

Abstract
Iron has played an important role in energy production since the beginning of life, as iron-catalyzed redox reactions are required for energy production. Oxygen, a highly efficient electron acceptor with high reduction potential, facilitates highly efficient energy production in eukaryotic cells. However, the increasing atmospheric oxygen concentration produces new threats to the organism, as oxygen reacts with iron and produces reactive oxygen species unless its levels are strictly regulated. As the size of multicellular organisms increases, these organisms must transport oxygen to the peripheral tissues and begin to employ red blood cells containing hemoglobin. This system is potentially a double-edged sword, as hemoglobin autoxidation occurs at a certain speed and releases free iron into the cytoplasm. Nrf2 belongs to the CNC transcription factor family, in which NF-E2p45 is the founding member. NF-E2p45 was first identified as a transcription factor that binds to the erythroid gene regulatory element NF-E2 located in the promoter region of the heme biosynthetic porphobilinogen deaminase gene. Human Nrf2 was also identified as a transcription factor that binds to the regulatory region of the β-globin gene. Despite these original findings, NF-E2p45 and Nrf2 knockout mice exhibit few erythroid phenotypes. Nrf2 regulates the expression of a wide range of antioxidant and detoxification enzymes. In this review article, we describe and discuss the roles of Nrf2 in various iron-mediated bioreactions and its possible coevolution with iron and oxygen.
Keywords: Nrf2, iron, heme, hemoglobin, oxidative stress
Roles of iron and oxygen in energy production
Organisms of all 3 major domains of life (i.e., bacteria, archaea and eukaryotes) produce energy by forming a redox reaction-coupled proton gradient, suggesting an ancestral origin of the energetic mechanism (1). The existence of iron in the evolutionarily conserved cellular respiration systems (i.e., iron sulfur proteins, such as Riske iron-sulfur proteins and heme binding proteins, such as cytochrome C) suggests that iron has had an important role in energy production since the beginning of life (2). As iron is a highly redox-active substance that displays high reactivity with molecular oxygen, the system is susceptible to oxidative stress unless the reactions of iron with oxygen and reactive oxygen species (ROS) are strictly regulated (3). Many antioxidant enzymes and iron storage proteins have evolved to cope with oxidative stress. One of the most important gene regulatory systems that protect against oxidative stress in higher eukaryotes is the Keap1-Nrf2 pathway (4). Here, we review the relationships between the Keap1-Nrf2 system and iron, oxygen.
CNC family transcription factors and Nrf2
Nrf2 belongs to the Cap'n'collor (CNC) subfamily of basic leucine zipper type transcription factors; the founding member of which is NF-E2p45 (5) (Figure 1). NF-E2 designates binding activity to the NF-E2 binding site in the gene regulatory regions of various heme biosynthetic genes, such as the porphobilinogen deaminase (PBGD) gene, and the enhancer region of the β-globin gene in electrophoretic mobility shift assays (EMSAs). The CNC family includes Nrf1, Nrf2, Nrf3, Bach1 and Bach2 in addition to NF-E2p45. Of these CNC family proteins, Bach1 and Bach2 act as transcriptional repressors and the other proteins act as transcriptional activators. Nrf2 was first identified in humans as a protein that recognizes the NF-E2 binding site of the human β-globin gene (6). We independently cloned chicken Nrf2 (originally named as ECH; Erythroid cell–derived protein with CNC Homology) by screening a peripheral red blood cell (RBC) cDNA library (RBC4) obtained from chickens with phenylhydrazine-induced hemolytic anemia using sequence homology to the mouse NF-E2p45 cDNA (7). Phenylhydrazine is known to induce oxidative stress and cause hemolytic anemia (8). Interestingly, the chicken homolog of NF-E2p45 was not obtained in the screen, but ECH represents a major CNC family protein expressed in peripheral blood RBCs from anemic chickens. The expression of the ECH gene is induced upon the differentiation of chicken erythroid cell line HD3, suggesting that chicken Nrf2 has important roles in maintaining the homeostasis of mature RBCs. Notably, among vertebrates, only mammalian RBCs lost their nuclei during the differentiation into the mature RBCs (i.e., transcription does not occur in mature RBCs). Unexpectedly, mice lacking CNC transcription family proteins exhibit few erythroid phenotypes. Neonatal NF-E2p45 knockout (KO) mice suffer from lethal bleeding due to the lack of platelets caused by abnormal megakaryocyte differentiation (9). Mice who have survived the neonatal lethality exhibit only mild hypochromic anemia with reticulocytosis and splenomegaly. Nrf2 KO mice are fertile and exhibit no signs of anemia (10), although their teeth are whitish brown due to the defect of iron deposition on the tooth surface (11) (discussed later). Furthermore, mice lacking both NF-E2p45 and Nrf2 did not exacerbated phenotype of NF-E2p45 KO mice (12). KO mice of other CNC transcription factors did not show phenotypes related to erythrocyte function (5).
