

Abstract
The use of replication-competent viruses as oncolytic agents is rapidly expanding, with several oncolytic viruses approved for cancer therapy. As responses to therapy are highly variable, understanding the dynamics of therapy is critical for optimal application of virotherapy in practice. Although mathematical models have been developed to understand the dynamics of tumor virotherapy, a scarcity of in vivo data has made difficult parametrization of these models. To tackle this problem, we studied the in vitro and in vivo spread of two oncolytic measles viruses that induce expression of the sodium iodide symporter (NIS) in cells. NIS expression enabled infected cells to concentrate radioactive isotopes that could be reproducibly and quantitatively imaged using SPECT/CT. We observed a strong linear relationship in vitro between infectious virus particles, viral N and NIS gene expression, and radioactive isotope uptake. In vivo radioisotope uptake was highly correlated with viral N and NIS gene expression. Similar expression patterns between viral N and NIS gene expression in vitro and in vivo implied that the oncolytic virus behaved similarly in both scenarios. Significant titers of viable virus were consistently isolated from tumors explanted from mice that had been injected with oncolytic MV. We observed a weaker but positive in vivo relationship between radioisotope uptake and the viable virus titer recovered from tumors; this was likely due to anisotropies in the viral distribution in vivo. These data suggest that methods that enable quantitation of in vivo anisotropies are required for continuing development of oncolytic virotherapy.
INTRODUCTION
The introduction of oncolytic viruses for cancer therapy has provided a novel avenue of therapy that is showing increasingly promising results (1–3). Oncolytic viruses have been engineered to selectively infect and spread within the tumor cell population leading to cancer cell death due to a variety of mechanisms including direct cell lysis, cell-to-cell fusion, expression of therapeutic genes or enzymes that leads to the activation of toxic molecules, and stimulation of the immune system with breakdown of immune tolerance (4–8). The first proof of principle demonstrating successful therapy of a disseminated malignancy (multiple myeloma) with a single, systemic injection of an engineered oncolytic measles virus (MV-NIS) was recently reported (9). Oncolytic viruses are already approved for the therapy of head and neck cancer and malignant melanoma (10), and other indications likely will soon follow (11). Successful tumor therapy with oncolytic viruses is based on the premise that the virus gains access to the tumor and then selectively proliferates within the tumor cell population leading to its destruction (12). However, the outcomes in animal models and human studies have been variable. Oncolytic viruses that reliably eliminate tumor cell line populations in vitro give variable results in vivo when the same cells are used to generate tumor xenografts (13, 14). It is critical to understand the dynamics of the virus within the tumor if we want to make sense of the outcome of therapy and optimize the use of these novel therapeutic agents. Mathematical models of tumor virotherapy have provided important insights into the dynamics of tumor therapy with viruses (15–21). However, given the scarcity of in vivo data available, approaches that enable the in vivo quantitation of the tumor and/or virus populations would be highly beneficial to allow for proper parameterization of such models. In this regard, strategies that enable repeated and non-invasive monitoring of the biodistribution of the oncolytic virus and reliable estimates of the in vivo virus and infected cell populations are critical for successful translation of these novel therapeutics. MV-NIS is a unique oncolytic virus in this respect since expression of the sodium iodide symporter (NIS) by infected tumor cells may enable visualization of the in vivo biodistribution of the oncolytic virus, and allow for a quantitative determination of virus population dynamics within the tumor (9, 22, 23). In this work, we report our results on inferring the oncolytic virus population within a tumor in vitro and in vivo using molecular imaging techniques. Although our approach works well both in vitro and in vivo, additional complexities in vivo likely require a combination of imaging techniques to enable an accurate estimation of virus populations.
MATERIALS AND METHODS
Cell lines
All cell lines were obtained from the ATCC (Manassas, VA) and maintained in their respective media at 37°C in an environment with 5% carbon dioxide. Vero cells (ATCC# CRL-1586) were grown in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, Grand Island, NY) with 5% fetal bovine serum (FBS). The human pancreatic adenocarcinoma cell line BxPC3 (ATCC# CRL-1687) was cultured in RPMI with 10% FBS. The ATCC guarantees the authenticity of the cell lines. The cell lines are kept in culture after thawing for a maximum of 2 months (10 passages) and after that a new vial is thawed to maintain a low passage. The cells are routinely monitored for mycoplasma infection by polymerase chain reaction (IDEXX BioResearch, Columbia, MO) and were consistently negative. Prior to injection in animals, all cells were washed in sterile cold phosphate buffered saline (PBS) and suspended at a concentration of 1 × 107 cells/100μL.
Viruses
The oncolytic virus MV-NIS has been described previously (22, 24). It is a recombinant virus based on the Edmonston vaccine strain of measles virus (MV-Edm) with the gene for the human sodium iodide symporter (NIS) inserted downstream of the viral hemagglutinin gene.
MV-eGFP-NIS was generated by digestion of plasmids p(+)MV-eGFP and p(+) MV-NIS with NotI and Sac II (New England Biolabs, Ipswich, MA). The smaller fragment from p(+)MV-eGFP (5712 bp) and the larger fragment from p(+)MV-NIS (16125 bp) were isolated by gel electrophoresis and ligated overnight at room temperature. The recombinant plasmid was amplified in E coli (Top 10) and used to rescue the recombinant virus by transfection in 293–346 cells together with pEMCLa (1 ug) using calcium phosphate precipitation as previously described (22, 25). After 48 hours, the transfected cells were overlaid on Vero cells and rescue inferred by the formation of syncytia that were green under ultraviolet light.
MV-I98A-NIS is a recombinant virus that expresses both the enhanced green fluorescent protein (eGFP) and NIS. In addition has a mutation in the viral hemagglutinin (H) gene where isoleucine 98 is replaced by alanine (I98A) (25). The result is a virus that does not lead to significant cell-to-cell fusion as described previously (25). MV-I98A-NIS was generated by substitution of the wild type H gene with the mutant gene using Pac1 and Spe1 to digest pCG-H-I98A and p(+)MV-eGFP-NIS. The recombinant plasmid was used to rescue the virus as described above (22, 25).
