

Abstract
Background:
Kaposi sarcoma (KS) is a vascular tumor initiated by infection of endothelial cells (ECs) with KS-associated herpesvirus (KSHV). KS is dependent on sustained pro-inflammatory signals provided by intra-lesional leukocytes and continued infection of new ECs. However, the sources of these cytokines and infectious virus within lesions are not fully understood. Here, mast cells (MCs) are identified as pro-inflammatory cells within KS lesions that are permissive for, and activated by, infection with KSHV.
Methods:
Three validated MC lines were used to assess permissivity of MC to infection with KSHV and to evaluate MC activation following infection. Biopsies from 31 AIDS-KS cases and 11 AIDS controls were evaluated by immunohistochemistry for the presence of MCs in KS lesions and assessment of MC activation state and infection with KSHV. Plasma samples from 26 AIDS-KS, 13 classic KS, and 13 healthy adults were evaluated for levels of MC granule contents tryptase and histamine.
Results:
In culture, MCs supported latent and lytic KSHV infection, and infection induced MC degranulation. Within KS lesions MCs were closely associated with spindle cells. Furthermore, MC activation was extensive within KS patients, reflected by elevated circulating levels of tryptase and a histamine metabolite. One patient with clinical signs of extensive MC activation was treated with antagonists of MC pro-inflammatory mediators, which resulted in a rapid and durable regression of AIDS-KS lesions.
Discussion:
Using complimentary in vitro and in vivo studies we identify MCs as a potential long-lived reservoir for KSHV and a source of pro-inflammatory mediators within the KS lesional microenvironment. Additionally, we identify MC antagonists as a promising novel therapeutic approach for KS.
INTRODUCTION
Kaposi’s sarcoma (KS) is a unique, highly inflammatory “hemorrhagic sarcoma” initiated by infection of endothelial cells (ECs) by KS-associated herpesvirus (KSHV; also known as human herpesvirus 8). There are four epidemiologic types of KS: Classic KS, which typically occurs in elderly men in Mediterranean regions and is relatively indolent; Endemic KS, occurring in persons in sub-Saharan Africa; Iatrogenic KS, occurring in immunosuppressed persons undergoing transplantation; and Epidemic KS, occurring in HIV/AIDS patients. Independent of form, histologically, lesions are characterized by proliferating spindle-shaped ECs and infiltrating leukocytes (1). Spindle cells carry latent KSHV, defined by limited viral gene expression and no progeny production, but cells explanted from KS lesions and grown in culture rapidly lose the KS genome (2). Thus, a source of infectious KSHV virions within lesions capable of infecting new ECs is thought to be essential. Furthermore, while latently-infected ECs secrete some inflammatory mediators, the levels are insufficient to drive survival and growth of spindle cells in culture. Thus paracrine inflammatory signaling is thought to be essential for KS lesion development. The interplay between infection and inflammation allows for the enhanced survival of spindle cells even prior to malignant transformation.
Previous reports have described mast cells (MCs) in KS lesions but their significance has not been established (3). MCs are long-lived, tissue-resident cells that, like ECs, develop from bone marrow-derived CD34+ progenitor cells (4). Consistent with their role in sensing pathogens and allergens, MCs are most prevalent in mucosal tissues where they interface directly with the external environment, as well as adjacent to microvessels where they monitor both blood and lymph circulation (5). As immune surveillance cells (6), they have essential roles in both acute and chronic inflammation (5,7). Given the shared ontogeny of ECs and MCs, as well as their close association within all vascularized tissues, it is not surprising that these cell types cross-regulate each other. For example, chemical mediators from MCs promote EC activation, which is required for the regulated recruitment of leukocytes to inflamed tissues (8 ,9). There is evidence that MC activation of ECs is key to the pathophysiology of vascular compromise observed in severe dengue (10,11) and influenza infections (12), and MCs are known to be permissible to infection by multiple viruses. Furthermore, there is accumulating evidence that MC activation is a promising target for adjunctive therapy in multiple cancer types. MCs are found in a wide range of malignancies, often associated with poor prognosis, increased metastasis, and reduced survival, including melanoma, prostate, pancreatic adenocarcinoma, and squamous cell carcinoma [reviewed in (13)]. MC-cancer links are illustrated in the clinical observation that patients with mastocytosis, which is defined by pathologically increased numbers of MCs, have increased risk of developing both myeloid and lymphoid neoplasms (14). MCs are key effectors in the development and progression of small bowel cancer (15), malignant pleural effusion (MPE) (16) and primary cutaneous lymphoma (17), and are pathologically associated with several other B cell cancers including Hodgkin lymphoma (18 ,19), diffuse large B cell lymphoma (20) and B-cell chronic lymphocytic leukemia (18).
Here we provide the first evidence of a central role of MCs in the pathogenesis of KS. Specifically, we demonstrate that MCs are permissive for KSHV infection and that infection leads to activation and degranulation of MCs. We demonstrate also that MCs are abundant within KS lesions, that they express both latent and lytic viral proteins, and that they are predominantly activated. We also demonstrate the potential of anti-MC medications as a promising novel therapeutic approach for KS. Collectively, our data identify MCs as potentially central mediators of KS pathophysiology, and further evaluation of the therapeutic potential of MC-targeted treatment is warranted.
Methods
Cell Culture
BCBL-1 PEL cells were maintained in RPMI medium 1640 containing 10% FBS, 1% penicillin-streptomycin (PS), and 0.05 M beta-mercaptoethanol. LAD2 (a kind gift from Arnold Kirshenbaum, NIH, Bethesda) and LUVA (a kind gift from John Steinke, University of Virginia, Charlottesville) MCs were cultured in serum-free media (StemPro-34 SFM, Life Technologies, Gaithersburg, MD). HMC-1 clone 5C6 MCs were cultured in Iscove’s medium (Gibco) and authenticated by the University of Arizona genetics core by Promega PowerPlex assay and STR results compared to genomic databases. Because LAD2 and LUVA cell sequences are not currently available, these cells were authenticated by FLOW cytometry using MC specific receptor staining, CD117 (ThermoFisher, Clone 104D2) and FcεRI (ThermoFisher, clone AER-37). MC media was supplemented with 2 mM L-glutamine, 100 U/ml penicillin, 50 μg/ml streptomycin. 100 ng/ml SCF was added to LAD2 cultures. LAD2 cells were fed by hemi-depletion of media twice a week. Primary human umbilical vein endothelial (HUVEC) cells were purchased from Lonza. Cultures were expanded in EBM-2 media (Lonza) supplemented with the EGM-2 bullet kit in 6-well tissue culture plates coated with 0.1 % (w/v) gelatin in PBS and used between passage 2 and 5 for experiments. Cells were maintained at 37 °C in 5% CO2. All cell lines were tested every 1–2 month(s) for mycoplasma contamination using the MycoProbe Mycoplasma detection kit (R&D Systems, Minneapolis, MN). Cell lines were thawed and used for approximately 2 months before being discarded and a fresh aliquot thawed for use in experiments with the exception of LAD2 cells that were cultured continuously as per instructions.
Virus production and quantification
KSHV was obtained from cultures of BCBL-1 cells that harbor latent KSHV. Lytic reactivation of 5 × 105 BCBL-1 cells/ml was induced by addition of 0.4 mM valproic acid (Sigma) to cultures for 7 days. On day 7, cell free virus–containing supernatant was harvested from cultures and concentrated by centrifugation at 10,000g for 2 h. The viral pellet was resuspended in EBM-2 media, aliquotted, and frozen at −80 ºC. To determine the viral titer, viral preparations were treated with DNAse I (InVitrogen). KSHV DNA was extracted (Qiagen QiaAmp DNA Mini kit) and copy number quantified by real-time DNA PCR using primers that amplify the latency-associated nuclear antigen (LANA, also known as ORF 73 gene, see supplementary table 1 for sequence). The KSHV ORF 73 gene cloned in the pCR2.1-TOPO vector (InVitrogen) was used for the external standard. Known amounts of ORF 73 plasmid (107, 106, 105, 104, 103, 102, 10 and 1 copies) were used to generate a standard curve. Reaction conditions for 2-step PCR were performed on a Bio-Rad iCycler, using 42 cycles of 95 for 10 sec, then 60 for 30 sec. The Ct values were used to plot the standard graph and to calculate the copy numbers of viral DNA.
Titering of KSHV
Quantification of infectious virus was assessed on 1 ×105 cells/well HUVEC monolayers plated on gelatin-coated 6-well plates with glass coverslips. The next day monolayers were inoculated with KSHV at different dilutions in the presence of 8 μg/ml Polybrene (Sigma) for 2h at 37ºC followed by spinoculation at 2000 rpm for 15 min. 48 h post-infection (pi) HUVECs were washed with PBS, fixed in 4% paraformaldehyde for 10 min at room temperature (RT), and stained with a rabbit anti-LANA (1:1000) antibody and visualized with goat anti-rabbit 488 (Molecular Probes) as previously described (21). Infectious units were determined by counting the number of LANA dots/cell in 5 fields of view with one LANA dot equivalent to 1 infectious viral particle, as previously described (22).
In vitro KSHV infection of MCs
4×106 HMC-1 or LAD2 MCs were infected with KSHV (MOI 1–3) for 3h at 37°C. Cells were mixed gently by hand every 20 min during the 3 h incubation time. Post-infection, cells were washed two times with 100 volumes of warm media to remove any unbound virus, set up at 5×105 cells/ml, and cultured for indicated times. For some experiments UV inactivation of virus was carried out. For ultraviolet inactivation (UV), KSHV was exposed to 5×1200 μJ UV in a UV Stratalinker (23).
