

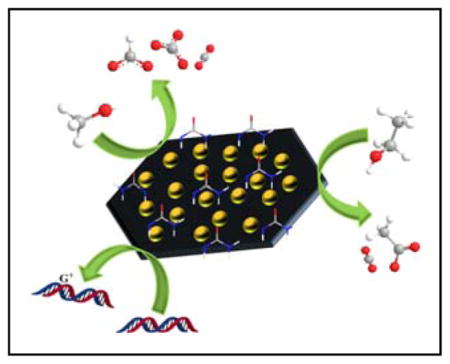
Abstract
Gold (Au) is chemically stable and resistant to oxidation. Although bulk Au is catalytically inert, nanostructured Au exhibits unique size-dependent catalytic activity. When Au nanocatalysts are supported on conductive carbon (denoted as Au@C), Au@C becomes promising for a wide range of electrochemical reactions such as electrooxidation of alcohols and electroreduction of carbon dioxide. In this mini-review, we summarize Au@C nanocatalysts with specific attention on the most recent achievements including findings in our own laboratories, and show that Au nanoclusters (AuNCs, < 2 nm) on nitrided carbon are excellent electrocatalysts for oxidation of organic molecules including guanines in DNA. State-of-the-art synthesis and characterization of these nanomaterials are also documented. Synergistic interactions among Au-containing multicomponents on carbon supports and their applications in electrocatalysis are discussed as well. Finally, challenges and future outlook for those emerging and promising nanomaterials are envisaged.
Keywords: Gold nanocatalysts, “soft” nitriding, electrocatalysis, DNA oxidation
Graphical abstract
Recent advances in synthesizing gold nanocatalysts on carbon materials are reviewed, including applications to electrocatalytic oxidation.
1. Introduction
Gold (Au) is chemically stable and resistant to oxidation. When it has a particle size in the low nanometer range, nanostructured Au with unique size-dependent catalytic properties that differs from those of bulk counterpart has attracted considerable attention in catalysis.1–4 Relevant to this review, early examples reported by Haruta and others showed small Au nanoparticles (AuNPs, < 5 nm) supported on a number of metal oxides were extremely active for low-temperature CO oxidation,5–7 CO2 hydrogenation,8 catalytic combustion of methanol,9 NO reduction,10 selective epoxidation of propene11 and low-temperature water gas shift reactions,12 etc. All those results point out that AuNPs with small size (< 5 nm) are usually more catalytically active compared to larger ones (>10 nm).13 Atomically precise alkylthiol-protected Au nanoclusters (AuNCs, e.g., Au25, Au38 and Au144) with excellent size control have been synthesized later via wet-chemical reduction. This further allows quantifying size-dependent catalytic performance.14, 15
In addition to the intrinsic size effect, the activity of Au nanocatalysts can be influenced by the nature and morphology of supports.16, 17 The primary role of the support is to anchor nanoparticles (NPs) onto its surface, preventing the NPs from sintering and agglomerating during catalytic reactions. Supported Au nanocatalysts can be very stable and resistant from corrosion in the harsh environments like strong acid/base and high temperature. There are many different types of supports used for adsorption and stabilization of AuNPs, such as metal oxide powder,18–21 magnetic microspheres,22, 23 polymer nanospheres or nanofibers,24–26 mesoporous silica27, 28 and carbonaceous supports,29–33 etc. Low-cost carbon supports have shown to be superior compared to other supports in terms of their conductivity, the cost of raw materials, resistance to strong acid or base and the possibility to control porosity and surface chemistry.34, 35 A good example of the influence of the support was shown by Benkó et al., where carbon-supported Au nanocatalysts (denoted Au@C) outperformed other Au nanocatalysts supported on TiO2, SiO2 and CeO2 in CO oxidation and glucose oxidation reactions.35
Various kinds of carbon materials have been extensively studies in electrocatalysis,36, 37 including carbon nanotubes (CNTs)38 and carbon nanofibers,39 two-dimensional graphene oxide/reduced graphene oxide (GO/RGO),40 mesoporous carbon and hollow carbon nanospheres (HCNS),41, 42 and metal-organic framework (MOF)-derived carbonaceous materials,43 etc. The merits of simple synthesis and superior conductivity have enormously enriched the practical applications of carbon supported noble metal nanocatalysts in electrocatalysis.44–46 On the other hand, due to the relatively inert nature of carbon and the weak interaction between Au and support, AuNPs tend to overgrow and aggregate. Thus, the modification and functionalization of the carbon surface, such as the introduction of hydroxyl groups and heteroatom doping, become useful tools to enhance the interaction of Au and carbon and thus to stabilize AuNPs supported on carbon. In addition, the synergistic effect of Au/support may vary the charge state of AuNPs, resulting in the electron-rich NP surface. In a recent study, we showed that electron-rich AuNCs supported on nitrided carbon supports favors the adsorption of electrophile reactants like CO2; therefore, this can promote the activity and selectivity for CO2 electroreduction in comparison to AuNCs on pristine carbon, due to the strong electronic interaction of Au/support.47 This will be discussed in detail in Section 2.3.
Au-based multicomponent nanocatalysts have attracted much attention and benefited from the controllable synthesis in bimetallic or multimetallic NPs. So, a second metal, such as Ag, Co, Cu, Ni, Pd, Pt and Sn, etc., have been introduced in the syntheses to form Au-based binary or multicomponent nanocatalysts. It has been an efficient strategy to reduce noble metal loading and show remarkable enhancement of electrocatalytic activity due to the multifunctionality synergistic effects.48, 49 As briefly summarized in Table 1, a variety of heterogeneous reactions can be catalyzed by using multicomponent nanocatalysts.30, 32, 50–65 For example, incorporation of Pt and Pd with Au usually can promote electrochemical reactions like methanol oxidation and oxygen reduction. Meanwhile, the presence of Au could retain effective CO tolerance during the catalytic reaction.57, 64
Table 1.
Summary of catalytic reactions using carbon-supported Au-based multicomponent nanocatalysts.
| Catalyst composition | Carbonaceous type | Catalytic reaction | Ref. |
|---|---|---|---|
| AuCs | activated carbon | acetylene hydrochlorination | 50 |
| PdxAu1-x | Vulcan XC-72 | borohydride oxidation | 30 |
| AuPd | Vulcan XC-72 | CO and H2 oxidation | 51 |
| AuPd | activated carbon | toluene oxidation | 52 |
| AuPd | RGO | O2 reduction | 53 |
| AuPd | Vulcan XC-72 | H2, CO oxidation | 54 |
| Pd1Au24(SC12H25)18 | CNTs | benzyl alcohol oxidation | 55 |
| AuPd4 | Vulcan XC-72 | ethanol oxidation | 58 |
| Pd@Au | Vulcan XC-72 | ethanol oxidation | 62 |
| AuPt | graphene | O2 reduction, methanol oxidation | 32 |
| AuPt | Vulcan XC-72 | methanol oxidation | 56 |
| AuPt | Vulcan XC-72R | O2 reduction | 57 |
| AuPt | Vulcan XC-72R | CO oxidation | 61 |
| AuPt | carbon black | methanol oxidation | 64 |
| Pd-Co-M(M=Pt, Au, Ag) | Vulcan XC-72R | O2 reduction | 59 |
| Au-Co(III)-Cu(II) | spherical activated carbon (SAC) | acetylene hydrochlorination | 60 |
| NiPdAu | Vulcan XC-72R | methanol oxidation | 63 |
| PtxMyAuz(M=Ni, Cu, Co) | Vulcan XC-72 | O2 reduction | 65 |
The remainder of this review will discuss typical synthetic routes for Au@C nanocatalysts. Next, we will focus on their fascinating applications for various electrocatalytic oxidative reactions. At the end of the article, we analyze current problems and future perspectives in this area. We hope that this review will help readers to gain insight into the design and applications of well-defined Au nanocatalysts for electrocatalysis.