Figure 1.

Members of the Maf and CNC transcription factor families in mammals. Maf transcription factors are classified into large Maf proteins, consisting of c-Maf, MafA, MafB, and NRL, and small Maf proteins (sMafs), comprising MafF, MafG, and MafK. The CNC family comprises NF-E2p45, Nrf1-3, Bach1, and Bach2. Only mammalian family members are shown in this figure. CLS, cytoplasmic localization signal. This figure is modified from reference (4).
Antioxidant responsive elements (AREs) or electrophile responsive elements (EpREs) are regulatory DNA elements that mediate the gene induction in response to electrophiles and are present in the regulatory regions of genes encoding various phase 2 detoxifying enzymes, such as electrophile-conjugating enzymes (13). Based on the similarity between the NF-E2 binding sequence and AREs, we previously showed that Nrf2 regulates ARE-mediated gene induction in response to electrophiles by generating Nrf2 KO mice (10). Currently, Nrf2 is known to regulate the expression of a wide range of genes related to 1) electrophile detoxification 2) antioxidant synthesis and antioxidant proteins, as well as 3) proteasome subunits, etc. (please refer to the comprehensive reviews (4, 14) for additional details). Nrf2 is mainly regulated by the Keap1- and ubiquitin/proteasome-mediated pathway. Nrf2 is constitutively degraded by the pathway, but upon exposure to electrophiles or oxidative stress, Keap1-mediated Nrf2 degradation is attenuated, leading to Nrf2 stabilization and nuclear accumulation (4, 14) (Figure 2). The extracellular administration of hemin also activates Nrf2 (15). In contrast, Bach1-mediated transcriptional repression is relieved by increasing heme concentration (16). Bach1 negatively regulates the expression of the β-globin and heme oxygenase 1 (HO-1) genes, and probably other ARE-containing genes to lesser extent (17, 18). Both Bach1 and Bach2 possess heme-binding CP (Cysteine-Proline) motifs and heme inactivates the Bach1-mediated transcriptional repression. Bach1 nuclear export and degradation are regulated by heme.
Figure 2.

Role of Nrf2 in heme and iron metabolism. Nrf2 is a master regulator of the antioxidant response induced by thiol-reactive substances, such as electrophilic reagents and heavy metals. In unstressed cells, Nrf2 is constitutively ubiquitinated by the Keap1-Cullin 3 ubiquitination complex and degraded by the 26S proteasome. Keap1 oxidation induces Nrf2 stabilization, nuclear translocation, heterodimerization with sMafs, and the transcriptional activation of target genes by binding to antioxidant response elements (AREs). An Nrf2 target gene, HO-1, degrades free heme into biliverdin, carbon monoxide and iron. HO-1 transcription is repressed by Bach1. Heme binding induces the nuclear export and degradation of Bach1. Free iron released from heme also regulates ROS production. Nrf2 induces the transcription of ferritin heavy chain (FTH1) and light chain (FTL) to store iron in the oxidized state and of ferroportin 1 (FPN1) to export intracellular labile iron.
The protective effects of Nrf2 on oxidative stress and infection/inflammation are mediated by iron regulation
Nrf2 regulates a wide range of cytoprotective responses and protects cells against various diseases and toxicities (14). In this review, we will focus on the Nrf2-mediated cytoprotective response achieved by iron regulation and detoxification.