Quantitative PCR
Total RNA from KSHV infected mast cell cultures was isolated using the RNeasy Mini Kit (QIAGEN) with DNase treatment performed on column with the Qiagen RNase-free DNase according to the manufacturer’s protocols. The quality and concentration of RNA was assessed using the ND1000 spectrophotometer (Nanodrop). 0.5 to 1 μg of RNA was reverse transcribed using a Qiagen Quantitect Reverse Transcription kit according to the manufacturer’s instructions. qPCR using Biorad iQ SYBR Green Supermix was run on a Bio-Rad iCycler, using a 2-step program of 95/60ºC for 45 cycles. All reactions were performed in duplicate. Samples were normalized and expressed as ΔCt of the internal control gene (GAPDH) and target gene using the ΔCt method. All samples had a no RT control and NTC to control for DNA contamination. Each “n” represents an experiment executed independently, representing a different RNA sample.
RT-PCR
Total RNA was isolated and converted into cDNA as described in the quantitative PCR section. The primer sequences used in the RT-PCR analyses are listed in supplementary Table 1. For RT-PCR, cDNA was diluted 1:4 and 2 μl used in subsequent PCR reactions with KSHV gene-specific primers. All samples had a no RT control and NTC to control for DNA contamination. BCBL-1 cells induced with 0.4mM Valproic acid for 2 days served as a positive control for lytic viral gene expression. PCR was carried out using Promega Go-TaqGreen Master mix in a Bio-Rad thermocycler. PCR products were run on 2% agarose-ethidium bromide gels and viewed on a Bio-Rad geldoc.
Limiting dilution qPCR
To determine the frequency of cells carrying the KSHV genome limiting dilution qPCR was performed on serial dilutions of MCs infected with KSHV at 24 hpi using previously published methods (24). At 24 hpi MCs were washed 2 times with 10 and 100x volume of 5% FCS Iscoves before being resuspended in isotonic buffer (150 mM KCl, 10 mM Tris-HCl [pH 7.5], 1.5 mM MgCl2). Serial ten-fold and two-fold dilutions of MCs were made in an isotonic buffer ranging from 1000 test cells to 1 test cell per PCR. 10–12 PCRs were analyzed per cell concentration. Five-microliter cell dilutions were added to PCR tubes containing 5 μl of lysis buffer (10 mM Tris-HCl [pH 8.5], 1.5 mM MgCl2, 1% Nonidet P-40, 1% Tween 20, 0.2 mg/ml proteinase K) and lysed overnight at 56°C. Proteinase K was inactivated at 95°C for 15 min, and 15 μl of Biorad iQ SYBR Green Supermix master mix containing LANA primers were added directly to each cell lysate and run on a Bio-Rad IQ5 I cycler, using a 2-step program of 95/60 for 45 cycles. Negative uninfected cells, and positive BCBL-1 controls were included for each set of PCRs performed. Poisson distribution was used to calculate the percentage of MCs infected.
KSHV Infection Assay
HMC-1 cells were infected with KSHV at MOI 1–3 as described above for 24–72 h. Following inoculation MCs were washed twice with 100 volumes of 5% FCS Iscoves and set up at 5×105 cells/ml. At 48 hpi cell-free supernatants were harvested and used directly to infect semi-confluent monolayers of primary HUVECs as described above. Cell-free supernatants from mock-infected or UV-inactivated virus inoculated cultures were used as controls for LANA specific staining. At 48 hours post-treatment KSHV infection was assessed by LANA microscopy as described above (21).
β-hexosaminidase release assay (Degranulation assay)
LAD2 and LUVA cells (1 ×106 cells/ml) were incubated for 60 min at 37°C in the absence or presence of increasing concentrations of live or UV-inactivated KSHV. Mock-treated cultures were used to indicate baseline β-hexosaminidase release. Cultures were centrifuged at 200 g for 10 min at 4 °C. After collection of supernatants, the pellets were resuspended at 1 ×106 cells/ml in buffer and disrupted by 3 rounds of snap freeze-thaw. Supernatants and pellets were frozen at −80°C for future analysis. β-hexosaminidase assay was carried out as described (25). The net percent β-hexosaminidase release was calculated as follows: [(β-hexosaminidase in supernatant)/(β-hexosaminidase in supernatant + β-hexosaminidase in pellet)] × 100.
Histology
De-identified tissue from 31 cases of AIDS-KS (20 dermal, 1 oral mucosa, 6 lymph node, 2 lung, and 2 intestinal) and 10 control non-KS samples from 10 HIV-positive cases (2 dermal, 2 lung, 3 lymph node, and 3 intestinal) were obtained from the AIDS and Cancer Specimen Resource (ACSR/NCI) and the Tissue Archives, Department of Pathology, College of Medicine, The Ohio State University. Dermal biopsies from one patient with AIDS-KS was obtained as part of routine clinical care in the Infectious Diseases clinic at SUNY Upstate Medical University. Hematoxylin and Eosin (H&E) and Prussian Blue Iron (Potassium Ferrocyanide 2%) stains were performed using standard protocols at the Pathology laboratory in Ohio State University or at SUNY Upstate. IHC following steam and citrate antigen retrieval was performed to identify HHV-8 LANA (Novocastra, clone 13B10 at 1:100 dilution); tryptase (Dako, clone AA1 at 1:100 dilution); HHV-8 K8.1a (Advanced Biotechnologies, clone 228 at 1:500 dilution); histamine (Lifespan Biosciences, polyclonal at 1:2000 dilution) and CD117 (Dako, polyclonal at 1:2000 dilution). Stained sections were digitized at 20 X using Aperio XT digital imaging system (Aperio, Vista, CA) and reviewed by Leona Ayers, in the Department of Pathology, Ohio State, or by Ethel Cesarman at Weill Cornel Medical College. Representative images were selected for publication presentation.
Tryptase and N-methlyhistamine ELISA
Total MC tryptase (Cusabio, China) and the histamine metabolite N-methylhistamine (IBL International, Canada) were measured by ELISA on archived plasma samples from 25 HIV+ and 13 HIV− KS patients obtained from the ACSR. Control samples were obtained from 13 healthy adults from the university community at SUNY Upstate. The assays were carried out according to the manufacturer’s instructions.
Statistics
Difference in percent β-hexosaminidase release was analyzed by using 1-way ANOVA, followed by Bonnferroni multiple comparison test. Differences in tryptase and N-methylhistamine levels between KS patients and healthy comparators were assessed by 1-way ANOVA, followed by Mann-Whitney two-tailed nonparametric test. To test the relationship between HIV VL and CD4+ T cell count and MC activation products Spearman nonparametric correlation analysis was performed. Value was considered significant if p <0.05. All statistical analyses were performed with Prism 6 software (GraphPad, La Jolla, California).
Study approval
Human studies were approved a prori by the Institutional Review Board at SUNY Upstate Medical University. All tissues and plasma samples were obtained for testing following signed Material Transfer Agreements based on SUNY Upstate Medical University and Ohio State IRB approvals. Normal comparators were obtained following signed consent and SUNY Upstate Medical University approval. The case study patient was included upon signed consent at the Infectious Disease clinic, SUNY Upstate Medical University.
RESULTS
Mast cells are permissive to KSHV infection in vitro and in vivo
Because MCs are permissive to infection by other viruses we assessed the ability of two extensively validated human MC lines, HMC-1 5C6 and LAD2, for permissivity to infection with KSHV. MCs were inoculated at an MOI of 1–3, and RNA was then isolated at 6, 24, 48 and 72 hpi. We assessed expression of KSHV latent genes (ORF71, ORF72, ORF73, and K12) and lytic genes of all three temporal classes (including immediate early, ORF50; early, ORF27, K2, K4, K5, K6; and late, ORF64 and K8.1) and found robust expression at all time points analyzed, suggesting that MCs support KSHV gene expression (Figure 1A). No viral gene expression was observed in mock- or UV-irradiated virus treated MCs or in the no RT controls. Valproic acid treated BCBL-1 cells served as a positive control for KSHV lytic gene expression. Overall these data demonstrate MCs support both latent and lytic KSHV gene expression.
Figure 1. Mast cells support KSHV infection in vitro and in patient lesions.
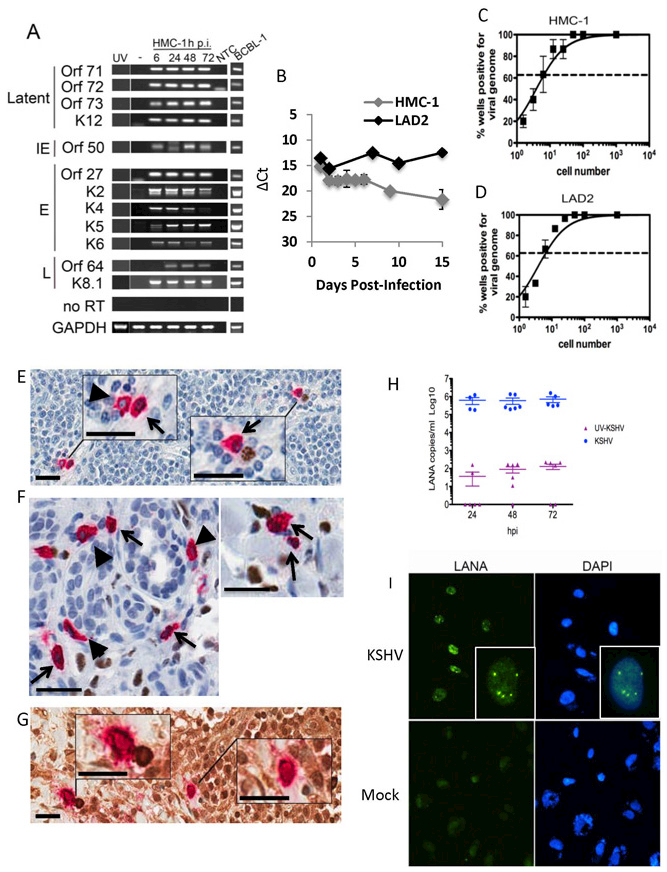
(A-D) HMC-1 and LAD2 cells were infected with KSHV and cultured for indicated times. (A) Mast cells express abundant KSHV genes. GAPDH was used as a loading control. BCBL-1 cells induced with valproic acid were used as a positive control for viral lytic gene expression. NTC- no template control. Data are representative of two separate experiments. (B) QRT-PCR gene expression indicated MCs express LANA in growing cultures over the entire culture period. Data are expressed as CT values normalized to GAPDH. Data are representative of three independent experiments performed in triplicate. (C-D) Limiting dilution qPCR analysis of infected (C) HMC-1 and (D) LAD2 demonstrated approximately 1 in 7 MCs were KSHV genome+ at 24 hpi.. Data are expressed as mean ±SEM, n=3. (E-G) HIV/KS tissue double stained for KSHV LANA (nucleus-brown) and MC-specific tryptase (cytoplasm-red) demonstrate: (E) LN shows (left upper inset) two tryptase+ MCs, one LANA− (blue nucleus, arrow head) and the other LANA+ (brown nucleus, black arrow), (right lower inset) tryptase+ LANA+ MC (brown nucleus, black arrow) paired with a LANA+ plasma cell (brown nucleus); (F) dermal lesion shows tryptase+ LANA+ MCs (black arrows) and tryptase+ LANA− MCs (arrow heads); (G) HIV-KS LN double stained for MC-specific tryptase (cytoplasm-red) and KSHV lytic antigen K8.1a (cytoplasm-brown) shows (left upper inset) a tryptase+ K8.1a+ MC paired with a KSHV K8.1a+ stained plasma cell cytoplasm and (right lower inset) a solo tryptase+ K8.1+ MC cytoplasm (brown). Scale 25 μm (inset scale, 10 μm). (H-I) HMC-1 cells were infected as described in the methods. (H) KSHV-infected MCs produce encapsulated, DNAse resistant, virus during infection. Data are from two independent experiments with three experimental replicates and two technical replicates each and expressed as mean ± SEM. Mock infected cultures gave no CTs. (I) Mast cell-derived-KSHV establishes latency in primary human endothelial cells. Cell-free supernatants were isolated from MCs uninfected or infected with KSHV at 24h p.i. and used to treat primary human ECs. 48 h post-treatment, primary ECs were fixed and stained as indicated in the Methods; LANA positive nuclei-localized staining demonstrated establishment of latency. Dapi was used to visualize nuclei. Magnification x630. Data are representative of 3 independent experiments.
