in settings where ablation of these proteins is desired compared to inhibition of select enzymatic activities or domains. ASOs have demonstrated clinical activity for the treatment of multiple diseases, including diabetes, hyperlipidemias, cardiovascular diseases, neurodegenerative diseases, and cancer; those currently approved for therapeutic use include Kynamro®, Spinraza™, and Exondys 51™ (13). Herein, a novel ASO strategy targeting EZH2 and AR was shown to be effective in inhibiting CRPC growth in vivo, suggesting that targeting this axis may provide clinical benefit in patients that have developed resistance to established therapies.
MATERIALS AND METHODS
Cell culture
LNCaP, C4–2B, and CWR-R1 prostate cancer cell lines were grown in RPMI 1640 (Invitrogen) supplemented with 10% fetal bovine serum (FBS) (Invitrogen) in a 37°C incubator with 5% CO2. The LNCaP-abl (14) and LNCaP-EnzR (15) cell lines were propagated as previously described. LNCaP cells were obtained from ATCC. C4–2B cells were generously provided by Evan Keller, Ph.D., at the University of Michigan, CWR-R1 and LNCaP-EnzR cell lines were kindly provided by Donald Vander Griend, Ph.D., at the University of Chicago, and the LNCaP-abl cell line was kindly provided by Myles Brown, Ph.D., at Harvard University. Cells were genotyped to confirm identity at the University of Michigan Sequencing Core and tested routinely for Mycoplasma contamination.
Epigenetic inhibitors screen
C4–2B cells were seeded in 96-well plates at a density of 2000 cells/well in a total volume of 50 μL media. Each epigenetic inhibitor (see Supplementary Table S1) in the customized panel (Selleck) was added at 1 μM concentration to an individual well containing either enzalutamide or DMSO vehicle (final concentration 2 μM). Cells were incubated for five days before quantification with CellTiter-Glo reagent (Promega), per manufacturer’s instructions. CellTiter-Glo analysis was performed after transferring 100 μL of CellTiter-Glo solution from each well into a Costar 96-well clear flat bottom plate for reading with a Tecan Infinite M1000 PRO reader. CellTiter-Glo luminescent signal was normalized to negative control wells (without epigenetic inhibitor); raw values are included in Supplementary Table S2.
Prostate tumor xenograft model and drug studies
Six-week-old male CB17 severe combined immunodeficiency (SCID) mice were used for experiments. C4–2B prostate cancer cells (1 X 106 cells) in 50% Matrigel (BD Biosciences) were injected subcutaneously into bilateral dorsal flanks. When xenografts reached an average size of 50–100 mm3, animals were randomized into groups (each containing 10 animals) subsequently treated with one of the following: PBS, Vehicle, Enzalutamide (30 mg/kg), ASO-Ctrl (50 mg/kg), ASO-AR (25 mg/kg), ASO-EZH2–65 (50 mg/kg), or ASO combo (25 mg/kg ASO-AR, 50 mg/kg ASO-EZH2–65). For experiment in supplemental data, treatment cohorts were as follows: ASO-Ctrl (75 mg/kg), ASO-AR (25 mg/kg) + ASO-Ctrl (50 mg/kg), ASO-EZH2–65 (50 mg/kg) + ASO-Ctrl (25 mg/kg), or ASO combo (25 mg/kg ASO-AR, 50 mg/kg ASO-EZH2–65). All reagents were dosed five days per week for three weeks. Enzalutamide was delivered by gavage and ASOs by subcutaneous injection. Tumor volume was calculated from digital caliper measurements made at the study outset, then twice weekly. Tumors were resected and weighed at the end of the time course. Animal experiments were approved by the University of Michigan Institution Animal Care and Use Committee (IACUC).
Chou-Talalay combination index for Loewe additivity
Loewe additivity is a dose-effect model which states that additivity occurs in a two-drug combination if the sum of the ratios of the dose vs. the median effect for each individual drug is 1. In this model, combination index (CI) scores estimate the interaction between the two drugs. If CI < 1, the drugs have a synergistic effect and if CI > 1, the drugs have an antagonistic effect. CI = 1 means the drugs have additive effect. The CI coefficients were computed based on the Chou-Talalay median effect model as implemented in CalcuSyn v2.11 (http://www.biosoft.com/w/calcusyn.htm).
Antibodies and immunoblot analyses
Antibodies used in the immunoblotting assays are as follows: AR (Millipore, Cat. # 06–680), PSA (DAKO, Cat. # A0562), EZH2 (Cell Signaling, Cat. # 5246S), H3K27Me3 (Diagenode, Cat. # C15410069), Histone H3 (Cell Signaling, Cat. # 9715S), GAPDH (Cell Signaling, Cat. # 3683S). All antibodies were employed at dilutions suggested by the manufacturers. For Western blot analysis, 200 μg total protein extract was boiled in sample buffer and 10–20 μg aliquots were separated by SDS-PAGE and transferred onto Polyvinylidene Difluoride membrane (GE Healthcare). The membrane was incubated for one hour in blocking buffer [Tris-buffered saline, 0.1% Tween (TBS-T), 5% nonfat dry milk] followed by incubation overnight at 4°C with primary antibody. Following a wash with TBS-T, the blot was incubated with horseradish peroxidase-conjugated secondary antibody, and signals were visualized by enhanced chemiluminescence system as per manufacturer’s protocol (GE Healthcare).