All recombinant viruses were propagated by infection of Vero cells at an MOI of 0.03 and cell associated virus was harvested in Opti-MEM (Gibco) when 80% of the cells were infected. The cell-associated virus was released by freeze-thawing of the cell preparation three times in liquid nitrogen followed by centrifugation to precipitate cellular debris. The virus containing supernatant was harvested and the viral titer was determined by serial logarithmic dilutions of the virus containing supernatant that were used to infect Vero cells in a 96 well plate. The 50% tissue culture infective dose (TCID50/ml) was determined 4 days later using the method of Spearman and Karber (26–28). Virus stocks were maintained at −80°C until used for in vitro or in vivo experiments.
In vitro iodide uptake studies
In vitro iodide uptake studies were performed as previously described (22, 29). Cell lines were infected with MV-NIS, MV-I98A-NIS or MV-Edm (control) at an MOI of 0.03, 0.1, 1 and 3 and iodide uptake determined at specific time intervals by washing the infected cells with Hank’s balanced salt solution (HBSS) supplemented with HEPES (10mM), pH 7.3, and cold potassium iodide (100μM). In half of the wells, the specific NIS inhibitor potassium perchlorate (100μM) was added and in all wells Na125I was added (activity of 1 × 105 cpm/0.1mL) and incubated for 45 minutes at 37°C. Subsequently, the cells were washed twice with ice cold HBSS and the retained activity measured in a gamma counter (Isodata 20/10 Gamma Counter, ICN Biomedicals, Inc., Fresno, CA). Uninfected cells served as controls. All experiments were performed in triplicate and set up simultaneously. At each specific time point, triplicate samples were selected for the in vitro iodide uptake studies as well as viral titer and gene expression measurements.
Q-RT-PCR
RNA was extracted from control (uninfected) or infected cell lines (5 ×106) using the RNEasy kit (Qiagen, Valencia, CA) as recommended by the manufacturer. RNA from tumor samples harvested from animals at the time of autopsy was isolated using the same approach although the tumor samples were immediately placed in RNAlater (Qiagen) and stored according to the manufacturer’s recommendations prior to RNA isolation. Five hundred nanogram RNA was used for reverse transcription (RT) reaction using random hexamers and SuperScript III reverse transcriptase (Invitrogen) in a 30 μl reaction. Five microliter of the resulting cDNA was used for real-time PCR using TaqMan gene expression assay for NIS or Measles N according to the manufacturer’s instructions.
Generation of RNA for standard curve: A plasmid encoding the measles virus N gene, under control of a T7 promoter, pTM1-MV-N was obtained from Dr. R. Cattaneo (Mayo Clinic, Rochester, MN). This plasmid was linearized by digestion with XhoI (New England Biolabs) in CutSmart Buffer for 90 minutes at 37°C in 20μl reaction volume. 40 μl of 100% ethanol was added to the reaction and it was placed at −20°C for a minimum of 20 minutes. The DNA was centrifuged at 14,000 RPM for 15 minutes. The supernatant was removed and the pelleted DNA was re-suspended in RNAse free water. In vitro transcription was performed using the linearized pTM1-MV-N as a template according to the protocol for Life Technologies MEGAscript T7 in vitro transcription Kit (AM133). DNA was removed by addition of DNAturbo. The generated RNA was purified using a MEGAclear kit (Life Technologies AM1908) and quantified using a Nanodrop and aliquoted to avoid multiple free thaw cycles. All samples were stored at −20°C and used to generate a standard curve.
Quantitative reverse transcriptase polymerase chain reaction (Q-RT-PCR) was performed as previously described (23) using the Roche Master Hydrolysis Probes kit (Catalog # 4991885001). Primers to detect NIS were as follows: forward primer 5`-GCCCCGGGATTGTTGTGG-3` and reverse primer 5`CCAGTGGGGAGTTCTT CAGGAC-3` and probe labeled 5`-/56-FAM-/GGATGGCCA/ZEN/CTTCTTCTT TGG/3IABkFQ/−3` manufactured by IDT. Primers to detect MV-N RNA were forward primer 5`-AGAAGCCAGGGAGAGCTACAGA-3` and reverse primer 5`GGGTGTGC CGGTTGGA-3’ and probe 5`-/56-FAM/TGGGCAG CT/ZEN/CTCGCATCACTT GC/3IABkFQ/−3`. The reaction was run on Roche LightCycler 480 II Real Time PCR Instrument in 96 well plates.
In vivo studies
All procedures involving animals were approved by the Institutional Animal Care and Use Committee of Mayo Foundation. BxPC-3 human pancreatic carcinoma cells (106) were implanted in the flank of nude mice (6 week old females BALB/c nu/nu mice, Taconic) (30). Each group consisted of 6 mice: 2 served as controls for background correction and 4 were injected with either virus (MV-NIS or MV-I98A-NIS). When tumors reached approximately 5 mm in diameter, the mice were injected with Opti-MEM (control), MV-NIS or MV-I98A-NIS (1×107 TCID50/ml) via the tail vein. On days 3, 5, 8, 10, 12, 15, 17 and 19 after virus injection, mice from the control and virus injected groups were imaged with SPECT/CT (U-SPECT II, MILabs, Utrecht, The Netherlands) 1 hour after injection of 99mTc (314μCi) (11.6 MBq, range: 10.73 – 12.65MBq) under general anesthesia with inhaled isoflurane (1%) (n=6 per group). The residual activity in each syringe used to inject isotope in the mice was measured: median residual activity was 10.06 μCi (0.37 MBq, range: 0.13 – 0.78 MBq). Immediately afterwards, the mice were euthanized, the tumors excised, weighed and isotope activity measured with a Capintec CRC127 dose calibrator (Capintec, Florham Park, New Jersey). The tumors were divided into parts for histologic analysis and RNA extraction. NIS gene expression was determined by Q-RT-PCR and immunohistochemistry, while viral genome copy number was determined by Q-RT-PCR for the nucleoprotein gene (N) as well as NIS. Image analysis using region of interest (ROI) was performed using AMIDE (0.6) (amide.sourceforge.net) with background correction across all imaging planes as previously described (31, 32). Two independent scientists analyzed the imaging data, blinded to each other’s results.