KSHV-infected ECs lose the viral episome as they divide and eventually die (2,26). To evaluate the ability of MCs to maintain KSHV infection over a longer time period and to evaluate the levels of latent ORF73/LANA quantitatively, we measured gene expression in cultured MCs for up to 15 days pi by qPCR. Expression of ORF73 was robust throughout the culture period in both HMC-1 and LAD2 MCs (Figure 1B), suggesting maintenance of viral infection long-term. To evaluate the number of infected cells in our cultures we carried out limiting dilution qPCR; Poisson distribution demonstrated 14% of HMC-1 (Figure 1C) and 11% of LAD2 (Figure 1D) MCs in culture harbored LANA+ KSHV genomes at 24 hpi suggesting both LAD2 and HMC-1 cells supported similar levels of KSHV infection. To investigate whether MCs support infection in KS lesions, biopsies of AIDS-KS were examined by double staining for MC-specific tryptase and either the KSHV latent LANA/ORF73 (nuclear) or lytic K8.1A (cytoplasmic) proteins. We observed subpopulations of MCs that expressed LANA (Figure 1E and 1F) or K8.1A protein (Figure 1G). Figure 1E, left inset box, shows two tryptase+ MCs, one which has a brown LANA+ nucleus (black arrow); the right inset box shows a tryptase+ LANA+MC paired with a LANA+ plasma cell (as demonstrated by the whirled nucleus stain). Latent KSHV infection of MCs was not restricted to lymph nodes (LNs) as subpopulations of MCs in dermal lesions also showed evidence of LANA expression (black arrows are LANA+, black arrow heads are LANA− (Figure 1F). The localization of LANA to the nucleus demonstrates that the KSHV latency program was established in these MCs and, as expected, within their companion plasma cell in vivo. To quantify the number of LANA+ MCs we counted 5 high-powered fields of view from 5 different dermal lesions and found 12% ± 3% (mean ± SD) of MCs were LANA+. We also identified MCs expressing cytoplasmic lytic K8.1A protein in LNs, consistent with our in vitro infection data; Figure 1G, left inset box demonstrates tryptase+K8.1A+ MCs seen paired with K8.1A+ plasma cell while the right inset box shows a single tryptase+K8.1A+ MC. The strongly cytoplasmic localization of late lytic K8.1A suggests that these cells clearly support lytic infection in vivo. K8.1A was not assessed in all lesion locations and is currently under investigation. The close association of MCs with lytic infected plasma cells presents a potential in vivo mechanism for KSHV infection of MCs within the lesion microenvironment. We were unable to visualize both lytic K8.1A and latent LANA expression in tryptase+ MCs due to technical constraints that prevented us from determining the ratio of latent to lytic MCs within KS lesions. Taken together, our data clearly demonstrated that MCs are permissive for KSHV infection and maintain viral gene expression long-term, positioning MCs as possible KSHV reservoirs.
Mast cells support productive infection with KSHV
Because we observed lytic gene expression from all temporal classes in MC cultures up to 72 hpi we evaluated whether MCs complete the lytic cycle and produce progeny virus. To test this, HMC-1 cells were infected with live and UV-irradiated KSHV and cell-free supernatants obtained at various times pi were evaluated for the presence of encapsidated virus particles. Supernatants were treated with DNAse to digest any naked viral DNA, proteinase K treated to remove the DNAse, and then DNA was extracted. Quantitative-PCR to detect KSHV LANA-positive genomes demonstrated approximately 106 particles/ml in culture medium isolated from cells infected with live KSHV as early as 24hpi (Figure 1H). Cultures treated with UV-inactivated virus had approximately 102 particles/ml controlling for amount of residual virus left after extensive wash. The data suggest MCs can produce encapsidated viral genomes.
Herpesviruses are known to produce defective, non-infectious progeny (27); therefore, we evaluated whether MC-derived KSHV virions were infectious to ECs using cell-free supernatants harvested from infected MCs at 24hpi. LANA-positive ECs are evident 48h post-infection by antibody-specific staining indicating that ECs were infected and had established latency with the encapsidated cell-free virus present in MC cultures (Figure 1I, top panels). No staining was observed in mock-infected (Figure 1I, bottom panels) or UV-treated cultures (data not shown). QRT-PCR analysis of HUVEC cultures treated for 3 days with cell-free supernatants from MCs infected with live virus confirmed LANA expression in these cells. Two independent experiments, in triplicate and run in duplicate, gave an average of 12.69 ±1.91 CTs for GAPDH expression and 30.50 ± 8.60 CTs for LANA expression (mean± SD). UV-virus treated HUVECs, mock treated and no RT controls gave no amplification. Together, our in vitro and in vivo studies provide consistent evidence to affirm our hypothesis that MCs support KSHV infection and may represent a reservoir for latent KSHV.
KSHV induces activation of mast cells in vitro
MCs mediate inflammation by releasing a wide variety of mediators including β-hexosaminidase, tryptase, histamine, and heparin. To determine if KSHV activates MCs to release stored mediators we evaluated β-hexosaminidase release by LUVA and LAD2 MCs. Within 1h of treatment KSHV induced a significant, dose-dependent release of β-hexosaminidase by both MC lines into cultures with up to 40% (LAD2, Figure 2A) and 25% (LUVA, Figure 2B) of total enzyme released at the highest concentration of virus compared to mock. Degranulation was dependent on live virus, as MC infected with UV-inactivated virus remained quiescent. These data demonstrate robust activation with degranulation of MCs following infection with KSHV, suggesting that MCs are capable of modulating the local inflammatory environment in KS lesions though release of pre-formed mediators.
Figure 2. KSHV virus activates mast cells in vitro and in vivo. (A-B).
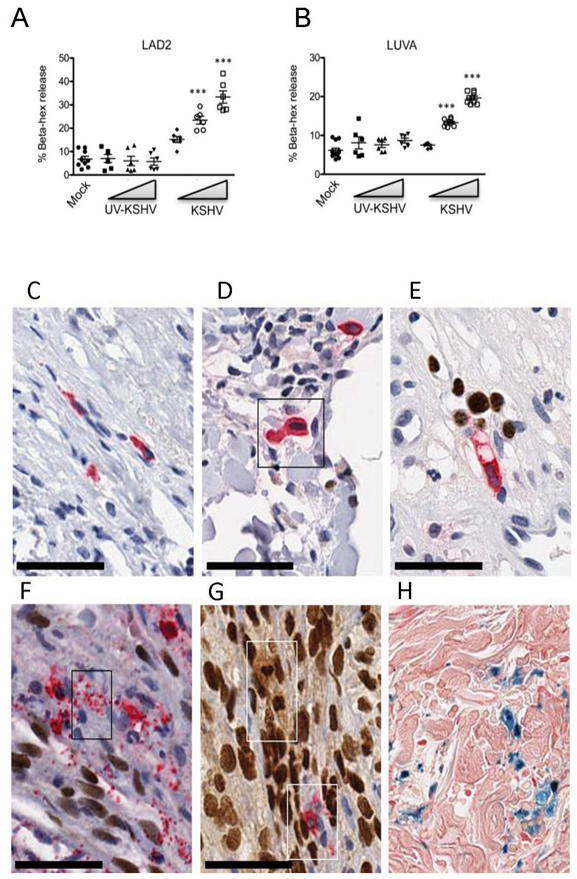
Live KSHV virus induces a dose-dependent increase in β-hexosaminidase release from LAD2 (A) and LUVA (B) cells within 1 hr (n=3, mean ±SEM). Difference in percent β-hexosaminidase release was analyzed by using 1-way ANOVA, followed by Bonnferroni multiple comparison test. **p<0.01, ***p<0.001. (C-H) Activated, degranulating and degranulated MCs are associated with KSHV LANA+ KS cell nuclei and EC nuclei in vivo; HIV+ KSHV LANA+ KS tissues double stained for MC-specific tryptase (cytoplasm-red) and KSHV LANA (nucleus-brown) demonstrate: (C) A resting MC from HIV+ control tissue is shown for size comparison; (D) HIV+ early KS skin patch lesion demonstrates an enlarged, activated MC showing abundant tryptase filled cytoplasm and enlarged nucleus compared to (C); (E) MC directional tryptase degranulation. An activated, elongated and vacuolated MC produces a narrow stream of discrete tryptase+ granules (red) from the MC tip directed toward the enlarged intravascular KSHV LANA infected EC nuclei (brown); (F) MC mass tryptase granule degranulation (box) within a tissue bundle of proliferating KSHV infected spindle cells. Discrete tryptase+ granules flood the infected spindle cell bundle. (G) Partial and complete MC degranulation between compact KS spindle cells in developing KS nodular lesion. Note the MC ‘ghost’ lobulated LANA+ nucleus and foamy cytoplasm (upper box) and a partially degranulated MC (lower box); (H) Prussian blue hemosiderin granules are a persistent feature of MC degranulation in KS lesions. Scale bar 50 μm.