Overview of systemic iron regulation
Systemic iron homeostasis is maintained by multiple mechanisms (19). Humans contain ~3,500–4,000 mg of iron, and two-thirds of the iron pool is sequestered in the hematopoietic organs such that RBC contains ~2,400–2,500 mg iron (20). Most of the remaining iron is present in the liver (~1,000 mg). Of the iron used for hemoglobin synthesis, 20–25 mg per day of iron is reutilized for RBC production in the spleen. Systemic iron homeostasis is regulated by the liver peptide hormone hepcidin. Hepcidin expression is induced under iron-overloaded conditions. Ferroportin (FPN1, also referred as SLC40A1) is the sole iron exporter in mammalian cells and is expressed at high levels in the basolateral membrane of the small intestine. Hepcidin degrades ferroportin and inhibits iron absorption from the intestine. Macrophages in reticuloendothelial systems scavenge circulating heme and hemoglobin and engulf aged or stressed erythrocytes (erythrophagocytosis) to degrade heme into biliverdin, carbon monoxide and iron via HO-1 activity. Liberated iron is stored with ferritin or exported via FPN1 for utilization in other cells (21) (Figure 2). In particular, HO-1 and FPN1 expressed in splenic macrophages regulate iron reutilization from senescent RBCs for de novo erythropoiesis.
Nrf2 regulates ferritin and ferrroportin1 gene expression
Since free iron readily transforms between Fe2+ and Fe3+ and is involved in the production of the highly reactive hydroxyl radical (HO•) from hydrogen peroxide (H2O2) (Fenton reaction), the labile iron pool is tightly regulated by multiple mechanisms, including Nrf2-mediated ferritin and ferroportin expression. Excess labile iron is stored in ferritin, which consists of 24 subunits of heavy chain (FTH1) and light chain (FTL) in various ratios and having differing functions. FTH1 possesses ferroxidase activity and stores iron in the stable ferrihydrite form (Figure 2). According to Pietsch et al., Nrf2 activator, β-naphthoflavone and dithiolethione induce FTH1 and FTL expression in wild-type mouse embryonic fibroblasts (MEFs) but not in Nrf2 KO MEFs, and an ARE located 4 kb upstream of the FTH1 transcriptional start site is responsible for the induction by Nrf2 (22). FTH1 and FTL expression are regulated not only by transcription but also by translation; an iron-responsive element (IRE) located in the 5′-untranslated region of target mRNAs is bound to iron regulatory proteins (IRPs) and negatively regulates its translation in the absence of iron (23, 24). Marro et al. clarified that heme-induced FPN1 transcription is mediated by Nrf2 activation and Bach1 inactivation in the RAW264.7 murine macrophage cell line (25). The authors identified an ARE motif present 7 kb upstream of the FPN1 transcription start site that is responsible for Nrf2- and Bach1-mediated FPN1 gene regulation. Bach1 forms a heterodimer with sMaf and competes with Nrf2 for ARE binding in the absence of heme binding. FPN1 translation is regulated by IRE and IRPs (25), similar to FTH1 and FTL. Furthermore, FPN1 is degraded by the hepcidin-mediated lysosomal pathway. Theurl et al. identified the liver is the primary tissue responsible for disease-associated stressed erythrocyte clearance by transiently differentiated macrophages displaying Nrf2-dependent FPN1 expression (26). The monocytes ingesting stressed erythrocytes are attracted by the chemokines Ccl2 and Ccl3 produced in the liver. Monocytes transiently differentiate into FPN1+Tim-4neg macrophages induced by colony stimulating factor 1 (Csf1) produced by Kupffer cells in the liver challenged with stressed erythrocytes. Csf1 induces FPN1 expression in wild-type mice, but not in Nrf2 KO mice, while Csf2 produced in the spleen antagonizes FPN1 induction. Ccr2 and Ccr5 antagonists block the aforementioned response and result in increased labile plasma iron levels and liver injury.