Isotope quantification was performed with the U-SPECT system according to the manufacturer’s recommendations. A known volume with a known activity of each isotope that was utilized is scanned for a designated amount of time with its respective collimator. The data was reconstructed with parameters that were specific to the reconstruction that were used for the animal imaging studies. A volume of interest (VOI) was created and the corresponding statistics were used to determine the total sum voxel value. The quantification factor was determined by the activity in the vessel, corrected for time and decay, the total voxel sum and the specific voxel size. This factor was then tested by filling a second vessel with an exact volume, activity and time of activity measurement and scanned using the same parameters used for animal imaging. Statistics were generated from the VOI and the reported activity/volume was compared to the known vessel concentration. The estimated activity based on SPECT imaging was required to be within 5% of the known concentration.
Statistical considerations
All comparisons between groups were performed using the Kruskall-Wallis and Mann-Whitney tests with p<0.05 considered to be statistically significant. Correlations between quantitative data sets were performed with the Spearman’s correlation coefficient. All statistical studies were performed using GraphPad Prism Version 5 (La Jolla, CA).
RESULTS
Measles virus infection and NIS expression in vitro
Initially, we wanted to determine the relationship between oncolytic MV infection, expression of virus encoded proteins and isotope uptake in vitro. BxPC3 cells were infected at several multiplicities of infection (MOI of 0.03, 0.1, 1 and 3) with MV-NIS or MV-I98A-NIS. Starting 24 hours after infection, iodide uptake studies were performed as described in the Materials and Methods and continued daily. In parallel, cells were lysed and cell associated virus was released by freeze-thawing of the cells in liquid nitrogen 3 times and virus titers were determined. Similar cell conditions were used to isolate total cellular RNA to determine virus encoded gene expression using Q-RT-PCR. All experimental conditions for NIS mediated isotope uptake, viral titer and RNA extraction (to quantitate NIS and N gene expression) were in triplicate at each time point. We determined by Q-RT-PCR that BxPC-3 cells do not express endogenous NIS (3.15 × 10−3 copies/ng RNA). In contrast, cells infected (MOI = 1.0) with recombinant MV that include the NIS gene expressed the reporter gene (1.17 × 102 copies/ng for MV-NIS and 2.19 × 101/ng for MV-I98A-NIS respectively at 48 hours post infection, p < 0.0015 for all comparisons). As can be seen from Figure 1A, isotope uptake peaked at 48 hours after infection in the case of MV-NIS but the peak was not reached at 120 hours with MV-I98A-NIS (n=3 per time point per condition). The peak at 48 hours for MV-NIS also coincided with the highest levels of expression for the measles virus ‘N’ and ‘NIS’ (Figure 1B) gene expression (data for MV-NIS shown). The correlation between in vitro isotope uptake (μCi) and MV mediated NIS gene expression as measured by Q-RT-PCR was excellent – Spearman’s rho ρ = 0.979, 95% CI: 0.89, 0.99, R2 = 0.96, p < 0.0001 (Figure 1C). A high correlation between isotope uptake (activity) and viable virus as measured by TCID50/ml was also observed (Figure 1D).
Figure 1. In vitro dynamics of virus replication, spread and isotope uptake.
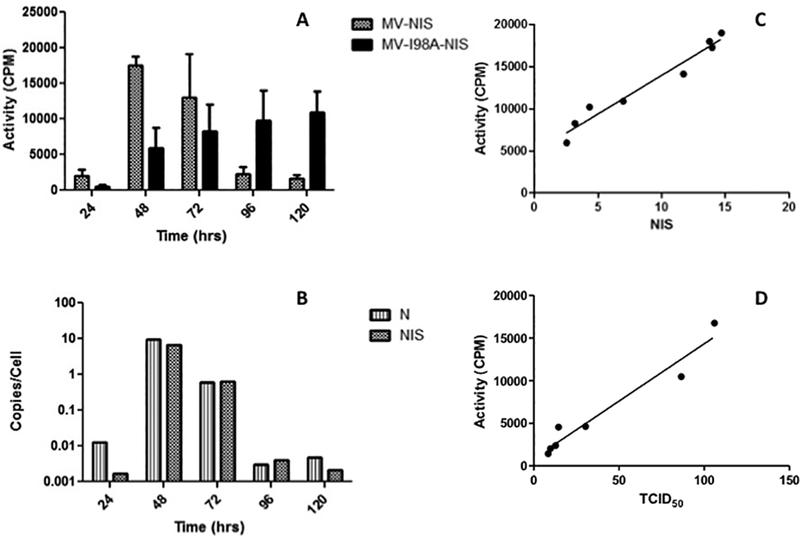
(A) After infection of BxPC3 tumor cells with MV-NIS or MV-I98A-NIS, pertechnetate uptake studies were performed serially. Isotope uptake peaked at 48 after MV-NIS infection but the peak with MV-I98A-NIS was later. Uninfected controls did not take up any significant isotope. (B) Q-RT-PCR on RNA isolated from cells in (A) quantifying MV induced ‘N’ and ‘NIS’ expression paralleled isotope uptake by the cells (data for MV-NIS shown). (C) Radioactivity isolated from the same cells correlated with NIS gene expression levels as well as viable virus produced by the infected cells (D).
We used this time dependent data to correlate the levels of virus encoded ‘N’ and ‘NIS’ genes with the titer of infectious virus (TCID50/ml). As can be seen from Figure 2A, the levels of expression of both genes correlated well with the virus titer (R2 = 0.93 and R2 = 0.99 for ‘N’ and ‘NIS’ respectively). The coefficient for ‘N’ gene copy number as a function of virus titer (TCID50/ml) was higher (0.096, 95% CI 0.0826, 0.109) compared to ‘NIS’ (0.076, 95% CI 0.0713, 0.0802). This is expected given that the gene encoding ‘N’ is upstream of ‘NIS’ in the genome and therefore is transcribed at a higher level than the NIS gene (33, 34). We also correlated NIS and N gene expression by Q-RT-PCR at all specific time points in vitro (Figure 2B) and established that there is a linear relationship between the two (correlation coefficient, ρ = 0.96, R2 = 0.93, 95% CI: 0.91, 0.99, p < 0.0001) and this relationship is independent of the MOI used to initiate infection. In addition, the linear relationship between NIS and N gene expression, as expected showed that the level of NIS gene expression is lower than N (slope: 0.7392 ± 0.048, 95% CI: 0.638, 0.841, p < 0.0001). This is in keeping with the above reported relationship between viral gene expression and titer and also compatible with the known biology of the virus (33, 34).