Activated MCs are associated with spindle cells in vivo
MC location and activation state was assessed in 20 HIV+ KS dermal biopsies and were compared to 2 control HIV+ tissues without KS lesions. Compared to resting tissue MCs (Figure 2C) in normal skin, in early patch stage lesions, activated MCs had pronounced tryptase+ cytoplasmic granules and enlarged nuclei (Figure 2D) consistent with an activated phenotype. MCs were found to align directionally with infected ECs, vacuolate and release granules in the direction of the enlarged, infected LANA+ ECs even before full tissue invasion occurs (Figure 2E). In more advanced KS nodular lesions showing increased tissue KS spindle cell density, massive (28) degranulation of MCs was observed with discrete tryptase filled granules flooding the infected spindle cells (Figure 2F, red granules, see inset box). In tumor tissues with confluent spindle cell density, partial and fully degranulated MCs were observed (Figure 2G). The MC nuclei in some of the activated MC cells (Figure 2G, upper inset box) showed nuclear staining with anti-LANA antibody supporting KSHV infection in a subpopulation of these effector skin MCs. Degranulated “ghost” or “phantom” MCs were plasma membrane c-kit/CD117+ (prototypical marker of MCs, data not shown) confirming the presence of a granule-depleted MC. Images representative of single antibody staining for both KSHV LANA and the MC markers tryptase, histamine, and CD117, are shown in Supplementary Figure 1. In later stage KS lesions, MC degranulation was less obvious. However, the presence of significant extravasated erythrocytes and hemosiderin deposition suggested ongoing local heparin anticoagulation and bleeding related to release of heparin-stabilized tryptase (Figure 2H). MC association with LANA+ spindle cells as well as increased density, activation, and degranulation were observed in all cutaneous KS lesions examined.
Increased MC density, activation, and degranulation were not limited to cutaneous KS. We also examined 11 tissue sections from lung, gut, and LN KS along with 11 corresponding HIV+ control tissues. Similar to active KS lesions in the dermis (Figure 3A), lung (Figure 3B), LN (Figure 3C), and gut (Figure 3D) lesions demonstrated association of activated MCs with KSHV LANA+ spindle cells. These KS lesion-associated MCs demonstrated the same tryptase+ granulated, hypogranulated, and degranulated MC forms irrespective of KS lesion location. In overall comparison of KS lesion tissue to control HIV+ tissues (Figure 3 E-H), we consistently observed large and activated tryptase+ MCs within and around KS lesions that was not seen in our HIV+KS− control tissues. Together these data provide in vivo evidence for the accumulation, infection, activation, and degranulation of MCs within KSHV-LANA+ in diverse tissues. These data suggest MCs are a key component of KS lesions capable of supporting infection and as a potent source of inflammatory mediators necessary to help drive the establishment and progression of lesions.
Figure 3. MCs associate with KSHV LANA+ KS spindle cells and are activated with degranulation of tryptase+ discrete granules within developing KS lesions.
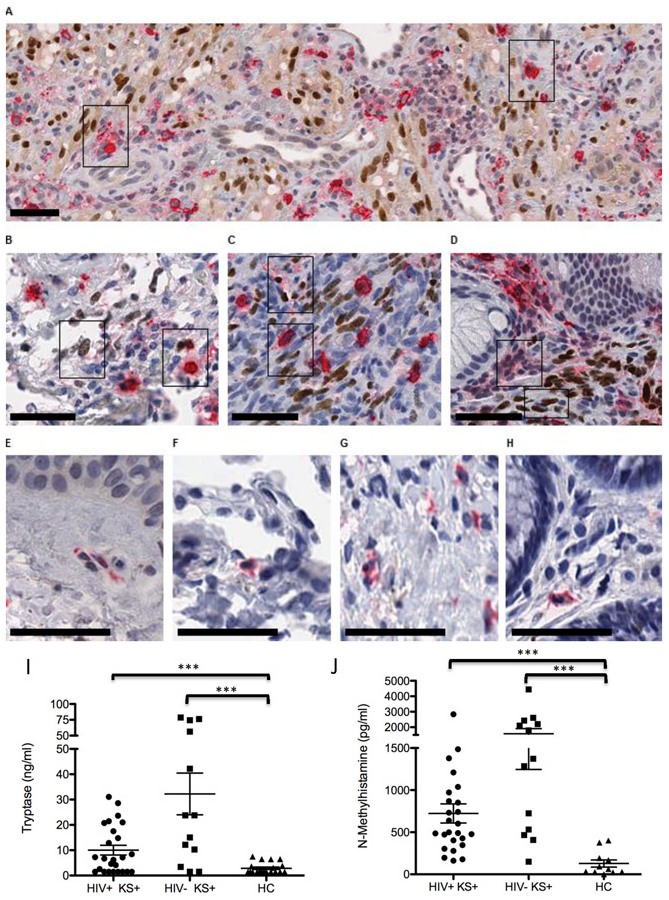
(A-D) HIV+ patients with Kaposi’s sarcoma herpesvirus (KSHV) lesions double stained for MC-specific tryptase (cytoplasm-red) and KSHV LANA (nucleus-brown). (A) Skin- KS lesions with abundant LANA+ECs and activated tryptase+ MCs. A subset of activated MCs are LANA+ (right inset box). (B) Lung-LANA+ spindle cell nuclei and associated tryptase+ MCs. (C) lymph node- LANA+ spindle cell proliferation and associated tryptase+ activated, enlarged, MCs, and (D) gut with LANA+ spindle cell nuclei and abundant activated MCs with tryptase degranulation. (E-H) Control HIV+ KS negative tissues show small dendritic shaped MCs with tryptase+ cytoplasm. Note the broad distribution of MCs and their obscure appearance in control HIV+ tissues. Scale bar, 50μm. (I-J) Elevated mast cell tryptase (I) and N-methlyhistamine (J) in plasma samples from 26 AIDS-associated HIV+ KS patients, 13 classic HIV− KS patients and 13 healthy comparators (HC). Data are expressed as mean ± SEM. Differences in tryptase and N-methylhistamine levels between KS patients and healthy comparators were assessed by 1-way ANOVA followed by Mann-Whitney two-tailed nonparametric test, ***p<0.001.
Mast cell-specific tryptase and N-methylhistamine are elevated in the blood of KS patients
Our complementary in vitro and in vivo data suggest MCs are key cellular players in the KS lesion microenvironment, promoting and supporting disease in a multi-faceted way including by intense activation and inflammation. In some diseases, MC activation is so wide-spread or intense that the response can measured in blood. We hypothesized that the extensive KSHV-mediated activation of MCs in KS would result in circulating levels of MC-specific, granule-stored, mediators. Increased tryptase (Figure 3I) and N-methylhistamine, a metabolite of histamine (Figure 3J), were found in plasma samples obtained from 26 patients with extensive HIV associated KS (HIV+KS+) and in 13 HIV negative Classic KS (HIV−KS+) patients as compared to 13 healthy comparators (HC). These data confirm significant in vivo MC activation and degranulation (29 ,30). Corresponding patient characteristics and assay results are given in Table 1. The mean age of HIV− KS+ patients was 71 y and 11 of 13 were male; for the HIV+ KS+ patients the mean age was 36 y and 18 of 26 were male, with a mean CD4+ T cell count of 205.8. Tryptase levels in classic HIV− KS+, AIDS –associated HIV+ KS+ vs HCs were significantly elevated, 32.25 ± 8.24 vs. 9.98±1.83 vs. 1.64±0.69 ng/ml, p=0.0053. Significant increases were also observed in N-methylhistamine levels in KS patients, 1578 ± 333.10 vs. 723.10±114.10 vs. 101.10±31.96 pg/ml, p<0.001, mean ± SEM. Seven classic HIV− KS+ patients (54%) and 6 AIDS –associated HIV+ KS+ patients (23%) had tryptase levels higher than 20 ng/ml (levels observed in MC activation disease and in mastocytosis). No correlation was found between levels of MC activation products and either HIV viral load or CD4+ T cell count (HIV VL vs. tryptase r = 0.359, p = 0.092; CD4+ T cell count vs. tryptase r = −0.031, p = 0.886; HIV VL vs. N-methylhistamine r = −0.188, p = 0.391; CD4+ T cell count vs. N-methylhistamine r = −0.061, p = 0.778). In addition, no correlation was found between age or sex of individual and levels of MC activation products tryptase (age vs. tryptase r = 0.297, p = 0.118; sex vs. tryptase r = 0.143, p = 0.451; age vs. N-methylhistamine r = 0.117, p = 0.484; sex vs. N-methylhistamine r = 0.005, p = 0.974). The corresponding significant increase in circulating histamine in KS patients supports active MC degranulation in these patients that is independent of the degree of immune suppression, age, or sex.
Table 1.