2. Synthesis of AuNPs supported on carbon materials
The intrinsic physicochemical properties of AuNPs are largely influenced by their size and morphology. Considerable effort has been devoted to the precise control over size, shape, stability, and functionality of monodispersed AuNPs supported on carbon through the following two methods. First, one can synthesize AuNPs primarily through wet-chemistry reduction of precursors, then assembling pre-synthesized AuNPs onto carbon surface. This is also known as “self-assembly” method. It allows to predesign the size, shape and chemical composition of AuNPs using well-developed colloidal synthesis method. While the catalytic performance and stability of these prepared AuNPs often suffer from: i) surface ligands that block the electron transfer for electrocatalysis; and ii) the weak interaction with carbon support that destabilizes AuNPs and thus leads to sintering during reaction. Later on, the “direct growth” method has been developed as a means to solve those problems. However, the direct growth of AuNPs on carbon does not offer any control over the size of AuNPs. To resolve the large dispersity of AuNPs in the “direct growth” method, the doping techniques of carbon with various heteroatoms is developed to enhance the interaction between AuNPs and carbon. For example, “soft nitriding” method which effectively modifies the surface of carbon with N emerged recently as demanded. As such, the nucleation rate of Au is significantly enhanced on nitrided carbon resulting in the formation of ultrasmall AuNCs with diameter < 2 nm. The doping techniques to modify the carbon surface also ensure the strong interaction between Au and carbon to stabilize AuNPs in the course of electrocatalysis. In this section, we will summarize and discuss the synthetic strategies of Au supported on carbon nanomaterials.
2.1. Self-assembly method
The self-assembly of AuNPs on carbon supports involves synthesis of Au colloids in solution and subsequent assembly of as-resultant AuNPs on functionalized carbon support. It allows to predesign the nanostructures of AuNPs using various synthetic methods developed up to date. The self-assembly of AuNPs on carbon is often triggered by the non-covalent interactions, such as electrostatic attraction, hydrophobic interactions, hydrogen-bonding interaction and coordination. For example, CNTs or GO treated with concentrated HNO3 or a H2SO4-HNO3 mixture can produce carboxylic acid and hydroxyl groups on the surface of carbon.66 The negatively charged carbon surface is able to physically adsorb AuNPs capped with positively charged ligands through electrostatic attraction. This method has been widely used to assemble AuNPs on GO due to the negatively charged GO surface.67–70 Through the self-assembly approach, AuNPs with size of 3.5, 9 and 25 nm prepared via the chemical reduction of NaBH4 can uniformly self-assemble on GO sheets.69 Surface charge of carbon can be introduced through layer-by-layer technique. For instance, positively charged AuNPs (a zeta potential of +32.4 mV, pH 7) can self-assemble on functionalized multiwall carbon nanotubes (MWCNTs) coated with negatively charged poly(diallyldimethylammonium chloride)/poly(sodium 4-styrenesulfonate) (PDAC/PSS) bilayer (a zeta potential of −46.2 mV, pH 7) via the electrostatic interaction as reported by Bumsu et al.71 By tailoring the surface of carbon to be positively charged by adsorbing cationic polyelectrolytes, negatively charged AuNPs can be adsorbed on the CNTs as well.72–74
Another example by Han and co-workers shows the pre-synthesized 2 nm AuNPs capped with decanethiol (DT) monolayer shell assembled on the CNTs in the presence of mediating linker 11-mercaptoundecanoic acid/1,9-nonanedithiol.75 A combination of hydrophobic interaction and hydrogen bonding interactions between the alkyl chains/carboxylic groups of capping/linking agents and surface functional groups of CNTs drove the assembly of AuNPs on CNTs.
Huang and co-workers demonstrated high-density self-assembly of AuNPs on the surface of MWCNTs without any pretreatment of carbon support.76 They used 1-pyrenemethylamine as the linker where the alkylamine substituent of the pyrene bound to AuNPs; meanwhile, the pyrene fluorophore bound to MWCNTs via π-π stacking. Shi et al. established a simple method to disperse AuNPs on CNTs in aqueous solution by sonication without any acid oxidation or functionalization of CNTs (Fig. 1). A small amount of ethanol was added into the aqueous suspension of CNTs to reduce the interfacial tension between CNTs and water, thereby changing the wettability of hydrophobic CNTs and enhancing the interaction between CNTs and AuNPs.77
Fig 1.

Transmission electron microscopy (TEM) images of Au@CNTs with loading ratio of (A) 5 wt% and (B) 10 wt%. Reprinted with permission from ref 77. Copyright 2009, Elsevier.
In addition to monometallic AuNPs, Au-based bimetallic NPs were reported by many groups to self-assemble on the surface of carbon. For example, Han et al. reported that Au-Pd alloy NPs capped by poly(vinyl alcohol) (PVA) adsorbed on g-C3N4 by simple mixing process(Fig. 2).78 Lin’s group reported the loading of 3.3 nm Au-Pt alloy NPs on graphene nanosheets and XC-72 carbon black with the assistance of poly(diallyldimethylammonium chloride) (PDDA).79 As-synthesized Ag@Au and Fe@Au NPs attached onto p-aminothiophenol (PATP)-functionalized GO sheets to obtain the bimetal-graphene nanocomposites were also prepared by Gupta and coworkers.80, 81
Fig 2.

High-resolution TEM images of the as-prepared AuPd/g-C3N4 (a–c) and unsupported AuPd NPs samples (d). Reprinted with permission from ref 78. Copyright 2015, Elsevier.
2.2. Direct growth method
The direct growth of metal NPs or nanoclusters on supports is a classical route to highly dispersed supported nanocatalysts. Taking AuNPs supported on GO as example, in situ reduction of gold salts on support materials by reducing agents like BH4−, sodium citrate and ascorbic acid can result in the growth of AuNPs attached on GO. The growth of AuNPs on graphene has been reported by Goncalves82 and Pocklanova83 where sodium citrate was used as a reductant for AuCl4− in the presence of RGO. However, the produced AuNPs usually have a broad size distribution and relatively large size (a few to tens of nanometers). Zhang and co-workers developed a one-pot synthesis of 7 nm AuNPs on RGO using sodium citrate as reductant and stabilizer simultaneously.84 Sodium citrate can reduce both GO and Au precursors, and prevent the formation of agglomerates/overgrowth of AuNPs. Other than graphene nanosheets, activated carbon is also a choice for the in situ growth of AuNPs. Yan et al. reported that AuNPs in the range of 2 to 16 nm supported on activated carbon were prepared by rapid reduction of AuCl4− on the surface of activated carbon with KBH4 in the presence of polyvinylpyrrolidone (PVP) (Fig. 3).85
Fig 3.

(a) Schematic illustration of the synthesis of Au@C nanocatalyst; (b) TEM image of Au@C nanocatalyst. Reprinted (adapted) with permission from ref 85. Copyright 2011, American Chemical Society.
Those direct growth syntheses suffer from poor control over the Au size and morphology, and particularly limit the applications of Au@C as nanocatalysts, since AuNPs with size > 5 nm are less active. To control the size of Au, various stabilizers and surfactants are required to precisely control the size and shape of AuNPs, enhance the interaction between the NPs and supports, and prevent overgrowth of AuNPs.2, 86, 87 Huang et al. developed an efficient synthetic method for in situ growth of 1-octadecanethiol (ODT) capped AuNCs on GO nanosheets by photoirradiation.88 AuNCs with a size of 1.2 ± 0.3 nm were distributed in an ordered pattern where the distance between the particle chains was ~4.4 nm. This was attributed to the linear thiol ODT self-assembled along the <100> direction on graphene surfaces.
Surfactants, however, cover the surface of AuNPs that inevitably blocks the surface catalytically active sites and essentially slows or shuts down the electron transfer. This is detrimental for electrocatalytic performance.89, 90 Therefore, surfactant-free synthetic methods are highly desirable to overcome such challenge. For example, 12.8 ± 2.5 nm AuNPs on GO sheets can be synthesized using hydrothermal reduction of HAuCl4 in an aqueous NaOH/GO mixture at 180 °C for 12 h.91 Another example by Shao et al. showed well-dispersed AuNPs on GO with a mean size of 5.2 ± 0.2 nm via H2 reduction of HAuCl4 and GO, even though some aggregates of AuNPs were also seen.92 Tang’s group developed a “clean” method to grow Au and other metal (Pt, Pd) nanoclusters with average diameter of 1.8 nm on RGO using sonication without any additional surfactants (Fig. 4).93 The tunable reduction of GO sheets by adding various amount of hydrazine was the key to give the possibility for the reduction of AuNCs by sonication. Similar result was reported that single-wall carbon nanotubes (SWNTs) could reduce HAuCl4 to generate 7 nm AuNPs in the absence of additional reductants.94
Fig 4.

Electron microscopy characterization of as-synthesized Au/RGO (a–d). Scheme of the formation mechanism of Au/RGO hybrids (e). Reprinted (adapted) with permission from ref 93. Copyright 2012, American Chemical Society.
