

Summary
Lipoic acid is a cofactor required for intermediary metabolism that is either synthesized de novo or acquired from environmental sources. The bacterial pathogen Staphylococcus aureus encodes enzymes required for de novo biosynthesis, but also encodes two ligases, LplA1 and LplA2, that are sufficient for lipoic acid salvage during infection. S. aureus also encodes two H proteins, GcvH of the glycine cleavage system and the homologous GcvH-L encoded in an operon with LplA2. GcvH is a recognized conduit for lipoyl transfer to α-ketoacid dehydrogenase E2 subunits, while the function of GcvH-L remains unclear. The potential to produce two ligases and two H proteins is an unusual characteristic of S. aureus that is unlike most other Gram positive Firmicutes and might allude to an expanded pathway of lipoic acid acquisition in this microorganism. Here, we demonstrate that LplA1 and LplA2 facilitate lipoic acid salvage by differentially targeting lipoyl domain-containing proteins; LplA1 targets H proteins and LplA2 targets α-ketoacid dehydrogenase E2 subunits. Furthermore, GcvH and GcvH-L both facilitate lipoyl relay to E2 subunits. Altogether, these studies identify an expanded mode of lipoic acid salvage used by S. aureus and more broadly underscore the importance of bacterial adaptations when faced with nutritional limitation.
Keywords: lipoic acid, ligase, S. aureus, metabolic adaptation, virulence, nutrient acquisition
Graphical Abstract
Lipoic acid is a metabolic cofactor required for intermediary metabolism. Staphylococcus aureus acquires lipoic acid through either de novo biosynthesis or salvage. We show that S. aureus adapts to lipoic acid limitation through an expanded salvage pathway including two lipoic acid ligases with divergent targets; intrinsic lipoamidase activity that extracts inaccessible lipoic acid from peptide-bound sources; and an accessory lipoyl-protein that serves as an additional source of lipoic acid for transfer to metabolic enzyme subunits.
Introduction
Staphylococcus aureus is an opportunistic pathogen that causes disease in a wide range of tissues including skin, kidney, bone, lung, and heart (Tong et al., 2015, Liu et al., 2011). Each infectious niche poses unique challenges to S. aureus survival due to significant differences in nutrient bioavailability at each site. In order to thrive in such a wide range of environments, S. aureus harnesses key adaptive traits that permit trace metal/nutrient acquisition, maintenance of metabolic homeostasis, and survival in nutritionally deficient or otherwise inhospitable conditions (Cassat & Skaar, 2012, Hood & Skaar, 2012, Richardson et al., 2015, Grim et al., 2017, Kelliher et al., 2018, Beasley & Heinrichs, 2010, Mazmanian et al., 2003). Aside from a relatively well-developed understanding of trace metal acquisition, the mechanisms used by S. aureus to acquire other scarce nutrients remains comparatively understudied.
In addition to the necessity for trace metal scavenging for optimal metabolic activity and bacterial survival, S. aureus also requires the essential cofactor lipoic acid (Zorzoli et al., 2016, Grayczyk et al., 2017). Lipoic acid, a derivative of the medium-chain fatty acid octanoic acid, is a sulfur-containing cofactor that is covalently attached to conserved lysine residues in multi-subunit enzyme complexes needed for aerobic metabolism, fatty acid biosynthesis, glycine detoxification, and maintenance of redox homeostasis (Spalding & Prigge, 2010, Cronan, 2016). Five lipoylated enzyme complexes are used in bacteria: pyruvate dehydrogenase (PDH) catalyzes the oxidative decarboxylation of pyruvate to acetyl CoA; 2-oxoglutarate dehydrogenase (OGDH) converts α-ketoglutarate to succinyl-CoA; branched-chain 2-oxoacid dehydrogenase (BCODH) degrades branched chain amino acids to make a branched chain CoA intermediate needed for fatty acid biosynthesis; acetoin dehydrogenase (AoDH) is similar to the PDH complex and also catalyzes the conversion of pyruvate to acetyl CoA; and the glycine cleavage system (Gcs) catalyzes the reversible decarboxylation of glycine (Cronan, 2016, Spalding & Prigge, 2010, Perham, 2000). S. aureus encodes PDH, OGDH, and BCODH α-ketoacid dehydrogenase complexes, as well as the Gcs, but not AoDH (Spalding & Prigge, 2010, Zorzoli et al., 2016). The α-ketoacid dehydrogenases are comprised of multiple copies of three different subunits referred to as E1, E2, and E3, whereas the glycine cleavage system uses subunits referred to as P (pyridoxal phosphate-containing protein), H (hydrogen carrier protein), T (tetrahydrofolate-containing protein), and L (lipoamide dehydrogenase) proteins (Douce et al., 2001, Kikuchi et al., 2008). The lipoic acid cofactor is attached by an amide bond to a conserved lysine residue on E2 and H subunits and acts to channel substrates through the active sites of the enzyme complexes (Douce et al., 2001, Perham, 2000). The necessity of lipoic acid for the activity of the α-ketoacid dehydrogenase complexes and glycine cleavage system demands a strategy to synthesize and/or salvage lipoic acid from the environment, as well as a method to attach lipoic acid to the conserved lysine within the lipoyl domains of E2 and H subunits.
A growing body of literature suggests that lipoic acid biosynthesis and salvage can have a major role in facilitating the pathogenesis of microorganisms (Spalding & Prigge, 2010). Lipoic acid salvage in parasites such as Plasmodium falciparum, the causative agent of malaria, is crucial for survival and growth of the parasite during the blood and liver stage (Afanador et al., 2014, Gunther et al., 2009, Storm & Muller, 2012, Falkard et al., 2013). In addition, disruption of the lipoic acid biosynthesis and salvage pathway attenuates Burkholderia pseudomallei virulence in an intranasal mouse infection model (Pilatz et al., 2006). In Pseudomonas aeruginosa, a functional lipoylated PDH enzyme complex is important for the expression of the type three secretion system (Dacheux et al., 2002). Furthermore, disruption of dlaT, a gene encoding the E2 PDH subunit in Mycobacterium tuberculosis, results in increased susceptibility to macrophage killing and oxidative stress (Bryk et al., 2013, Bryk et al., 2002). Listeria monocytogenes, a lipoic acid auxotroph, uses one of two lipoic acid salvage enzymes to facilitate acquisition of lipoic acid within the intracellular environment (Christensen et al., 2011a, Keeney et al., 2007, O’Riordan et al., 2003). Overall, these examples demonstrate the range of effects that lipoic acid acquisition and lipoic acid-dependent enzyme activities have on promoting pathogen survival within an infected host. Our studies with S. aureus suggest niche specific requirements for de novo biosynthesis and salvage during infection, where de novo biosynthesis is required to infect the heart and salvage is necessary for optimal infection of the kidney, highlighting the relevance of both pathways during infection (Zorzoli et al., 2016). In addition, we have identified a novel activity for lipoyl-E2-PDH in the extracellular environment where it acts to blunt macrophage activation by toll-like receptor ligands, further expanding the relevance of protein lipoylation in the context of infection (Grayczyk et al., 2017).
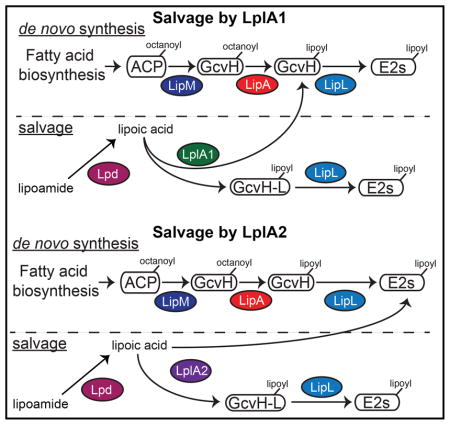
The mechanisms of lipoic acid acquisition and synthesis can vary considerably among bacteria, fungi, and protozoans (Cronan, 2016). Many of these organisms either encode a de novo biosynthesis pathway, salvage pathway, or both. Each confers a range of acquisition strategies that presumably have evolved to satisfy the unique lipoic acid requirements of that particular microorganism within its replicative niche. Most studies of lipoic acid biosynthesis and salvage in bacteria were conducted in Escherichia coli, Bacillus subtilis, and L. monocytogenes (Cronan, 2016, Christensen & Cronan, 2010, Christensen et al., 2011a, Christensen et al., 2011b, Hermes & Cronan, 2009, Jordan & Cronan, 2003, Martin et al., 2011, Miller et al., 2000, Morris et al., 1995, Vanden Boom et al., 1991, Zhao et al., 2003). These studies highlight the diversity of lipoic acid biosynthesis and salvage strategies between bacterial species. Recently, our lab used a genetic approach to define the lipoic acid acquisition pathways of S. aureus (Zorzoli et al., 2016). This work determined that S. aureus has a complex lipoic acid biosynthesis and salvage pathway that bears similarities to that of B. subtilis and L. monocytogenes combined. S. aureus encodes the three characteristic enzymes needed for de novo biosynthesis in B. subtilis, LipM, LipA, and LipL, as well as iron sulfur cluster biogenesis proteins, SufCDSUB and SufT, needed for incorporation of sulfur into the lipoic acid molecule (Christensen & Cronan, 2010, Martin et al., 2011, Christensen et al., 2011b, Mashruwala et al., 2016, Roberts et al., 2017). However, it also encodes two lipoic acid ligases, LplA1 and LplA2, needed for salvage during infection and two H proteins, GcvH and GcvH-Like (GcvH-L), which we suspect may be used as a source of lipoyl protein for subsequent transfer to E2 subunits by an amidotransferase (Christensen et al., 2011a, Zorzoli et al., 2016, Cao et al., 2018).
As with B. subtilis, lipoic acid synthesis in S. aureus begins with transfer of octanoic acid, a medium chain fatty acid, from its acyl carrier protein (ACP) to GcvH by the octanoyl transferase, LipM (Christensen & Cronan, 2010, Zorzoli et al., 2016). The lipoyl synthetase, LipA, then converts the octanoyl moiety to lipoic acid, followed by transfer of the lipoyl group from GcvH to the E2 subunits of PDH, BCODH, and OGDH by LipL (Zorzoli et al., 2016, Christensen et al., 2011b, Martin et al., 2011). S. aureus contains two additional enzymes that have activities associated with lipoic acid salvage from the host environment, LplA1 and LplA2 (Zorzoli et al., 2016). This is reminiscent of L. monocytogenes, which also encodes two lipoic acid ligases, but unlike S. aureus, has no de novo biosynthesis enzymes (Christensen et al., 2011a, Keeney et al., 2007). In L. monocytogenes, LplA1 is integral for lipoic acid acquisition in vitro and in vivo, whereas LplA2 only facilitates acquisition in vitro (Keeney et al., 2007). In a similar vein, S. aureus LplA1, but not LplA2, was found to be crucial for the salvage of lipoic acid in vitro (Zorzoli et al., 2016). However, in vivo studies indicate that either LplA1 or LplA2 is sufficient to promote kidney infection during murine systemic infection. Because the LplA2 ligase does not have a discernable function in vitro - unless bacteria are cultured in the presence of fetal bovine serum (FBS) - its exact mode of action in lipoic acid salvage is unknown (Zorzoli et al., 2016). Furthermore, LplA2 is encoded in an operon containing an ADP-ribosyltransferase, macrodomain protein, luciferase-like monooxygenase, and the H subunit-like protein GcvH-L (Zorzoli et al., 2016, Rack et al., 2015). This operon is postulated to participate in lipoic acid-dependent maintenance of redox homeostasis, which suggests that LplA2 may have roles that extend beyond lipoic acid salvage (Rack et al., 2015). Nonetheless, the presence of a potential alternative H protein encoded within this operon may indicate an unusual layer of complexity should lipoyl GcvH-L indeed act as a substrate in lipoic acid transfer (Cao et al., 2018). Based on this information, we hypothesized that LplA1 and LplA2 in S. aureus use alternative lipoylated substrates and/or the enzymes have different lipoylation targets, thereby providing S. aureus with an increased degree of flexibility in its mechanisms of lipoic acid acquisition in lipoic acid-limiting environments.
In this study, we used a biochemical approach to interrogate the activity of both LplA1 and LplA2 of S. aureus. We found that LplA1 attaches lipoic acid primarily to the H subunit, GcvH, and the H subunit-like protein, GcvH-L, whereas LplA2 attaches lipoic acid to E2 subunits, as well as operon-linked GcvH-L. Each ligase functions independently, with limited overlap in protein targets. Furthermore, both ligases have the capacity to use octanoic acid as a substrate for attachment to GcvH and GcvH-L. Neither enzyme uses lipoyl tripeptides as a substrate unless co-incubated with crude lysates of S. aureus, which harbors lipoamidase activity. Furthermore, GcvH and GcvH-L both provide a source of lipoyl protein for subsequent transfer to E2 subunits, despite a postulated role for GcvH-L in orchestrating resistance to host-derived redox stress (Rack et al., 2015). Taken together, these studies highlight the importance of the divergent functions of LplA1 and LplA2, as well as the contributions of both GcvH and GcvH-L to protein lipoylation and likely explains why S. aureus thrives so well when faced with low levels of free lipoic acid during host infection.