RNA isolation, quantitative real-time PCR, and RNA-seq
Total RNA was isolated from cells using Direct-zol kit (Zymo), and cDNA was synthesized from 1,000 ng total RNA using Maxima First Strand cDNA Synthesis Kit for RT-qPCR (Thermo Fisher Scientific). Quantitative real-time PCR was performed in triplicate using standard SYBR green reagents and protocols on a StepOnePlus Real-Time PCR system (Applied Biosystems). The target mRNA expression was quantified using the ΔΔCt method and normalized to HMBS expression. All primers were designed using Primer 3 (http://frodo.wi.mit.edu/primer3/) and synthesized by Integrated DNA Technologies (Coralville). Primer sequences are listed in Supplementary Table S3. RNA-seq was performed using the Illumina HiSeq 2000 in paired-end mode, as previously described (16). Detailed description of GSEA analysis is provided in the Supplementary Materials and Methods.
Chromatin immunoprecipitation (ChIP)-seq
ChIP assays for AR were performed using HighCell ChIP kit (Diagenode) according to manufacturer’s protocol, using AR antibody (Millipore, Cat. # 06–680). For AR ChIP-seq experiments with EPZ-6438 and EZH2 ASOs, C4–2B cells were treated with 1 μM EPZ-6438 or ASO-EZH2–65 for 5 days. Next, cells were cross-linked for 10 minutes with 1% formaldehyde. Cross-linking was terminated by the addition of 1/10 volume 1.25 M glycine for five minutes at room temperature followed by cell lysis and sonication (Bioruptor, Diagenode). This resulted in an average chromatin fragment size of 200 base pairs. A chromatin equivalent of 5×106 cells was used for each assay. ChIP DNA was isolated (IPure Kit, Diagenode) from samples by incubation with antibody at 4°C overnight followed by wash and reversal of cross-linking. The ChIP-seq sample preparation for sequencing was performed according to manufacturer’s instructions (Illumina). ChIP-enriched DNA samples (1–10 ng) were converted to blunt-ended fragments using T4 DNA polymerase, E. coli DNA polymerase I large fragment (Klenow polymerase), and T4 polynucleotide kinase (New England BioLabs, NEB). A single A-base was added to fragment ends by Klenow fragment (3′ to 5′ exo minus; NEB) followed by ligation of Illumina adapters (Quick ligase, NEB). The adapter-modified DNA fragments were enriched by PCR using the Illumina Barcode primers and Phusion DNA polymerase (NEB). PCR products were size selected using 3% NuSieve agarose gels (Lonza) followed by gel extraction using QIAEX II reagents (Qiagen). Libraries were quantified with the Bioanalyzer 2100 (Agilent) and sequenced on the Illumina HiSeq 2000 Sequencer (100 nucleotide read length). Detailed description of ChIP-seq data analysis is provided in the Supplementary Materials and Methods.
Assay for Transposase-Accessible Chromatin using sequencing (ATAC-seq)
ATAC-seq was performed as previously described (17). Briefly, C4–2B cells were treated with 1 μM EPZ-6438 or DMSO control for 5 days (or ASO-Ctrl and ASO-EZH2), then trypsinized and pelleted. 25,000 cells were washed in cold PBS and resuspended in lysis buffer (CER-I of NE-PER kit, Invitrogen, Cat. # 78833) on ice for 10 minutes with occasional pipetting. The lysate was centrifuged at 1300g for five minutes at 4°C. Nuclei were resuspended in 2X TD buffer, then incubated with Tn5 enzyme for 30 minutes at 37°C (Nextera DNA Library Preparation Kit, Cat. # FC-121–1031). Samples were immediately purified by Qiagen minElute column and PCR amplified with the NEBNext High-Fidelity 2X PCR Master Mix (NEB, Cat. # M0541L). qPCR was utilized to determine the optimal PCR cycles to prevent over-amplification. The amplified library was further purified by Qiagen minElute column and SPRI beads (Beckman Coulter, Cat. # A63881). ATAC-seq libraries were sequenced on the Illumina HiSeq 2500, paired-end, 50 cycles. Detailed description of ATAC-Seq data analysis is provided in the Supplementary Materials and Methods.
RNA in situ hybridization (RNA-ISH)
The RNAscope 2.5 HD BROWN assay (Advanced Cell Diagnostics, Cat. #322300) was performed using target probes to EZH2 or AR according to the manufacturer’s instructions. EZH2 RNA probes (Cat. #405491, accession #NM_001203248.1, targeting 197 – 1337) and AR RNA probes (Cat. #40049, accession # NM_000044.3, targeting 5604 – 6660) are complementary to the target mRNA. Probes Hs-PPIB (human peptidylprolyl isomerase B) and DapB (bacterial dihydrodipicolinate reductase) were used as positive and negative controls, respectively. Paraffin sections from formalin-fixed tumor samples were incubated for one hour in a 60°C drying oven, before deparaffinization in xylene, brief incubation in 100% ethanol, and air drying. Samples were permeabilized with Protease Plus, then subjected to probe hybridization with two-hour incubation in HybEZ at 40°C. After washing, slides were processed by standard signal amplification steps. Chromogenic detection was performed using DAB, followed by counterstaining with 50% Gill’s hematoxylin I (Fisher, Cat. #26801–01).
All slides were examined for EZH2 or AR ISH signals in morphologically intact cells and scored by a blinded independent pathologist. ISH signal was defined as brown, punctate dots, and expression level scored as described in (18): 0 = no staining or less than one dot per 10 cells, 1 = one to three dots per cell, 2 = four to nine dots per cell (few or no dot clusters), 3 = 10 to 15 dots per cell (less than 10% in dot clusters), and 4 = greater than 15 dots per cell (more than 10% in dot clusters). Cumulative ISH product score was calculated for each evaluable slide as the sum of the individual products of the expression level (0–4) and percentage of cells [0–100; i.e., (A% × 0) + (B% × 1) + (C% × 2) + (D% × 3) + (E% × 4); total range = 0 to 400].