Figure 2. In vitro and in vivo correlations.
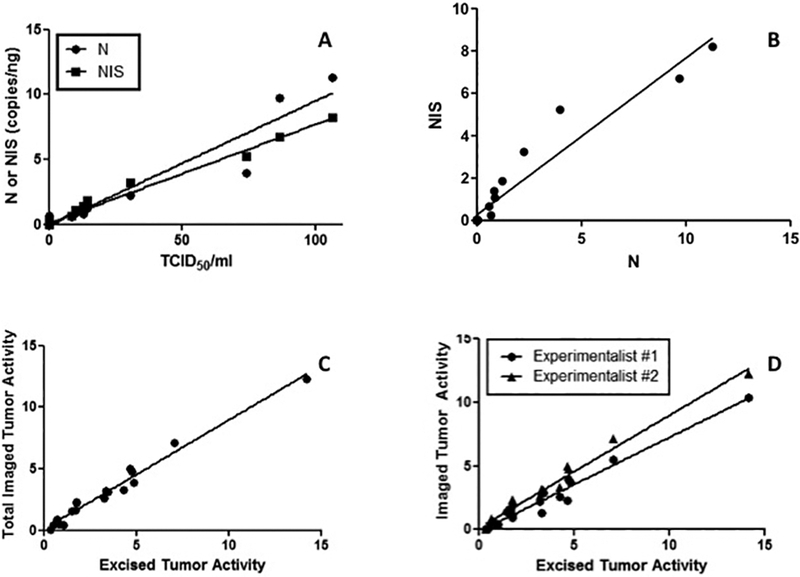
(A) Both ‘N’ and ‘NIS’ gene expression correlated well with the viable virus produced by the infected cells. (B) NIS and N gene expression were well correlated with each other. (C) The intratumoral radioactivity based on SPECT/CT imaging using ROI analysis correlated well with the activity measured in the explanted tumor xenografts (both axes have units of μCi/g). (D) In vivo activity based on SPECT/CT image analysis is reproducible and can be accurately measured even by independent observers (both axes have units of μCi/g) (D).
In vivo imaging and iodide uptake studies
A key component for reliable estimation of in vivo viral titer using molecular imaging is the need for objective and reproducible analysis of the imaging data. To address this issue, we implanted BxPC3 tumors in nude mice (see Methods) and when the tumors reached ~0.5cm in diameter, the mice were injected with MV-NIS or MV-I98A-NIS and imaged serially using SPECT/CT. The mice were euthanized immediately after imaging, the tumors excised and weighed and the intra-tumoral isotope activity determined (See Methods). The activity injected in each mouse across all days of the experiment was determined and found not to be significantly different across groups (median 314μCi or 11.6MBq, range: 10.73, 12.65 MBq, p>0.11 across all time points). The median activity retained in the syringe after injection was 3.2% or 10.06μCi (0.37MBq: range 0.13 – 0.78MBq) - across all experiments. Two independent investigators evaluated the in vivo imaging studies to determine the activity within the tumor using the Region of Interest (ROI) analysis approach after correction for background activity outside and inside each mouse. The data were individually correlated with the activity measured using the radiation counter. As can be seen from Figure 2C, there was a linear relationship between the measured activity using imaging and the actual activity found in the excised tumor (ρ = 0.89, R2 = 0.98, 95% CI: 0.81, 0.96). As expected, the activity estimated from imaging was consistently less than the actual activity likely due to attenuation and losses due to collimation. The two independent investigators also agreed on the estimate of the intra-tumoral isotope activity (ρ1 = 0.82, R2 = 0.97, 95% CI: 0.72, 0.92 and ρ2 = 0.88, R2 = 0.98, 95% CI: 0.77, 0.98) (p = 0.677, paired Mann Whitney test) (Figure 2D).
In vivo imaging showed hardly any background activity in tumors not injected with a NIS expressing virus (Figure 3Ai) compared to mice injected with MV-NIS or MV-I98A-NIS (Figure 3Aii, iii). The patchy distribution of the infected foci is also evident.
Figure 3. In vivo isotope uptake studies after infection.
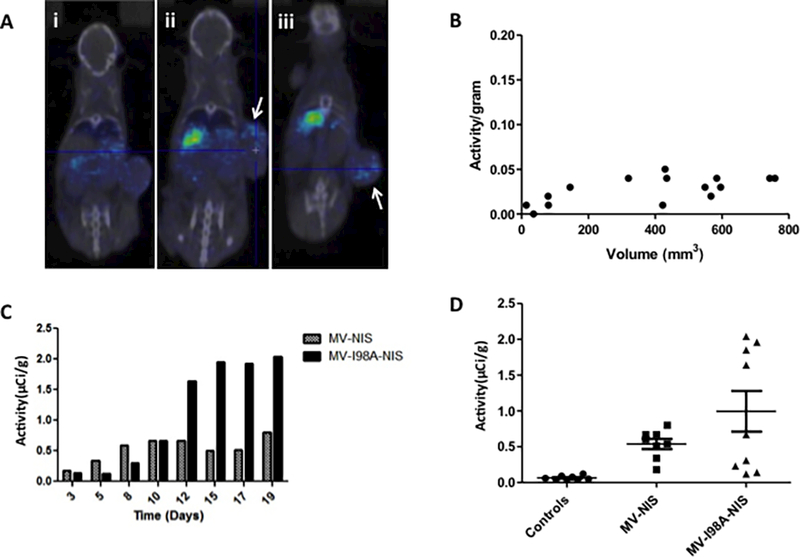
(A) In vivo imaging of isotope uptake in tumors. In (i) a control tumor has no isotope activity, whereas two representative tumors (ii and iii) infected with MV expressing NIS concentrate the isotope above background. The patchy distribution of the isotope is evident (arrows). (B) Pancreatic tumor xenografts that do not express NIS do not concentrate the radioactive isotope. (C) Tumor xenografts in mice injected with MV-NIS or MV-I98A-NIS concentrated the isotope and the activity in such tumors varies as a function of time. MV-I98A-NIS mediated isotope uptake persists for longer suggesting ongoing virus replication and spread. (D) Tumors infected with NIS expressing viruses concentrated isotope significantly higher than controls (p=0.0009, ANOVA).