Results
S. aureus LplA1 promotes lipoic acid salvage and stimulates bacterial replication in broth culture
Our genetic analyses of lipoic acid biosynthesis and salvage in S. aureus have led to a model whereby the enzymes LipM, LipA, and LipL comprise a functional de novo lipoic acid biosynthesis pathway that acts in vitro and within certain tissues in vivo (Figure 1A) (Zorzoli et al., 2016). In addition, S. aureus produces two lipoic acid salvage enzymes, LplA1 and LplA2, implying potential for flexibility in lipoic acid salvage. However, biochemical studies to date suggest the activity of LplA2 is limited to lipoylation of GcvH-L (Figure 1A) (Rack et al., 2015). In contrast, our genetic analyses indicate that LplA1 and LplA2 are both sufficient to promote survival of a lipoic acid auxotroph during infection, implying some degree of functional overlap between LplA1 and LplA2, at least in the host environment (Zorzoli et al., 2016). To begin to interrogate the ligase activities of LplA1 and LplA2, we conducted a series of growth curves using S. aureus strains harboring deletions of lipA and one or both lipoic acid ligases, followed by complementation with lplA1 or lplA2 driven by a constitutive promoter. When inoculated into medium lacking free lipoic acid (RPMI) or supplemented with the de novo biosynthesis precursor octanoic acid (OA), only the wild type strain was able to replicate (Figure 1B–C). In contrast, supplementation of the medium with lipoic acid restored wild type growth characteristics to ΔlipA ΔlplA2 and ΔlipA ΔlplA1 ΔlplA2 + lplA1 strains, while ΔlipA ΔlplA1 and ΔlipA ΔlplA1 ΔlplA2 + lplA2 strains were unable to replicate (Figure 1D). These data support our prior studies and suggest that LplA1, but not LplA2 scavenges free lipoic acid to support bacterial replication (Zorzoli et al., 2016).
Figure 1. LplA1, but not LplA2, restores growth to lipoic acid auxotrophs in vitro.

(A) Model of the lipoic acid biosynthesis and salvage pathways in S. aureus. S. aureus synthesizes lipoic acid via de novo biosynthesis using the enzymes LipM- octanoyl transferase; LipA – lipoyl synthethase; and LipL – amidotransferase. During lipoic acid salvage, S. aureus is thought to use two lipoic acid ligases, LplA1 and LplA2. Current evidence suggests LplA1 lipoylates GcvH, while LplA2 lipoylates the GcvH-Like protein, GcvH-L. (B–D) Growth (OD550) of the indicated lipoic acid auxotrophs in (B) RPMI, (C) RPMI + 25 μM octanoic acid (OA), and (D) RPMI + 5 μM lipoic acid (LA). Strain designations are as follows: Wildtype (WT), ΔlipA ΔlplA1 (ΔA ΔA1), ΔlipA ΔlplA2 (ΔA ΔA2), ΔlipA ΔlplA1 ΔlplA2 (ΔA ΔA1 ΔA2), ΔlipA ΔlplA1 ΔlplA2 + lplA1 (ΔA ΔA1 ΔA2 + A1), and ΔlipA ΔlplA1 ΔlplA2 + lplA2 (ΔA ΔA1 ΔA2 + A2).
Both LplA1 and LplA2 are sufficient to promote lipoylation of E2 and H subunits
The fact that LplA2 is unable to support replication of lipoic acid auxotrophs in the presence of lipoic acid was unusual given our observation that LplA2 is sufficient to promote kidney infection by ΔlplA1 ΔlplA2 and ΔlipA ΔlplA1 ΔlplA2 + lplA2 strains (Zorzoli et al., 2016). Furthermore, a ΔlipA ΔlplA1 ΔlplA2 + lplA2 strain constitutively expresses high levels of LplA2 in broth culture as determined by immunoblot with polyclonal rabbit α-LplA2 antibody (Figure 2A). Thus, although restoration of growth was not observed, we reasoned that we might still detect ligase activity for LplA2 by monitoring protein lipoylation. To this end, we tested whether or not the ΔlipA ΔlplA1 ΔlplA2 + lplA2 strain is capable of lipoylating E2 and H proteins despite the inability of LplA2 to restore growth in broth culture. WT, ΔlipA ΔlplA1 ΔlplA2, ΔlipA ΔlplA1 ΔlplA2 + lplA1, ΔlipA ΔlplA1 ΔlplA2 + lplA2 strains were grown in medium containing the branched chain fatty acid precursors isobutyric acid, isovaleric acid, and 2-methylbutyric acid (BCFA), to bypass the requirement of lipoic acid for growth (Martin et al., 2011, Martin et al., 2009). The medium was supplemented with either lipoic acid or octanoic acid, followed by immunoblot of whole cell lysates to ascertain LplA1 and LplA2-dependent protein lipoylation. Contrary to its inability to grow in the presence of free lipoic acid, the ΔlipA ΔlplA1 ΔlplA2 + lplA2 strain resembled a ΔlipA ΔlplA1 ΔlplA2 + lplA1 strain in its lipoylation of E2-PDH, E2-OGDH, and GcvH with modest reductions in the lipoylation on E2-BCODH (Figure 2B and Figure S1). Lipoylation of these same subunits when grown in BCFA medium or BCFA supplemented with octanoic acid was only observed for the wild type strain (Figure 2B and Figure S1). In summary, LplA1 and LplA2-dependent salvage of lipoic acid occurs in broth culture.
Figure 2. Constitutively expressed LplA1 and LplA2 promote lipoylation in medium containing free lipoic acid.

(A) Immunoblot with α-LplA2 antibody of whole cell lysates derived from the indicated strains grown in RPMI supplemented with branched chain carboxylic acids (10 mM isobutyric acid, 9 mM 2-methylbutyric acid, 9 mM isovaleric acid, and 10 mM sodium acetate - BCFA) in order to bypass the requirement of lipoic acid for replication. (B) Immunoblot with α-lipoic acid antibody of whole cell lysates derived from the indicated strains grown in the same medium as (A) and supplemented with 5 μM lipoic acid (LA) or 150 μM octanoic acid (OA). The positions of the four lipoyl proteins in S. aureus (E2-PDH, E2-OGDH, E2-BCODH, and GcvH) are indicated. Strain designations are as follows: wildtype (WT), ΔlipA ΔlplA1 ΔlplA2 (ΔA ΔA1 ΔA2), ΔlipA ΔlplA1 ΔlplA2 + lplA1 (ΔA ΔA1 ΔA2 + A1), and ΔlipA ΔlplA1 ΔlplA2 + lplA2 (ΔA ΔA1 ΔA2 + A2).
Interrogating the functional activities of LplA1 and LplA2
Thus far, our data indicate that LplA1 and LplA2 behave as lipoic acid ligases. However, their exact biochemical function and protein targets remain unclear. In E. coli, the lipoic acid ligase is a promiscuous enzyme and attaches lipoic acid or octanoic acid to most H proteins as well as E2 subunits from a variety of species (Green et al., 1995, Morris et al., 1994). In contrast, lipoic acid attachment in B. subtilis and L. monocytogenes occurs on the H subunit of the glycine cleavage system followed by transfer to E2 subunits by the amidotransferase LipL (Christensen et al., 2011a, Christensen et al., 2011b). These enzymes do not appreciably target E2 subunits in vitro (Christensen et al., 2011a, Christensen et al., 2011b). The diversity of functions in the ligase family of enzymes argues that the biochemical functions, protein targets, or substrates could vary between LplA1 and LplA2 (Cronan, 2016). Therefore, to interrogate the activities of the ligases in greater detail, we purified recombinant S. aureus LplA1 and LplA2 from E. coli for use in in vitro lipoylation assays with purified S. aureus E2-PDH, E2-OGDH, E2-BCODH, GcvH, and GcvH-L (Figure 3A).
Figure 3. Targeting of GcvH and GcvH-L by LplA1 and LplA2.
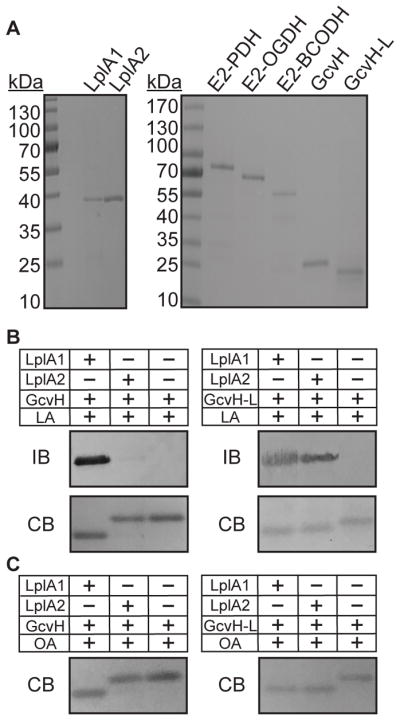
(A) GelCode Blue stained SDS-PAGE gels containing 2 μg of the salvage enzymes, LplA1 and LplA2, and lipoyl domain containing subunits E2-PDH, E2-OGDH, E2-BCODH, GcvH, and GcvH-L. (B–C) LplA1 and LplA2 attachment of (B) lipoic acid (2.4 mM) and (C) octanoic acid (2.4 mM) to GcvH and GcvH-L. Lipoylation was assessed by conducting an immunoblot (IB) with rabbit α-lipoic acid antibody. Parallel 12% SDS PAGE gels were stained with GelCode Blue (CB). Octanoylation was visualized as a shift in apparent molecular weight after resolving proteins on a 12% SDS-PAGE gel and staining with GelCode Blue (CB).
LplA1 and LplA2 targeting of GcvH and GcvH-L
S. aureus contains a canonical GcvH protein important for glycine cleavage, but also encodes a GcvH-Like protein (GcvH-L), with 29% amino acid identity (54% similarity) to GcvH. GcvH-L is encoded in a putative operon with the lplA2 gene - the last gene in the operon (Zorzoli et al., 2016). Our genetic evidence supports a model where, in addition to its role in glycine cleavage, GcvH acts as a major conduit for transfer of lipoic acid to E2 subunits by the amidotransferase LipL (Zorzoli et al., 2016). GcvH-L, like LplA2, does not appear to be expressed in broth culture, precluding direct assessment of its activity (Zorzoli et al., 2016, Rack et al., 2015, Nobre & Saraiva, 2013). Based on their proposed assignment as H proteins, we reasoned that both GcvH of the glycine cleavage system and GcvH-L are targets for lipoylation by one or both S. aureus ligases. Support from this notion comes from Rack et al, who demonstrated that a 1:1 to 1:2 molar ratio of S. aureus LplA1 or LplA2 and GcvH-L is sufficient to promote lipoylation (Rack et al., 2015). To begin to decipher the biochemical mechanisms of lipoic acid salvage in S. aureus, we first tested the ability of LplA1 and LplA2 to attach free lipoic acid to one of these two H proteins. Lipoylation assays conducted with LplA1 or LplA2 and GcvH (molar ratio of enzyme to substrate 1:20) with free lipoic acid showed complete lipoylation of GcvH by LplA1, as evidenced by immunoblot with α-lipoic acid antibody and a shift in apparent molecular weight presumably due to a loss of positive charge at the conserved lysine of the lipoyl domain after attachment (Figures 3B and Figure S2A, left) (Martin et al., 2011, Posner et al., 2009). In contrast, no lipoylation was detected for reactions containing LplA2 and GcvH. When LplA1 and LplA2 were used in lipoylation reactions with GcvH-L, lipoylation by immunoblot and shifts in apparent molecular weight were observed regardless of the ligase used (Figure 3B and Figure S2A, right) (Rack et al., 2015). When octanoic acid was used in place of lipoic acid, we found that LplA1 could target both GcvH and GcvH-L for octanoic acid attachment, whereas LplA2 only targeted GcvH-L (Figure 3C and Figure S2B). Taken together, these data indicate that LplA1 is indiscriminate in its ability to attach lipoic acid and octanoic acid to S. aureus H proteins, whereas LplA2 attaches lipoic acid and octanoic acid to GcvH-L.