2.3. Soft nitriding method
Although numerous effort has been devoted to decorating ultrafine metal NPs on carbon supports, the weak interaction between noble metals and carbon surface is still problematic. This may lead to the leaching of metal NPs in the catalytic process, resulting in the loss of catalytic performance. According to previous studies, through doping electron-rich heteroatoms like N,95, 96 P,97, 98 and S99 elements into carbon nanomaterials, the electronic characteristics can be altered, thus producing more active sites and unexpected electrical and catalytic properties, such as high stability.100 Zhang et al. proposed the synthesis of nanoporous carbon materials derived from glucose with N-containing additives by hydrothermal carbonization (HTC) process, which was used as supports to confine Pd NPs of 5.9 nm.101 They indicated that the incorporated N atoms in the carbon contributed to more structure defects which enhanced the adsorption of Pd. The interaction of surface N atoms and Pd also altered the electronic density of Pd species, which improved the reaction rate. A number of studies by Yin,93 Koo102 and Xie103 showed that surface N atoms embedded in GO could act as initial nucleation sites for metal NPs. Nitrided carbon materials not only serve as reducing agents for metal precursors, but also increase the numbers of anchoring force to adsorb metal ions and NPs.
Recently, in situ growth of ligand-free ultrasmall (< 2 nm) noble metal nanoclusters (e.g., Au, Pd and Pt) onto carbon supports was developed by our group (Fig. 5).104–106 N-doped carbon supports with abundant nitrogen sites were synthesized by annealing with urea at 300 °C. Urea decomposed into NH3 and HCNO, efficient to integrate surface N atoms on nearly any commercial carbon. In our synthesis, the surface N content can reach 19 at% in the form of pyridine/graphitic N into the graphene framework and ureido groups on the surface of carbon, respectively. The presence of those surface N species enhanced the affinity to metal ions and prevented aggregation or overgrowth of Au. AuNCs of 1.6 nm with narrow size distribution were synthesized on the surface of nitrided carbon (denoted as AuNCs@NC) through a rapid chemical reduction of HAuCl4 with NaBH4. Moreover, the generality of the soft nitriding was confirmed using seven different carbonaceous supports where the growth of AuNCs was independent of the initial properties of carbon supports. For soft nitriding method, the size of AuNCs can be readily tuned by pH of the solution. Larger AuNPs with an average diameter of 6–10 nm were grown at pH < 4 or > 10. The faster nucleation of AuCl4− occurred at lower pH. AuNCs nucleated and grew in aqueous solution rather than on carbon surface at pH < 4. At pH > 10, the insufficient affinity of AuCl4− to the carbon led to the overgrowth of AuNPs.
Fig 5.

Morphology characterization of AuNCs@NC: (a) bright-field TEM; (b) high-angle annular dark-field scanning TEM (HAADF-STEM); (c) high-resolution TEM images and (d) scanning TEM-energy dispersive X-ray spectroscopy mappings of AuNCs@NC: Au, N, C, and O were given in (d1–d4), respectively; (e) high-resolution X-ray photoelectron spectrum (XPS) Au 4f spectrum of AuNCs@NC (top) and Au thin film (bottom); (f) the average diameter and distribution of AuNCs. Reprinted with permission from ref 104. Copyright 2016, American Chemical Society.
More recently, we extended this synthetic method to grow Au-Pd alloy NPs on nitrided carbon using AuNCs@NC as seeds (Fig. 6a, b).107 When employing ascorbic acid and 4-mercaptobenzoic acid (4-MBA) as mild reductant and surfactant, respectively, Au-Pd core-shell NPs (2–5 nm) with different compositions of Au and Pd formed firstly (Fig. 6c–f), then converted to Au-Pd alloy NPs after thermal activation at 250 °C for 1 h. The size of Au-Pd alloy NPs is uniform and highly controllable depending on the amount of the second precursors during the growth of the shell. Remarkably, the Au-Pd alloy NPs retained the uniform size without any aggregation or sintering after calcination. This confirms the strong affinity of Au-Pd alloy NPs on nitrided carbon support.
Fig 6.

Representative TEM images of Au-2@NC (a–b), Au@Au-5@NC (c–d) and Au@Pd-5@NC (e–f). Reprinted (adapted) with permission from ref 107. Copyright 2018, Elsevier.
3. Electrocatalytic properties of Au@C nanocatalysts
3.1. Catalytic oxidation of methanol
Methanol as a feedstock for direct methanol fuel cells (DMFCs) is promising for clean energy technology alternative to fossil fuels. DMFCs have higher energy density and less pollutant/byproducts when using new anode catalysts.108, 109 The complete methanol oxidation reaction (MOR) involves six electrons and one water molecule or adsorbed residue (adsorbed OH−), as follows:110
| (1) |
However, the six-electron reaction is slow and relatively complex, involving the formation of several accumulated intermediates such as CO, formaldehyde and formic acid other than CO2 as the final product. The default highly efficient anode catalysts are Pt and Pt-based nanomaterials. However, those Pt-containing anode catalysts are expensive and susceptible to poisons like adsorbed CO and acid intermediates that potentially cause the fast decay in activity. Those factors largely limit the lifetime and overall cost in DMFCs.
Being strongly resistant to the adsorption of CO-like intermediates, the reactivity of Au catalysts has attracted great attention in the applications of DMFCs, especially in alkaline media.111–113 Several studies have suggested that AuNPs are excellent anode catalysts for electrocatalytic MOR.114–117 Previous literatures suggest that the MOR pathway catalyzed by Au is independent in two potential regions.118, 119 At lower potentials before the formation of Au oxide layer, the chemical adsorption of OH− and possible preoxidation of Au surface occurs. Methanol is mainly oxidized to formates with four-electron exchange, as given:115, 120
| (2) |
At higher potentials, surface gold oxide monolayer restrains the chemiadsorption of OH−, thus inhibiting the formation of formates. Methanol is oxidized to carbonates with six electrons directly, as given below:104, 107
| (3) |
A typical example is the demonstration of methanol electrooxidation by AuNPs supported on activated carbon (6.7 nm) by Yan et al.85 The synthesis was described in Section 2.2. AuNPs stabilized by PVP showed high activity for MOR in 0.1 M KOH with 5 M CH3OH, reaching 48.6 mA mg−1Au at 0.355 V vs. Hg/HgO (Fig. 7a, b). In the low potential range (0.025–0.4 V), the catalytic activity of Au@C depended on the amount of adsorbed OH− anions ( ), thus the onset potential of MOR shifted negatively with the increasing concentration of metahnol and/or KOH (Fig. 7c, d). Yan and coworkers proposed that the species were beneficial to the acceleration of the reaction rate on the Au@C catalysts. The hydrogen-bonding interaction between adsorbed OH− and methanol was suggested as the rate-determining step, as depicted:
| (4) |
Fig 7.

Electrocatalytic oxidation of methanol in deoxygenated KOH solution: (a) Cyclic voltmmograms (CVs) on Au@C nanocatalyst in 0.1 M KOH with different concentration of methanol; (b) Tafel plots for electrooxidation of methanol on Au@C nanocatalyst as a function of methanol concentration; (c) CVs on Au@C nanocatalyst in deoxygenated 5 M CH3OH with different concentrations of KOH solution; (d) Tafel plots for electrooxidation of methanol on Au@C nanocatalyst as a function of KOH concentration. Reprinted with permission from ref 85. Copyright 2011, American Chemical Society.
Subsequently, the same group further investigated the effect of the size and loading amount of AuNPs on MORs. They suggested that the better activity was obtained on smaller AuNPs with more active sites on the corners and edges.121
Given the slower mass and electron transfer caused by surface ligands and the unique size-dependent catalytic activity of Au, our group demonstrated the efficient oxidation of methanol using ligand-free ultrasmall AuNCs (1.6±0.3 nm) supported on nitrided carbon.104 The mass activity (current normalized to unit mass) was 1.2 A/mgAu for AuNCs@NC at 1.11 V vs. RHE comparable with 1.9 A/mgPd at 0.8 V vs. RHE for Pd nanoclusters supported on nitrided carbon (Fig. 8a, c). The ultrasmall AuNCs expectedly exhibited much higher electroactivity compared to Au-6 nm@NC (obtained at pH 3). In the absence of surface ligands or surfactants, a low charge transfer resistance (1291 Ω) and a fast electron transfer rate (~0.02 cm/s) for ligand-free AuNCs@NC were revealed in Fig. 8b. Ligand-free AuNCs@NC were threefold more active than that for AuNCs@NC capped with 3-mercaptopropionic acid (MPA) and dodecanethiol (DT).
Fig 8.

Electrocatalytic oxidation of methanol using AuNCs@NC: (a) CVs of the NC, AuNCs@NC, AuNCs@NC-MPA, AuNCs@NC-DT and Au-6 nm@NC in 0.1 M NaOH with 1 M methanol; (b) electrochemical impedance spectroscopy (EIS) behaviors of different nanocatalysts to MOR with a frequency range from 0.1 to 100000 Hz. The inset was the best fitted circuit diagram; (c) mass activities of different nanocatalysts at the peak potentials. Reprinted (adapted) with permission from ref 104. Copyright 2016, American Chemical Society.