FPN1 is not only important for systematic iron homeostasis, but also involved in certain anti-inflammatory mechanisms. It is previously reported that inflammatory cytokines, such as LPS repress FPN1 gene transcription in splenic macrophages (27). Furthermore, hepcidin is increased by the inflammatory cytokine, such as IL-6 in the liver and degrades FPN1 in macrophages (28). This FPN1 suppression is proposed to contribute to the anemia of chronic disease by sequestering iron in the macrophages and accumulated iron in the macrophage may accelerates the inflammatory response (29). Furthermore, it is reported that the translation of inflammatory cytokine, such as TNFα and IL-6 in macrophage requires iron (30) and that TRAM/TRIF pathway downstream of TLR4 (31) or TLR4 localization to lipid raft is specifically sensitive to iron depletion (32). Consistently, FPN1 overexpression lowered LPS-induced IL-6 production in J774 macrophages (30). As shown in our previous study, Nrf2 activation induces the expression of FPN1 and another ion transporter, Nramp1, in wild-type bone marrow-derived macrophages (BMDMs), but not in BMDMs from Nrf2 KO mice (33). Although an LPS treatment suppresses FPN1 transcription in macrophages, Nrf2 activation reversed the FPN1 suppression in human peripheral blood-derived macrophages and murine peritoneal macrophages (33). Therefore, we proposed that Nrf2 plays an anti-inflammatory role by antagonizing the inflammation-mediated suppression of FPN1 gene expression. On the other hand, FPN1 is also important for immune response, as iron is required for microbial proliferation. Nairz et al. clarified that Nrf2 is involved in nitric oxide (NO)-induced FPN1 expression and iron efflux to prevent intracellular pathogen proliferation (34). NO is a gaseous radical produced by inducible NO synthase (NOS2 or iNOS) as mediator of the immune response, as well as a toxic effector against infected pathogens. Both in the NOS2−/− spleen and primary peritoneal macrophages derived from NOS2−/− mice, FPN1 expression is decreased and cellular iron content is increased compared to wild-type mice, which are associated with decreased Nrf2 activation. NO donor treatment or Salmonella enterica infection induces both Nrf2 activation and FPN1 expression, and the latter is suppressed by a NOS2 inhibitor. NOS2−/− peritoneal macrophages are susceptible to salmonella infection, but rescued by the overexpression of Nrf2 or FPN1. Interestingly, in vivo Salmonella enterica infection experiment demonstrated that iron chelation rescues the susceptible phenotypes observed in NOS2−/−, such as increased bacterial colony formation and reduced inflammatory cytokine production (TNFα, IL-12, and IFNγ) (34).
Sirtuin family proteins belong to the class III histone deacetylases and deacetylate substrate proteins using NAD+ as a cofactor. There are 7 sirtuins (Sirts) in mammals and Sirt2 is a cytoplasmic enzyme that regulates various processes including aging, apoptosis and cellular response (35). Yang et al reported that non-heme iron content is decreased in MEFs of Sirt2 KO mice and Sirt2 knockdown HepG2 cells, which is associated with increased FPN1 gene expression (36). They showed that Sirt2 deacetylates Nrf2 and renders Nrf2 susceptible to the proteasomal degradation. In Sirt2 KO livers, hyperactivated Nrf2 causes FPN1 overexpression and this increase was restored in Sirt2/Nrf2 double KO mice. Sirt2 KO hepatocytes are susceptible to iron deficiency and Sirt2 activity is negatively correlated with iron content in liver biopsies from human fatal neonatal hemochromatosis patients. Thus, the Nrf2-FPN1 axis participates in the cytoprotective effects on iron-mediated oxidative stress and in the host immune response to intracellular pathogens.