LplA1 and LplA2 targeting of E2 proteins
Our prior genetic data and H protein lipoylation studies presented here suggest that S. aureus LplA1 and LplA2 functions diverge in such a way that each has different targets for lipoyl attachment (Zorzoli et al., 2016). A scenario such as this would increase the likelihood of sufficient lipoylation in the absence of de novo biosynthesis, allow for fine-tuning of protein lipoylation in cases where requirements for exogenous lipoic acid are high, or facilitate lipoylation of a particular enzyme complex over another. To test the range of E2 protein targets of LplA1 and LplA2, we conducted lipoic acid attachment assays with purified lipoyl domain-containing E2 proteins of S. aureus - E2-PDH, E2-OGDH, and E2-BCODH. In a reaction containing E2-PDH and LplA2, a prominent lipoyl protein band was detected after immunoblot with α-lipoic acid antibody (Figure 4A and Figure S3A). Modest, but detectable lipoylation over that of the control reaction containing only E2-PDH and lipoic acid was observed in reactions with LplA1. In reactions containing E2-OGDH, lipoylation was seen in the presence of LplA1 and LplA2 although LplA2-dependent attachment was more robust than that caused by LplA1 (Figure 4B and Figure S3B). In contrast, lipoic acid attachment to E2-BCODH was only observed in the presence of LplA2, but not LplA1 (Figure 4C and Figure S3C). Although a faint band was observed in lanes from reactions with LplA1, this band was also present in control wells containing only E2-BCODH and lipoic acid (Figure 4C). These data suggest that LplA1-mediated lipoic acid attachment to E2 subunits is limited, whereas LplA2 is promiscuous and targets all three E2 subunits for lipoic acid attachment.
Figure 4. Targeting of E2 subunits by LplA1 and LplA2.

(A–C) LplA1 and LplA2 attachment of lipoic acid (2.4 mM) to (A) E2-PDH, (B) E2-OGDH, and (C) E2-BCODH. Lipoyl attachment was assessed by conducting immunoblots (IB) with rabbit α-lipoic acid antibody. A parallel 12% SDS-PAGE gels was stained with GelCode Blue (CB).
Use of lipoyl peptides as a source of lipoic acid requires lipoamidase activity and LplA1 or LplA2
Thus far, our data indicate that the lipoic acid ligases of S. aureus are capable of attaching free lipoic acid to E2 and H protein subunits with different efficiencies. However, free lipoic acid is limiting within the host environment and is likely to be present attached to lysine residues within conserved lipoyl domains via an amide bond. Previous studies with L. monocytogenes suggest that lipoamide can be used as a source of lipoic acid to stimulate growth, by virtue of the activity of an L. monocytogenes-encoded lipoamidase (Christensen et al., 2011a). We wondered whether or not peptide-bound lipoic acid might facilitate growth of S. aureus lipoic acid auxotrophs. We first tested if supplementation of RPMI medium with porcine PDH was sufficient to stimulate growth of gene deletion mutants lacking one or both ligases in a ΔlipA mutant background. Addition of PDH was unable to promote growth of any mutant strain tested (Figure 5A). In contrast, when PDH was first co-incubated with proteinase K agarose beads prior to supplementation, the ΔlipA ΔlplA2 and ΔlipA ΔlplA1 ΔlplA2 + lplA1 strains grew identically to the WT strain (Figure 5B). Because commercial porcine PDH is unlikely to be uniformly pure, we synthesized a series of tripeptides [Asp-Lys-Ala (DKA), Asp-LysLipoyl-Ala (DKLA), Asp-Lys-Thr (DKT), and Asp-LysLipoyl-Thr (DKLT)] that resemble the minimum peptides generated by digestion of porcine PDH with proteinase K (Keeney et al., 2007). These tripeptides are sufficient to stimulate the growth of L. monocytogenes in the absence of free lipoic acid (Christensen et al., 2011a, Keeney et al., 2007). We monitored the growth of the indicated S. aureus strains after supplementation with DKA, DKLA, DKT, and DKLT into RPMI medium. Growth was restored for the ΔlipA ΔlplA2 and ΔlipA ΔlplA1 ΔlplA2 + lplA1 strains, but only when the peptide containing the lipoyl moiety was present (Figure 5C–F). Consistent with prior observations with lipoic acid, lipoyl peptides were unable to promote growth of ΔlipA ΔlplA1 and ΔlipA ΔlplA1 ΔlplA2 + lplA2 strains (Figure 5C–F).
Figure 5. LplA1, but not LplA2, restores growth to lipoic acid auxotrophs in the presence of lipoyl peptides.

(A–F) Growth (OD550) of the indicated lipoic acid auxotrophs in RPMI supplemented with (A) 2.5 mg ml−1 undigested pyruvate dehydrogenase (PDH), (B) 2.5 mg ml−1 proteinase K digested PDH, (C) 100 μM DKA tripeptide, (D) 100 μM DKLA tripeptide, (E) 100 μM DKT tripeptide, and (F) 100 μM DKLT tripeptide. Strain designations are as follows: Wildtype (WT), ΔlipA ΔlplA1 (ΔA ΔA1), ΔlipA ΔlplA2 (ΔA ΔA2), ΔlipA ΔlplA1 ΔlplA2 (ΔA ΔA1 ΔA2), ΔlipA ΔlplA1 ΔlplA2 + lplA1 (ΔA ΔA1 ΔA2 + A1), and ΔlipA ΔlplA1 ΔlplA2 + lplA2 (ΔA ΔA1 ΔA2 + A2).
Stimulation of growth after supplementation with lipoyl peptides suggests that S. aureus is capable of acquiring lipoic acid from its lysine-bound state. The ability to do so would either require intrinsic amidotransferase activity within LplA1 or LplA2, or the presence of an enzyme with lipoamidase activity. To test whether or not LplA1 or LplA2 harbors amidotransferase activity, we conducted biochemical assays with recombinant LplA1 or LplA2 with GcvH, GcvH-L, E2-PDH, E2-OGDH, E2-BCODH and DKLA. Neither LplA1 nor LplA2 was able to use DKLA to lipoylate H or E2 subunits (Figure 6A and Figure S4A–B). This finding suggests that like L. monocytogenes, S. aureus must also produce an enzyme with lipoamidase activity in order to liberate free lipoic acid for subsequent ligation by LplA1 and LplA2. To test this possibility, we set up a series of lipoylation reactions with DKLA, LplA1, LplA2, GcvH, or GcvH-L in the presence or absence of crude lysates derived from a ΔlipA ΔlipM ΔlipL ΔlplA1 ΔlplA2 mutant lacking all lipoic acid de novo biosynthesis and salvage enzymes. Indeed, GcvH and GcvH-L were now partially lipoylated by LplA1 (Figure 6B and Figure S5A), while GcvH-L was partially lipoylated by LplA2 in the presence of crude lysates (Figure 6C and Figure S5B). Altogether, these data indicate that LplA1 and LplA2-dependent utilization of lipoyl peptides requires the presence of an endogenous lipoamidase produced by S. aureus.
Figure 6. S. aureus crude lysates harbor lipoamidase activity that permits the use of DKLA as a source of lipoic acid for attachment by LplA1 and LplA2.

(A) LplA1 and LplA2 attachment of DKLA tripeptide-derived lipoic acid (2.4 mM) to GcvH and GcvH-L. Lipoylation was assessed by conducting immunoblots (IB) with rabbit α-lipoic acid antibody. A parallel 12% SDS-PAGE gels was stained with GelCode Blue (CB). (B) LplA1-dependent attachment of DKLA tripeptide-derived lipoic acid (2.4 mM) to GcvH, GcvH-L and (C) LplA2 attachment of DKLA tripeptide-derived lipoic acid (2.4 mM) to GcvH-L in the presence of S. aureus whole cell lysates derived from a ΔlipA ΔlipM ΔlipL ΔlplA1 ΔlplA2 mutant. Lipoylation was assessed by conducting immunoblots (IB) with rabbit α-lipoic acid antibody. A parallel 12% SDS-PAGE gel was stained with GelCode Blue (CB). The positions of GcvH and GcvH-L are indicated.
GcvH-L is sufficient to restore growth to a lipoic acid auxotroph in the absence of free lipoic acid
LplA1 and LplA2 both attach lipoic acid to GcvH-L, while LplA1 only lipoylates GcvH. Although GcvH-L has been implicated in promoting resistance to redox stress during infection, its classification as a lipoyl domain H protein and recent evidence for evolutionary maintenance of H proteins as lipoyl reservoirs suggests that it may also promote lipoic acid biosynthesis and salvage (Rack et al., 2015, Cao et al., 2018). In Gram-positive Firmicutes, the H subunit of the glycine cleavage system is a critical conduit for the transfer of lipoic acid to E2 subunits, an activity that is mediated by the LipL amidotransferase (Christensen et al., 2011a, Christensen et al., 2011b, Martin et al., 2011). A ΔgcvH mutant is unable to replicate in the absence of free lipoic acid due to an inability to shuttle lipoic acid from GcvH to E2 proteins (Zorzoli et al., 2016, Christensen et al., 2011b). To test whether or not GcvH-L is sufficient to restore growth to a ΔgcvH mutant in the absence of free lipoic acid – and hence restore lipoyl transfer to E2 proteins – we generated ΔgcvH + gcvH, ΔgcvH + gcvH-L, and ΔgcvH + gcvH-L* (containing a single mutation K56A to remove the conserved lysine of the lipoyl domain (Rack et al., 2015)) strains, wherein the expression of gcvH, gcvH-L, and gcvH-L* is driven by the constitutive PHELP promoter. We subsequently monitored growth on solidified RPMI medium, growth in RPMI liquid culture, and lipoylation of E2 and H proteins in cell lysates. Expression of either GcvH or GcvH-L permitted growth on solid agar plates lacking lipoic acid, while expression of GcvH-L* displayed poor growth (Figure 7A). Furthermore, ΔgcvH + gcvH and ΔgcvH + gcvH-L strains grew to similar final optical densities in broth culture, although the ΔgcvH + gcvH-L strain had a slightly reduced growth rate compared to WT and ΔgcvH + gcvH strains (Figure 7B). In contrast, the ΔgcvH + gcvH-L* strain grew poorly and resembled the ΔgcvH mutant. Supplementation of lipoic acid into the culture medium restored WT growth characteristics to all strains due to limited lipoic acid attachment to E2-OGDH and subsequent transfer to other E2 subunits as seen in our prior studies (Figure 7C and (Zorzoli et al., 2016)). Immunoblots of whole cell lysates derived from the ΔgcvH + gcvH strain in BCFA medium lacking lipoic acid yielded prominent bands corresponding to lipoylation of all E2 proteins (Figure 7D, left and Figure S6). Similarly, lysates derived from the ΔgcvH + gcvH-L strain contained a dominant lipoyl E2-PDH band with detectable, albeit reduced, levels of lipoylation on E2-OGDH and E2-BCODH (Figure 7D, left). Lysates derived from the ΔgcvH + gcvH-L* strain had low levels of lipoylation on E2 subunits that were similar to the background levels seen in a ΔgcvH mutant (Figure 7D, left). Lipoylation of all E2 subunits was observed when the BCFA medium was supplemented with exogenous lipoic acid (Figure 7D, right and Figure S6). A band corresponding to lipoyl GcvH-L was not detected in any condition after immunoblot (Figure 7D). Identical results were obtained when experiments were repeated with a ΔgcvH ΔgcvH-L double mutant and ΔgcvH ΔgcvH-L + gcvH or ΔgcvH ΔgcvH-L + gcvH-L strains (Figure S7) Together, these data indicate that both GcvH and GcvH-L are sufficient to promote lipoyl transfer in S. aureus with important implications for both de novo biosynthesis and salvage of lipoic acid since H proteins are required for de novo biosynthesis, but are also direct targets of LplA1 and LplA2 (Figure 3).
Figure 7. GcvH and GcvH-L both participate in lipoyl relay to E2 subunits.

(A) Growth of the indicated strains on RPMI agar lacking free lipoic acid. (B–C) Growth (OD550) of the indicated strains in (B) RPMI and (C) RPMI + 5 μM lipoic acid (LA). (D) α-lipoic acid immunoblot of whole cell lysates derived from the indicated strains grown in RPMI supplemented with branched chain carboxylic acids (10 mM isobutyric acid, 9 mM 2-methylbutyric acid, 9 mM isovaleric acid, and 10 mM sodium acetate - BCFA) in order to bypass the requirement of lipoic acid for replication with or without lipoic acid (LA). The positions of the four lipoyl proteins in S. aureus (E2-PDH, E2-OGDH, E2-BCODH, and GcvH) are indicated.
Discussion
Microbial pathogens depend on a wide range of adaptive traits to promote their survival in nutrient-limited conditions. Trace nutrient acquisition pathways constitute a key bacterial survival strategy during infection. In this study, we used a biochemical approach to dissect the functional overlap between two lipoic acid ligases and two H proteins of S. aureus as it relates to flexibility in lipoic acid acquisition. Our findings indicate that LplA1 and LplA2 indeed harbor canonical lipoic acid ligase activity, but differ in their protein targets as outlined in Figure 8. LplA1 primarily lipoylates H proteins, whereas LplA2 has the capacity to target a wider range of proteins including the three E2 subunits of PDH, OGDH, and BCODH (Figure 8A–B). Neither of these ligases harbors secondary amidotransferase activity that permits the use of substrates other than free lipoic acid. However, S. aureus appears to make a soluble lipoamidase that allows use of peptide-bound lipoic acid by these ligases (Figure 8A–B). Furthermore, the ability to produce two H proteins – one that is expressed in culture and one that is posited to be upregulated in vivo or during periods of redox stress – provides an additional source of protein bound lipoic acid for transfer to E2 subunits. Together, these findings expand our understanding of lipoic acid salvage in S. aureus, and suggest an adaptive strategy that makes this pathogen extremely adept at lipoic acid acquisition.