Although Au exhibits excellent resistance to the poisonous species, the activity of monometallic Au towards MOR is lower compared to Pt-based nanocatalysts. The oxidation potential of methanol on Au nanocatalysts is 0.3 V higher than that on Pt catalysts. Alloying Pt or Pd with Au is a promising strategy to attain the superior activity accompanying with anti-poisoning capacity. Zeng et al. designed the synthesis of core-shell Au-Pt NPs loaded on carbon which was used to catalyze MOR in acidic media.112 The electron exchange between Au core and thin Pt shell played an important role in promoting the formation of active oxygen species on Pt, thus facilitating the removal of the accumulated carbonaceous intermediates. AuPt and AuPd nanoalloys dispersed on Vulcan XC-72 carbon also gave the excellent MOR activity with long-term stability as demonstrated by Hu32 and Lu.122, 123
3.2. Catalytic oxidation of ethanol and other alcohols
Although DMFCs have been developing for many decades, there are some disadvantages using methanol for fuel cells, e.g. toxicity of methanol, non-renewable sources and high methanol cross-over through membranes. As attractive alternatives, ethanol and other less toxic alcohols easily produced in large quantities from biomass have been considered lately.110, 124, 125 The oxidation of ethanol is much more complex. Twelve electrons are involved in the complete oxidation of ethanol, leading to the sluggish reaction kinetics and numerous carbonaceous intermediates, as depicted:
| (5) |
The generally studied reaction route is that ethanol is oxidized to acetate/acetic acid through four-electron oxidation in alkaline media, as given:126, 127
| (6) |
It usually involves four consecutive steps on metal@C catalysts where the rate determining step is depicted in eq (8) similar to MOR.128, 129
| (7) |
| (8) |
| (9) |
| (10) |
The nanostructured AuNPs were shown to be catalytically active in ethanol oxidation.130, 131 For instance, Yang et al. developed a rapid, template-free methodology to prepare hierarchical nanostructured Au nanoflowers (AuNFs) on carbon fiber cloth (CFC) via potentiostatic electrodeposition.132 The open channels on the CFC support enabled the even distribution of AuNFs and efficient mass transport. The results of ethanol electrooxidation reaction (EOR) showed that AuNFs exhibited 4 times higher catalytic activity and much less poisoning in contrast to AuNPs and bare gold electrodes. These features can be ascribed to the unique nanostructures such as the sharp edges or tips, which produced larger surface area and more active sites. The size and shape driven activity of Au nanocatalysts in various electrochemical reactions including oxidation of organic molecules, CO oxidation and oxygen reductions were also reported by Bikash,133 Qin,134 and Li.135
Another example of alcohol electrooxidation on Au@C nanocatalysts was demonstrated by Yan et al. They used PVP-capped AuNPs supported on activated carbon to electrooxidize methanol, ethanol and ethylene glycol.136 The mass-specific current densities on AuNPs peaked at 442 mA mg−1Au at 0.35 V vs. Hg/HgO in 0.1 M KOH solution with 2 M ethanol. In addition, unlike methanol and ethanol where the alcohol concentration influenced the mass activity, the mass activity of Au@C nanocatalysts was almost independent on the ethylene glycol concentration (Fig. 9a). However, the steady-state current densities of alcohols after 4000 s maintained ~40% of the initial (Fig. 9b). The incorporation of heteroatoms into the carbon frameworks or doping metal with non-metal elements has emerged as an effective strategy to enhance the interactions between AuNPs and supports. Recently, Li et al. synthesized a series of carbon supported Au-phosphorus (AuP@C) catalysts by hot-reflux method using white phosphorus (P4) as a reductant and a dopant.137 The mass activity of AuP@C (3.7 nm) was 7.83-fold higher than that of the undoped Au@C (5.6 nm) prepared by NaBH4 reduction (Fig. 9c). This is attributed to the highly uniform distribution of ultrafine AuP NPs and the altered electronic structure of Au by interactions with P. The initial oxidation potential of the EOR on the AuP@C nanocatalyst shifted negatively about 50 mV compared to that of Au@C catalyst, indicating better electrocatalytic performance at a lower potential. The lower d-band center caused by the P doping further weakened the adsorption of the intermediates on Au surface, thus enhancing the poisoning resistance and attaining a relatively high steady-state current density in comparison with Au@C catalyst as shown in Fig. 9d. It further verified the AuP@C catalyst displayed a better stability for ethanol oxidation.
Fig 9.

(a) CVs on the Au@C catalyst in deoxygenated 0.1 M KOH solution with different concentrations of ethylene glycol; (b) chronoamperograms on the Au@C catalyst in deoxygenated 0.1 M KOH solution at the potential of 0.25 V vs. Hg|HgO with different alcohols. Adapted with permission from ref 136. Copyright 2013, Elsevier; (c) mass and specific normalized CVs and (d) chronoamperometry curves of the AuP@C and Au@C catalysts in N2-saturated 0.5 M KOH and 1.0 M C2H5OH mixing solution at a potential of 0.20 V vs. SCE. Adapted with permission from ref 137. Copyright 2017, Elsevier.
Considering the synergetic effects between the two components of bimetallic NPs, there are a number of reports on supported Au-bimetallic NPs for EOR catalysis.138–140 Our group confirmed that the synergies between Au and Pd could effectively enhance long-time stability and accelerate charge transfer in comparison with monometallic NPs.107 In our recent work, the specific activity of AuPd NPs supported on nitrided carbon (AuPd NPs@NC) was approximately 5 times more than commercial Pd/C (5 wt% loading) when the Au/Pd ratio was 45:55. Moreover, the chronoamperometric response showed that the current decay for AuPd NPs@NC (45:55) was much slower and maintained the highest steady-state current density, 2.9 times higher than that of commercial Pd/C, which indicated that alloying Au with Pd not only enhanced the electrocatalytic activity, but also improved the stability of catalysts. Additionally, alloying with a small amount of Au (10%) significantly enhanced the oxidation current density and resulted in the highest peak current density ratio (If/Ib) of the forward (If) to the backward scan (Ib) up to 5.8 compared to all other catalysts with different compositions of Au and Pd, indicating the improved poisoning tolerance by the introduction of Au into Pd.
3.3. Oxidation of DNA and amines
The guanine (G) and adenine (A) bases in the DNA molecular skeleton are easy to be electrooxidized.141–143 A single electron oxidation potential of G and A base is higher than +1.2 V vs. NHE. And, the oxidization of cytosine (C) and thymine (T) bases requires a higher potential.144, 145 The detection of DNA or DNA damage by direct oxidation current of nucleic acid bases can reach the sensitivity of nanomole.146 However, the electrochemical oxidation of electrodes and/or decomposition of solvent molecules (e.g., water) is often caused by the positive base oxidation potential during testing, thus resulting in a very high background current and interfering the DNA oxidation signals. The common method is to use indirect electrochemical signal conversion through charge mediators. For instance, when coupling Ru(bpy)32+ with DNA molecules on the electrode surface, the redox reaction of Ru2+ can mediate the oxidation of guanine. This method has little effect on the electrolytic background current of water and the non-specific adsorption of target DNA molecule. The electrochemical detection of label-free target DNA molecule was successfully achieved.147 On the other hand, tripropylamine (TprA) is an important trialkylamine as an efficient co-reactant for electrochemiluminescent (ECL) in biosensor technology.148 Since the electrochemical oxidation of TprA occurs at about +0.85 V (vs. Ag/AgCl), the oxidation of TprA is a useful probe to examine the oxidative damage of biomolecules during detection sequence or DNA analysis.149
As reported by our group recently, the AuNCs@NC catalysts were incorporated into films of architecture {poly(diallyldimethylammonium) (PDDA)/AuNCs@NC}n by using layer-by-layer (LBL) assembly with oppositely charged PDDA on pyrolytic graphite (PG) electrodes for the electrocatalytic oxidation of double-stranded (ds)-DNA and TprA.105 Ligand-free AuNCs@NC in these films exhibited excellent electrocatalytic oxidation activity for ds-DNA and TprA. The oxidation peak potential of DNA showed a negative shift by 140 mV and the peak current was enhanced by three times on the {PDDA/AuNCs@NC}2/PDDA film electrode compared to blank electrode (Fig. 10A). Similarly, the oxidation peak current density of TprA on the {PDDA/AuNCs@NC}3 film electrode increased dramatically to 4.7 A mg−1Au with a negative shift (~200 mV) of peak potential compared to the control films (without AuNCs) (Fig. 10B). Meanwhile, 86% of catalytic current was still retained after 100 scanning cycles, and the resulting {PDDA/AuNCs@NC}3 film displayed a superior stability. A strong electrochemical response of DNA adsorbed on AuNPs is usually difficult to be present and measured due to the overlap with Au oxidation peak.150, 151 However, in our case, the highly dispersed ligand-free ultrasmall AuNCs exhibited a lower oxidation potential and the unexpected capacity for amplifying the detection signal. The supported AuNCs may provide a promising approach for the practical applications in biotechnology.