Data availability
The raw RNA-seq, ChIP-seq (AR) and ATATC-seq data have been deposited at SRA(SRP157942).
RESULTS
EZH2 inhibitors sensitize prostate cancer cells to enzalutamide
Since past studies associated epigenetic changes with CRPC development, we postulated that inhibition of key epigenetic regulators may sensitize CRPC cells to antiandrogen therapy, such as enzalutamide (5–7). An unbiased epigenetic inhibitor screening experiment was conducted, subjecting CRPC-derived C4–2B cells to treatment with a library of 92 epigenetic regulator inhibitors (Supplementary Table S1) (Fig. 1A). This screen found that EZH2 inhibitors markedly enhanced the inhibitory effect of enzalutamide on cell proliferation and ranked the highest among the compounds assessed (Fig. 1A). The experiment was repeated with the 10 most efficacious epigenetic inhibitors and found that six targeted EZH2 (Fig. 1B). Immunoblot analysis demonstrated that these six reduced H3K27Me3 protein content, indicating diminution of EZH2 function (Fig. 1B).
Figure 1. Identification of EZH2 as a key target for enzalutamide sensitization.
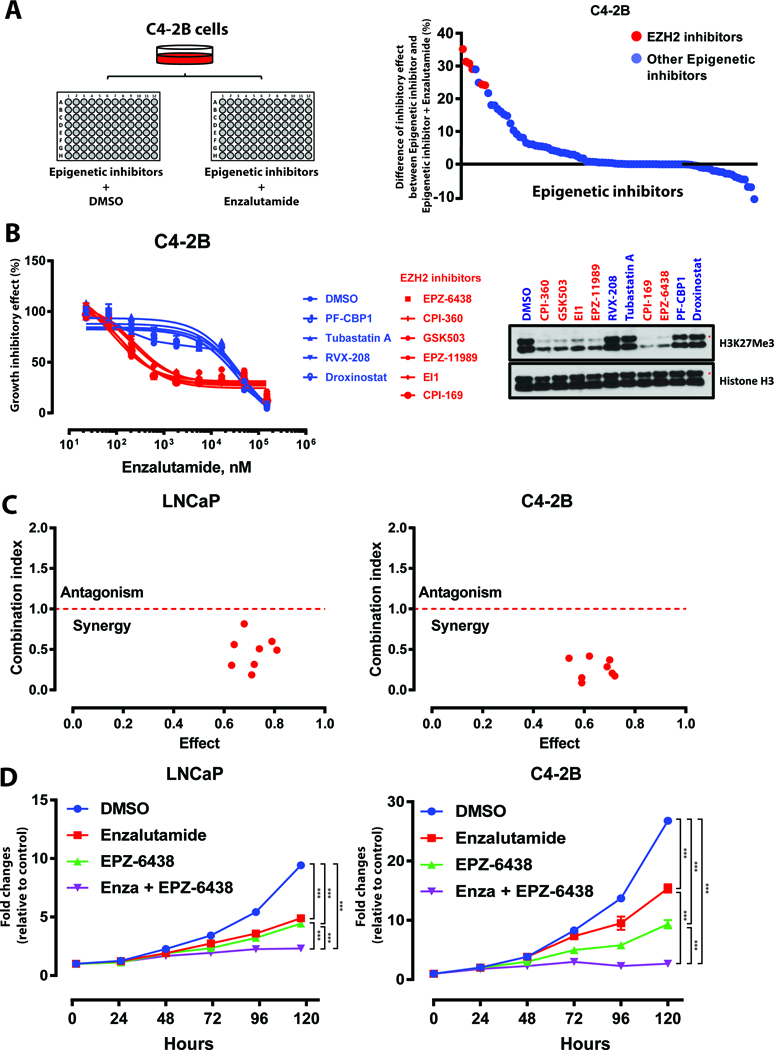
A. Diagram depicting screen performed in C4–2B cells treated with epigenetic inhibitor panel plus enzalutamide (2 μM) or DMSO control. Degree of proliferation inhibition for each individual inhibitor (1 μM) plus enzalutamide versus each inhibitor alone is graphed (all normalized to DMSO).
B. Dose response curves of C4–2B cells treated with enzalutamide in combination with each of the 10 most effective epigenetic inhibitors (1 μM). Data represent mean ± standard error (n = 3) from one of three independent experiments. Immunoblot analysis of H3K27Me3 levels in C4–2B cells treated with each epigenetic inhibitor demonstrated EZH2 inhibition with red compounds.
C. Combination index analysis (CI) for the combination of EPZ-6438 and enzalutamide. Circles represent experimentally determined CI values (Chou-Talalay method).
D. Growth curves of LNCaP and C4–2B cells treated with DMSO, enzalutamide (Enza, 2 μM), EPZ-6438 (1 μM), and combination of enzalutamide plus EPZ-6438. Data represent mean ± standard error (n = 6) from one of three independent experiments.
The experiment was conducted in biological triplicate. When not otherwise indicated, data shown as representative experiments, with each point representing the mean (± SD) of technical duplicates (t test). ***, P < 0.001.
These findings were validated by employing EPZ-6438, a small molecule EZH2 inhibitor that is effective in treating hematological malignancies and solid tumors (19). Combination index analysis indicated that co-treatment with EPZ-6438 and enzalutamide produced synergistic antiproliferative effects in C4–2B and androgen-dependent LNCaP cells (Fig. 1C). Co-treatment yielded the slowest growth curve compared to treatment by DMSO or single agent and resulted in near-complete inhibition of cell proliferation (Fig. 1D). We further confirmed that EPZ-6438 was able to sensitize cells to enzalutamide using an enzalutamide-resistant LNCaP model, LNCap-EnzR (15) (Supplementary Fig. S1). Selective disruption of EZH2 with CRISPR/Cas9 further established that loss of EZH2 enhanced the growth inhibitory effect of enzalutamide in prostate cancer cells and in an additional model, CWR-R1 cells (Supplementary Fig. S2A-D and Supplementary Fig. S3A-C).