Figure 8. Model of lipoic acid salvage by LplA1 and LplA2.

(A–B) de novo biosynthesis of lipoic acid occurs via an octanoic acid precursor attached to acyl carrier protein (ACP). The octanoyl group is transferred to the H subunit of GcvH by the octanoyl transferase LipM. The octanoyl moiety is converted to lipoic acid by LipA and transferred to the lipoyl domains of E2-PDH, E2-OGDH, or E2-BCODH by the amidotransferase LipL. (A) LplA1 scavenges free lipoic acid and primarily attaches the cofactor to the H subunits of GcvH and GcvH-L followed by transfer to E2 subunits by LipL. (B) LplA2 also scavenges free lipoic acid, but instead attaches the cofactor to the H subunit of GcvH-L as well as the E2 subunits of PDH, OGDH, and BCODH. The activity of both LplA1 and LplA2 requires free lipoic acid. However, S. aureus harbors lipoamidase (Lpd) activity that can hydrolyze the amide bond of lipoamide, providing an additional source of free lipoic acid for use by these ligases.
The lipoic acid ligase of B. subtilis, LplJ, targets GcvH but not other E2 subunits, while LplA of E. coli is indiscriminate in its recognition of lipoyl domain lysines (Christensen et al., 2011a, Christensen et al., 2011b, Green et al., 1995, Morris et al., 1994). This function for LplA in E. coli is logical because prior lipoyl attachment to GcvH and subsequent transfer to E2 subunits is not required for synthesis of lipoic acid (Cronan, 2016). In B. subtilis and L. monocytogenes on the other hand, GcvH serves as an important conduit for lipoic acid transfer to E2 subunits due to the amidotransferase activity of LipL (Christensen et al., 2011a, Christensen et al., 2011b, Martin et al., 2011). Direct lipoylation of H subunits in this scenario allows GcvH to serve as a source of lipoic acid for the bacterial cell. However, herein lies a potential bottleneck for these Gram positive species during periods of lipoic acid starvation; salvage and transfer to E2 subunits depends on passing through a GcvH intermediate, rather than direct lipoylation of the target E2 protein. In S. aureus, this limitation is eliminated by LplA2, which lipoylates all E2 subunits without the requirement of first passing through GcvH. However, S. aureus still maintains the LplA1 ligase, which behaves similarly to LplJ of B. subtilis and incidentally has greater amino acid sequence identity (57% compared to 39% for LplA2). The presence of two ligases with different lipoyl targets likely confers a level of added flexibility that may allow S. aureus to overcome severe limitations in bioavailable lipoic acid by directing trace amounts of the cofactor to the most critical complexes. A similar scenario may be true in L. monocytogenes, which also encodes two lipoic acid ligases, though technical difficulties in the purification of LplA2 have prevented an in-depth assessment of its biochemical activity (Christensen et al., 2011a).
An added layer of complexity in our understanding of how LplA1 and LplA2 promote lipoic acid salvage exists in the varied gene expression of the two enzymes. lplA1 is expressed both in vitro and in vivo as evidenced by: (1) the ability of a ΔlipA ΔlplA2 mutant to grow in the presence of exogenous lipoic acid in broth culture; and (2) the fact that ΔlplA2 and ΔlipA ΔlplA1 ΔlplA2 + lplA1 strains infect the kidneys of mice at similar levels to WT S. aureus, while ΔlplA1 ΔlplA2 and ΔlipA ΔlplA1 ΔlplA2 strains are markedly attenuated (Zorzoli et al., 2016). This is not true for a ΔlipA ΔlplA1 strain, which is defective for growth and lipoylation in broth culture even in the presence of free lipoic acid – unless cultured in the presence of FBS (Zorzoli et al., 2016). When lplA2 expression is driven by a constitutive promoter, lipoylation in the presence of exogenous lipoic acid is observed, although the levels of lipoylation on the E2 subunit of BCODH appear to be slightly lower when compared to complementation with lplA1 (Figure 2). Together, these observations strongly suggest that lplA2 is not normally expressed under standard growth conditions. In contrast, a ΔlplA1 mutant is fully virulent in vivo, while a ΔlplA1 ΔlplA2 mutant is significantly attenuated, indicating that lplA2 is expressed and sufficient to promote salvage in vivo (Zorzoli et al., 2016). These prior observations, coupled with our new understanding of LplA1 and LplA2 targeting, suggest that variations in both gene regulation and lipoyl protein targeting are likely to greatly expand the breadth and efficiency of lipoic acid attachment in appropriate environmental conditions. Identification of an in vitro growth condition that leads to high expression of the operon containing lplA2 has yet to be uncovered despite prior suggestion that redox stress is an inducing signal (Nobre & Saraiva, 2013). Although limited LplA2 activity is observed in the presence of FBS, this growth condition may eventually prove useful in the characterization of the gene regulatory programs of the lplA2 operon. Current work in the laboratory aims to decipher how gene regulation and the newly identified divergent activities of LplA1 and LplA2 interface with one another to promote diversification in lipoic acid acquisition.
In this study, we determined that S. aureus harbors lipoamidase activity and permits the salvage of lysine-bound lipoic acid. An acquisition strategy such as this is likely imperative to an infectious microbe such as S. aureus, which can reside in host environments that have negligible free lipoic acid levels (Zorzoli et al., 2016, Akiba et al., 1998), but instead contain significant amounts of the cofactor covalently attached to enzyme complex subunits. Enterococcus faecalis is the only bacterium with a fully characterized lipoamidase that is required for removal of lipoic acid from covalently attached lysines (Spalding & Prigge, 2009, Jiang & Cronan, 2005). BLAST of this sequence for similar proteins in S. aureus yielded no hits. Interestingly, Cristea and colleagues have determined that sirtuins of mammalian cells and bacteria can harbor lipoamidase activities that allow them to fine-tune the activity of major metabolic enzyme complexes (Rowland et al., 2017, Mathias et al., 2016, Mathias et al., 2014). It stands to reason that such a sirtuin might also exist in S. aureus. In a recent study, the E. coli sirtuin, CobB, a previously characterized deacetylase, was shown to have lipoamidase activity that can regulate the function of PDH and OGDH (Rowland et al., 2017). SrtN, the sirtuin homolog in B. subtilis also acts as a lipoamidase (Rowland et al., 2017). A homolog of this sirtuin exists in S. aureus (SAUSA300_2157), but deletion of this gene has no effect on the ability of a S. aureus ΔlipA strain to grow in the presence of DKLA, suggesting that another, or more than one, lipoamidase exists to carry out this activity (Teoh, unpublished).
One unusual outcome of this study was the observation that expression of lplA2 via a constitutive promoter restored lipoylation to lipoic acid auxotrophs in the presence of free lipoic acid, but was insufficient to restore bacterial growth in the presence of free lipoic acid. A similar phenotype is observed in yeast where supplementation of the growth medium with free lipoic acid does not restore aerobic growth to a LIP5 (LipA homolog) mutant, but is sufficient to promote lipoylation of target proteins (Sulo & Martin, 1993, Hermes & Cronan, 2013). At present, we suggest two possible explanations for this outcome. First, the observation that lipoylation of E2-BCODH is reduced in broth culture after complementation with lplA2 compared to WT suggests deficiencies in E2-BCODH activity may hamper the generation of branched chain fatty acids important for S. aureus membrane fluidity and replication. Indeed, supplementation of RPMI medium with BCFA restores growth to the ΔlipA ΔlplA1 and ΔlipA ΔlplA1 Δlpla2 + lplA2 strains in vitro, supporting the notion that perturbations in branched chain fatty acid metabolism are at least partially responsible for the growth deficit observed in our study (Figure S8). Our own efforts to generate an e2-BCODH mutant in S. aureus have been unsuccessful suggesting that the activity of this enzyme is critical for bacterial replication (Zorzoli et al., 2016). This is further supported by the poor growth of a transposon insertion mutant that disrupts BCODH activity (Mashruwala et al., 2016). The growth of this BCODH null strain is improved by supplementing BCFA to the culture medium. Other experimental perturbations that reduce the demand for lipoic acid-dependent enzyme function - oxygen depletion or mutation of clpC – are likely to yield the same outcome (Mashruwala et al., 2016). Alternatively, an inability of LplA2 to complement replication defects in vitro may suggest an unappreciated role for the other enzymes in the lplA2-encoding operon for restoration of growth. It is interesting to note that GcvH-L is also encoded in this operon where it is posited to have functions related to redox homeostasis that are regulated by lipoylation-dependent ADP-ribosylation by an operon-encoded sirtuin (Rack et al., 2015). Certainly, a deeper understanding of this locus and its biological activities is warranted and we are currently exploring the relationship between its primary activities and how they relate to lipoic acid salvage. Nonetheless, our data clearly indicate that despite the putative functions of GcvH-L and LplA2 that may be related to redox homeostasis and ADP-ribosylation, GcvH-L can also participate in lipoyl relay to E2 subunits, thereby expanding the functional lipoic acid biosynthesis and salvage pathways of S. aureus. Recent studies from Cao et al. corroborate this notion and provide evidence that many H and H-Like proteins, even those whose activities may not be directly involved in glycine cleavage, have evolved in an unusual way to participate in lipoyl relay (Cao et al., 2018).
Altogether, this study has expanded our understanding of the lipoic acid salvage strategies of S. aureus and highlights a unique adaptive strategy that is likely to contribute significantly to survival in nutrient deficient environments. Our prior demonstration that LplA1 and LplA2 can compensate for one another during murine infection underscores the biological significance of this point (Zorzoli et al., 2016). Further, the work herein places this observation within a mechanistic framework by highlighting the divergent targets of these two enzymes and the expanded acquisition pathways imparted by lipoamidase and GcvH-L. Thus, expansion of lipoic acid salvage mechanisms by S. aureus represents a critical adaptive trait likely used for survival in a wide range of nutrient limited environments, including host tissues.
Experimental Procedures
Bacterial strains and growth conditions
All bacterial strains used in this manuscript are listed in Table 1. E. coli strains were routinely grown in Lysogeny Broth (LB) (Amresco) with antibiotics added as necessary. S. aureus strains were grown in either rich medium, Tryptic Soy Broth (TSB) (Criterion), or in defined medium, Roswell Park Memorial Institute medium (RPMI) (Corning) supplemented with 1% casamino acids (Amresco). All strains were grown overnight at 37°C at a 45° angle with shaking unless stated otherwise. For growth curves, S. aureus overnight cultures were grown in RPMI containing branched chain carboxylic acids (10 mM isobutyric acid, 9 mM 2-methylbutyric acid, 9 mM isovaleric acid) and 10 mM sodium acetate (Sigma) in order to bypass the requirement of lipoic acid or octanoic acid for replication (Sigma) (Martin et al., 2011, Martin et al., 2009). When needed, cultures were supplemented with the following agents for selection: 100 μg ml−1 ampicillin (Amp), 3 μg ml−1 erythromycin (Erm), 10 μg ml−1 chloramphenicol (Cam), 50 μg ml−1 kanamycin (Kan), 50 μg ml−1 neomycin (Neo), and 0.2 mM cadmium chloride.
Table 1.