Fig 10.

A: CVs of the {PDDA/AuNCs@NC}2/PDDA/DNA (a), {PDDA/NC}2/PDDA/DNA (b) and {PDDA/AuNCs@NC}2 (c) electrodes in 0.01 M pH 7.4 phosphate buffers; B: CVs of the bare PG (a), {PDDA/NC}3 (b), {PDDA/AuNCs@NC}3 (c) electrodes in 0.01 M TprA solution, and {PDDA/AuNCs@NC}3 electrode (d) in the absence of 0.01 M TprA. Adapted with permission from ref 105. Copyright 2016, John Wiley and Sons.
The research works mentioned in the third section have been summarized in Table 2. Various Au@C nanocatalysts exhibited excellent electrocatalytic performance for the methanol, ethanol, TprA and DNA moleules. Among those nanocatalysts, AuNCs@NC exhibited superior mass activity (1190 mA/mgAu) and AuP@C owned the highest specific activity (3.53 mA/cm2) toward MOR and EOR, respectively. 104, 137 Ligand-free AuNCs@NC assembled in {PDDA/AuNCs@N-C}n films exhibited unexpected activity for electrooxidation of DNA and TprA with reduced oxidation potentials. This makes it possible to facilitate their applications in biosensors.105
Table 2.
Electrocatalytic performances of Au@C nanocatalysts for methanol, ethanol, DNA and TprA oxidation reaction.
| Catalyst composition | Preparation method | Average diameter (nm) | Catalyticreaction | Mass activity(mA/mg) | Specific activity(mA/cm2) | If/Ib | Ref |
|---|---|---|---|---|---|---|---|
| PtAu@graphene | electrodeposition | 150–200 | MOR | 394 | - | 1.25 | 32 |
| Au@C | rapid reduction | 6.7 | MOR | 48.6 | - | - | 85 |
| AuNCs@NC | soft nitriding | 0.7–2 | MOR | 1190 | - | - | 104 |
| AuPt NPs@C | seed-mediated growth | 4–10 | MOR | - | 1.65 | - | 112 |
| Au@Ca | rapid reduction | 4.73 | MOR | 47.06 | - | - | 121 |
| Au2Pt1@C | capping agent-free | 3.4 | MOR | - | - | 1.84 | 122 |
| Pd2Au@GCb | hydrothermal | 11.42 | MOR | 491.84 | 1.09 | - | 123 |
| AuPd NPs@NC | seed-mediated growth | 2–5 | EOR | 430 | 1.11 | 5.8 | 107 |
| Au NFsc@CFCd | electrodeposition | - | EOR | - | - | 0.29 | 132 |
| AuNPs@C | deposition reduction | 4.65 | MOR, EOR | 69.5(MOR)442(EOR) | - | - | 136 |
| Au-P@C | hot-reflux | 3.7 | EOR | 642.33 | 3.53 | - | 137 |
| AuNCs@NC | soft nitriding | 1.6 | DNA and TprA | - | - | - | 105 |
Au@C catalyst with 20 wt% Au;
GC: glassy carbon;
NFs: nanoflowers;
CFC: carbon fiber cloth.
4. Outlook
We summarized the most recent studies on Au nanocatalysts supported on conductive carbon as anode catalysts for electrooxidation of alcohols, DNA and TprA. Au as a chemically stable catalyst shows unique size-dependent catalytic properties. Although numerous effort has been devoted to designing Au@C nanocomposites, there are still unmet challenges in the synthesis and applications of Au@C as catalysts. One of obvious problems in the use of Au@C is the cost of catalysts, since the price of Au is as expensive as that of Pt. The apparent solution is to decrease the size of AuNPs that potentially exposes more surface atoms for catalysis. However, due to the large surface energy, how to stabilize those AuNPs on carbon supports become critical. The new methods for surface modification and functionalization of carbon by integrating heteroatoms as binding sites of AuNPs should be further investigated for low cost and scale up production. Other carbon supports such as mesoporous carbon, and MOF-derived nanomaterials can potentially be used for Au@C to offer high surface area and adjustable pore size, as well as restrain the overgrowth of Au nanostructures during synthesis. Using carbon with high N content, it is possible to prepare single-atom Au catalyst where atomic Au can be well dispersed and stabilized through the coordination of multiple N atoms.152 It will be of interest to study the electrocatalytic performance of single-atom Au catalyst since the atom efficiency of Au can reach 100%.
Understanding the electronic interaction between Au and doped carbon is an attractive alternative to develop Au@C as highly selective catalysts for heterogeneous reactions. Through changing surface electron density of AuNPs, Au has been used to catalyze selective hydrogenation of C=C bonds153 and oxidation of C=C bonds.106 Heteroatoms, like N and P, can induce the electronic perturbation at the Au-carbon interface where the catalytic selectivity usually relies on. The electron-rich Au nanocatalysts also tend to favor the binding of electrophiles, like CO2, to promote the reaction selectivity.
Other than the electronic interactions of Au and doped carbon, surface ligands can potentially modulate the reaction pathway as well.154 Unlike surface ligands with long alkyl chains which block the electron transfer, short ligands can potentially play a critical role in electrocatalysis. Chang and Yang demonstrated the N-heterocyclic carbene-functionalized AuNPs selectively reduced CO2 to CO with a Faradaic efficiency of 83%, since carbene showed a strong σ-donation with AuNPs.155 More attention on the use of carbon support and surface ligands as a means to control the surface electronic properties of Au nanocatalysts should be drawn in future studies.
Supplementary Material
Acknowledgments
J.F.R. is grateful for financial support of the National Institute of Environmental Health Sciences (NIH, Grant No. ES03154). J.H. thanks support from the National Science Foundation (CBET 1705566). Z.C. thanks the support from the China Scholarship Council for her visiting at the University of Connecticut. Y.Y. thanks the support from the China Scholarship Council and Nanjing University of Science and Technology for her visiting at the University of Connecticut.
Footnotes
Notes
The authors declare no competing financial interest.