Nrf2-dependent regulation of heme oxygenase 1 and heme toxicity
Heme toxicity is an important pathophysiology manifested by intravascular hemolysis. Sickle cell disease (SCD) is inherited disorder caused by a missense mutation in the β-globin gene (hemoglobin S). A homozygous β-globin gene mutation causes hemoglobin polymerization under hypoxic conditions that deforms red blood cells into a sickle-like morphology (37). Patients with SCD develop chronic intravascular hemolysis, anemia and inflammation, as well as increased vascular occlusion whereas the thalassemia patients, another prevalent hemoglobinopathy, demonstrate intrasplenic destruction of RBCs. It is reported that methemoglobin A or more vigorously hemoglobin S releases free heme (38). Furthermore, it is reported that free heme is increased in sickle cell disease model mouse (39). The free heme released by hemolysis and ischemia-reperfusion injury induces ROS production and subsequently multiple tissue injuries. Townes and colleagues generated mice harboring human α-globin and β-globin genes with or without mutations (βA and βS, respectively) as SCD models (40). It is reported that Keap1-flox allele is hypomorphic and mice with this allele (KeapF/−) show significant Nrf2 activation and target gene induction, but escape the postnatal lethality of Keap1−/− mice (41). Keleku-Kukwete et al. evaluated the effect of Nrf2 activation on SCD by crossing SCD mice with hypomorphic Keap1 heterozygotes (Keap1F/−) (39). Nrf2 activation alleviates liver injury and lung inflammation in SCD mice. Nrf2 activation decreases the serum concentrations of heme, indicating an impact of HO-1 on heme breakdown (Figure 3). In contrast, Nrf2 activation fails to attenuate hemolysis and splenomegaly, a hallmark of stress-induced erythropoiesis in SCD mice (39). The authors also showed that the administration of the Nrf2 activator CDDO-Im improves liver and lung pathology and inflammation in SCD mice, indicating that Nrf2 is a pharmacological target for SCD therapy (39). Hydroxyurea (HU) is currently utilized in SCD therapy to induce the production of γ-globin, a fetal hemoglobin subunit produced during infant liver erythropoiesis, to replace βS-globin in adult hemoglobin (42). Although Nrf2 activation by Keap1F/− fails to induce significant γ-globin expression in the spleen, Krishnamoorthy et al. showed that the Nrf2 activator dimethyl fumarate (DMF) induces γ-globin expression in peripheral blood mononuclear cells derived from patients with SCD (43). DMF not only improves the RBC count and morphology but also attenuates inflammatory cytokine production and adhesion molecule expression. Moreover, DMF and HU co-administration induces a further increase in γ-globin expression, and DMF effectively induces fetal hemoglobin production in the cynomolgus monkey (43). Based on these findings, Nrf2 activation exerts beneficial effects by eliminating heme via HO-1 induction and increases the number of erythrocytes containing fetal hemoglobin by inducing γ-globin expression (Figure 3).
Figure 3.
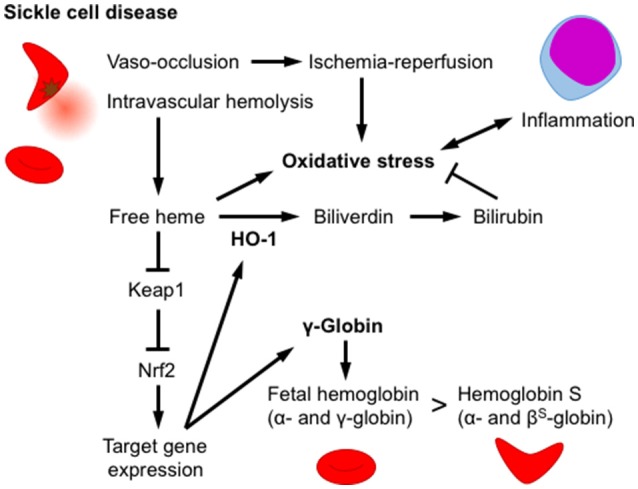
Nrf2/HO-1 counteracts heme toxicity. In sickle cell disease, erythrocytes are deformed by the polymerization of hemoglobin S, and oxidative stress subsequently develops, inducing intravascular hemolysis and vaso-occlusion. An increase in the free heme concentration activates Nrf2 and induces HO-1 expression. HO-1 degrades heme into biliverdin, which is subsequently reduced into the antioxidant bilirubin. Nrf2 activation increases fetal hemoglobin levels by inducing the expression of the γ-globin gene and alleviates hemoglobin S production in erythroid progenitor cells. Nrf2 activation attenuates damage to multiple tissues, as well as the production of inflammatory cytokines.