List of strains used in this study.
| USA300 LAC | S. aureus USA300 Strain LAC (AH-1264). Plasmid cured. | AH-LAC (WT) | (Boles et al., 2010) |
| DC10B | E. coli strain for recombinant pJC plasmids (dcm-) | DC10B | (Monk et al., 2012) |
| lysY/Iq | E. coli lysY/Iq for expression of LplA1 and LplA2 | lysY/Iq | NEB |
| FA-E1344 | E. coli lysY/Iq ΔlipA::kan for expression of H and E2 subunits | lysY/Iq ΔlipA::kan | (Grayczyk et al., 2017) |
| RN4220 | Restriction deficient S. aureus for plasmid passage | RN4220 | (Fairweather et al., 1983) |
| RN9011 | RN4220 + pRN7203 expressing SaPI integrase | RN9011 | (Chen et al., 2014) |
| FA-S1249 | AH-LAC with in-frame deletions of lipA and lplA1 | ΔlipAΔlplA1 | (Zorzoli et al., 2016) |
| FA-S1180 | AH-LAC with in-frame deletions of lipA and lplA2 | ΔlipAΔlplA2 | (Zorzoli et al., 2016) |
| FA-S1178 | AH-LAC with in-frame deletions of lipA, lplA1, and lplA2 | ΔlipAΔlplA1ΔlplA2 | (Zorzoli et al., 2016) |
| FA-S1200 | FA-S1178 complemented with pJC1111-lplA1 | ΔlipAΔlplA1ΔlplA2+lplA1 | (Zorzoli et al., 2016) |
| FA-S1212 | FA-S1178 complemented with pJC1111-lplA2 | ΔlipAΔlplA1ΔlplA2+lplA2 | (Zorzoli et al., 2016) |
| FA-S953 | AH-LAC ΔlipL::kan | ΔlipL::kan | (Zorzoli et al., 2016) |
| FA-S698 | JE2 lipM::erm transposon insertion mutant (NE1334) | lipM::erm | (Zorzoli et al., 2016) |
| FA-S1476 | AH-LAC ΔlipA ΔlplA1 ΔlplA2 ΔlipL::kan lipM::erm | ΔlipAΔlipMΔlipLΔlplA1ΔlplA2 | This work |
| FA-S1038 | AH-LAC with gene replacement of gcvH | ΔgcvH::kan | (Zorzoli et al., 2016) |
| FA-S1637 | AH-LAC ΔgcvH::kan containing pJC1111-gcvH | ΔgcvH::kan+gcvH | This work |
| FA-S1582 | AH-LAC ΔgcvH::kan containing pJC1111-gcvH-L | ΔgcvH::kan+gcvH-L | This work |
| FA-S1636 | AH-LAC ΔgcvH::kan containing pJC1111-gcvH-L (K56A) | ΔgcvH::kan+gcvH-L* | This work |
| FA-S1435 | AH-LAC with in-frame deletion of gcvH-L | ΔgcvH-L | This work |
| FA-S1498 | AH-LAC ΔgcvH-L containing pJC1111-gcvH-L | ΔgcvH-L + gcvH-L | This work |
| FA-S1645 | AH-LAC with gene replacement of gcvH and in-frame deletion of gcvH-L | ΔgcvH::kan ΔgcvH-L | This work |
| FA-S1684 | AH-LAC ΔgcvH::kan ΔgcvH-L containing pJC1111-gcvH | ΔgcvH::kan ΔgcvH-L + gcvH | This work |
| FA-S1686 | AH-LAC ΔgcvH::kan ΔgcvH-L containing pJC1111-gcvH-L | ΔgcvH::kan ΔgcvH-L + gcvH-L | This work |
| FA-E1357 | E. coli lysY/Iq ΔlipA::kan + pET15b-6x-His-gcvH | 6x-His-GcvH | This work |
| FA-E1383 | E. coli lysY/Iq ΔlipA::kan + pET15b-6x-His-gcvH-L | 6x-His-GcvH-L | This work |
| FA-E1359 | E. coli lysY/Iq ΔlipA::kan + pET15b-6x-His-e2-pdh | 6x-His-E2-PDH | This work |
| FA-E1363 | E. coli lysY/Iq ΔlipA::kan + pET15b-6x-His-e2-ogdh | 6x-His-E2-OGDH | This work |
| FA-E1367 | E. coli lysY/Iq ΔlipA::kan + pET15b-6x-His-e2-bcodh | 6x-His-E2-BCODH | This work |
| FA-E1284 | E. coli lysY/Iq + pET15b-6x-His-lplA1 | 6x-His-LplA1 | This work |
| FA-E1278 | E. coli lysY/Iq + pET15b-6x-His-lplA2 | 6x-His-LplA2 | This work |
Molecular genetic techniques
When necessary for validation of mutant strains, chromosomal DNA was isolated from S. aureus using the Wizard Genomic DNA purification kit (Promega) following the manufacturer’s protocol with minor modifications. Overnight cultures were started in 5 ml TSB and 1.5 ml was pelleted by centrifugation at 21,000 x g for 3 min the following day. Bacterial pellets were resuspended in 200 μl of TSM (50 mM Tris, 0.5 M Sucrose, 10 mM MgCl2, pH 7.5). In order to disrupt the cell wall, 2.5 μl of lysostaphin (2 mg ml−1 in 0.5 Tris, pH 8.0) was added to the resuspended cell pellet and incubated for 15 min at 37°C. Following incubation, the bacteria were pelleted at 21,000 x g for 3 min and the supernatant was discarded. The remaining steps to purify genomic DNA from S. aureus were completed using the manufacturer’s protocol. Recombinant plasmids were extracted using QIAGEN mini and midi prep kits with the following modifications for plasmid isolation from S. aureus when necessary. An overnight culture of S. aureus was grown in 5 ml TSB and pelleted at 3,000 x g for 10 min the following day. The bacterial pellet was resuspended in 400 μl TSM (50 mM Tris, pH 7.5, 0.5 M Sucrose, 10 mM MgCl2) followed by the addition of 20 μl of lysostaphin. This mixture was incubated for 10 min at 37°C to digest the cell wall and then pelleted at 16,000 x g for 2 min after which the supernatant was discarded. The remaining procedures were completed as suggested by the manufacturer’s protocol. Polymerase chain reaction (PCR) products were either gel-extracted or purified using QIAGEN QIAquick gel extraction and PCR purification kits. All PCRs were conducted using Phusion high-fidelity DNA polymerase (NEB).
E. coli competent cell preparation
An overnight culture of E. coli was grown in 3 ml LB at 37°C with shaking. The next day, the bacteria were subcultured 1:55 into a 250 ml flask and grown for an additional ~2.5 hours at 37°C with shaking until the culture reached an optical density at 600 nm (OD600) of 0.3–0.4. The following steps were all completed on ice. Cultures were aliquoted into 50 ml conical tubes and chilled on ice for 10 min. Afterwards, bacteria were pelleted by centrifugation at 3000 x g for 10 min and the bacterial pellet was washed twice with 10 ml TFB-1 (20 mM KOAc, 100 mM RbCl2, 10 mM CaCl2, 50 mM MnCl2, 15% glycerol, adjusted to pH 5.8 using 0.2 M Acetic acid) and incubated on ice for 10 min between washes. Bacteria were then pelleted at 3,000 x g for 10 min and resuspended in 1/25 of the original culture volume in TFB-2 (10 mM MOPS, 75 mM CaCl2, 10 mM RbCl2, 15% glycerol, adjusted to pH 6.5 using KOH). 100 μl of the competent cells were aliquoted into 1.5 mL tubes and stored at −80°C.
E. coli heat transformation
5 μl of ligation mix or 1 μl of purified plasmid were added to 50 μl of competent E. coli. The mixture was incubated on ice for 30 min, heat shocked at 42°C for 45 seconds, and then incubated for an additional 2 minutes on ice. 250 μl of super optimal broth with catabolite repression (SOC) medium (0.5% tryptone, 0.5% yeast extract, 0.05% NaCl, and 250 mM KCl adjusted to pH 7.0 with 5 M NaOH followed by addition of 20 mL 1M glucose) was added and the bacteria were incubated for 2 hours at 37°C with shaking. 100 μl of the bacterial suspension was plated onto the appropriate selection medium and incubated overnight at 37°C.
Preparation of S. aureus electrocompetent cells
An overnight culture of S. aureus was grown in 5 ml TSB at 37°C with shaking. The following day the bacteria were subcultured 1:100 in 30 ml TSB and incubated for an additional 3 hours at 37°C until the culture reached an OD600 of 0.5. The bacterial culture was pelleted by centrifugation at 3,000 x g for 10 min. All subsequent steps were carried out on ice. After pelleting, the bacteria were washed three times by resuspending in 30 ml ice cold 10% glycerol and pelleted by centrifugation at 3,000 x g for 10 min. After the last wash, the bacteria were suspended in 3 ml 10% glycerol, aliquoted into 1.5 ml tubes, and stored at −80°C.
Transformation of S. aureus
Frozen competent cells were thawed at room temperature for 5 min. 2 μl of plasmid DNA was then added to 50 μl of S. aureus LAC competent cells and incubated at room temperature for 30 min. The competent cell mixture was transferred to sterile 2 mm electroporation cuvettes and pulsed at 1800 V, 10 μF, and 600 Ω. After electroporation, the bacteria were resuspended in 750 μl of TSB or TSB + BCFA and incubated at 37°C or 30°C for 1.5 hours. After incubation, the bacteria were pelleted at 10,000 x g for 2 min and resuspended in 100 μl of TSB or TSB + BCFA, plated on TSA or TSA + BCFA plates containing antibiotic and incubated at 37°C or 30°C for 1–2 days.
Generation of a ΔlipA ΔlipM ΔlipL ΔlplA1 ΔlplA2 mutant
Strain FA-S1178 (ΔlipA ΔlplA1 ΔlplA2) was used as the recipient strain for transductions with generalized transducing phage φ11 harboring marked mutations in lipL (FA-S953 - ΔlipL::kan) and lipM (NE1334 – lipM::erm). In order to package φ11 with donor DNA, a 3 ml overnight culture of the marked donor strain (FA-S953 or NE1334) was started in TSB +/− BCFA:LB (1:1) supplemented with 5 mM CaCl2 and 5 mM MgSO4 and grown overnight with shaking at 37°C. The following day, the overnight strain was subcultured 1:100 into 10 ml TSB +/− BCFA:LB (1:1) supplemented with 5 mM CaCl2 and 5 mM MgSO4 and grown for 2.5 – 4 hours with shaking at 37°C until the culture reached an OD600 of 0.3 to 0.9. 500 μl of the bacterial culture was incubated with 10-fold serial dilutions of φ11 phage stock in TMG (10 mM Tris pH 7.5, 5 mM MgCl2, 0.01% gelatin (v/v)), vortexed gently, and incubated at room temperature for 30 minutes. After 30 minutes, tubes containing the bacteria and phage dilutions were mixed with 3 ml CY Top agar (casamino acids 3 g L−1, yeast extract 3 g L−1, NaCl 6 g L−1, 7.5 g L−1 agar, +/− BCFA as needed) supplemented with 5 mM CaCl2 and 5 mM MgSO4, cooled to 55°C, and poured onto TSA +/− BCFA plates. After the top agar solidified, plates were incubated at 30°C overnight face up. The next day the top agar from 2–3 plates with confluent plaques was scraped off the plate using a sterile scoopula and resuspended in 2 ml TMG buffer per plate followed by extensive vortexing. The top agar was pelleted by centrifugation at 16,000 x g for 15 minutes. The supernatant containing φ11 phage was filtered twice using a 0.2 μm filter and then an additional two times with a 0.45 μm filter. All phage stocks were stored at 4°C.
To transduce marked mutations, the recipient strain FA-S1178 (ΔlipA ΔlplA1 ΔlplA2) was cultured overnight with shaking at 37°C in 20 ml TSB+BCFA:LB (1:1) supplemented with 5 mM CaCl2. The following day, FA-S1178 was pelleted by centrifugation at 16,000 x g for 15 min and resuspended in 3 ml of TSB+BCFA:LB (1:1) supplemented with 5 mM CaCl2. 500 μl of the recipient bacteria were serial diluted and incubated with 100 μl of the φ11 phage (108–109 PFU) collected as described above or 100 μl of TMG buffer as an uninfected control for 30 min at room temperature, inverting the tubes every 10 min. After 30 min the bacteria/phage suspension was supplemented with 40 mM sodium citrate and incubated for an additional 30 min, inverting the tubes every 10 min. The bacteria were pelleted by centrifugation at 16,000 x g for 3 min and washed twice with 500 μl TSB+BCFA:LB (1:1) supplemented with 10 mM sodium citrate. Washed bacterial pellets were resuspended in 250 μl of TSB+BCFA:LB (1:1) supplemented with 10 mM sodium citrate and 200 μl was plated on TSA + BCFA containing 10 mM sodium citrate supplemented with erythromycin or kanamycin/neomycin as needed. Plates were incubated at 37°C for 24–48 hours until bacterial colonies were detected. All mutants were verified by PCR.
Generation of ΔgcvH-L and ΔgcvH::kan ΔgcvH-L mutants
Regions of homology corresponding to 498 bp upstream and 492 bp downstream of gcvH-L were amplified using primer pairs gcvH-L-SOE1 and gcvH-L-SOE2 or gcvH-L-SOE3 and gcvH-L-SOE4 (Table 2). The amplicons corresponding to the upstream and downstream regions of homology flanking gcvH-L were used as template in a splicing by overlap extension PCR reaction with primers gcvH-L-SOE1 and gcvH-L-SOE4 followed by sub-cloning the resulting amplicon into the allelic replacement plasmid pIMAY to generate pIMAY-gcvH-L (Monk et al., 2012). pIMAY-gcvH-L was introduced into WT AH-LAC and mutagenesis was carried out as previously described to obtain an in-frame deletion of gcvH-L that removed the entire coding region (Zorzoli et al., 2016). A marked ΔgcvH::kan mutation was subsequently transduced into the ΔgcvH-L strain, as described above, to generate a ΔgcvH::kan ΔgcvH-L double mutant.
Table 2.