We measured the isotope activity in the excised tumors and normalized it to their mass (Activity/gram, μCi/g). The results were compared using the Mann Whitney test. The median activity in the control tumors which were not injected with the virus and therefore do not express NIS was 0.04μCi/gram (0.01 – 0.05) (Figure 3B). This did not vary with the mass of the tumor as a function of time. In contrast, we observed variable intratumoral isotope uptake due to viral infection and NIS expression with different kinetics for the two viruses (Figure 3C). The median activity in tumors infected with MV-NIS was 1.72μCi/gram (0.92 – 7.17) (0.636MBq/g, range: 0.34 – 2.65MBq/g) and 3.93μCi/g (1.03 – 6.22) (1.45MBq/g, range: 0.381 – 2.3MBq/g) for mice injected with MV-I98A-NIS (Figure 3D). Therefore, the tumors that were infected with the recombinant viruses expressed NIS and concentrated the isotope at a minimum of 7.7 fold higher compared to background. The difference between controls and infected tumors was highly statistically significant (p=0.0009) for both viruses. This provides evidence that a radionuclide signal above background is a reliable indicator of MV infection and virus mediated NIS expression within the tumor cell population.
We quantitated the level of isotope uptake in the tumor as a function of time after MV-NIS or MV-I98A-NIS injection. As can be seen, from Figure 3C, isotope uptake was different for tumors infected with different viruses. MV-NIS spread rapidly and by inducing cell death, led to a rapid loss of isotope signal. In contrast, NIS expression due to MV-I98A-NIS increased with time, suggesting spread of the infection with higher levels of NIS expression and a larger population of the virus and virus infected cells within the tumors. As expected, tumor cells infected with MV-I98A-NIS generally had higher levels of isotope uptake, due to higher levels of NIS expression. Moreover, since the cells infected with MV-I98A-NIS remain viable for longer (25), this also likely enhances isotope uptake and retention within the tumor (ρ1 = 11.5, R2 = 0.79, ρ2 = 14.4, R2 = 0.94, for MV-NIS and MV-I98A-NIS respectively). Therefore, radionuclide imaging is sensitive enough to detect subtle differences in the behavior of two viruses based on the same platform (MV) but with different abilities to spread from cell to cell (25) and different rates of cell killing.
In vivo correlations
We performed Q-RT-PCR for MV encoded NIS and N genes on RNA isolated from the excised tumors after in vivo imaging. N and NIS gene expression levels mirrored intra-tumoral activity (Figure 4A). Moreover, at most time points the levels of expression of N were higher than NIS. We found that the correlation coefficient between NIS and N in vivo was similar to the in vitro observations with ρ = 0.93, R2 = 0.86, 95% CI: 0.64, 0.99, p = 0.0009 for MV-NIS and ρ = 0.96, 95% CI: 0.77, 0.99, R2 = 0.92, p = 0.0002 for MV-I98A-NIS. Linear fitting of NIS versus N results in a slope of 0.68 ± 0.11, 95% CI: 0.41, 0.95, R2 = 0.86, p = 0.0009) (Figure 4B). The difference in the slopes of the correlations in vitro and in vivo is ~ 5% and not statistically significant (p = 0.06). This result suggests that viral gene expression kinetics and virus replication dynamics in vivo are similar to what was observed in vitro.
Figure 4. In vivo correlations between MV infection, isotope and gene expression.
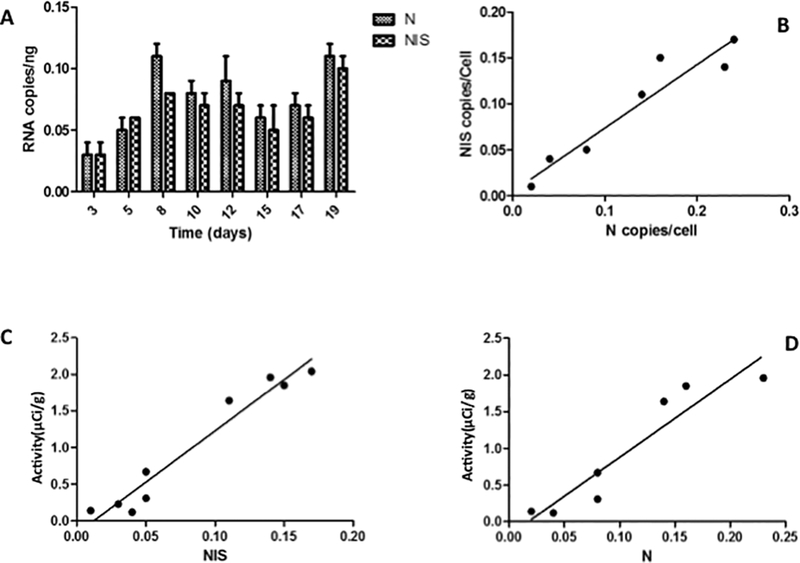
(A) Q-RT-PCT for MV ‘N’ and ‘NIS’ gene expression (data for MV-I98A-NIS shown) illustrate the parallel expression of the two viral genes and how it mirrors isotope uptake (c.f. Figure 3B). (B) NIS and N gene expression in vivo were highly correlated. (C and D) Similar to the in vitro scenario, intratumoral pertechnetate activity was well correlated with viral ‘NIS’ and ‘N’ gene expression.