Rodent specific Nrf2 function in iron transport of the enamel organ
The incisors of Nrf2 KO mice are decolorized by defects in iron transport during enamel organ development (11). In the early maturation stage of mouse incisors, FTH1 is expressed in both the papillary cell layer and ameloblasts, and iron accumulates in the ameloblast cytosol. In the late maturation stage, iron accumulation shifts to the plasma membrane on the enamel side and is finally deposited onto the tooth surface, which is characterized by brownish-yellow color. Nrf2 KO mice show decreased levels of the FTH1 protein in the papillary cell layer and reduced iron accumulation in ameloblasts at the early maturation stage. Furthermore, marked degeneration of Nrf2 KO ameloblasts is observed at the late maturation stage, leading to the iron transport defect and the loss of the acid resistance of the teeth. Although the precise mechanisms underlying the tooth phenotype have not been clarified, this finding suggested the susceptibility of Nrf2 KO mice in which iron metabolism is sufficiently proficient to cause oxidative stress.
Revisiting the roles of NF-E2p45 and Nrf2 in RBCs
Roles of NF-E2p45 and Nrf2 in red cell homeostasis
Mature RBCs (mRBCs) are particularly susceptible to oxidative stress, as oxygen-bound hemoglobin is oxidized by autoxidation (44, 45) (Figure 4B). During autoxidation, oxygen bound to ferrous-hemoglobin is reduced by iron and generates superoxide. Superoxide is dismutated to hydrogen peroxide through both enzymatic and non-enzymatic processes and reacts with oxygen-bound hemoglobin to generate ferryl-hemoglobin [HbFe(IV) = O2, Figure 5A]. The ferryl-hemoglobin subsequently reacts with hydrogen peroxide and is degraded to ferric iron and porphyrin. An estimated 3% of the total oxygenated hemoglobin is oxidized to methemoglobin per day. However, mature RBCs (mRBCs) contain ~1% of methemoglobin, as NADH-dependent cytochrome reductase converts methemoglobin to Fe2+-hemoglobin. The liberated ferric iron reacts with superoxide in Haber-Weiss reaction and generates hydroxyl radicals that are toxic to RBCs, mainly due to membrane lipid peroxidation (46) (Figure 4B). mRBCs are equipped with a meticulous network of antioxidant systems to combat these highly oxidative environments (46). Glutathione peroxidase, catalase and membrane localized peroxiredoxin 2 are mainly responsible for detoxifying hydrogen peroxide in RBCs. Furthermore, ferroportin was recently reported to be abundantly expressed in mRBCs and has an important antioxidant function (47). As shown in the study by Zhang et al. of erythroblast-specific FPN1 KO mice, the labile iron pool in erythroblasts is increased, leading to increased hemolysis (47). This observation illustrates the degradation of heme in hemoglobin to ferrous iron in erythroblasts and even in erythrocytes (48).
Figure 4.
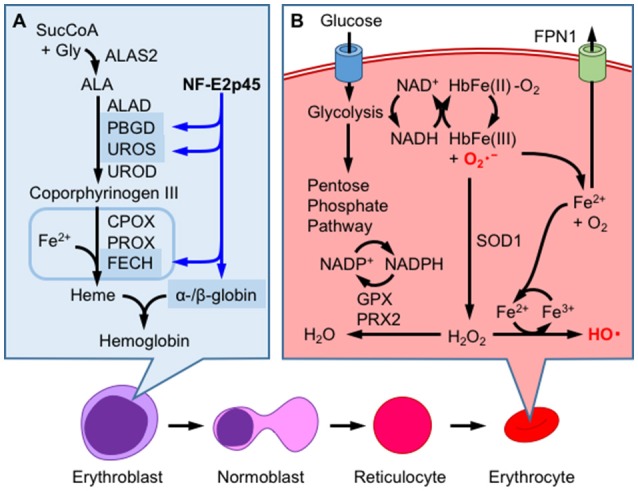
Hemoglobin production and redox homeostasis in erythrocytes. (A) NF-E2p45 participates in the transcriptional activation of α- and β-hemoglobin, as well as heme-synthetic enzymes, such as porphobilinogen deaminase (PBGD), uroporphyrinogen III synthase (UROS), and ferrochelatase (FECH), in erythroblasts. After enucleation, erythrocytes combat oxidative stress inducined by hemoglobin autoxidation through a mechanism that is not regulated by gene transcription. (B) Glucose metabolism supplies NADH, which is utilized for methemoglobin reduction. Superoxide anions () are eliminated by superoxide dismutase 1 (SOD1), which converts these ions into hydrogen peroxide (H2O2), or are converted into oxygen through heme degradation. Ferric iron reacts with by Haber-Weiss reaction to generate highly reactive hydroxyl radical (HO•) and oxygen molecule. H2O2 is further reduced to water by the consumption of NADPH by glutathione peroxidase (GPX) and peroxiredoxin 2 (PRX2); NADPH is produced by the pentose phosphate pathway. FPN1 exports cellular labile iron to prevent ROS production.