References
- 1.Cuenya BR, Baeck SH, Jaramillo TF, McFarland EW. J Am Chem Soc. 2003;125:12928–12934. doi: 10.1021/ja036468u. [DOI] [PubMed] [Google Scholar]
- 2.Daniel MC, Astruc D. Chem Rev. 2004;104:293–346. doi: 10.1021/cr030698+. [DOI] [PubMed] [Google Scholar]
- 3.Panigrahi S, Basu S, Praharaj S, Pande S, Jana S, Pal A, Ghosh SK, Pal T. J Phys Chem C. 2007;111:4596–4605. doi: 10.1021/jp065740u. [DOI] [PubMed] [Google Scholar]
- 4.Sardar R, Funston AM, Mulvaney P, Murray RW. Langmuir. 2009;25:13840–13851. doi: 10.1021/la9019475. [DOI] [PubMed] [Google Scholar]
- 5.Haruta M. J New Mater Electrochem Syst. 2004;7:163–172. [Google Scholar]
- 6.Haruta M, Kobayashi T, Sano H, Yamada N. Chem Lett. 1987;16:405–408. [Google Scholar]
- 7.Haruta M, Yamada N, Kobayashi T, Iijima S. J Catal. 1989;115:301–309. [Google Scholar]
- 8.Sakurai H, Tsubota S, Haruta M. Appl Catal A-Gen. 1993;102:125–136. [Google Scholar]
- 9.Sakurai H, Haruta M. Catal Today. 1996;29:361–365. [Google Scholar]
- 10.Ueda A, Oshima T, Haruta M. Appl Catal B-Environ. 1997;12:81–93. [Google Scholar]
- 11.Uphade B, Tsubota S, Hayashi T, Haruta M. Chem Lett. 1998;27:1277–1278. [Google Scholar]
- 12.Haruta M. Cattech. 2002;6:102–115. [Google Scholar]
- 13.Ma C, Xue W, Li J, Xing W, Hao Z. Green Chem. 2013;15:1035–1041. [Google Scholar]
- 14.Abroshan H, Li G, Lin J, Kim HJ, Jin R. J Catal. 2016;337:72–79. [Google Scholar]
- 15.Zhao JB, Jin RC. Nano Today. 2018;18:86–102. [Google Scholar]
- 16.Haruta M. Catal Today. 1997;36:153–166. [Google Scholar]
- 17.Liu XY, Wang A, Zhang T, Mou CY. Nano Today. 2013;8:403–416. [Google Scholar]
- 18.Comotti M, Li WC, Spliethoff B, Schüth F. J Am Chem Soc. 2006;128:917–924. doi: 10.1021/ja0561441. [DOI] [PubMed] [Google Scholar]
- 19.Nie X, Qian H, Ge Q, Xu H, Jin R. ACS Nano. 2012;6:6014–6022. doi: 10.1021/nn301019f. [DOI] [PubMed] [Google Scholar]
- 20.Yan W, Mahurin SM, Pan Z, Overbury SH, Dai S. J Am Chem Soc. 2005;127:10480–10481. doi: 10.1021/ja053191k. [DOI] [PubMed] [Google Scholar]
- 21.Liu X, Liu MH, Luo YC, Mou CY, Lin SD, Cheng H, Chen JM, Lee JF, Lin TS. J Am Chem Soc. 2012;134:10251–10258. doi: 10.1021/ja3033235. [DOI] [PubMed] [Google Scholar]
- 22.Yue Q, Zhang Y, Wang C, Wang X, Sun Z, Hou XF, Zhao D, Deng Y. J Mater Chem A. 2015;3:4586–4594. [Google Scholar]
- 23.Zeng T, Zhang XL, Niu HY, Ma YY, Li WH, Cai YQ. Appl Catal B-Environ. 2013;134–135:26–33. [Google Scholar]
- 24.Miao S, Zhang C, Liu Z, Han B, Xie Y, Ding S, Yang Z. J Phys Chem C. 2008;112:774–780. [Google Scholar]
- 25.Han J, Liu Y, Li L, Guo R. Langmuir. 2009;25:11054–11060. doi: 10.1021/la901373t. [DOI] [PubMed] [Google Scholar]
- 26.Han J, Li L, Guo R. Macromolecules. 2010;43:10636–10644. [Google Scholar]
- 27.Villa A, Schiavoni M, Prati L. Catal Sci Technol. 2012;2:673–682. [Google Scholar]
- 28.Rocha M, Fernandes C, Pereira C, Rebelo SLH, Pereira MFR, Freire C. RSC Adv. 2015;5:5131–5141. [Google Scholar]
- 29.Ma X, Li X, Lun N, Wen S. Mater Chem Phys. 2006;97:351–356. [Google Scholar]
- 30.Simões M, Baranton S, Coutanceau C. J Phys Chem C. 2009;113:13369–13376. [Google Scholar]
- 31.Dong Z, Le X, Liu Y, Dong C, Ma J. J Mater Chem A. 2014;2:18775–18785. [Google Scholar]
- 32.Hu Y, Zhang H, Wu P, Zhang H, Zhou B, Cai C. Phys Chem Chem Phys. 2011;13:4083–4094. doi: 10.1039/c0cp01998d. [DOI] [PubMed] [Google Scholar]
- 33.Yang M, Zhou M, Zhang A, Zhang C. J Phys Chem C. 2012;116:22336–22340. [Google Scholar]
- 34.Demirel S, Kern P, Lucas M, Claus P. Catal Today. 2007;122:292–300. [Google Scholar]
- 35.Benkó T, Beck A, Geszti O, Katona R, Tungler A, Frey K, Guczi L, Schay Z. Appl Catal A-Gen. 2010;388:31–36. [Google Scholar]
- 36.Fang B, Kim M, Yu JS. Appl Catal B-Environ. 2008;84:100–105. [Google Scholar]
- 37.Bang JH. Electrochim Acta. 2011;56:8674–8679. [Google Scholar]
- 38.Planeix J, Coustel N, Coq B, Brotons V, Kumbhar P, Dutartre R, Geneste P, Bernier P, Ajayan P. J Am Chem Soc. 1994;116:7935–7936. [Google Scholar]
- 39.Bezemer GL, Bitter JH, Kuipers HP, Oosterbeek H, Holewijn JE, Xu X, Kapteijn F, van Dillen AJ, de Jong KP. J Am Chem Soc. 2006;128:3956–3964. doi: 10.1021/ja058282w. [DOI] [PubMed] [Google Scholar]
- 40.Xue T, Jiang S, Qu Y, Su Q, Cheng R, Dubin S, Chiu CY, Kaner R, Huang Y, Duan X. Angew Chem. 2012;124:3888–3891. doi: 10.1002/anie.201108400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu B, Yao HQ, Daniels RA, Song WQ, Zheng HQ, Jin L, Suib SL, He J. Nanoscale. 2016;8:5441–5445. doi: 10.1039/c6nr00604c. [DOI] [PubMed] [Google Scholar]
- 42.Liu B, Jin L, Zheng HQ, Yao HQ, Wu Y, Lopes A, He J. ACS Appl Mater Interfaces. 2017;9:1746–1758. doi: 10.1021/acsami.6b11958. [DOI] [PubMed] [Google Scholar]
- 43.Chaikittisilp W, Ariga K, Yamauchi Y. J Mater Chem A. 2013;1:14–19. [Google Scholar]
- 44.Prati L, Rossi M. J Catal. 1998;176:552–560. [Google Scholar]
- 45.Figueiredo JL. J Mater Chem A. 2013;1:9351–9364. [Google Scholar]
- 46.Ma Z, Dai S. Nano Res. 2011;4:3–32. [Google Scholar]
- 47.Jin L, Liu B, Wang P, Yao HQ, Achola L, Kerns P, Lopes A, Yang Y, Ho J, Moewes A, Pei Y, He J. Nanoscale. 2018 doi: 10.1039/c8nr04322a. [DOI] [PubMed] [Google Scholar]
- 48.Xu Y, Chen L, Wang X, Yao W, Zhang Q. Nanoscale. 2015;7:10559–10583. doi: 10.1039/c5nr02216a. [DOI] [PubMed] [Google Scholar]
- 49.Wang A, Liu XY, Mou CY, Zhang T. J Catal. 2013;308:258–271. [Google Scholar]
- 50.Zhao J, Zhang T, Di X, Xu J, Xu J, Feng F, Ni J, Li X. RSC Adv. 2015;5:6925–6931. [Google Scholar]
- 51.Gallo IB, Carbonio EA, Villullas HM. ACS Catal. 2018;8:1818–1827. [Google Scholar]
- 52.Kesavan L, Tiruvalam R, Ab Rahim MH, bin Saiman MI, Enache DI, Jenkins RL, Dimitratos N, Lopez-Sanchez JA, Taylor SH, Knight DW. Science. 2011;331:195–199. doi: 10.1126/science.1198458. [DOI] [PubMed] [Google Scholar]
- 53.Raghavendra P, Vishwakshan Reddy G, Sivasubramanian R, Sri Chandana P, Subramanyam Sarma L. Int J Hydrogen Energ. 2018;43:4125–4135. [Google Scholar]
- 54.Schmidt T, Jusys Z, Gasteiger H, Behm R, Endruschat U, Boennemann H. J Electroanal Chem. 2001;501:132–140. [Google Scholar]