KS patient characteristics and assay results.
| Case ID |
Year | Age | Gender | HIV | HIV VLa | CD4 + T cell |
Tryptase ng/ml |
N-Methylhistamine pg/ml |
|---|---|---|---|---|---|---|---|---|
| 1 | 2004 | 48 | M | + | − | 200 | 6.20 | 162 |
| 2 | 2003 | 52 | M | + | 56 | 257 | 4.13 | 178 |
| 3 | 2003 | 26 | M | + | >750000 | 56 | 6.72 | 404 |
| 4 | 2004 | 42 | M | + | − | − | 2.57 | 197 |
| 5 | 2003 | 43 | M | + | >750000 | 474 | 4.62 | 304 |
| 6 | 2007 | 40 | M | + | 93893 | 4 | BDc | 347 |
| 7 | 2007 | 26 | F | + | 319498 | 260 | 7.25 | 491 |
| 8 | 2008 | 25 | F | + | 134796 | 116 | 23.66 | 500 |
| 9 | 2008 | 39 | M | + | 2103 | 586 | BD | 1378 |
| 10 | 2008 | 54 | M | + | 183825 | 169 | 28.59 | 602 |
| 11 | 2008 | 33 | F | + | 398614 | 144 | 20.76 | 732 |
| 12 | 2008 | 28 | M | + | 718772 | 102 | BD | 1038 |
| 13 | 2008 | 22 | M | + | 166517 | 432 | BD | 869 |
| 14 | 2008 | 22 | F | + | >750000 | 125 | 21.46 | 428 |
| 15 | 2008 | 36 | M | + | 239967 | 609 | 8.40 | 972 |
| 16 | 2008 | 39 | M | + | 487195 | 104 | 14.79 | 852 |
| 17 | 2008 | 34 | F | + | 49860 | 112 | 31.05 | 1486 |
| 18 | 2008 | 40 | M | + | 367543 | 26 | BD | 486 |
| 19 | 2009 | 42 | F | + | 118665 | 174 | 17.52 | 278 |
| 20 | 2009 | 39 | M | + | 720640 | 12 | 21.00 | 638 |
| 21 | 2009 | 40 | M | + | 533368 | 387 | BD | 473 |
| 22 | 2009 | 33 | F | + | 40213 | 103 | BD | 2836 |
| 23 | 2009 | 32 | M | + | 693204 | 10 | 7.13 | 741 |
| 24 | 2009 | 31 | M | + | 578202 | 118 | BD | 1210 |
| 25 | 2010 | 21 | F | + | 187011 | 146 | 12.88 | 478 |
| 26 | 2017 | 46 | M | + | 40121 | 419 | 8.80 | ndd |
| 27 | 2002 | 79 | F | − | nab | na | 23.80 | 725 |
| 28 | 2003 | 60 | M | − | na | na | 10.27 | 466 |
| 29 | 2004 | 79 | M | − | na | na | 23.80 | 1372 |
| 30 | 2004 | 80 | M | − | na | na | BD | 408 |
| 31 | 2005 | 82 | M | − | na | na | 78.65 | 1795 |
| 32 | 2005 | 72 | M | − | na | na | 74.31 | 532 |
| 33 | 2006 | 59 | M | − | na | na | 42.20 | 2431 |
| 34 | 1999 | 76 | M | − | na | na | BD | 2200 |
| 35 | 2010 | 82 | M | − | na | na | 76.27 | 2605 |
| 36 | 2010 | 62 | M | − | na | na | 3.39 | 150 |
| 37 | 2011 | 88 | F | − | na | na | 12.16 | 1285 |
| 38 | 2012 | 47 | M | − | na | na | 56.32 | 4446 |
| 40 | 2013 | 57 | M | − | na | na | 15.03 | 2105 |
VL, viral load
na, not applicable
BD, below detection
nd, not done
Antagonism of MC inflammatory mediators is associated with durable regression of AIDS-KS lesions
A 46-year-old HIV-positive man with acutely progressing KS presented to establish care in our Infectious Diseases clinic. Since his HIV diagnosis 14 years prior he had been intermittently adherent to his anti-retroviral medications (ARVs). At diagnosis he had KS lesions on the distal lower extremities bilaterally, but his lesions had been stable in number, size, and color for years. Five weeks prior to presentation to our clinic, he underwent an emergency appendectomy. Following surgery he developed worsening of long-standing, but previously unrecognized, symptoms consistent with MC activation (31), including intermittent vertigo, headaches, gastroesophageal reflux (GERD), nausea, watery diarrhea, arthralgias, myalgias, and fatigue. Coincident with worsening of these symptoms he noted development of significant bilateral lower extremity edema, bilateral tinnitus (also symptoms of significant MC activation), and an increase in the size and a darkening pigmentation of all lower extremity lesions, in particular the lesions on his left foot. Additionally, he noted that a new lesion developed on his abdomen. Biopsies of the lower extremity and abdominal lesions were obtained. H & E staining demonstrated abnormal microvascular structures with extravasated erythrocytes and significant infiltration of mononuclear cells (Figure 4A). IHC staining for the KSHV-specific LANA (brown) confirmed that the lesions were KS (Figure 4B). Serial sections stained for MC markers tryptase (Figure 4C, brown) and CD117 (Figure 4D, red) demonstrated extensive MC infiltration. Notably, the distribution of tryptase in many cells was extracellular, indicating that the potent pro-inflammatory effects of MC degranulation were active in the patient’s tumors.
Figure 4. Regression of cutaneous KS lesions in a HIV+KS+ patient treated with MC-specific anti-inflammatory therapy.
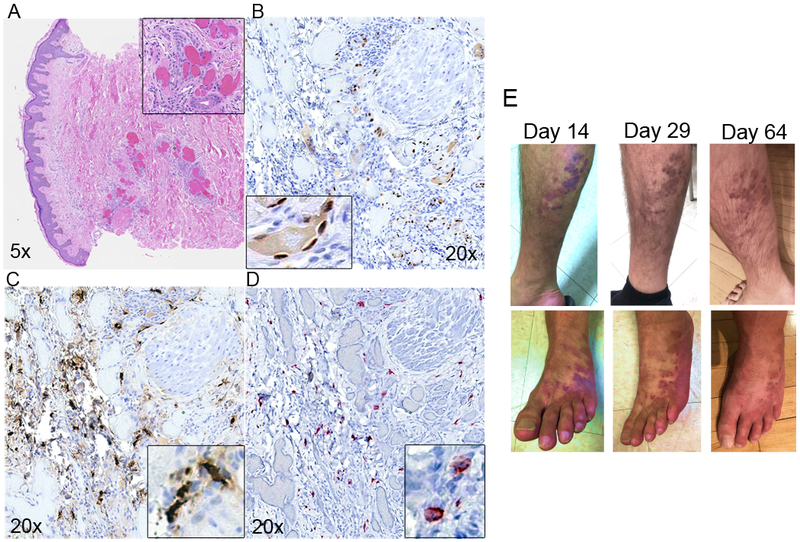
Serial sections of lesion biopsy were stained (A-D); A) H&E, (B) IHC LANA staining indicating presence of KSHV, (C) MC specific tryptase shows extensive infiltration and activation, (D) MCs in lesions are CD117/c-Kit positive. (E) Visible lesion regression on right leg (top row) and left foot (bottom row) at Day 14, 29 and 64 post-initiation of anti-MC treatment.
We hypothesized that his worsening constitutional symptoms and progression of his previously stable KS both resulted from extensive MC activation triggered by his recent inflammatory appendicitis and abdominal surgery. Appendicitis is characterized by enhanced MC localization and activation (32). We sought to interrupt the pro-inflammatory response driven by ongoing MC degranulation with agents that block the target effects of MC products. Therefore, we prescribed cetirizine (a type 1 histamine receptor antagonist, 20 mg daily), ranitidine (a type 2 histamine receptor antagonist, 300 mg twice a day), montelukast (a leukotriene receptor antagonist, 10 mg daily), and vitamin C (a MC stabilizer, 1 g daily).
At a follow up appointment two weeks later his weight was down 4.5 kg, and he demonstrated significantly reduced lower extremity edema. Additionally, he reported resolution of many of his worsening constitutional symptoms, including fatigue, GERD, diarrhea, tinnitus, and vertigo. Notably, he thought that the KS lesions on his legs and abdomen had lightened and shrunk. Upon examination his lesions had become lighter in color and were visibly smaller (Figure 4E). The top 3 images are of the right leg, the bottom three images are of the left foot; both series demonstrate dramatic reduction in lesion pigmentation and size over the course of treatment. Over the course of 2 months of treatment targeting MC-specific mediators we observed that his lesions continued to shrink, becoming smaller and macular; the lesion on his abdomen had fully resolved (not shown). The patient also reported feeling the best he had in more than 10 years.
These dramatic changes occurred in the absence of HIV control. At presentation to our clinic he reported variable adherence to his previously prescribed Elvitegravir, Cobicistat, Emtricitabine, and Tenofovir. His HIV viral load was 40,121 RNA copies/ml and CD4+T cell count was 419. Viral resistance testing was sent at the initial visit and subsequently demonstrated that his viral isolate was in fact resistant to Elvitegravir, Emtricitabine, and Tenofovir. It is significant, therefore, that his dramatic regression of KS lesions was associated with initiation of anti-MC medications rather than suppression of his viral load. When his resistance testing was available, his ARVs were changed to Duranavir, Ritonavir, Dolutegravir, and Etravirine. One month after changing his ARV regimen his viral load was 162 and CD4+T cell count was 577. Thus, viral suppression was achieved after KS lesion regression was observed.
Discussion
Progression of KS lesions is dependent on the expression of both latent and lytic viral genes, repeated rounds of new infection, and secretion of pro-inflammatory cytokines. Given that the majority of ECs within the KS lesional microenvironment are latently infected, and explanted KS cells rapidly lose viral infection upon culture, non-EC cell types are likely involved in the production of progeny virus. Likewise, explanted KS cells require the provision of exogenous inflammatory cytokines for growth. Therefore, the known autocrine and paracrine signaling pathways active in KS cells are not sufficient to drive tumor progression. Based on our complementary in vitro and in vivo observations we propose that MCs may play a role in KS pathogenesis by acting as a long-lived reservoir for KSHV and by chronic release of pro-inflammatory compounds. A proposed model for the role of MCs in KS is shown in Figure 5.