List of oligonucleotides used in this study.
| Name | Sequence |
|---|---|
| His-LplA1-F | ATAT-CATATG(NdeI)-AAATTCATTAGTAATAATAATATT |
| His-LplA1-R | ATAT-GGATCC(BamHI)-TTATGACATTAATCTAATTAATT |
| His-LplA2-F | ATAT-CATATG(NdeI)-TACTTAATAGAACCGATTAG |
| His-LplA2-R | ATAT-GGATCC(BamHI)-TTAACTTAAAATCATATCCAC |
| His-GcvH-F | ATAT-CATATG(NdeI)-GCAGTACCAAATGAATTGAA |
| His-GcvH-R | ATAT-GGATCC(BamHI)-TTATTCACCAATCATTTCTGA |
| His-GcvH-L-F | ATAT- CATATG (NdeI)-AAAAAGTTAGCCAATTATTTAT |
| His-GcvH-L-R | ATAT- GGATCC(BamHI)-TTAAGCCTCCGGTAATGC |
| His-E2PDH-F | ATAT-CATATG(NdeI)-GCATTTGAATTTAGATTACCC |
| His-E2PDH-R | ATAT-GGATCC(BamHI)-TTACCCCTCCATTAATAATAA |
| His-E2OGDH-F | ATAT-CATATG(NdeI)-CCAGAGGTTAAAGTTCCAG |
| His-E2OGDH-R | ATAT-GGATCC(BamHI)-TTAAGATTCTAATAATAAGTCTT |
| His-E2BCODH-F | ATAT-CATATG(NdeI)-GAAATAACAATGCCTAAGTTA |
| His-E2BCODH-R | ATAT-GGATCC(BamHI)-CTAATATATATTTGTATTTTCTAA |
| UniCompSOE1 | ATAT-CTGCAG(PstI)-ATCCCATTATGCTTTGGCA |
| gcvH-L-CompSOE2 | CCCATAAATAATTGGCTAACTTTTTCATGGGTTTCACTCTCCTTCTA |
| gcvH-L-CompSOE3 | TAGAAGGAGAGTGAAACCCATGAAAAAGTTAGCCAATTATTTATGGG |
| gcvH-L-CompSOE4 | ATAT-GAGCTC(SacI)-TGCCGGTCTGACATTTGC |
| gcvH-CompSOE2 | TGGTACTGCCAAGGGTTTCACTCTCCTTCTA |
| gcvH-CompSOE3 | TAGAAGGAGAGTGAAACCCTTGGCAGTACCA |
| gcvH-CompSOE4 | ATAT-GAGCTC(SacI)-AATAGCTAAATTCATTATTTCCATCATTCTTGA |
| gcvH-L-K56AF | [Phos]CGAAGCATCGGCAACGGTCATT |
| gcvH-L-K56AR | [Phos]ATACTCACAATTTCATCATCCACTTTAACTTC |
| gcvH-L-SOE1 | ATAT-GGTACC(KpnI)-GGTATAGGGCTTTCTGTTGGAACA |
| gcvH-L-SOE2 | CTCGCTTTATTTGATTTTAACGTTTCCATCTTATCTTCATCCTTTCTCTTC |
| gcvH-L-SOE3 | GAAGAGAAAGGATGAAGATAAGATGGAAACGTTAAAATCAAATAAAGCGAG |
| gcvH-L-SOE4 | CAACA-GAGCTC(SacI)-CTTTGCTGACAAATTATATCCACGTG |
Generation of ΔgcvH::kan + gcvH, ΔgcvH::kan + gcvH-L, ΔgcvH-L + gcvh-L, ΔgcvH::kan ΔgcvH-L + gcvH, and ΔgcvH::kan ΔgcvH-L + gcvH-L strains
ΔgcvH::kan + gcvH, ΔgcvH::kan + gcvH-L, ΔgcvH-L + gcvh-L, ΔgcvH::kan ΔgcvH-L + gcvH, and ΔgcvH::kan ΔgcvH-L + gcvH-L complementation strains were generated using plasmid pJC1111, which stably integrates into the SaPI-1 site of the S. aureus chromosome after passage through S. aureus strain RN9011 containing pRN7203 expressing the SaPI-1 integrase, leading to single-copy stable integration of the plasmid (Chen et al., 2014). We first generated pJC1111 plasmids that drive expression of the gcvH or gcvH-L genes under the control of the constitutive PHELP promoter obtained from allelic replacement plasmid pIMAY (Monk et al., 2008, Monk et al., 2012, Riedel et al., 2007). The oligonucleotides listed in Table 2 were used to amplify the PHELP promoter and the open reading frames corresponding to gcvH and gcvH-L including ~150 nucleotides downstream of the stop codon. The PHELP amplicon was combined with either the gcvH or gcvH-L amplicon and the two fragments were joined by splicing by overlap extension PCR. The resulting PHELP-gcvH and PHELP-gcvH-L amplicons were subsequently purified and cloned into pJC1111 after digestion with restriction endonucleases PstI and SacI and subsequent ligation into similarly digested pJC1111, generating pJC1111-PHELP-gcvH and pJC1111- PHELP-gcvH-L.
Both pJC1111-PHELP-gcvH and pJC1111- PHELP-gcvH-L were propagated in E. coli DC10B, followed by isolation and subsequent electroporation into S. aureus RN9011 and plating on TSA plates supplemented with chloramphenicol and CdCl2. Plasmid integrants were used as donors to package and transduce the complementation allele into the AH-LAC ΔgcvH::kan, ΔgcvH-L, and ΔgcvH::kan ΔgcvH-L strain backgrounds. Final complementation strains were selected based on their CdCl2 resistance and validated via PCR. This resulted in the generation of the strains designated ΔgcvH::kan + gcvH, ΔgcvH::kan + gcvH-L, ΔgcvH-L + gcvh-L, ΔgcvH::kan ΔgcvH-L + gcvH, and ΔgcvH::kan ΔgcvH-L + gcvH-L.
Generation of ΔgcvH::kan + gcvH-L*
Using the complementation plasmid pJC1111-PHELP-gcvH-L as template, a K56A substitution was introduced into GcvH-L. K56 corresponds to the conserved lysine residue of the GcvH-L lipoyl domain that is targeted for lipoylation. The mutation was generated by introducing a two-nucleotide change in the center of the 5′-phosphorylated forward primer gcvH-L-K56A–F (Table 2). The 5′ phosphorylated reverse primer gcvH-L-K56A–R was designed so that the 5′ ends of the two primers anneal back-to-back. After exponential amplification with Phusion® High-Fidelity DNA Polymerase (NEB), the PCR reaction was treated with the methylation-dependent restriction enzyme DpnI (NEB) for one hour at 37°C to eliminate the original, non-mutated plasmid template. The blunt-end PCR products were then ligated with T4 DNA ligase (NEB) at room temperature for two hours and the reaction was transformed into DC10B competent cells and plated on LB agar with 100 μg ml−1 ampicillin (Monk et al., 2012). Plasmids were purified from transformants and sequenced with the external primers used to generate PHELP-gcvH-L (Table 2) to validate the substitution (K56A). The resultant pJC1111-PHELP-gcvH-L* was introduced into the ΔgcvH::kan strain as described above, yielding ΔgcvH::kan+gcvH-L*
Whole cell lysate preparation
Wild type and mutant S. aureus strains were grown overnight at 37°C with shaking in 15 mL conical tubes containing 5 mL of RPMI + BCFA medium. 50 μL of these overnight cultures was inoculated into 15 mL conical tubes containing 5 mL of RPMI + BCFA; RPMI + BCFA + 5 μM α-lipoic acid, or RPMI + BCFA + 150 μM octanoic acid. Samples were incubated with shaking at 200 rpm for 9 hours and bacterial growth was determined by measuring OD600 using a Genesys 10S UV-Vis spectrophotometer (Thermo Scientific). The remaining culture volume was centrifuged at 3,000 x g for 15 minutes, the supernatant was discarded, and the bacterial pellets were suspended in 250 μL of PBS and transferred to screw cap microcentrifuge lysing tubes (Fisher Scientific) containing 250 μL of 0.1 mm glass cell disruption beads (Scientific Industries, Inc.). Cells were lysed using a Fast Prep-24 5G (MP Biomedicals) bead disruption system in two sequential steps, at 5.0 speed for 20 seconds and at 4.5 speed for 20 seconds, each separated by a 5 minute incubation period on ice. After cell disruption, samples were centrifuged at 19,000 x g for 15 minutes. 45 μL of the supernatant were collected in microcentrifuge tubes containing 15 μL of 6X SDS sample buffer and subsequently boiled for 10 minutes prior to storage at −20°C.
Determination of protein lipoylation and LplA2 levels in whole cell lysates
Protein samples from OD-normalized whole cell lysates were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) in 12% polyacrylamide gels at 120 volts for approximately 4 hours. Gel staining was performed to evaluate protein patterns and equivalent loading of samples using GelCode Blue stain reagent (Thermo Scientific) with PageRuler Prestained Protein Ladder (Thermo Scientific) used as a molecular weight marker. Protein lipoylation and LplA2 levels were assessed via immunoblot. Briefly, resolved proteins were transferred from polyacrylamide gels to 0.2 μm PVDF membranes (Immobilon, Roche) at 70 V for 1 hour and 15 minutes in a Trans-Blot® Cell (Bio-Rad). After transfer, membranes were incubated for 1 hour with TBST (0.1% Tween-20 in Tris Buffered Saline) supplemented with 5% bovine serum albumin (BSA) at room temperature. A 1:3,000 dilution of rabbit polyclonal α-lipoic acid antibody (Calbiochem) or 1:50,000 dilution of rabbit polyclonal α-LplA2 antibody (Pacific Immunology) was added to the membrane followed by incubation for 1 hour and three subsequent washes in ~20 mL of TBST. AP-conjugated Goat anti-Rabbit IgG (H+L) (Invitrogen) was then added at a 1:5000 dilution in TBST for 45 minutes followed by three 15 minute washes in ~20 mL of TBST. Membranes were developed with BCIP/NBT (5-bromo-4-chloro-3-indoyl-phosphate/nitro blue tetrazolium) color development substrate (VWR).
Preparation of Proteinase K agarose beads and digestion of porcine PDH
Proteinase K agarose beads and porcine pyruvate dehydrogenase (PDH) were purchased from Sigma. The Proteinase K beads were activated by suspending 40 mg beads in 1 ml activation buffer (20 mM Tris-HCl, 1 mM CaCl2, pH 7.4) and incubated for 2 hours at room temperature. Before protein digestion, the beads were washed three times by centrifugation for 3 min at 400 x g followed by resuspension in 800 μl activation buffer. PDH (16.0 mg ml−1) was exchanged from its storage buffer into 20 mM Tris-HCl, 1 mM CaCl2 pH 7.4. Briefly, 2 ml PDH solution was diluted into 18 ml of 20 mM Tris-HCl, 1 mM CaCl2 pH 7.4 followed by concentration to 250 μl using a 10 kDa cut-off Amicon Ultra-15 spin column (Milipore). This step was repeated 5 times. After buffer exchange, 2.5 mg ml−1 PDH was digested overnight with 400 μl Proteinase K agarose bead suspension. The following morning, the beads were removed from the digestion reaction by centrifugation at 16,000 x g for 10 min and the supernatant was used in subsequent growth curves.
Growth curves
Overnight cultures were grown in 200 μl of RPMI + BCFA in a 96-well plate at 37°C with shaking. The next day, the strains were pelleted for 10 min at 2,000 x g at 4°C. The strains were washed three to five times with 200 μl of RPMI in order to remove any remaining BCFA that might stimulate growth. Each strain was grown in RPMI supplemented with 5 μM lipoic acid (Sigma), 25 μM octanoic acid (Sigma), 0.25 mg ml−1 PDH (Sigma) or the volume equivalent of proteinase K digested PDH, and 100 μM DKLA or 100 μM DKA tripeptides (Anaspec). All growth curves were conducted in a 96-well plate at 37°C for 8–10 hours until reaching stationary phase. Changes in turbidity were recorded every hour by measuring OD550 on a BioTek plate reader.
Generation of 6x-Histidine tagged protein expression plasmids
The coding sequences of lplA1, lplA2 e2-PDH, e2-OGDH, e2-BCODH, gcvH, and gcvH-L, were amplified using the following primer pairs: His-LplA1-F/His-LplA1-R (lplA1), His-LplA2-F/His-LplA2-R (lplA2), His-E2PDH-F/His-E2PDH-R (e2-PDH), His-E2OGDH-F/His-E2OGDH-R (e2-OGDH), His-E2BCODH-F/His-E2BCODH-R (e2-BCODH), His-GcvH-F/His-GcvH-R (gcvH), and His-GcvH-L-F/His-GcvH-L-R (gcvH-L) (See Table 2). The resulting amplicons were sub-cloned into protein expression vector pET-15b using NdeI and BamHI restriction endonucleases to generate expression plasmids containing each gene with an N-terminal 6x-Histidine tag. Plasmids containing 6x-His-lplA1 and 6x-His-lplA2 were transformed into lysY/Iq E. coli and plasmids containing 6x-His-e2-PDH, 6x-His-e2-OGDH, 6x-His-e2-BCODH, 6x-His-gcvH, and 6x-His-gcvH-L were transformed into ΔlipA::kan lysY/Iq E. coli to ensure each subunit would be purified in its apo form (Grayczyk et al., 2017).