Antisense oligonucleotides (ASOs) represent a novel technique to target AR and EZH2 in prostate cancer cells
AR is central to prostate cancer development, progression, and drug resistance (2). Among the many AR signaling perturbations in CRPC, AR mutant forms or ligand-independent variants (AR-Vs) are not effectively inhibited by currently approved therapies, such as enzalutamide (20,21). Similarly, most inhibitors of EZH2, including EPZ-6438, target the SET domain of EZH2 responsible for methyltransferase activity. Studies have, however, shown that the oncogenic activity of EZH2 in CRPC can occur independently of its Polycomb-repressive function, and targeting the non-PRC2 (polycomb repressive complex-2) function of EZH2 may have therapeutic efficacy (8). ASOs optimized for in vivo delivery present a novel solution by targeting all forms of EZH2 and AR at the mRNA level, leading to ablation of the protein itself rather than inhibition of enzymatic function.
In collaboration with Ionis Pharmaceuticals, we have tested clinical-grade ASOs targeting AR and EZH2. ASOs were delivered to the cells via free uptake, as previously described (22). As shown in Fig. 2A and Supplementary Fig. S4A-B, ASOs inhibited their targets and downstream activities efficiently, as indicated by respective dose-dependent reductions in EZH2/H3K27Me3 and AR/PSA levels. We compared the growth inhibitory effects of EZH2 ASOs to EPZ-6438 in LNCaP-abl cells, an androgen-independent cell line which relies on the PRC2-independent functions of EZH2 (8). As shown in Supplementary Fig. S5A, EPZ-6438 and EZH2 ASOs both inhibited LNCaP-abl cell growth, but EZH2 ASOs exhibited enhanced efficacy. Furthermore, ASO-AR treatment was able to decrease growth of enzalutamide-resistant LNCaP-EnzR cells (Supplementary Fig. S5B), suggesting that ablation of EZH2 and AR by ASOs may indeed be advantageous to enzymatic inhibition in certain settings.
Figure 2. EZH2- and AR-targeting ASOs synergistically inhibit prostate cancer growth.
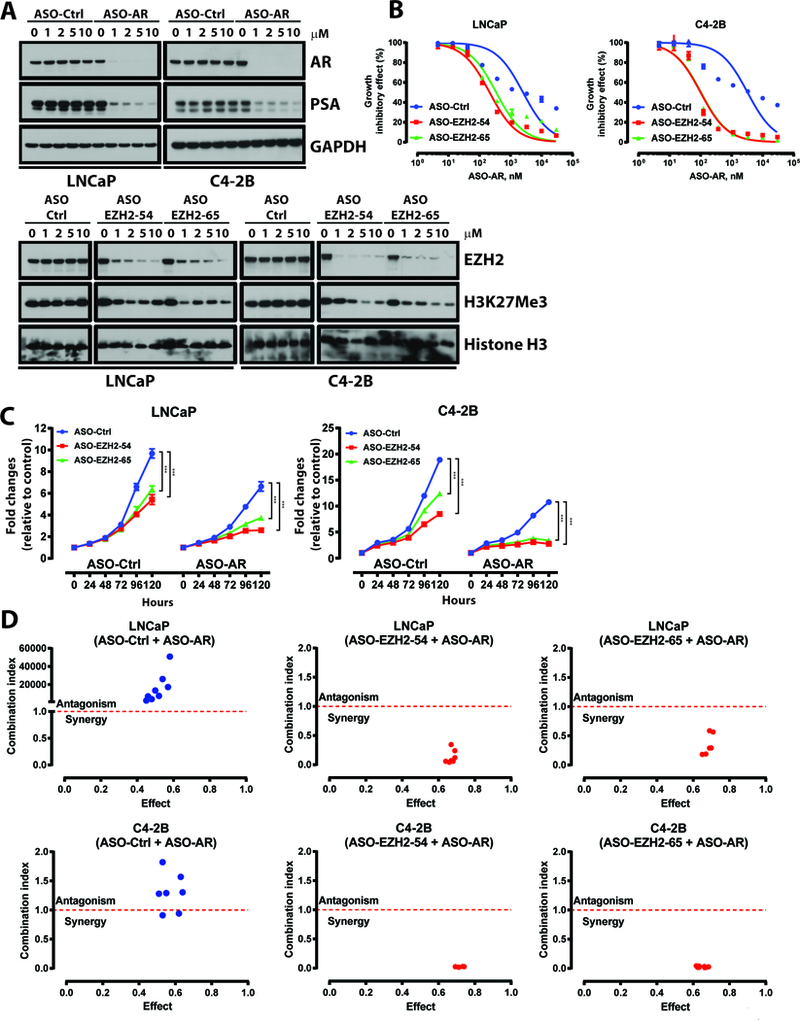
A. Top, immunoblot analysis of AR and PSA in LNCaP and C4–2B cells treated with ASO-Ctrl or ASO-AR. ASOs were delivered to cells in all experiments via free uptake. Bottom, immunoblot analysis of EZH2 and H3K27Me3 in LNCaP and C4–2B cells treated with ASO-Ctrl and EZH2 ASOs (EZH2–54 and EZH2–65).