We also compared the intra-tumoral radioisotope activity to MV related viral NIS and N gene expression (Figure 4C, D). Again, high-level correlations between intra-tumoral radioisotope activity and viral NIS and N gene expression was found (ρNIS = 0.9733, 95% CI: 0.87, 0.99, R2 = 0.95, p < 0.0001; ρN = 0.94, 95% CI: 0.65, 0.99, R2 = 0.89, p = 0.0015). Our results therefore suggest a linear correlation between in vivo isotope uptake and viral mediated NIS and N gene expression. In addition, we showed that both in vitro and in vivo, viral N and NIS gene expression levels correlate well with each other with a similar ratio. The levels of both genes also correlated well with the titer of viable virus in vitro. Therefore, assuming that the relationship between viral N or NIS gene expression and TCID50/ml also holds in vivo, we can use the in vivo quantitation of isotope uptake to estimate the virus population in vivo.
Finally, although our studies were not aimed to evaluate for tumor growth control, Figure 5A shows that both oncolytic viruses slowed down the rate of growth of the pancreatic adenocarcinoma tumor xenografts. Except for the early time points, the size of the treated tumors was smaller than controls (2 way ANOVA p < 0.002 for both MV-NIS and MV-I98A-NIS compared to controls).
Figure 5. Efficacy of MV on tumor control and in vivo correlations.
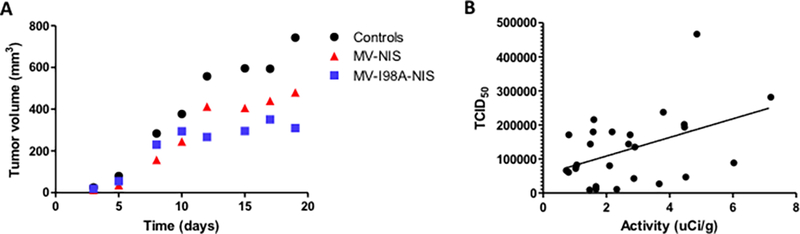
(A)Tumor growth was measured by serial caliper data. Untreated tumor xenografts grew the fastest. MV-NIS did slow down tumor growth but the effect was maximal for MV-I98A-NIS showing that at least in this model, faster replication may not result in a better outcome. (B) A positive correlation (ρ = 0.4475, p = 0.02) between viable virus isolated from excised tumors and the intratumoral isotope activity determined by imaging data from 26 mice is illustrated.
In vivo isotope uptake and virus population
Although viral gene expression such as ‘N’ and ‘NIS’ are surrogates for the presence of the virus in the tumor, we wanted to determine whether there is a linear relationship between NIS mediated isotope uptake and the virus population within the tumors. Therefore, after pancreatic tumor xenografts were established in nude mice, they were injected with MV-NIS and after pertechnetate injection (see Methods), the mice were imaged using SPECT/CT and immediately afterwards, they were euthanized and the tumors extracted. Isotope activity in the tumors was measured immediately after using a radiation counter. Each tumor was divided into 2 parts and weighed. One half of the sample was used to determine viral N and NIS gene expression and the other half was used to isolate virus which was then titrated by infection on Vero cells (see Methods).
Oncolytic virus was detectable in all mice injected with the oncolytic viruses but in none of the controls. The median titer of MV-NIS was 2.6 × 104 TCID50/gram (8.3 × 103−9.2 × 105) of tumor versus no titer in the controls (p = 0.0007). Similarly, we could not detect the N and NIS gene in control tumors but we detected high levels of virus specific RNA expression in mice injected with the oncolytic viruses: NC = 1.5 versus NMV = 76.9, p = 0.001; NISC = 4.2 versus NISMV = 50.3, p = 0.0078. The higher levels of N gene expression compared to NIS is again consistent with the known biology of the virus and our prior results. We also confirmed a strong positive correlation between the intratumoral isotope activity with MV mediated N and NIS gene expression (ρN = 0.68, p = 0.007; ρNIS = 0.75, p = 0.003). We subsequently correlated the intratumoral isotope concentration based on imaging with the virus titer isolated from the explanted tumors (Figure 5B). When we take all the mice studied (n = 42), we found a positive correlation between isotope uptake and viable virus titer: ρ = 0.4545 (range: 0.1747, 0.6665), p = 0.0025. If the analysis was restricted to mice with intratumoral activity > 1μCi/g, (n = 31) (to exclude a possible threshold effect), we found a similar correlation (ρ = 0.425, range: 0.083, 0.6774, p = 0.0172). Another analysis that excluded the high outliers (>10μCi/g) with a sample size of n = 37, gave us similar results (ρ = 0.4662, range: 0.1674, 0.6866, p = 0.0036). Finally, an analysis restricted to mice with an intratumoral isotope concentration >1μCi/g and <10μCi/g (n = 26), also led to a similar correlation (ρ = 0.4475, range: 0.0726, 0.712, p = 0.0219) (Figure 5B). We note that the range for the correlation coefficient is quite large and likely due to inter sample variability, anisotropic distribution of the virus within the tumor, or the virus levels were below threshold for accurate determination of activity by SPECT/CT.
Discussion
Cancer therapy has seen a revolution in the last decade with the development of many novel therapeutics including small molecules, monoclonal antibodies and now oncolytic viruses that can selectively infect, replicate and kill tumor cells. Proof of principle that an oncolytic virus can lead to long-term disease control for disseminated malignancy was also recently reported when one patient with disseminated relapsed and refractory multiple myeloma experienced meaningful tumor cytoreduction after a single dose of MV-NIS and remained in remission for almost 2 years (9). Other viruses have shown similar activity even in immunocompetent animal models of disease (35). However, tumor virotherapy is different from almost every other type of cancer therapy (since the target is expected to amplify the therapy itself (the only other known exception is CAR-T cell therapy) that ultimately leads to the destruction of the tumor. As a result, the outcome of oncolytic virotherapy depends on the complex dynamic interactions between the tumor, the virus and the immune system, and understanding these interactions is critical for their optimal use (1, 15–21, 36). Mathematical modeling can greatly assist in these optimization approaches, but an accurate estimation of the virus population as it propagates in time will likely facilitate this strategy, and in particular can identify potential reasons for therapeutic failures and methods to improve the outcome. For this reason, MV-NIS was generated, and in this work, we tested the hypothesis that NIS mediated isotope uptake in the tumor can be used to estimate the viral load in the tumor population as a function of time.