Figure 5. Model for the role of mast cells in KSHV-induced Kaposi’s sarcoma.
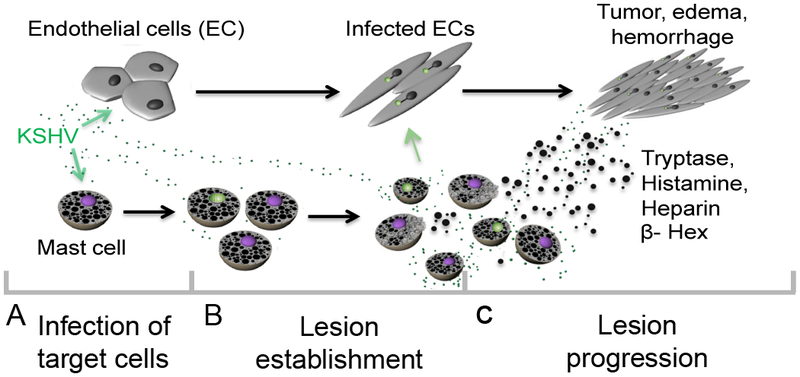
MCs support both latent and lytic KSHV infection in vivo; in vitro our data suggest completion of the cycle with release infectious virus able to establish latency in primary infected ECs. By contrast, primary KSHV infection of EC results in the establishment of viral latency soon after infection and is characterized by very limited viral gene expression and no viral progeny production. Infection induces major EC actin cytoskeleton changes resulting in spindle formation. Spindle cells proliferate in response to local inflammatory mediators and during proliferation some ECs lose the viral episome and eventually die. KS lesion maintenance and expansion must involve both a source of infectious virus capable of “re-seeding” the lesion, and a potent source of required paracrine inflammatory effectors. KSHV virus induces significant MC activation, both in culture and in vivo, with release of pro-tumourigenic, pro-inflammatory and pro-angiogenic granule contents, including the highly abundant tryptase, histamine and heparin, and the lysosomal enzyme β-hexosaminidase, into the cellular environment promoting oncogenesis via proliferation and survival of the latently infected ECs that compose the bulk of the tumor. Furthermore, concomitant release of heparin upon release of tryptase likely induces the edema and hemorrhage that are prominent characteristics of KS lesions.
MC support productive infection
Prior to lesion development, KSHV infection of target cells takes place (Figure 5A). Like ECs, MCs are fully permissive to KSHV infection in vitro (Figure 1). Expression of viral transcripts was rapid and sustained following infection of MC lines. MCs expressed viral transcripts by 6 hpi, similar to what was observed following primary EC infections (Figure 1A) (33). In contrast to the rapid establishment of latency in ECs following infection, however, MCs maintained expression of lytic transcripts at 72 hpi, while previous data suggest that virtually all lytic genes declined or were undetectable by 8 hpi in ECs (34). We also found that MCs maintained infection in vitro for at least 15 days (Figure 1B). This appears to be in contrast to latent infected primary ECs that lose the viral episome over time in continuous cultures, accompanied by a dramatic decline in LANA gene expression by 9 dpi with complete loss by 11 dpi (2,26). As early as 24 hpi MCs produced progeny virions capable of establishing latency in EC (Figure 1H and I) similar to what has been observed during primary infection of activated B cells (35) and in an in vivo in a model of acute cytomegalovirus infection (36). Similar to what has been observed in acute infection of B cells, we found MCs released maximal virus by 24hpi. We did not see an increase over time likely reflecting an equilibrium between those infected cells releasing virions, with subsequent infection occurring (perhaps at lower levels given the large volume the suspension cells are cultured in) and likely influenced by continual degradation in the culture culminating to yield a relatively constant viral titer. Together with identification of MCs expressing both latent and lytic viral proteins in KS biopsy samples (Figure 1E-G), these data demonstrate that MCs support productive viral infection and provide evidence that MCs may help maintain a reservoir of KSHV within lesions. How MCs become infected is unknown, though our observations support direct interactions of MCs and K8.1+ plasma cells (Figure 1E). These data suggest a possible route of infection via MC expression of α3β1 (CD49c/29) integrins (37), and/or via xCT (38), known KSHV entry receptors (39,40).
MCs accumulate in KS tissue
Immunohistochemical analysis of KS biopsies taken from diverse tissues and from lesions at different stages of development consistently demonstrated an enrichment of MCs in KS lesions compared with normal tissue (Figures 2 and 3). This accumulation is likely driven by both recruitment of bone marrow-derived CD34+ MCs and local proliferation of tissue resident MCs. KS lesions are rich in MC chemotactic factors, including platelet-derived growth factor-AB, vascular endothelial cell growth factor (VEGF, which is also present in MC granules), basic fibroblast growth factor, stromal derived factor-1 (41 ,42), stem cell factor (SCF), and CCL5. Moreover, KSHV encodes a homolog of cellular CCL2, K4; CCL2 is essential for recruitment and activation of MCs in adenocarcinoma-associated malignant pleural effusion (16). Our data demonstrate that in vitro infected MCs express K4 (Figure 1A). In addition, lesions are rich in COX-2 derived lipid mediators including PGE2 and LTB4, known to be produced by both activated MCs (43–45) and KSHV-infected ECs (46). LTB4 is a potent chemoattractant for immature MCs and bone marrow derived MC progenitors (45) and PGE2 induces the release of CCL2 and VEGF (44). Thus, infection with KSHV may induce MC chemotaxis to the developing lesions via both direct viral gene expression and by induction of potent chemotactic signals (Figure 5B).
KSHV activates MCs
MCs initiate inflammatory responses, in part, by degranulation, whereby pre-formed inflammatory mediators are rapidly discharged into the local environment. Infection of MCs in vitro induced a rapid, dose-dependent release of one granule-associated substance β-hexosaminidase (Figure 2A and B). MC degranulation appears to depend on expression of viral genes, as there was no detectable increase in release of β-hexosaminidase following infection with UV-inactivated virus. By IHC we also found widespread activation of MC in KS biopsies (Figures 2, 3, and 4). In plasma samples from patients with AIDS-associated and Classic KS we detected significantly higher systemic levels of two other granule products (tryptase and the histamine metabolite N-methylhistamine) compared with healthy controls (Figure 3 I and J). The plasma levels of these granule products in patients with KS are similar to those seen in patients with dengue fever (47), mastocytosis, anaphylaxis (48), and MC activation disease (31). These MC products have very short half-lives (i.e., tryptase 2h; histamine 1 min). Therefore, identification of anaphylaxis-level plasma concentrations of these inflammatory mediators in randomly acquired samples suggests that patients with KS have robust and sustained activation of MC.
The known effects of MC granule products are consistent with some of the key histological features of KS lesions. Histamine, for example, is a potent vasodilator, and along with dysregulation of EC adhesion molecules, may contribute to the vascular leak and tissue edema in KS lesions. Histamine is also known to induce proliferation of malignant melanoma (49). And despite an abundance of the pro-coagulant tissue factor within KS lesions there is minimal activation of the clotting cascade demonstrated by limited deposition of fibrin (50). Heparin, one of the most abundant granule products, is a potent anti-coagulant and likely contributes to the “hemorrhagic sarcoma” phenotype of KS. Heparin is also able to bind secreted growth factors and viral particles, possibly limiting diffusion out of lesional tissue. Sustained release of these heparin-bound products by the enzymatic activity of tryptase, including the MC granule component VEGF, may contribute to ongoing proliferation of spindle cells and EC available for further rounds of viral infection. It is possible, therefore, that the combined release of progeny virions and granule contents contributes to lesion progression (Figure 5C). Though not directly assessed in this study, there is tight overlap between the known effects of MC granule components and the composition of the KS lesional microenvironment. Studies are underway to evaluate the effects of individual MC granule components on EC infected with KSHV.
Antagonism of MC granule contents has anti-KS effects
Our model of MC contribution to the establishment and progression of KS lesions is supported by our demonstration of elevated plasma levels of MC granule contents and the rapid and durable regression of KS lesions in a patient with AIDS-associated KS treated with MC antagonists (Figure 4). While this is the first demonstration of an association between MCs and KS, MCs are known to contribute to the pathogenesis of myriad tumor types, including MPE, primary cutaneous lymphoma, papilloma virus-associated squamous cell carcinomas, and pancreatic β-cell tumors. Likewise, there is precedence for an anti-tumor response with agents that antagonize MC survival or degranulation. Sodium cromolyn, an MC stabilizer that inhibits degranulation, blocks maintenance and progression of pancreatic β-cell tumors. Treatment of mice with the c-kit inhibitor imatinib mesylate significantly limited MPE formation.
Along with granule content markers, the other proto-typical marker for MCs is C-kit/CD117. SCF binding triggers CD117 signaling, which is critical for the differentiation, migration, maturation, and survival of MCs (51). C-kit/CD117 activity is the target for treatment with the c-kit tyrosine kinase inhibitor imatinib mesylate and is used in the treatment of mastocytosis patients (52). Detection of CD117 staining identified both granulated MCs and ‘ghost’ or granule-depleted MCs within KS lesions independent of granule content. Importantly, fully degranulated MCs remain viable, slowly re-granulating over a period of days (53). These cells are then able to undergo the entire process of activation and degranulation again, making them particularly potent effectors. One may envisage a MC activation and degranulation response in KS disease, followed by re-granulation and subsequent degranulation thereby providing a continuous source of paracrine inflammatory mediators that help drive KS development and maintenance.