B. Dose response curves of LNCaP and C4–2B cells treated with ASO-Ctrl or ASO-EZH2 (EZH2–54 and EZH2–65, 1 μM), then treated with ASO-AR for five days. Data represent mean ± standard error (n = 3) from one of three independent experiments.
C. Growth curves of LNCaP and C4–2B cells treated with ASO-Ctrl, ASO-AR, ASO-EZH2 (EZH2–54 and EZH2–65, 1 μM), or combination of ASO-AR (1 μM) plus ASO-EZH2 (1 μM). Data represent mean ± standard error (n = 6) from one of three independent experiments.
D.Combination index analysis (CI) for the combination of ASO-AR and EZH2 ASOs. Circles represent experimentally determined CI values (Chou-Talalay method).
The experiment was conducted in biological triplicate. When not otherwise indicated, data shown as representative experiments, with each point representing the mean (± SD) of technical duplicates (t test). ***, P < 0.001.
Consequently, we determined whether the results generated in Fig. 1 could be recapitulated with ASOs to ablate EZH2 and AR. In corroboration with data in Fig. 1, EZH2 ASOs sensitized LNCaP and C4–2B cells to ASO-AR, lowering the IC50 of ASO-AR and shifting the growth inhibitory curve to the left (Fig. 2B). Furthermore, both CellTiter-Glo assays (Fig. 2C) and soft agar colony experiments (Supplementary Fig. S6A-B) demonstrated that ASOs co-targeting EZH2 and AR yielded the most potent cellular growth inhibition. Combination index analysis indicated that EZH2 and AR ASOs synergized in both LNCaP and C4–2B cells (Fig. 2D). Similar results were obtained using CRISPR/Cas9 to target EZH2 in the presence of ASO-AR (Supplementary Fig. S6C-D). Combination ASO-AR and ASO-EZH2 treatment was also tested in LNCaP-EnzR cells and shown to be more effective at inhibiting growth of this model than either agent alone (Supplementary Fig. S5B).
EZH2 inhibition activates AR signaling in prostate cancer cells
We sought to evaluate how EZH2 inhibition may sensitize CRPC cells to AR-targeted therapies. RNA-seq was employed to profile transcriptomic changes in C4–2B cells treated with EPZ-6438 or ASO-EZH2. Interestingly, gene set enrichment analysis (GSEA) revealed AR signaling to be the set most significantly activated by either agent (Fig. 3A-B). RT-PCR confirmed that multiple AR target genes were significantly upregulated upon treatment with EPZ-6438 or EZH2 ASOs in LNCaP and C4–2B cells (Fig. 3C).
Figure 3. EZH2 inhibition enhances AR signaling and genome-wide AR binding.
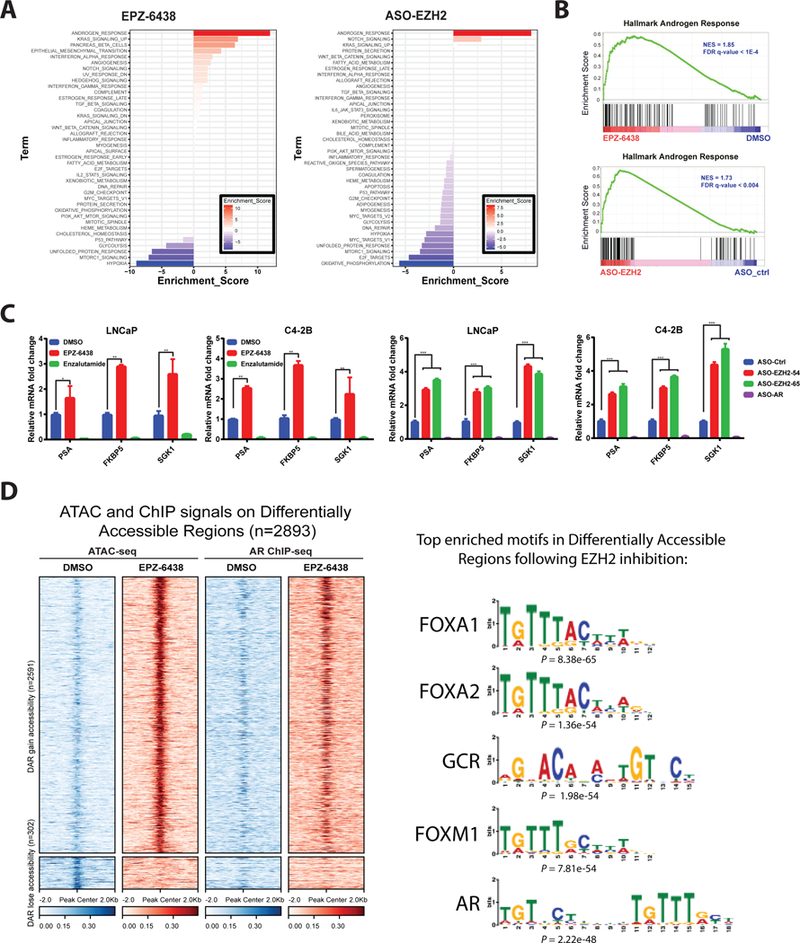
A. Transcriptomic changes induced in C4–2B cells by treatment with EPZ-6438 and ASO-EZH2, as determined by RNA-seq with GO analysis. GO enrichment p values and number of genes in each GO category are respectively indicated at the x-axis and next to the bar. “Androgen Response” was the top activated gene set by either agent.
B. EPZ-6438 and ASO-EZH2-mediated elevation of AR signaling as demonstrated by GSEA. Hallmark Androgen Response gene sets shown for C4–2B cells treated with EPZ-6438 or ASO-EZH2.