Our results show that in vitro, MV mediated N and NIS gene expression are tightly correlated and also correlate well with NIS mediated isotope uptake and virus titer. This suggested that the principle may work in vivo. However, the translation of this concept into an animal model has proved more challenging. We have shown that virus induced isotope uptake in vivo increased with time, presumably due to ongoing virus propagation and NIS expression. The virus can also be consistently isolated from explanted tumors and shown to be present in significant titers. There is no background NIS activity in control tumors. In vivo we still find an excellent correlation between expression of the two viral genes (N and NIS) which suggests that the oncolytic virus is behaving in the same way as in the in vitro environment. There is isotope uptake within tumors that is above background and this correlates well with the levels of viral N and NIS gene expression. We could also demonstrate a positive correlation between isotope uptake and the titer of the virus recovered from the tumors although there is considerable variability in the correlation between these two important variables. There are several potential explanations for these observations including (i) a threshold effect relationship between the virus titer in the tumor and the amount of isotope concentrated within the tumor and detectable by SPECT/CT imaging (31), (ii) anisotropies in the distribution of virus within the tumor that interfere with the correlation between isotope uptake and the virus population (32, 36). Although sampling variability could also contribute to this weak correlation, this is unlikely to be a reasonable explanation if enough tumors are studied. It is known that the initial distribution of the virus in the tumor is not uniform (32, 36, 37) and the tumor architecture may interfere with the spread of the oncolytic virus within a tumor (37). Our own preliminary studies in vitro using tumor cells as a monolayer versus a spheroid also show significant differences in the rate of spread of the same virus and the duration of infected cell viability (Dingli et al, unpublished observations). It is possible that the 3 dimensional structure of the in vivo tumor introduces new complexities in the model and dynamics that limit our ability to infer virus population from the in vivo imaging studies.
Despite the fact that the tumors did not regress (this was not an endpoint of the study), we could show that the tumors in mice treated with MV were smaller compared to controls at each time point. Moreover, despite the MV-I98A-NIS virus having slower infection kinetics (25), it controlled the tumors equally effectively in our studies. In addition, since this virus kills infected cells at a slower rate compared to MV-NIS, the infected cells retained the isotope at higher concentrations and this was detectable with SPECT/CT imaging and confirmed by radiation measurements on the explanted tumors. Future studies will focus on the use of this virus at higher concentrations to determine whether it is indeed possible to estimate the in vivo virus population from molecular imaging technologies. We are also developing recombinant viruses with which we can determine non-invasively the biodistribution of the virus at a single cell level in an attempt to dissect the impact of the virus biodistribution, spatial relationships and threshold of infected cells on our ability to quantitate the virus population in vivo.
Significance:
Findings address a fundamental gap in our knowledge of oncolytic virotherapy by presenting technology that gives insight into the behavior of oncolytic viruses in vivo.
Acknowledgements
Funding for the work was provided in part by R01 CA164241 from the National Cancer Institute, National Institutes of Health to DD.
This work is dedicated to the memory of Chetan P. Offord who died while this work was in progress.
Footnotes
Conflict of interest: The authors declare no potential conflicts of interest.
References
- 1.Kirn D, Martuza RL, Zwiebel J. Replication-selective virotherapy for cancer: Biological principles, risk management and future directions. Nat Med. 2001;7:781–7. [DOI] [PubMed] [Google Scholar]
- 2.Cattaneo R, Miest T, Shashkova EV, Barry MA. Reprogrammed viruses as cancer therapeutics: targeted, armed and shielded. Nat Rev Microbiol. 2008;6:529–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miest TS, Cattaneo R. New viruses for cancer therapy: meeting clinical needs. Nat Rev Microbiol. 2014;12:23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lichty BD, Breitbach CJ, Stojdl DF, Bell JC. Going viral with cancer immunotherapy. Nat Rev Cancer. 2014;14:559–67. [DOI] [PubMed] [Google Scholar]
- 5.Patel MR, Jacobson BA, Ji Y, Drees J, Tang S, Xiong K, et al. Vesicular stomatitis virus expressing interferon-beta is oncolytic and promotes antitumor immune responses in a syngeneic murine model of non-small cell lung cancer. Oncotarget. 2015;6:33165–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grote D, Cattaneo R, Fielding AK. Neutrophils contribute to the measles virus-induced antitumor effect: enhancement by granulocyte macrophage colony-stimulating factor expression. Cancer Res. 2003;63:6463–8. [PubMed] [Google Scholar]
- 7.Dey A, Zhang Y, Castleton AZ, Bailey K, Beaton B, Patel B, et al. The Role of Neutrophils in Measles Virus-mediated Oncolysis Differs Between B-cell Malignancies and Is Not Always Enhanced by GCSF. Mol Ther. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rajani K, Parrish C, Kottke T, Thompson J, Zaidi S, Ilett L, et al. Combination Therapy With Reovirus and Anti-PD-1 Blockade Controls Tumor Growth Through Innate and Adaptive Immune Responses. Mol Ther. 2016;24:166–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Russell SJ, Federspiel MJ, Peng KW, Tong C, Dingli D, Morice WG, et al. Remission of disseminated cancer after systemic oncolytic virotherapy. Mayo Clin Proc. 2014;89:926–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grigg C, Blake Z, Gartrell R, Sacher A, Taback B, Saenger Y. Talimogene laherparepvec (T-Vec) for the treatment of melanoma and other cancers. Semin Oncol. 2016;43:638–46. [DOI] [PubMed] [Google Scholar]