Purification of LplA1 and LplA2
LplA1 and LplA2 were purified using Ni2+ affinity chromatography. lysY/Iq E. coli strains containing pET15b-6x-His-lplA1, and pET15b-6x-His-lplA1 were grown in 5 ml LB with 100 μg ml−1 ampicillin at 37°C with shaking overnight. The following day, bacteria were subcultured 1:100 and allowed to grow for 3 hours at 37°C until reaching an OD600 of 0.25–0.3. Protein expression was induced upon addition of 0.1 mM IPTG followed by incubation for 16 hours at 16°C with shaking. The next day, bacteria were pelleted by centrifugation at 13,000 x g for 10 min at 4°C followed by storage at −80°C. To purify the recombinant proteins, bacterial pellets were thawed at 37°C and resuspended in lysis buffer (25 mM imidazole, 50 mM Tris-HCl, 300 mM NaCl, pH 8) supplemented with 1 mM dithiothreitol (DTT) and 1 mM phenylmethylsulfonyl fluoride (PMSF). The bacteria were lysed at a constant rate of 0.8 seconds per pulse and an output of 340 W in 20 second intervals for 15 min on ice using a Branson S-450A large tip sonicator. Debris from lysed bacteria was pelleted by centrifugation for 30 min at 16,000 x g followed by filtration of the clarified lysate using a 0.45 μm syringe filter. The lysate was then incubated with 1 ml nickel-NTA resin (Qiagen) while rocking for 1 hour at 4°C, washed with wash buffer (50 mM imidazole, 1 mM DTT, 50 mM Tris-HCl, 300 mM NaCl, pH 8.0), followed by elution of the bound protein using the same buffer containing 500 mM imidazole. Samples were dialyzed with 10 kDa molecular weight cut-off (MWCO) snakeskin dialysis tubing (Thermo Scientific) using the following scheme: the sample was first placed into 1 L buffer containing 100 mM imidazole, 50 mM Tris-HCl, 300 mM NaCl, pH 8 for 3 hours, then into buffer containing 25 mM imidazole, 50 mM Tris-HCl, 300 mM NaCl, pH 8 overnight, followed by an additional 4 hours in buffer containing 50 mM Tris-HCl, 300 mM NaCl, pH 8 the following day. The concentration of the purified protein was measured using a bicinchoninic acid (BCA) kit (Thermo Scientific) and stored at −80°C. Protein purity was confirmed by resolving ~2 μg of purified protein on SDS-PAGE gels followed by staining with GelCode Blue. Where necessary, proteins were further purified to homogeneity using fast protein liquid chromatography (FPLC) on a Superdex 100 Increase 3.2/300 size exclusion column.
Purification of E2 and H subunits of lipoylated enzyme complexes
The E2-subunits of PDH, BCODH, OGDH, GcvH, and GcvH-L were purified from a ΔlipA::kan lysY/Iq E. coli strain using Ni2+ affinity chromatography. ΔlipA::kan lysY/Iq E. coli strains containing pET15b-6x-His-e2-PDH, and pET15b-6x-His-E2-OGDH, pET15b-6x-His-e2-BCODH, pET15b-6x-His-gcvH, and pET15b-6x-His-gcvH-L were grown overnight in 30 ml LB with 100 μg ml−1 ampicillin at 37°C with shaking. The following day, strains were subcultured 1:100 into LB medium containing 100 μg ml−1 ampicillin and grown for 20 hours at 37°C with shaking. The next day, protein expression was induced with 0.5 mM IPTG for 4 hours at 37°C with shaking. After induction, the bacteria were pelleted by centrifugation at 13,000 x g for 10 min at 4°C and stored at −80°C overnight. The remaining purification of E2 and H subunits followed the same protocol used for LplA1 and LplA2 described above.
Lipoylation and octanoylation assays
Lipoylation assays were set up as described by Martin et al with some modifications (Martin et al., 2011). All assays were conducted in 50 μl reaction volumes in reaction buffer (50 mM Tris-HCl, 300 mM NaCl, pH 8.0) containing 6 mM ATP, 1 mM DTT, 1 mM MgCl2, 1 μM purified LplA1 or LplA2, and 20 μM substrate (E2-PDH, E2-OGDH, E2-BCODH, GcvH, or GcvH-L). The reactions were incubated with or without lipoic acid (2.4 mM), octanoic acid (2.4 mM), or DKLA (2.4 mM) for 2 hours at 37°C with shaking. After incubation, the reaction mixtures were resolved on parallel 12% SDS-PAGE gels at 120 V for approximately 3 hours. One gel was stained with GelCode Blue (Thermo Scientific) and the other was used for immunoblotting. The resolved reactions were transferred to 0.2 μM PVDF membranes at 1000 mA for 1 hour. After transfer, the membrane was blocked overnight at 4°C in Tris-buffered saline + 0.1% Tween (TBST) containing 5% bovine serum albumin (BSA). The following day, rabbit α-lipoic acid antibody was added to the membrane at a dilution of 1:7,500 in TBST + 5% BSA for 1 hour at room temperature with rocking. The membrane was washed three times in TBST followed by addition of goat α-rabbit IgG AP conjugate to the membrane at a 1:5,000 dilution in TBST + 5% BSA for 1 hour at room temperature. The membrane was washed three times in TBST at 15 min intervals followed by visualization of lipoylated proteins using colorimetric detection upon addition of 66 μl of nitro-blue tetrazolium (NBT) (50 mg NBT in 1 ml 70% dimethylformamide (DMF)/30% H2O) and 35 μl of 5-bromo-4-chloro-3′-indolyphosphate (BCIP) (50 mg BCIP in 1 ml DMF) to 10 ml AP Buffer (100 mM Tris, 100 mM NaCl, 5 mM MgCl2, pH 9.5) and incubating the membrane with this solution for ~2 min while rocking at room temperature. Development of the blot was stopped by washing the membrane with water and allowing it to dry at 37°C for 15 min.
Lipoamidase activity assay
Overnight cultures of the ΔlipA ΔlipM ΔlipL ΔlplA1 ΔlplA2 S. aureus strain lacking all enzymes involved in de novo biosynthesis and salvage of lipoic acid was grown in 5 ml RPMI + BCFA at 37°C with shaking. The following day, the strain was subcultured 1:100 in 6 ml of RPMI + BCFA and grown for 9 hours at 37°C with shaking. The bacteria were pelleted by centrifugation at 3,000 x g for 15 min, the supernatant discarded, and remaining pellet stored at −80°C. The following day, the bacterial pellet was thawed on ice for 10 min, resuspended in 250 μl 1X PBS, and transferred to microcentrifuge screw-cap tubes containing ~250 μl 0.1 mm glass cell disruption beads (Scientific Industries, Inc.). The bacteria were lysed using a Fast Prep-24 5G (MP Biomedicals) at speed 5.0 for 20 seconds followed by speed 4.5 for an additional 20 seconds with 5 min incubation on ice between lysis steps. After lysis, the samples were centrifuged at 18,000 x g for 15 min and clarified lysates were collected and immediately used in subsequent assays of amidotransferase activity.
Amidotransferase activity was assayed in 50 μl reaction volumes containing 6 mM ATP, 1 mM DTT, 1 mM MgCl2, 1 μM purified LplA1 or LplA2, 20 μM substrate (GcvH or GcvH-L) and 35 μl of ΔlipA ΔlipM ΔlipL ΔlplA1 ΔlplA2 mutant whole cell lysates, prepared as described above. The reactions were incubated with or without DKLA (2.4 mM) for 2 hours at 37°C with shaking. After incubation, proteins were resolved on parallel SDS-PAGE gels for staining with GelCode Blue (Thermo Scientific) and immunoblot with rabbit α-lipoic acid antibody as described above.
Reproducibility and experimental rigor
All growth curves were conducted three times in triplicate. Data shown are of a single representative growth curve conducted in triplicate with error bars reflecting the standard deviation between samples. When no error bars are shown, this reflects a data point where the error was smaller than the size of the symbol used to plot that point. All biochemical assays were conducted at least four times with two independent preparations of recombinant proteins to ensure reproducibility and rule out effects of any potential impurities. DKA, DKLA, DKT and DKLT tri-peptides were synthesized by Anaspec and purified by HPLC.
Supplementary Material
Acknowledgments
We thank members of the Alonzo laboratory for critically reading this manuscript. We are grateful to the laboratories of Karen Visick and Alan Wolfe for helpful discussions. This study was supported by National Institutes of Health R01 AI120994 to FA, and American Heart Association 17PRE33660173 to JPG.
Footnotes
Author contributions
IL, WPT, and FA conceived and designed components of the study; IL, WPT, SF, JPG, AZ, and FA assisted in the acquisition, analysis, and interpretation of the data; IL, WPT, and FA wrote the manuscript; all authors commented on the final version manuscript.
References
- Afanador GA, Matthews KA, Bartee D, Gisselberg JE, Walters MS, Freel Meyers ST, Prigge CL. Redox-dependent lipoylation of mitochondrial proteins in Plasmodium falciparum. Mol Microbiol. 2014;94:156–171. doi: 10.1111/mmi.12753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiba S, Matsugo S, Packer L, Konishi T. Assay of protein-bound lipoic acid in tissues by a new enzymatic method. Anal Biochem. 1998;258:299–304. doi: 10.1006/abio.1998.2615. [DOI] [PubMed] [Google Scholar]
- Beasley FC, Heinrichs DE. Siderophore-mediated iron acquisition in the staphylococci. Journal of inorganic biochemistry. 2010;104:282–288. doi: 10.1016/j.jinorgbio.2009.09.011. [DOI] [PubMed] [Google Scholar]
- Boles BR, Thoendel M, Roth AJ, Horswill AR. Identification of genes involved in polysaccharide-independent Staphylococcus aureus biofilm formation. PLoS One. 2010;5:e10146. doi: 10.1371/journal.pone.0010146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryk R, Arango N, Maksymiuk C, Balakrishnan A, Wu YT, Wong CH, Masquelin T, Hipskind P, Lima CD, Nathan C. Lipoamide channel-binding sulfonamides selectively inhibit mycobacterial lipoamide dehydrogenase. Biochemistry. 2013;52:9375–9384. doi: 10.1021/bi401077f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryk R, Lima CD, Erdjument-Bromage H, Tempst P, Nathan C. Metabolic enzymes of mycobacteria linked to antioxidant defense by a thioredoxin-like protein. Science. 2002;295:1073–1077. doi: 10.1126/science.1067798. [DOI] [PubMed] [Google Scholar]
- Cao X, Hong Y, Zhu L, Hu Y, Cronan JE. Development and retention of a primordial moonlighting pathway of protein modification in the absence of selection presents a puzzle. Proc Natl Acad Sci U S A. 2018;115:647–655. doi: 10.1073/pnas.1718653115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassat JE, Skaar EP. Metal ion acquisition in Staphylococcus aureus: overcoming nutritional immunity. Semin Immunopathol. 2012;34:215–235. doi: 10.1007/s00281-011-0294-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Yoong P, Ram G, Torres VJ, Novick RP. Single-copy vectors for integration at the SaPI1 attachment site for Staphylococcus aureus. Plasmid. 2014;76C:1–7. doi: 10.1016/j.plasmid.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen QH, Cronan JE. Lipoic acid synthesis: a new family of octanoyltransferases generally annotated as lipoate protein ligases. Biochemistry. 2010;49:10024–10036. doi: 10.1021/bi101215f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen QH, Hagar JA, O’Riordan MX, Cronan JE. A complex lipoate utilization pathway in Listeria monocytogenes. J Biol Chem. 2011a;286:31447–31456. doi: 10.1074/jbc.M111.273607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen QH, Martin N, Mansilla MC, de Mendoza D, Cronan JE. A novel amidotransferase required for lipoic acid cofactor assembly in Bacillus subtilis. Mol Microbiol. 2011b;80:350–363. doi: 10.1111/j.1365-2958.2011.07598.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronan JE. Assembly of Lipoic Acid on Its Cognate Enzymes: an extraordinary and essential biosynthetic pathway. Microbiology and molecular biology reviews : MMBR. 2016;80:429–450. doi: 10.1128/MMBR.00073-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dacheux D, Epaulard O, de Groot A, Guery B, Leberre R, Attree I, Polack B, Toussaint B. Activation of the Pseudomonas aeruginosa type III secretion system requires an intact pyruvate dehydrogenase aceAB operon. Infect Immun. 2002;70:3973–3977. doi: 10.1128/IAI.70.7.3973-3977.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douce R, Bourguignon J, Neuburger M, Rebeille F. The glycine decarboxylase system: a fascinating complex. Trends in plant science. 2001;6:167–176. doi: 10.1016/s1360-1385(01)01892-1. [DOI] [PubMed] [Google Scholar]