C. qRT–PCR analysis of AR target gene in LNCaP and C4–2B cells treated with DMSO, EPZ-6438, or enzalutamide for 72 h (left) or designated ASOs for 72h (right). Data represent mean ± SEM (n = 3) from one of three independent experiments.
D. Heatmaps of ATAC-seq and AR ChIP-seq signals around ± 2kb of peak center (left). Each row represents ± 2kb around the center of differentially accessible regions (DAR). Top panel represents 2591 DAR that gain accessibility upon EPZ-6438 treatment compared to DMSO. Bottom panel represents 302 DAR that lose accessibility upon EPZ-6438 treatment compared to DMSO. (Right) Motif analysis showing enriched motifs in open chromatin upon EPZ-6438 treatment.
As a subunit of PRC2, EZH2 associates with chromatin, inducing its compaction and limiting access of transcription factors and ATP-dependent remodeling machinery; therefore, EZH2 inhibition may yield global redistribution of transcription factor cistromes. Genome-wide Assay for Transposase-Accessible Chromatin using sequencing (ATAC-seq) was performed to profile chromatin accessibility changes resulting from EZH2 inhibition in C4–2B cells with either EPZ-6438 or ASO-EZH2. As shown in Fig. 3D and Supplementary Fig. S7A, both treatments dramatically increased chromatin accessibility. De novo transcription factor binding site motif analysis was performed on differential accessible chromatin regions and revealed binding sites for AR and associated pioneer factors to be the most enriched motifs upon EZH2 inhibition or ablation (Fig. 3D and Supplementary Fig. S7B). Genome-wide AR chromatin immunoprecipitation sequencing (ChIP-seq) was employed and integrated with ATAC-seq data to directly profile EPZ-6438 and ASO-EZH2-mediated AR occupancy changes. Consistent with motif analysis, these data showed that differentially accessible regions were highly associated with AR binding (Fig. 3D, Supplementary Fig. S7A and C). Combined, these data propose that inhibition of EZH2 may lead to reprogramming of the AR cistrome and upregulation of AR signaling. These alterations could increase dependence of prostate cancer cells on this pathway and their sensitivity to AR-targeted therapies.
EZH2 and AR ASO therapy additively inhibit prostate cancer xenograft growth
ASOs engineered for in vivo delivery are an emerging class of oncology therapeutics (13). An ASO-based strategy was employed to determine the effect of inhibiting AR and EZH2 in prostate cancer xenografts. The efficacy of AR-targeting ASO as an anti-tumor agent in C4–2B cells was first evaluated. Following a three-week time course, xenografts in both active agent groups had significantly lower volumes than those of controls (Fig. 4A). Xenograft volumes were significantly reduced in animals treated with ASO-AR versus enzalutamide, highlighting advantages of the ASO technology; xenograft weights confirmed volume analysis (Fig. 4A).
Figure 4. ASOs targeting AR and EZH2 additively inhibit prostate cancer xenograft growth.
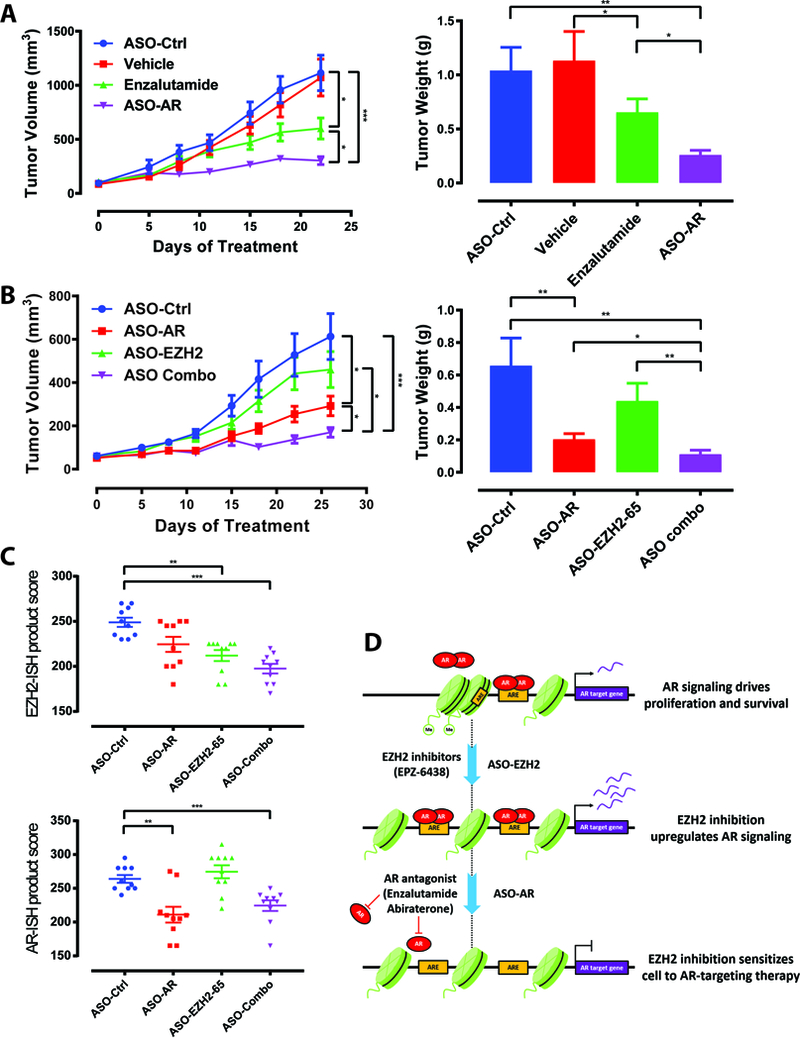
A. Tumor volume time course for subcutaneous C4–2B xenografts in CB17SCID mice. Treatment with vehicle, ASO-Ctrl, ASO-AR, or enzalutamide started at tumor volume 50–100 mm3. Tumor weights from respective treatment groups at day 21 were also determined. N=10 mice in all treatment groups for each panel.