Previous work demonstrated CD117 expression in vitro on latently infected dermal ECs (43,54) and on cells present in large numbers in KS lesions, with 93% of CD117+ lesions LANA+ (55). Clinical trials testing the efficacy of Gleevec/imatinib mesylate in the treatment of KS showed promise, with demonstrated clinical and histological regression of cutaneous KS lesions (56). Interestingly, all reports found no correlation with KSHV LANA and CD117/c-kit expression, suggesting that the main proliferating component of lesions, the KS LANA+ EC spindle cells were not the predominate source of the c-kit signal. In this study, we did not observe CD117+ staining on KS spindle cells using conditions that readily stained MCs. It is interesting to speculate that perhaps the reason why LANA positivity was not correlated with CD117 staining was that the CD117+ cells detected were MCs, a subset of which we have shown are KSHV LANA+. Activated MC can have an elongated phenotype and therefore some of the cKit-positive spindloid cells in KS biopsies may in fact be MCs rather than ECs. In comparison to the accumulated bundles of LANA+ KS EC spindle cells in later lesions, the signal from the CD117+ LANA+MC would be small.
Reconstitution of immune competence and suppression of viral load is the first goal of therapy for patients with KS. However, our study results suggest that modulation of MC activity could potently alter KS tumorigenesis irrespective of a significant immunological and virological response to ARVs. Safe, widely available MC stabilizing agents that are used to treat MC activation and allergic disease could provide a cost-effective approach to reducing both lesion inflammation and angiogenesis. Thus, modifying MC activation may represent a therapeutic strategy amenable for use in KS-prevalent, resource-constrained settings like sub-Saharan Africa.
Supplementary Material
{kind=link}
Statement of translational relevance.
Inflammation is a key driver of oncogenesis and modulation of inflammation in the tumor microenvironment is emerging as a viable therapeutic approach. Kaposi’s sarcoma (KS), one of the most commonly diagnosed and aggressive cancers in sub-Saharan Africa, is an unusual cancer, dependent on infection with KS herpesvirus (KSHV) and on inflammatory mediators. Despite extensive study the key cellular players and paracrine inflammatory mediators driving the disease are incompletely understood. Here we identify a previously unrecognized role for potent pro-inflammatory mast cells (MCs) in KS. MCs are localized, extensively activated, and infected by KSHV in KS tumors and their clinical importance is underscored by the successful treatment of a case of AIDS-KS with anti-MC therapy. These data strongly suggest a specific role for MCs in KSHV-driven oncogenesis and identify MC antagonists as a promising novel, pathogenesis-targeted therapeutic approach to KS. Our work adds to the growing body of knowledge on the role of MCs in oncogenesis, and demonstrates the efficacy of using widely available MC-directed therapies for treatment of KS.
Acknowledgements
The work was supported by Young Investigator Pilot Award from the AIDS and Cancer Specimen Resource NCI (ACSR) UM1 CA181255 sub award 7854SC to C King) and grant support from the NIH/NIAID (R03 AI122221to C King), by The Ohio State University (CCC-OSU 5OD160–201467 to L Ayers), by funding from the NIH (P30 AI027763, UM1 CA181255, U54 CA190153, R01 CA119903 to J Martin) and (R01 CA102667) to R Rochford. A Barbachano-Guerrero is a fellow of the Mexican National Council on Science and Technology (CONACYT). We thank David Nohle, David Kellough and Dr. Mamut Akgul for their expert technical assistance with this project.
Footnotes
The authors declare they have no conflicts of interest.
REFERENCES
- 1.Ganem D KSHV infection and the pathogenesis of Kaposi’s sarcoma. Annu Rev Pathol 2006;1:273–96 doi 10.1146/annurev.pathol.1.110304.100133. [DOI] [PubMed] [Google Scholar]
- 2.Grundhoff A, Ganem D. Inefficient establishment of KSHV latency suggests an additional role for continued lytic replication in Kaposi sarcoma pathogenesis. J Clin Invest 2004;113(1):124–36 doi 10.1172/JCI17803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hagiwara K, Khaskhely NM, Uezato H, Nonaka S. Mast cell “densities” in vascular proliferations: a preliminary study of pyogenic granuloma, portwine stain, cavernous hemangioma, cherry angioma, Kaposi’s sarcoma, and malignant hemangioendothelioma. Journal of Dermatology 1999;26(9):577–86. [DOI] [PubMed] [Google Scholar]
- 4.Maaninka K, Lappalainen J, Kovanen PT. Human mast cells arise from a common circulating progenitor. Journal of Allergy and Clinical Immunology 2013;132(2):463–9 e3 doi S0091–6749(13)00320–5 [pii] 10.1016/j.jaci.2013.02.011. [DOI] [PubMed] [Google Scholar]
- 5.Galli SJ, Nakae S, Tsai M. Mast cells in the development of adaptive immune responses. Nat Immunol 2005;6(2):135–42 doi ni1158 [pii] 10.1038/ni1158. [DOI] [PubMed] [Google Scholar]
- 6.Metz M, Maurer M. Mast cells--key effector cells in immune responses. Trends Immunol 2007;28(5):234–41 doi S1471–4906(07)00071–3 [pii] 10.1016/j.it.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 7.Metz M, Grimbaldeston MA, Nakae S, Piliponsky AM, Tsai M, Galli SJ. Mast cells in the promotion and limitation of chronic inflammation. Immunol Rev 2007;217:304–28 doi IMR520 [pii] 10.1111/j.1600-065X.2007.00520.x. [DOI] [PubMed] [Google Scholar]
- 8.Kunder CA, St John AL, Abraham SN. Mast cell modulation of the vascular and lymphatic endothelium. Blood 2011;118(20):5383–93 doi blood-2011–07-358432 [pii] 10.1182/blood-2011-07-358432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown MG, Hermann LL, Issekutz AC, Marshall JS, Rowter D, Al-Afif A , et al. Dengue virus infection of mast cells triggers endothelial cell activation. Journal of Virology 2011;85(2):1145–50 doi JVI.01630–10 [pii] 10.1128/JVI.01630-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.King CA, Anderson R, Marshall JS. Dengue virus selectively induces human mast cell chemokine production. J Virol 2002;76(16):8408–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.King CA, Marshall JS, Alshurafa H, Anderson R. Release of vasoactive cytokines by antibody-enhanced dengue virus infection of a human mast cell/basophil line. J Virol 2000;74(15):7146–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Graham AC, Hilmer KM, Zickovich JM, Obar JJ. Inflammatory response of mast cells during influenza A virus infection is mediated by active infection and RIG-I signaling. Journal of Immunology 2013;190(9):4676–84 doi jimmunol.1202096 [pii] 10.4049/jimmunol.1202096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maltby S, Khazaie K, McNagny KM. Mast cells in tumor growth: angiogenesis, tissue remodelling and immune-modulation. Biochim Biophys Acta 2009;1796(1):19–26 doi S0304–419X(09)00005–5 [pii] 10.1016/j.bbcan.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stoecker MM, Wang E. Systemic mastocytosis with associated clonal hematologic nonmast cell lineage disease: a clinicopathologic review. Arch Pathol Lab Med 2012;136(7):832–8 doi 10.5858/arpa.2011-0325-RS. [DOI] [PubMed] [Google Scholar]
- 15.Saadalla AM, Osman A, Gurish MF, Dennis KL, Blatner NR, Pezeshki A , et al. Mast cells promote small bowel cancer in a tumor stage-specific and cytokine-dependent manner. Proc Natl Acad Sci U S A 2018;115(7):1588–92 doi 10.1073/pnas.1716804115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giannou AD, Marazioti A, Spella M, Kanellakis NI, Apostolopoulou H, Psallidas I , et al. Mast cells mediate malignant pleural effusion formation. J Clin Invest 2015;125(6):2317–34 doi 10.1172/JCI79840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rabenhorst A, Schlaak M, Heukamp LC, Forster A, Theurich S, von Bergwelt-Baildon M , et al. Mast cells play a protumorigenic role in primary cutaneous lymphoma. Blood 2012;120(10):2042–54 doi 10.1182/blood-2012-03-415638. [DOI] [PubMed] [Google Scholar]
- 18.Glimelius I, Edstrom A, Fischer M, Nilsson G, Sundstrom C, Molin D , et al. Angiogenesis and mast cells in Hodgkin lymphoma. Leukemia 2005;19(12):2360–2 doi 10.1038/sj.leu.2403992. [DOI] [PubMed] [Google Scholar]
- 19.Mizuno H, Nakayama T, Miyata Y, Saito S, Nishiwaki S, Nakao N , et al. Mast cells promote the growth of Hodgkin’s lymphoma cell tumor by modifying the tumor microenvironment that can be perturbed by bortezomib. Leukemia 2012. doi leu201281 [pii] 10.1038/leu.2012.81. [DOI] [PubMed] [Google Scholar]
- 20.Sheng-Hsuan Chien Y-CL, Hong Ying-Chung,, Yang Ching-Fen, Liu Chun-Yu, Chiou Tzeon-Jye, Tzeng Cheng-Hwai, Liu Jin-Hwang, Gau Jyh-Pyng. Diffuse large B cell lymphoma coexistence with systemic mastocytosis. Journal of Cancer Research and Practice 2016;in press doi doi: 10.1016/j.jcrpr.2015.09.001. [DOI] [Google Scholar]