- Fairweather N, Kennedy S, Foster TJ, Kehoe M, Dougan G. Expression of a cloned Staphylococcus aureus alpha-hemolysin determinant in Bacillus subtilis and Staphylococcus aureus. Infect Immun. 1983;41:1112–1117. doi: 10.1128/iai.41.3.1112-1117.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkard B, Kumar TR, Hecht LS, Matthews KA, Henrich PP, Gulati S, Lewis RE, Manary MJ, Winzeler EA, Sinnis P, Prigge ST, Heussler V, Deschermeier C, Fidock D. A key role for lipoic acid synthesis during Plasmodium liver stage development. Cell Microbiol. 2013;15:1585–1604. doi: 10.1111/cmi.12137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grayczyk JP, Harvey CJ, Laczkovich I, Alonzo F., 3rd A Lipoylated metabolic protein released by Staphylococcus aureus suppresses macrophage activation. Cell Host Microbe. 2017;22:678–687e679. doi: 10.1016/j.chom.2017.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DE, Morris TW, Green J, Cronan JE, Jr, Guest JR. Purification and properties of the lipoate protein ligase of Escherichia coli. Biochem J. 1995;309(Pt 3):853–862. doi: 10.1042/bj3090853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grim KP, San Francisco B, Radin JN, Brazel EB, Kelliher JL, Parraga Solorzano PK, Kim PC, McDevitt CA, Kehl-Fie TE. The metallophore staphylopine enables Staphylococcus aureus to compete with the host for zinc and overcome nutritional immunity. mBio. 2017:8. doi: 10.1128/mBio.01281-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunther S, Matuschewski K, Muller S. Knockout studies reveal an important role of Plasmodium lipoic acid protein ligase A1 for asexual blood stage parasite survival. PLoS One. 2009;4:e5510. doi: 10.1371/journal.pone.0005510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermes FA, Cronan JE. Scavenging of cytosolic octanoic acid by mutant LplA lipoate ligases allows growth of Escherichia coli strains lacking the LipB octanoyltransferase of lipoic acid synthesis. J Bacteriol. 2009;191:6796–6803. doi: 10.1128/JB.00798-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermes FA, Cronan JE. The role of the Saccharomyces cerevisiae lipoate protein ligase homologue, Lip3, in lipoic acid synthesis. Yeast. 2013;30:415–427. doi: 10.1002/yea.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood MI, Skaar EP. Nutritional immunity: transition metals at the pathogen-host interface. Nat Rev Microbiol. 2012;10:525–537. doi: 10.1038/nrmicro2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Cronan JE. Expression cloning and demonstration of Enterococcus faecalis lipoamidase (pyruvate dehydrogenase inactivase) as a Ser-Ser-Lys triad amidohydrolase. J Biol Chem. 2005;280(3):2244–2256. doi: 10.1074/jbc.M408612200. [DOI] [PubMed] [Google Scholar]
- Jordan SW, Cronan JE., Jr The Escherichia coli lipB gene encodes lipoyl (octanoyl)-acyl carrier protein:protein transferase. J Bacteriol. 2003;185:1582–1589. doi: 10.1128/JB.185.5.1582-1589.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeney KM, Stuckey JA, O’Riordan MX. LplA1-dependent utilization of host lipoyl peptides enables Listeria cytosolic growth and virulence. Mol Microbiol. 2007;66:758–770. doi: 10.1111/j.1365-2958.2007.05956.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelliher JL, Radin JN, Grim KP, Parraga Solorzano PK, Degnan PH, Kehl-Fie TE. Acquisition of the phosphate transporter NptA enhances Staphylococcus aureus pathogenesis by improving phosphate uptake in divergent environments. Infect Immun. 2018:86. doi: 10.1128/IAI.00631-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi G, Motokawa Y, Yoshida T, Hiraga K. Glycine cleavage system: reaction mechanism, physiological significance, and hyperglycinemia. Proceedings of the Japan Academy Series B, Physical and biological sciences. 2008;84:246–263. doi: 10.2183/pjab/84.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Bayer A, Cosgrove SE, Daum RS, Fridkin SK, Gorwitz RJ, Kaplan SL, Karchmer AW, Levine DP, Murray BE, JRM, Talan HF, Chambers DA. Clinical practice guidelines by the infectious diseases society of america for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children: executive summary. Clin Infect Dis. 2011;52:285–292. doi: 10.1093/cid/cir034. [DOI] [PubMed] [Google Scholar]
- Martin N, Christensen QH, Mansilla MC, Cronan JE, de Mendoza D. A novel two-gene requirement for the octanoyltransfer reaction of Bacillus subtilis lipoic acid biosynthesis. Mol Microbiol. 2011;80:335–349. doi: 10.1111/j.1365-2958.2011.07597.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin N, Lombardia E, Altabe SG, de Mendoza D, Mansilla MC. A lipA (yutB) mutant, encoding lipoic acid synthase, provides insight into the interplay between branched-chain and unsaturated fatty acid biosynthesis in Bacillus subtilis. J Bacteriol. 2009;191:7447–7455. doi: 10.1128/JB.01160-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashruwala AA, Roberts CA, Bhatt S, May KL, Carroll RK, Shaw LN, Boyd JM. Staphylococcus aureus SufT: an essential iron-sulphur cluster assembly factor in cells experiencing a high-demand for lipoic acid. Mol Microbiol. 2016;102:1099–1119. doi: 10.1111/mmi.13539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathias RA, Greco TM, Cristea IM. Identification of Sirtuin4 (SIRT4) Protein Interactions: Uncovering Candidate Acyl-Modified Mitochondrial Substrates and Enzymatic Regulators. Methods Mol Biol. 2016;1436:213–239. doi: 10.1007/978-1-4939-3667-0_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathias RA, Greco TM, Oberstein A, Budayeva HG, Chakrabarti R, Rowland EA, Kang Y, Shenk T, Cristea IM. Sirtuin 4 is a lipoamidase regulating pyruvate dehydrogenase complex activity. Cell. 2014;159:1615–1625. doi: 10.1016/j.cell.2014.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazmanian SK, Skaar EP, Gaspar AH, Humayun M, Gornicki P, Jelenska J, Joachmiak A, Missiakas DM, Schneewind O. Passage of heme-iron across the envelope of Staphylococcus aureus. Science. 2003;299:906–909. doi: 10.1126/science.1081147. [DOI] [PubMed] [Google Scholar]
- Miller JR, Busby RW, Jordan SW, Cheek J, Henshaw TF, Ashley GW, Broderick JB, Cronan JE, Jr, Marletta MA. Escherichia coli LipA is a lipoyl synthase: in vitro biosynthesis of lipoylated pyruvate dehydrogenase complex from octanoyl-acyl carrier protein. Biochemistry. 2000;39:15166–15178. doi: 10.1021/bi002060n. [DOI] [PubMed] [Google Scholar]
- Monk IR, Gahan CG, Hill C. Tools for functional postgenomic analysis of Listeria monocytogenes. Appl Environ Microbiol. 2008;74:3921–3934. doi: 10.1128/AEM.00314-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monk IR, Shah IM, Xu M, Tan MW, Foster TJ. Transforming the untransformable: application of direct transformation to genetically manipulate Staphylococcus aureus and Staphylococcus epidermidis. mBio. 2012:3. doi: 10.1128/mBio.00277-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris TW, Reed KE, Cronan JE., Jr Identification of the gene encoding lipoate-protein ligase A of Escherichia coli. Molecular cloning and characterization of the lplA gene and gene product. J Biol Chem. 1994;269:16091–16100. [PubMed] [Google Scholar]
- Morris TW, Reed KE, Cronan JE., Jr Lipoic acid metabolism in Escherichia coli: the lplA and lipB genes define redundant pathways for ligation of lipoyl groups to apoprotein. J Bacteriol. 1995;177:1–10. doi: 10.1128/jb.177.1.1-10.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobre LS, Saraiva LM. Effect of combined oxidative and nitrosative stresses on Staphylococcus aureus transcriptome. Appl Microbiol Biotechnol. 2013;97:2563–2573. doi: 10.1007/s00253-013-4730-3. [DOI] [PubMed] [Google Scholar]
- O’Riordan M, Moors MA, Portnoy DA. Listeria intracellular growth and virulence require host-derived lipoic acid. Science. 2003;302:462–464. doi: 10.1126/science.1088170. [DOI] [PubMed] [Google Scholar]
- Perham RN. Swinging arms and swinging domains in multifunctional enzymes: catalytic machines for multistep reactions. Annual review of biochemistry. 2000;69:961–1004. doi: 10.1146/annurev.biochem.69.1.961. [DOI] [PubMed] [Google Scholar]
- Pilatz S, Breitbach K, Hein N, Fehlhaber B, Schulze J, Brenneke B, Eberl L, Steinmetz I. Identification of Burkholderia pseudomallei genes required for the intracellular life cycle and in vivo virulence. Infect Immun. 2006;74:3576–3586. doi: 10.1128/IAI.01262-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posner MG, Upadhyay A, Bagby S, Hough DW, Danson MJ. A unique lipoylation system in the Archaea. Lipoylation in Thermoplasma acidophilum requires two proteins. The FEBS journal. 2009;276:4012–4022. doi: 10.1111/j.1742-4658.2009.07110.x. [DOI] [PubMed] [Google Scholar]
- Rack JG, Morra R, Barkauskaite E, Kraehenbuehl R, Ariza A, Qu Y, Ortmayer M, Leidecker O, Cameron DR, Matic I, Peleg AY, Leys D, Traven A, Ahel I. Identification of a class of protein ADP-ribosylating sirtuins in microbial pathogens. Molecular cell. 2015;59:309–320. doi: 10.1016/j.molcel.2015.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson AR, Somerville GA, Sonenshein AL. Regulating the intersection of metabolism and pathogenesis in Gram-positive bacteria. Microbiology spectrum. 2015:3. doi: 10.1128/microbiolspec.MBP-0004-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedel CU, I, Monk R, Casey PG, Morrissey D, O’Sullivan GC, Tangney M, Hill C, Gahan CG. Improved luciferase tagging system for Listeria monocytogenes allows real-time monitoring in vivo and in vitro. Appl Environ Microbiol. 2007;73:3091–3094. doi: 10.1128/AEM.02940-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts CA, Al-Tameemi HM, Mashruwala AA, Rosario-Cruz Z, Chauhan U, Sause WE, Torres VJ, Belden WJ, Boyd JM. The Suf iron-sulfur cluster biosynthetic system is essential in Staphylococcus aureus, and decreased Suf function results in global metabolic defects and reduced survival in human neutrophils. Infect Immun. 2017:85. doi: 10.1128/IAI.00100-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland EA, Greco TM, Snowden CK, McCabe AL, Silhavy TJ, Cristea IM. Sirtuin lipoamidase activity is conserved in bacteria as a regulator of metabolic enzyme complexes. mBio. 2017:8. doi: 10.1128/mBio.01096-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spalding MD, Prigge ST. The amidase domain of lipoamidase specifically inactivates lipoylated proteins in vivo. PLoS One. 2009;4:e7392. doi: 10.1371/journal.pone.0007392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spalding MD, Prigge ST. Lipoic acid metabolism in microbial pathogens. Microbiology and molecular biology reviews : MMBR. 2010;74:200–228. doi: 10.1128/MMBR.00008-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm J, Muller S. Lipoic acid metabolism of Plasmodium--a suitable drug target. Curr Pharm Des. 2012;18:3480–3489. doi: 10.2174/138161212801327266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulo P, Martin NC. Isolation and characterization of LIP5. A lipoate biosynthetic locus of Saccharomyces cerevisiae. J Biol Chem. 1993;268:17634–17639. [PubMed] [Google Scholar]
- Tong SY, Davis JS, Eichenberger E, Holland TL, Fowler VG., Jr Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin Microbiol Rev. 2015;28:603–661. doi: 10.1128/CMR.00134-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Boom TJ, Reed KE, Cronan JE., Jr Lipoic acid metabolism in Escherichia coli: isolation of null mutants defective in lipoic acid biosynthesis, molecular cloning and characterization of the E. coli lip locus, and identification of the lipoylated protein of the glycine cleavage system. J Bacteriol. 1991;173:6411–6420. doi: 10.1128/jb.173.20.6411-6420.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Miller JR, Jiang Y, Marletta MA, Cronan JE. Assembly of the covalent linkage between lipoic acid and its cognate enzymes. Chem Biol. 2003;10:1293–1302. doi: 10.1016/j.chembiol.2003.11.016. [DOI] [PubMed] [Google Scholar]
- Zorzoli A, Grayczyk JP, Alonzo F., 3rd Staphylococcus aureus tissue infection during sepsis is supported by differential use of bacterial or host-derived lipoic acid. PLoS Pathog. 2016;12:e1005933. doi: 10.1371/journal.ppat.1005933. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.