- 55.Xie S, Tsunoyama H, Kurashige W, Negishi Y, Tsukuda T. ACS Catal. 2012;2:1519–1523. [Google Scholar]
- 56.Lu L, Nie Y, Wang Y, Wu G, Li L, Li J, Qi X, Wei Z. J Mater Chem A. 2018;6:104–109. [Google Scholar]
- 57.Selvarani G, Selvaganesh SV, Krishnamurthy S, Kiruthika G, Sridhar P, Pitchumani S, Shukla A. J Phys Chem C. 2009;113:7461–7468. [Google Scholar]
- 58.He Q, Chen W, Mukerjee S, Chen S, Laufek F. J Power Sources. 2009;187:298–304. [Google Scholar]
- 59.Mathiyarasu J, Phani KLN. J Electrochem Soc. 2007;154:B1100–B1105. [Google Scholar]
- 60.Zhang H, Dai B, Li W, Wang X, Zhang J, Zhu M, Gu J. J Catal. 2014;316:141–148. [Google Scholar]
- 61.Habrioux A, Vogel W, Guinel M, Guetaz L, Servat K, Kokoh B, Alonso-Vante N. Phys Chem Chem Phys. 2009;11:3573–3579. doi: 10.1039/b820668f. [DOI] [PubMed] [Google Scholar]
- 62.Zhu LD, Zhao TS, Xu JB, Liang ZX. J Power Sources. 2009;187:80–84. [Google Scholar]
- 63.Shang C, Hong W, Wang J, Wang E. J Power Sources. 2015;285:12–15. [Google Scholar]
- 64.Cao R, Xia T, Zhu R, Liu Z, Guo J, Chang G, Zhang Z, Liu X, He Y. Appl Surf Sci. 2018;433:840–846. [Google Scholar]
- 65.Lankiang SD, Baranton S, Coutanceau C. Electrochim Acta. 2017;242:287–299. [Google Scholar]
- 66.Lin Y, Taylor S, Li H, Fernando KAS, Qu L, Wang W, Gu L, Zhou B, Sun YP. J Mater Chem. 2004;14:527–541. [Google Scholar]
- 67.Hong W, Bai H, Xu Y, Yao Z, Gu Z, Shi G. J Phys Chem C. 2010;114:1822–1826. [Google Scholar]
- 68.Huang J, Zhang L, Chen B, Ji N, Chen F, Zhang Y, Zhang Z. Nanoscale. 2010;2:2733–2738. doi: 10.1039/c0nr00473a. [DOI] [PubMed] [Google Scholar]
- 69.Zhuo Q, Ma Y, Gao J, Zhang P, Xia Y, Tian Y, Sun X, Zhong J, Sun X. Inorg Chem. 2013;52:3141–3147. doi: 10.1021/ic302608g. [DOI] [PubMed] [Google Scholar]
- 70.Li F, Yang H, Shan C, Zhang Q, Han D, Ivaska A, Niu L. J Mater Chem. 2009;19:4022–4025. [Google Scholar]
- 71.Kim B, Sigmund WM. Langmuir. 2004;20:8239–8242. doi: 10.1021/la049424n. [DOI] [PubMed] [Google Scholar]
- 72.Jiang K, Eitan A, Schadler LS, Ajayan PM, Siegel RW, Grobert N, Mayne M, Reyes-Reyes M, Terrones H, Terrones M. Nano Lett. 2003;3:275–277. [Google Scholar]
- 73.Fang Y, Guo S, Zhu C, Zhai Y, Wang E. Langmuir. 2010;26:11277–11282. doi: 10.1021/la100575g. [DOI] [PubMed] [Google Scholar]
- 74.Balcioglu M, Rana M, Yigit MV. J Mater Chem B. 2013;1:6187–6193. doi: 10.1039/c3tb20992j. [DOI] [PubMed] [Google Scholar]
- 75.Han L, Wu W, Kirk FL, Luo J, Maye MM, Kariuki NN, Lin Y, Wang C, Zhong CJ. Langmuir. 2004;20:6019–6025. doi: 10.1021/la0497907. [DOI] [PubMed] [Google Scholar]
- 76.Ou YY, Huang MH. J Phys Chem B. 2006;110:2031–2036. doi: 10.1021/jp055920o. [DOI] [PubMed] [Google Scholar]
- 77.Shi Y, Yang R, Yuet PK. Carbon. 2009;47:1146–1151. [Google Scholar]
- 78.Han C, Wu L, Ge L, Li Y, Zhao Z. Carbon. 2015;92:31–40. [Google Scholar]
- 79.Zhang S, Shao Y, Liao HG, Liu J, Aksay IA, Yin G, Lin Y. Chem Mater. 2011;23:1079–1081. [Google Scholar]
- 80.Gupta VK, Yola ML, Atar N, Üstündağ Z, Solak AO. J Mol Liq. 2014;191:172–176. [Google Scholar]
- 81.Gupta VK, Atar N, Yola ML, Ustundag Z, Uzun L. Water Res. 2014;48:210–217. doi: 10.1016/j.watres.2013.09.027. [DOI] [PubMed] [Google Scholar]
- 82.Goncalves G, Marques PAAP, Granadeiro CM, Nogueira HIS, Singh MK, Grácio J. Chem Mater. 2009;21:4796–4802. [Google Scholar]
- 83.Pocklanova R, Rathi AK, Gawande MB, Datta KKR, Ranc V, Cepe K, Petr M, Varma RS, Kvitek L, Zboril R. J Mater Chem A. 2016;424:121–127. [Google Scholar]
- 84.Zhang Z, Chen H, Xing C, Guo M, Xu F, Wang X, Gruber HJ, Zhang B, Tang J. Nano Res. 2011;4:599–611. [Google Scholar]
- 85.Yan S, Zhang S, Lin Y, Liu G. J Phys Chem C. 2011;115:6986–6993. [Google Scholar]
- 86.Peng S, Lee Y, Wang C, Yin H, Dai S, Sun S. Nano Res. 2008;1:229–234. [Google Scholar]
- 87.Tsunoyama H, Ichikuni N, Sakurai H, Tsukuda T. J Am Chem Soc. 2009;131:7086–7093. doi: 10.1021/ja810045y. [DOI] [PubMed] [Google Scholar]
- 88.Huang X, Zhou X, Wu S, Wei Y, Qi X, Zhang J, Boey F, Zhang H. Small. 2010;6:513–516. doi: 10.1002/smll.200902001. [DOI] [PubMed] [Google Scholar]
- 89.Lopez-Sanchez JA, Dimitratos N, Hammond C, Brett GL, Kesavan L, White S, Miedziak P, Tiruvalam R, Jenkins RL, Carley AF. Nat Chem. 2011;3:551–556. doi: 10.1038/nchem.1066. [DOI] [PubMed] [Google Scholar]
- 90.Li D, Wang C, Tripkovic D, Sun S, Markovic NM, Stamenkovic VR. ACS Catal. 2012;2:1358–1362. [Google Scholar]
- 91.Zhao L, Gu W, Zhang C, Shi X, Xian Y. J Colloid Interf Sci. 2016;465:279–285. doi: 10.1016/j.jcis.2015.11.073. [DOI] [PubMed] [Google Scholar]
- 92.Shao L, Huang X, Teschner D, Zhang W. ACS Catal. 2014;4:2369–2373. [Google Scholar]
- 93.Yin H, Tang H, Wang D, Gao Y, Tang Z. ACS Nano. 2012;6:8288–8297. doi: 10.1021/nn302984x. [DOI] [PubMed] [Google Scholar]
- 94.Choi HC, Shim M, Bangsaruntip S, Dai H. J Am Chem Soc. 2002;124:9058–9059. doi: 10.1021/ja026824t. [DOI] [PubMed] [Google Scholar]
- 95.Gong K, Du F, Xia Z, Durstock M, Dai L. Science. 2009;323:760–764. doi: 10.1126/science.1168049. [DOI] [PubMed] [Google Scholar]
- 96.Qu L, Liu Y, Baek JB, Dai L. ACS Nano. 2010;4:1321–1326. doi: 10.1021/nn901850u. [DOI] [PubMed] [Google Scholar]
- 97.Liu B, Jin L, Zhong W, Lopes A, Suib SL, He J. Chem-Eur J. 2018;24:2565–2569. doi: 10.1002/chem.201705504. [DOI] [PubMed] [Google Scholar]
- 98.Hasegawa G, Deguchi T, Kanamori K, Kobayashi Y, Kageyama H, Abe T, Nakanishi K. Chem Mater. 2015;27:4703–4712. [Google Scholar]
- 99.Ji X, Lee KT, Holden R, Zhang L, Zhang J, Botton GA, Couillard M, Nazar LF. Nat Chem. 2010;2:286. doi: 10.1038/nchem.553. [DOI] [PubMed] [Google Scholar]
- 100.Li Y, Zhao Y, Cheng H, Hu Y, Shi G, Dai L, Qu L. J Am Chem Soc. 2011;134:15–18. doi: 10.1021/ja206030c. [DOI] [PubMed] [Google Scholar]
- 101.Zhang P, Gong Y, Li H, Chen Z, Wang Y. Nat Commun. 2013;4:1593. doi: 10.1038/ncomms2586. [DOI] [PubMed] [Google Scholar]
- 102.Koo HY, Lee H-J, Noh YY, Lee E-S, Kim Y-H, San Choi W. J Mater Chem. 2012;22:7130–7135. [Google Scholar]