- 11.Fukuhara H, Ino Y, Todo T. Oncolytic virus therapy: A new era of cancer treatment at dawn. Cancer Sci. 2016;107:1373–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol. 2012;30:658–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peng KW, TenEyck CJ, Galanis E, Kalli KR, Hartmann LC, Russell SJ. Intraperitoneal therapy of ovarian cancer using an engineered measles virus. Cancer Res. 2002;62:4656–62. [PubMed] [Google Scholar]
- 14.Peng KW, Hadac EM, Anderson BD, Myers R, Harvey M, Greiner SM, et al. Pharmacokinetics of oncolytic measles virotherapy: eventual equilibrium between virus and tumor in an ovarian cancer xenograft model. Cancer Gene Ther. 2006;13:732–8. [DOI] [PubMed] [Google Scholar]
- 15.Wodarz D Gene therapy for killing p53-negative cancer cells: use of replicating versus nonreplicating agents. Hum Gene Ther. 2003;14:153–9. [DOI] [PubMed] [Google Scholar]
- 16.Wu JT, Byrne HM, Kirn DH, Wein LM. Modeling and analysis of a virus that replicates selectively in tumor cells. Bull Math Biol. 2001;63:731–68. [DOI] [PubMed] [Google Scholar]
- 17.Wu JT, Kirn DH, Wein LM. Analysis of a three-way race between tumor growth, a replication-competent virus and an immune response. Bull Math Biol. 2004;66:605–25. [DOI] [PubMed] [Google Scholar]
- 18.Biesecker M, Kimn JH, Lu H, Dingli D, Bajzer Z. Optimization of virotherapy for cancer. Bull Math Biol. 2010;72:469–89. [DOI] [PubMed] [Google Scholar]
- 19.Dingli D, Cascino MD, Josic K, Russell SJ, Bajzer Z. Mathematical modeling of cancer radiovirotherapy. Math Biosci. 2006;199:55–78. [DOI] [PubMed] [Google Scholar]
- 20.Dingli D, Offord C, Myers R, Peng KW, Carr TW, Josic K, et al. Dynamics of multiple myeloma tumor therapy with a recombinant measles virus. Cancer Gene Ther. 2009;16:873–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rommelfanger DM, Offord CP, Dev J, Bajzer Z, Vile RG, Dingli D. Dynamics of melanoma tumor therapy with vesicular stomatitis virus: explaining the variability in outcomes using mathematical modeling. Gene Ther. 2012;19:543–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dingli D, Peng KW, Harvey ME, Greipp PR, O’Connor MK, Cattaneo R, et al. Image-guided radiovirotherapy for multiple myeloma using a recombinant measles virus expressing the thyroidal sodium iodide symporter. Blood. 2004;103:1641–6. [DOI] [PubMed] [Google Scholar]
- 23.Galanis E, Atherton PJ, Maurer MJ, Knutson KL, Dowdy SC, Cliby WA, et al. Oncolytic measles virus expressing the sodium iodide symporter to treat drug-resistant ovarian cancer. Cancer Res. 2015;75:22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dingli D, Kemp BJ, O’Connor MK, Morris JC, Russell SJ, Lowe VJ. Combined I-124 positron emission tomography/computed tomography imaging of NIS gene expression in animal models of stably transfected and intravenously transfected tumor. Mol Imaging Biol. 2006;8:16–23. [DOI] [PubMed] [Google Scholar]
- 25.Ennis MK, Hu C, Naik SK, Hallak LK, Peng KW, Russell SJ, et al. Mutations in the stalk region of the measles virus hemagglutinin inhibit syncytium formation but not virus entry. J Virol. 2010;84:10913–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Radecke F, Spielhofer P, Schneider H, Kaelin K, Huber M, Dotsch C, et al. Rescue of measles viruses from cloned DNA. EMBO J. 1995;14:5773–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spearman C The method of ‘right and wrong cases’ (‘constant stimuli’) without Gauss’s formulae. Br J Psychol. 1908;2:227–42. [Google Scholar]
- 28.Kaerber G Beitrag zur Kollektiven Behandlung Pharmakologischer Reihenversuche. Arch Exp Path Pharma. 1931;162:480–7. [Google Scholar]
- 29.Dingli D, Russell SJ, Morris JC 3rd. In vivo imaging and tumor therapy with the sodium iodide symporter. J Cell Biochem. 2003;90:1079–86. [DOI] [PubMed] [Google Scholar]
- 30.Penheiter AR, Wegman TR, Classic KL, Dingli D, Bender CE, Russell SJ, et al. Sodium iodide symporter (NIS)-mediated radiovirotherapy for pancreatic cancer. AJR Am J Roentgenol. 2010;195:341–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carlson SK, Classic KL, Hadac EM, Dingli D, Bender CE, Kemp BJ, et al. Quantitative molecular imaging of viral therapy for pancreatic cancer using an engineered measles virus expressing the sodium-iodide symporter reporter gene. AJR Am J Roentgenol. 2009;192:279–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Penheiter AR, Dingli D, Bender CE, Russell SJ, Carlson SK. Monitoring the initial delivery of an oncolytic measles virus encoding the human sodium iodide symporter to solid tumors using contrast-enhanced computed tomography. J Gene Med. 2012;14:590–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dowling PC, Blumberg BM, Menonna J, Adamus JE, Cook P, Crowley JC, et al. Transcriptional map of the measles virus genome. J Gen Virol. 1986;67 ( Pt 9):1987–92. [DOI] [PubMed] [Google Scholar]
- 34.Cattaneo R, Rebmann G, Baczko K, ter Meulen V, Billeter MA. Altered ratios of measles virus transcripts in diseased human brains. Virology. 1987;160:523–6. [DOI] [PubMed] [Google Scholar]
- 35.Naik S, Nace R, Barber GN, Russell SJ. Potent systemic therapy of multiple myeloma utilizing oncolytic vesicular stomatitis virus coding for interferon-beta. Cancer Gene Ther. 2012;19:443–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miller A, Suksanpaisan L, Naik S, Nace R, Federspiel M, Peng KW, et al. Reporter gene imaging identifies intratumoral infection voids as a critical barrier to systemic oncolytic virus efficacy. Mol Ther Oncolytics. 2014;1:14005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mok W, Stylianopoulos T, Boucher Y, Jain RK. Mathematical modeling of herpes simplex virus distribution in solid tumors: implications for cancer gene therapy. Clin Cancer Res. 2009;15:2352–60. [DOI] [PMC free article] [PubMed] [Google Scholar]