Figure 5.
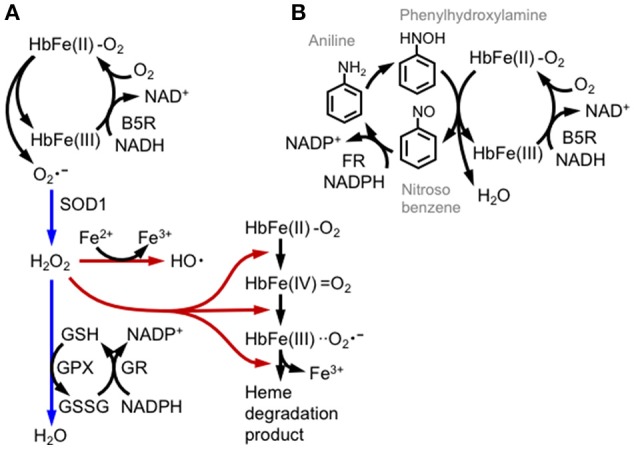
Hemoglobin oxidation and redox catastrophe. (A) Oxy-hemoglobin [HbFe(II)-O2] is partially converted into methemoglobin [HbFe(III)] and the superoxide anion () by autoxidation. Methemoglobin is then reduced by cytochrome b5 reductase (B5R) using NADH or by glutathione. Superoxide is detoxified by SOD1 and GPX activities (direction illustrated with blue arrows); otherwise hydrogen peroxide (H2O2) is converted into the hydroxyl radical (HO•) by the Fe2+ ion or is involved in series of hemoglobin degradation cascades (illustrated with brown arrows). (B) Aniline induces methemoglobinemia and hemolysis by repetitive redox cycles of hemoglobin oxidation. Aniline is oxidized by hepatic mixed-function oxidase into bioactive phenylhydroxylamine, which reacts with oxy-hemoglobin and generates nitrosobenzene and methemoglobin. Nitrosobenzene is reduced again by NADPH flavin reductase (FR). Although glycolytic metabolism supplies NADH and NADPH, the phenylhydroxylamine/nitrosobenzene redox cycle produces methemoglobin production and depletes cellular reductants.