- 21.King CA. Kaposi’s Sarcoma-Associated Herpesvirus Kaposin B Induces Unique Monophosphorylation of STAT3 at Serine 727 and MK2-Mediated Inactivation of the STAT3 Transcriptional Repressor TRIM28. Journal of Virology 2013;87(15):8779–91 doi JVI.02976–12 [pii] 10.1128/JVI.02976-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parsons CH, Adang LA, Overdevest J, O’Connor CM, Taylor JR Jr., Camerini D , et al. KSHV targets multiple leukocyte lineages during long-term productive infection in NOD/SCID mice. J Clin Invest 2006;116(7):1963–73 doi 10.1172/JCI27249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Punjabi AS, Carroll PA, Chen L, Lagunoff M. Persistent activation of STAT3 by latent Kaposi’s sarcoma-associated herpesvirus infection of endothelial cells. J Virol 2007;81(5):2449–58 doi JVI.01769–06 [pii] 10.1128/JVI.01769-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pollock JL, Presti RM, Paetzold S, Virgin HWt. Latent murine cytomegalovirus infection in macrophages. Virology 1997;227(1):168–79 doi S0042682296983038 [pii]. [DOI] [PubMed] [Google Scholar]
- 25.Schwartz LB, Austen KF, Wasserman SI. Immunologic release of beta-hexosaminidase and beta-glucuronidase from purified rat serosal mast cells. J Immunol 1979;123(4):1445–50. [PubMed] [Google Scholar]
- 26.Mutlu AD, Cavallin LE, Vincent L, Chiozzini C, Eroles P, Duran EM , et al. In vivo-restricted and reversible malignancy induced by human herpesvirus-8 KSHV: a cell and animal model of virally induced Kaposi’s sarcoma. Cancer Cell 2007;11(3):245–58 doi 10.1016/j.ccr.2007.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roizman B The Herpesviruses Wagner RR, editor. New York: Plenum Press; 1982. [Google Scholar]
- 28.Theoharides TC, Kempuraj D, Sant GR. Massive extracellular tryptase from activated bladder mast cells in interstitial cystitis. Urology 2001;58(4):605–6. [DOI] [PubMed] [Google Scholar]
- 29.Schwartz LB, Atkins PC, Bradford TR, Fleekop P, Shalit M, Zweiman B. Release of tryptase together with histamine during the immediate cutaneous response to allergen. J Allergy Clin Immunol 1987;80(6):850–5. [DOI] [PubMed] [Google Scholar]
- 30.Borer-Reinhold M, Haeberli G, Bitzenhofer M, Jandus P, Hausmann O, Fricker M , et al. An increase in serum tryptase even below 11.4 ng/mL may indicate a mast cell-mediated hypersensitivity reaction: a prospective study in Hymenoptera venom allergic patients. Clinical and Experimental Allergy 2011;41(12):1777–83 doi 10.1111/j.1365-2222.2011.03848.x. [DOI] [PubMed] [Google Scholar]
- 31.Molderings GJ, Brettner S, Homann J, Afrin LB. Mast cell activation disease: a concise practical guide for diagnostic workup and therapeutic options. J Hematol Oncol 2011;4:10 doi 1756–8722-4–10 [pii] 10.1186/1756-8722-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang Z, Esebua M, Layfield L. The role of mast cells in histologically “normal” appendices following emergency appendectomy in pediatric patients. Ann Diagn Pathol 2016;24:1–3 doi 10.1016/j.anndiagpath.2016.06.003. [DOI] [PubMed] [Google Scholar]
- 33.Sharma-Walia N, Naranatt PP, Krishnan HH, Zeng L, Chandran B. Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 envelope glycoprotein gB induces the integrin-dependent focal adhesion kinase-Src-phosphatidylinositol 3-kinase-rho GTPase signal pathways and cytoskeletal rearrangements. J Virol 2004;78(8):4207–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krishnan HH, Naranatt PP, Smith MS, Zeng L, Bloomer C, Chandran B. Concurrent expression of latent and a limited number of lytic genes with immune modulation and antiapoptotic function by Kaposi’s sarcoma-associated herpesvirus early during infection of primary endothelial and fibroblast cells and subsequent decline of lytic gene expression. J Virol 2004;78(7):3601–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rappocciolo G, Hensler HR, Jais M, Reinhart TA, Pegu A, Jenkins FJ , et al. Human herpesvirus 8 infects and replicates in primary cultures of activated B lymphocytes through DC-SIGN. J Virol 2008;82(10):4793–806 doi 10.1128/JVI.01587-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Becker M, Lemmermann NA, Ebert S, Baars P, Renzaho A, Podlech J , et al. Mast cells as rapid innate sensors of cytomegalovirus by TLR3/TRIF signaling-dependent and -independent mechanisms. Cell Mol Immunol 2015;12(2):192–201 doi 10.1038/cmi.2014.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tachimoto H, Hudson SA, Bochner BS. Acquisition and alteration of adhesion molecules during cultured human mast cell differentiation. Journal of Allergy and Clinical Immunology 2001;107(2):302–9 doi S0091–6749(01)07664–3 [pii] 10.1067/mai.2001.111930. [DOI] [PubMed] [Google Scholar]
- 38.Ghannadan M, Baghestanian M, Wimazal F, Eisenmenger M, Latal D, Kargul G , et al. Phenotypic characterization of human skin mast cells by combined staining with toluidine blue and CD antibodies. J Invest Dermatol 1998;111(4):689–95 doi 10.1046/j.1523-1747.1998.00359.x. [DOI] [PubMed] [Google Scholar]
- 39.Akula SM, Pramod NP, Wang FZ, Chandran B. Integrin alpha3beta1 (CD 49c/29) is a cellular receptor for Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8) entry into the target cells. Cell 2002;108(3):407–19 doi S0092867402006281 [pii]. [DOI] [PubMed] [Google Scholar]
- 40.Kaleeba JA, Berger EA. Kaposi’s sarcoma-associated herpesvirus fusion-entry receptor: cystine transporter xCT. Science 2006;311(5769):1921–4 doi 10.1126/science.1120878. [DOI] [PubMed] [Google Scholar]
- 41.Yao L, Salvucci O, Cardones AR, Hwang ST, Aoki Y, De La Luz Sierra M , et al. Selective expression of stromal-derived factor-1 in the capillary vascular endothelium plays a role in Kaposi sarcoma pathogenesis. Blood 2003;102(12):3900–5 doi 10.1182/blood-2003-02-0641 2003-02-0641 [pii] [DOI] [PubMed] [Google Scholar]
- 42.Lin TJ, Issekutz TB, Marshall JS. Human mast cells transmigrate through human umbilical vein endothelial monolayers and selectively produce IL-8 in response to stromal cell-derived factor-1 alpha. J Immunol 2000;165(1):211–20 doi ji_v165n1p211 [pii]. [DOI] [PubMed] [Google Scholar]
- 43.Moses AV, Jarvis MA, Raggo C, Bell YC, Ruhl R, Luukkonen BG , et al. Kaposi’s sarcoma-associated herpesvirus-induced upregulation of the c-kit proto-oncogene, as identified by gene expression profiling, is essential for the transformation of endothelial cells. J Virol 2002;76(16):8383–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nakayama T, Mutsuga N, Yao L, Tosato G. Prostaglandin E2 promotes degranulation-independent release of MCP-1 from mast cells. J Leukoc Biol 2006;79(1):95–104 doi 10.1189/jlb.0405226. [DOI] [PubMed] [Google Scholar]
- 45.Weller CL, Collington SJ, Brown JK, Miller HR, Al-Kashi A, Clark P , et al. Leukotriene B4, an activation product of mast cells, is a chemoattractant for their progenitors. J Exp Med 2005;201(12):1961–71 doi 10.1084/jem.20042407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sharma-Walia N, Paul AG, Bottero V, Sadagopan S, Veettil MV, Kerur N , et al. Kaposi’s sarcoma associated herpes virus (KSHV) induced COX-2: a key factor in latency, inflammation, angiogenesis, cell survival and invasion. PLoS Pathog 2010;6(2):e1000777 doi 10.1371/journal.ppat.1000777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Furuta T, Murao LA, Lan NT, Huy NT, Huong VT, Thuy TT , et al. Association of mast cell-derived VEGF and proteases in Dengue shock syndrome. PLoS Negl Trop Dis 2012;6(2):e1505 doi 10.1371/journal.pntd.0001505. PNTD-D-11-00422 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schwartz LB, Metcalfe DD, Miller JS, Earl H, Sullivan T. Tryptase levels as an indicator of mast-cell activation in systemic anaphylaxis and mastocytosis. N Engl J Med 1987;316(26):1622–6 doi 10.1056/NEJM198706253162603. [DOI] [PubMed] [Google Scholar]
- 49.Falus A, Hegyesi H, Lazar-Molnar E, Pos Z, Laszlo V, Darvas Z. Paracrine and autocrine interactions in melanoma: histamine is a relevant player in local regulation. Trends Immunol 2001;22(12):648–52. [DOI] [PubMed] [Google Scholar]
- 50.Zhang R, Tsai FY, Orkin SH. Hematopoietic development of vav−/− mouse embryonic stem cells. Proc Natl Acad Sci U S A 1994;91(26):12755–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Galli SJ, Tsai M, Wershil BK, Tam SY, Costa JJ. Regulation of mouse and human mast cell development, survival and function by stem cell factor, the ligand for the c-kit receptor. Int Arch Allergy Immunol 1995;107(1–3):51–3. [DOI] [PubMed] [Google Scholar]
- 52.Akin C, Fumo G, Yavuz AS, Lipsky PE, Neckers L, Metcalfe DD. A novel form of mastocytosis associated with a transmembrane c-kit mutation and response to imatinib. Blood 2004;103(8):3222–5 doi 10.1182/blood-2003-11-3816. [DOI] [PubMed] [Google Scholar]
- 53.Dvorak AM, Kissell S. Granule changes of human skin mast cells characteristic of piecemeal degranulation and associated with recovery during wound healing in situ. J Leukoc Biol 1991;49(2):197–210. [DOI] [PubMed] [Google Scholar]
- 54.Douglas JL, Whitford JG, Moses AV. Characterization of c-Kit expression and activation in KSHV-infected endothelial cells. Virology 2009;390(2):174–85 doi S0042–6822(09)00301–8 [pii] 10.1016/j.virol.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pantanowitz L, Schwartz EJ, Dezube BJ, Kohler S, Dorfman RF, Tahan SR. C-Kit (CD117) expression in AIDS-related, classic, and African endemic Kaposi sarcoma. Appl Immunohistochem Mol Morphol 2005;13(2):162–6 doi 00129039200506000–00009 [pii]. [DOI] [PubMed] [Google Scholar]
- 56.Koon HB, Bubley GJ, Pantanowitz L, Masiello D, Smith B, Crosby K , et al. Imatinib-induced regression of AIDS-related Kaposi’s sarcoma. Journal of Clinical Oncology 2005;23(5):982–9 doi JCO.2005.06.079 [pii] 10.1200/JCO.2005.06.079. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.