B. Tumor volume time course for subcutaneous C4–2B xenografts in CB17SCID mice. Treatment with ASO-Ctrl, ASO-AR, ASO-EZH2–65, or combination of ASO-AR and ASO-EZH2–65 started at tumor volume 50–100 mm3. Tumor weights from respective treatment groups at the termination of the study are also graphed.
C. EZH2 and AR expression (ISH product score) in indicated xenograft samples.
D. Schematic depicting proposed mechanism of EZH2 inhibition-mediated sensitization of prostate cancer cells to AR-targeted therapies. EZH2 inhibition increases activation of the AR signaling pathway, increasing their dependence on AR through reprogramming of the AR cistrome. In this setting, effectiveness of AR antagonists (e.g. enzalutamide) and ASO-AR are enhanced.
The experiment was conducted in data shown ± s.e.m. *, P < 0.05, **, P< 0.01, ***, P < 0.001.
The same experimental protocol was used to determine the impact of combining ASOs targeting AR and EZH2 on xenograft growth. As observed above, ASO-AR significantly inhibited xenograft growth, while ASO-EZH2 (50 mg/kg) had no effect (Fig. 4B). However, combination of ASO-AR and ASO-EZH2 yielded significant reduction in tumor volume versus ASO-AR alone. Measurements of xenograft weight paralleled those of volume (Fig. 4B), and a clear size decrement was appreciable in the combination treatment (Supplementary Fig. S8A). Quantitative RNA in situ hybridization (RNA-ISH) verified significant reductions in mRNA encoding EZH2 and AR (Fig. 4C and Supplementary Fig. S8B-C). Tumor growth and weights in the combination group were also significantly reduced when compared to single agent arms employing equal amounts of total ASOs (i.e. ASO-AR + ASO-Ctrl and ASO-EZH2 + ASO-Ctrl). This ensured that antitumor effects of combination treatment did not result from higher total ASO load (Supplementary Fig. S8D). These data demonstrate the ability of EZH2 inhibition to augment the growth inhibitory effect of AR inhibition in CRPC. Additionally, the findings highlight ASO therapy as a novel method of targeting both AR and EZH2 in vivo.
DISCUSSION
Although advanced prostate cancer often responds initially to therapies that suppress androgen-axis signaling, resistance inevitably develops, leading to the emergence of CRPC. The clinical efficacies of therapies targeting AR, such as abiraterone and enzalutamide, have confirmed that AR signaling remains an important driver of CRPC (3,4,23). Approaches to improve the duration of response and address key pathways of resistance are needed. The data presented here suggest that administration of therapies that inhibit EZH2 signaling may sensitize tumor cells to AR-targeting therapies in CRPC. EZH2 inhibitors lead to increased open chromatin and AR binding throughout the genome, upregulating AR signaling. Interestingly, this appears to increase dependence of CRPC cells on this pathway, as sensitivity to AR-targeting therapies is enhanced (Fig. 4D).
Past studies have associated epigenetic aberrations with prostate cancer progression and drug resistance and support targeting of epigenetic mediators as a potential therapeutic strategy. Our group previously found EZH2 to be overexpressed in prostate cancer and associated with progression and poor prognosis (7). Recent studies showed that EZH2 plays a critical role in NEPC development and suggested that inhibition of EZH2 is a promising therapeutic avenue for NEPC that could reactivate AR signaling and restore sensitivity to AR-targeting drugs (9–11). Here, we defined the role of EZH2 inhibition in CRPC adenocarcinoma models and demonstrated that EZH2 inhibition is a viable treatment strategy to restore sensitivity; results were similar in LNCaP cells, proposing EZH2 inhibition may also augment responses to AR-targeted drugs in androgen-dependent stages of prostate cancer. Interestingly, inhibition of EZH2 caused redistribution of AR binding and upregulated AR signaling, suggesting a greater dependence on this pathway underlies ADT sensitization. Recently, a phase 2 study showed that bipolar androgen therapy resulted in resensitization to enzalutamide in patients undergoing rechallenge (24). Although a different avenue of intervention, these data support our hypothesis that further activation of AR signaling may restore response to ADT.
Through exploration of alternative strategies to inhibit all functions of EZH2 and AR, we found ASOs, a rapidly expanding class of oncology therapeutic agents, as a novel treatment strategy for combinatorial targeting of these factors in CRPC (13). In reports involving other cancer lineages, AZD4785 (ASO targeting KRAS) has been shown to be an attractive therapeutic for treatment of KRAS-driven human cancers, and AZD9150 (ASO targeting STAT3) exhibited promising antitumor activity in patients with treatment-refractory lymphoma and non-small cell lung cancer (22,25). Previously, AR ASOs targeting both full-length AR and AR variants have been shown as a rational approach for treatment of AR-dependent CRPC (26). Our data demonstrate that combining AR-ASO with EZH2 ASOs enhances the antitumor effect in CRPC. Furthermore, tumors treated with ASO-AR alone exhibited enhanced growth inhibition compared to enzalutamide, another advantage of ASO technology in this setting. Finally, ASOs have the power to target previously undruggable proteins or disease-associated non-coding RNAs, such as AR variants or Polycomb-independent functions of EZH2 (8,20). Altogether, data from this study suggest that clinical studies investigating ASO-AR and ASO-EZH2 therapy in CRPC patients are warranted.