- 103.Xie X, Long J, Xu J, Chen L, Wang Y, Zhang Z, Wang X. RSC Adv. 2012;2:12438–12446. [Google Scholar]
- 104.Liu B, Yao H, Song W, Jin L, Mosa IM, Rusling JF, Suib SL, He J. J Am Chem Soc. 2016;138:4718–4721. doi: 10.1021/jacs.6b01702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yao H, Liu B, Mosa IM, Bist I, He J, Rusling JF. ChemElectroChem. 2016;3:2100–2109. doi: 10.1002/celc.201600283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Liu B, Wang P, Lopes A, Jin L, Zhong W, Pei Y, Suib SL, He J. ACS Catal. 2017;7:3483–3488. [Google Scholar]
- 107.Yang Y, Jin L, Liu B, Kerns P, He J. Electrochim Acta. 2018;269:441–451. [Google Scholar]
- 108.Ren X, Zelenay P, Thomas S, Davey J, Gottesfeld S. J Power Sources. 2000;86:111–116. [Google Scholar]
- 109.McGrath KM, Prakash GS, Olah GA. J Ind Eng Chem. 2004;10:1063–1080. [Google Scholar]
- 110.Lamy C, Lima A, LeRhun V, Delime F, Coutanceau C, Léger JM. J Power Sources. 2002;105:283–296. [Google Scholar]
- 111.Mikkelsen K, Cassidy B, Hofstetter N, Bergquist L, Taylor A, Rider DA. Chem Mater. 2014;26:6928–6940. [Google Scholar]
- 112.Zeng J, Yang J, Lee JY, Zhou W. J Phys Chem B. 2006;110:24606–24611. doi: 10.1021/jp0640979. [DOI] [PubMed] [Google Scholar]
- 113.Luo J, Njoki PN, Lin Y, Mott D, Wang L, Zhong CJ. Langmuir. 2006;22:2892–2898. doi: 10.1021/la0529557. [DOI] [PubMed] [Google Scholar]
- 114.Graf M, Haensch M, Carstens J, Wittstock G, Weissmüller J. Nanoscale. 2017;9:17839–17848. doi: 10.1039/c7nr05124g. [DOI] [PubMed] [Google Scholar]
- 115.Borkowska Z, Tymosiak-Zielinska A, Shul G. Electrochim Acta. 2004;49:1209–1220. [Google Scholar]
- 116.Luo J, Jones VW, Maye MM, Han L, Kariuki NN, Zhong CJ. J Am Chem Soc. 2002;124:13988–13989. doi: 10.1021/ja028285y. [DOI] [PubMed] [Google Scholar]
- 117.Zhong CJ, Maye MM. Adv Mater. 2001;13:1507–1511. [Google Scholar]
- 118.Bełtowska-Brzezinska M, Łuczak T, Holze R. J Appl Electrochem. 1997;27:999–1011. [Google Scholar]
- 119.Assiongbon K, Roy D. Surf Sci. 2005;594:99–119. [Google Scholar]
- 120.Zhang J, Liu P, Ma H, Ding Y. J Phys Chem C. 2007;111:10382–10388. [Google Scholar]
- 121.Yan S, Zhang S. Int J Hydrogen Energ. 2011;36:13392–13397. [Google Scholar]
- 122.Lu L, Nie Y, Wang Y, Wu G, Li L, Li J, Qi X, Wei Z. J Mater Chem A. 2018;6:104–109. [Google Scholar]
- 123.Luo LM, Zhang RH, Chen D, Hu QY, Zhang X, Yang CY, Zhou XW. Electrochim Acta. 2018;259:284–292. [Google Scholar]
- 124.Corti HR, Gonzalez ER. Direct alcohol fuel cells: materials, performance, durability and applications. Springer Science & Business Media; 2013. [Google Scholar]
- 125.Kamarudin M, Kamarudin SK, Masdar M, Daud WRW. Int J Hydrog Energy. 2013;38:9438–9453. [Google Scholar]
- 126.Shen S, Zhao T, Wu Q. Int J Hydrog Energy. 2012;37:575–582. [Google Scholar]
- 127.Tremiliosi-Filho G, Gonzalez E, Motheo A, Belgsir E, Leger JM, Lamy C. J Electroanal Chem. 1998;444:31–39. [Google Scholar]
- 128.Bianchini C, Shen PK. Chem Rev. 2009;109:4183–4206. doi: 10.1021/cr9000995. [DOI] [PubMed] [Google Scholar]
- 129.Liang Z, Zhao T, Xu J, Zhu L. Electrochim Acta. 2009;54:2203–2208. [Google Scholar]
- 130.Feng JJ, Li AQ, Lei Z, Wang AJ. ACS Appl Mater Inter. 2012;4:2570–2576. doi: 10.1021/am3002346. [DOI] [PubMed] [Google Scholar]
- 131.Stratakis M, Garcia H. Chem Rev. 2012;112:4469–4506. doi: 10.1021/cr3000785. [DOI] [PubMed] [Google Scholar]
- 132.Yang F, Cheng K, Ye K, Xiao X, Guo F, Yin J, Wang G, Cao D. Electrochim Acta. 2013;114:478–483. [Google Scholar]
- 133.Jena BK, Raj CR. Langmuir. 2007;23:4064–4070. doi: 10.1021/la063243z. [DOI] [PubMed] [Google Scholar]
- 134.Qin Y, Song Y, Sun N, Zhao N, Li M, Qi L. Chem Mater. 2008;20:3965–3972. [Google Scholar]
- 135.Li Y, Shi G. J Phys Chem B. 2005;109:23787–23793. doi: 10.1021/jp055256b. [DOI] [PubMed] [Google Scholar]
- 136.Yan S, Gao L, Zhang S, Zhang W, Li Y, Gao L. Electrochim Acta. 2013;94:159–164. [Google Scholar]
- 137.Li T, Fu G, Su J, Wang Y, Lv Y, Zou X, Zhu X, Xu L, Sun D, Tang Y. Electrochim Acta. 2017;231:13–19. [Google Scholar]
- 138.Wang ZL, Yan JM, Wang HL, Ping Y, Jiang Q. J Mater Chem A. 2013;1:12721–12725. [Google Scholar]
- 139.Feng YY, Liu ZH, Xu Y, Wang P, Wang WH, Kong DS. J Power Sources. 2013;232:99–105. [Google Scholar]
- 140.Ksar F, Ramos L, Keita B, Nadjo L, Beaunier P, Remita H. Chem Mater. 2009;21:3677–3683. [Google Scholar]
- 141.Paleček E, Boublíková P, Jelen F. Anal Chim Acta. 1986;187:99–107. [Google Scholar]
- 142.Brett AO, Serrano S. J Braz Chem Soc. 1995;6:97–100. [Google Scholar]
- 143.Fan Y, Huang KJ, Niu DJ, Yang CP, Jing QS. Electrochim Acta. 2011;56:4685–4690. [Google Scholar]
- 144.Steenken S, Jovanovic SV. J Am Chem Soc. 1997;119:617–618. [Google Scholar]
- 145.Crespo-Hernández CE, Close DM, Gorb L, Leszczynski J. J Phys Chem B. 2007;111:5386–5395. doi: 10.1021/jp0684224. [DOI] [PubMed] [Google Scholar]
- 146.Paleček E. Electroanal. 1996;8:7–14. [Google Scholar]
- 147.Armistead PM, Thorp HH. Anal Chem. 2000;72:3764–3770. doi: 10.1021/ac000051e. [DOI] [PubMed] [Google Scholar]
- 148.Bard AJ. Electrogenerated chemiluminescence. CRC Press; 2004. [Google Scholar]
- 149.Miao W, Choi JP, Bard AJ. J Am Chem Soc. 2002;124:14478–14485. doi: 10.1021/ja027532v. [DOI] [PubMed] [Google Scholar]
- 150.Ferapontova EE, Domínguez E. Electroanal. 2003;15:629–634. [Google Scholar]
- 151.Palecek E, Bartosik M. Chem Rev. 2012;112:3427–3481. doi: 10.1021/cr200303p. [DOI] [PubMed] [Google Scholar]
- 152.Zhou X, Shen Q, Yuan K, Yang W, Chen Q, Geng Z, Zhang J, Shao X, Chen W, Xu G, Yang X, Wu K. J Am Chem Soc. 2018;140:554–557. doi: 10.1021/jacs.7b10394. [DOI] [PubMed] [Google Scholar]
- 153.Wu B, Huang H, Yang J, Zheng N, Fu G. Angew Chem. 2012;124:3496–3499. doi: 10.1002/anie.201108593. [DOI] [PubMed] [Google Scholar]
- 154.Jin L, Liu B, Duay SS, He J. Catalysts. 2017;7:44. [Google Scholar]
- 155.Cao Z, Kim D, Hong D, Yu Y, Xu J, Lin S, Wen X, Nichols EM, Jeong K, Reimer JA, Yang PD, Chang CJ. J Am Chem Soc. 2016;138:8120–8125. doi: 10.1021/jacs.6b02878. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.