Supplementary Material
Statements of significance.
Simultaneous targeting of lysine methyltransferase EZH2 and the androgen receptor with antisense oligonucleotides proves a novel and effective therapeutic strategy in castration-resistant prostate cancer patients.
ACKNOWLEDGMENTS
We thank S.M. Dhanasekaran, C. Kumar, M. Cieślik, D. Robinson, Y-M. Wu, S. Shankar, and S. Kregel for helpful discussions. We also thank S. Ellison for assistance in writing and editing of the manuscript. This work was supported in part by a NCI Prostate SPORE (P50CA186786). A.M. Chinnaiyan is a Howard Hughes Medical Institute Investigator, A. Alfred Taubman Scholar, and American Cancer Society Professor as well as is supported by the Prostate Cancer Foundation. L. Xiao is supported by a Department of Defense (DoD) Postdoctoral Award (W81XWH-16–1-0195). J.C. Tien and Y. Qiao are supported by the PCF Young Investigator Award. A. Parolia is supported by the DoD Early Investigator Research Award (W81XWH-17–1-0130). Y. Zhang is also supported by the DoD Early Investigator Research Award (W81XWH-17–1-0134).
Footnotes
Conflict of interest statement: Y.K. and A.R.M. are employees of Ionis Pharmaceuticals, which developed the ASOs against EZH2 and AR that were used in this study.
REFERENCES
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin 2018;68:7–30. [DOI] [PubMed] [Google Scholar]
- 2.Ferraldeschi R, Welti J, Luo J, Attard G, de Bono JS. Targeting the androgen receptor pathway in castration-resistant prostate cancer: progresses and prospects. Oncogene 2015;34:1745–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med 2012;367:1187–97. [DOI] [PubMed] [Google Scholar]
- 4.Beer TM, Armstrong AJ, Rathkopf DE, Loriot Y, Sternberg CN, Higano CS, et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med 2014;371:424–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R, et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 2014;510:278–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coleman DJ, Van Hook K, King CJ, Schwartzman J, Lisac R, Urrutia J, et al. Cellular androgen content influences enzalutamide agonism of F877L mutant androgen receptor. Oncotarget 2016;7:40690–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002;419:624–9. [DOI] [PubMed] [Google Scholar]
- 8.Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis RT, et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science 2012;338:1465–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med 2016;22:298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dardenne E, Beltran H, Benelli M, Gayvert K, Berger A, Puca L, et al. N-Myc induces an EZH2-mediated transcriptional program driving neuroendocrine prostate cancer. Cancer Cell 2016;30:563–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017;355:78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Puca L, Bareja R, Prandi D, Shaw R, Benelli M, Karthaus WR, et al. Patient derived organoids to model rare prostate cancer phenotypes. Nat Commun 2018;9:2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bennett CF, Baker BF, Pham N, Swayze E, Geary RS. Pharmacology of antisense drugs. Annu Rev Pharmacol Toxicol 2017;57:81–105. [DOI] [PubMed] [Google Scholar]
- 14.Culig Z, Hoffmann J, Erdel M, Eder IE, Hobisch A, Hittmair A, et al. Switch from antagonist to agonist of the androgen receptor bicalutamide is associated with prostate tumour progression in a new model system. Br J Cancer 1999;81:242–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kregel S, Chen JL, Tom W, Krishnan V, Kach J, Brechka H, et al. Acquired resistance to the second-generation androgen receptor antagonist enzalutamide in castration-resistant prostate cancer. Oncotarget 2016;7:26259–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cieslik M, Chugh R, Wu YM, Wu M, Brennan C, Lonigro R, et al. The use of exome capture RNA-seq for highly degraded RNA with application to clinical cancer sequencing. Genome Res 2015;25:1372–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 2013;10:1213–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang L, Harms PW, Palanisamy N, Carskadon S, Cao X, Siddiqui J, et al. Age and gender associations of virus positivity in merkel cell carcinoma characterized using a novel RNA in situ hybridization assay. Clin Cancer Res 2017;23:5622–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Knutson SK, Kawano S, Minoshima Y, Warholic NM, Huang KC, Xiao Y, et al. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol Cancer Ther 2014;13:842–54. [DOI] [PubMed] [Google Scholar]
- 20.Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med 2014;371:1028–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCrea E, Sissung TM, Price DK, Chau CH, Figg WD. Androgen receptor variation affects prostate cancer progression and drug resistance. Pharmacol Res 2016;114:152–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hong D, Kurzrock R, Kim Y, Woessner R, Younes A, Nemunaitis J, et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci Transl Med 2015;7:314–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med 2011;364:1995–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Teply BA, Wang H, Luber B, Sullivan R, Rifkind I, Bruns A, et al. Bipolar androgen therapy in men with metastatic castration-resistant prostate cancer after progression on enzalutamide: an open-label, phase 2, multicohort study. Lancet Oncol 2018;19:76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ross SJ, Revenko AS, Hanson LL, Ellston R, Staniszewska A, Whalley N, et al. Targeting KRAS-dependent tumors with AZD4785, a high-affinity therapeutic antisense oligonucleotide inhibitor of KRAS. Sci Transl Med 2017;9:1–13. [DOI] [PubMed] [Google Scholar]
- 26.Yamamoto Y, Loriot Y, Beraldi E, Zhang F, Wyatt AW, Al Nakouzi N, et al. Generation 2.5 antisense oligonucleotides targeting the androgen receptor and its splice variants suppress enzalutamide-resistant prostate cancer cell growth. Clin Cancer Res 2015;21:1675–87. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw RNA-seq, ChIP-seq (AR) and ATATC-seq data have been deposited at SRA(SRP157942).