The oxidation of hemoglobin to methemoglobin is enhanced by various drugs, as observed in subjects with hemolytic anemia caused by a glucose-6-phosphate dehydrogenase (G6PD) deficiency (49, 50). In G6PD-deficient patients, NADPH production is substantially decreased, as mRBCs largely depend on the pentose phosphate pathway for NADPH production. We illustrate how drugs induce hemolytic anemia using the redox active compound aniline (Figure 5B). Aniline is firstly metabolized to phenylhydroxylamine to induce hemoglobin oxidation. Electrons from both ferrous iron and phenylhydroxylamine are transferred to oxygen, generating nitrosobenzene, water and methemoglobin. Nitrosobenzene is reduced back to aniline by flavin reductase using NADPH, or by GSH. Thus, in this reaction, methemoglobin formation is associated with the depletion of GSH and NADPH and therefore generates oxidative stress. Drug-induced hemolytic anemia is prevalent in patients with G6PD deficiencies, and the drugs include the anti-malaria drug primaquine and dapsone (49, 50). Dapsone is diaminodiphenyl sulfone that is metabolized to dapsone hydroxylamine and causes methemoglobin formation and oxidative stress (51). Although RBCs do not regulate transcription, they can determine the basal levels of proteins before enucleation. Indeed, Ter119+ RBCs derived from the bone marrow or spleen of NF-E2p45 KO mice accumulate high levels of ROS, and methemoglobin formation is increased after exposure to hydrogen peroxide (52). Methemoglobin formation is enhanced in NF-E2p45 KO mice after the intraperitoneal injection of dapsone, and NF-E2p45 KO mice are more susceptible to phenylhydrazine-induced hemolytic anemia. Furthermore, RBC longevity is reduced in NF-E2p45 KO mice. Interestingly, the expression of GST-Yc and catalase are decreased in the Ter119+ RBCs from NF-E2p45 KO mice compared to cells from wild-type mice (52). Selenoproteins are polypeptides that contain selenocysteines and are proficient in redox biochemistry. Importantly, glutathione peroxidase 1 and thioredoxin reductase (which is important for thioredoxin function) are selenoproteins that are mainly responsible for detoxifying ROS in RBCs. In the macrophage- and liver-specific selenocysteine tRNA KO mice, the levels of selenoproteins are decreased as their corresponding mRNAs are degraded by non-sense mediated decay, and conversely Nrf2 is activated to compensate for the oxidative stress in the mice (53). Kawatani et al. extended this observation and showed that Nrf2 and selenoproteins cooperate in the defense mechanism against oxidative stress in RBCs (54). The authors generated compound transgenic mice with a selenocysteine tRNA inducible KO and Nrf2 KO and showed that ROS production was specifically increased in erythroid lineages, but not other hematopoietic lineages, compared to inducible KO mice for the selenocysteine tRNA alone, strongly indicating that Nrf2 regulates RBC redox homeostasis when oxidative stress activates Nrf2, such as in the selenocysteine tRNA KO mice. According to Diederich et al., oxidative stress decreases the deformability of RBCs, and NO ameliorates this defect. Interestingly, the detrimental effect of tert-butylhydroperoxide is enhanced in Nrf2 KO RBCs (55). Thus, we surmise that NF-E2p45 regulates the basal expression of antioxidant enzymes and Nrf2 augments the response when Nrf2 is activated in erythroid lineages in response to oxidative stress.
Roles of Nrf2 and CNC family transcription factors in heme metabolism and globin gene expression—thoughts on evolutionary aspects
Heme-containing proteins, including cytochrome C, hemoglobin, and heme oxygenase, are ancient proteins whose homologs can be identified in bacteria (56, 57). Prototypic hemoglobin may have existed since the evolution of the last eukaryotic ancestor, and several types of hemoglobin or myoglobin-related proteins act as oxygen sensor proteins in bacteria or archaea (58). In multicellular organisms, hemoglobin in RBCs sequesters free oxygen and maintains a low oxygen concentration in tissues. Thus, hemoglobin not only delivers oxygen to peripheral tissues but is also considered a powerful antioxidant, as hemoglobin sequesters oxygen at high blood oxygen pressures (i.e., pVO2).
As mentioned above, NF-E2p45 was initially identified as a transcription factor that binds to the NF-E2 site of the PBGD gene promoter. Although only a subtle erythroid phenotype of NF-E2 p45 KO mice was initially described (9), the susceptibility of NF-E2p45 KO RBCs to oxidative stress was subsequently reported, as mentioned above (52). Furthermore, abnormal erythroid differentiation is observed in NF-E2p45 KO mice (59). Based on evidence from multiple in vitro experiments, NF-E2p45 is an important regulator of the differentiation to the erythroid lineage because it coordinately regulates genes involved in both globin and heme synthesis (see Ref. (60) for a comprehensive review). For example, mouse erythroleukemia (MEL) cells that lack NF-E2p45 (named CB3) do not express globin after the induction of differentiation, but the reintroduction of NF-E2p45 rescues the β-globin deficit (61). Furthermore, the induction of β-globin transcription in MEL cells is associated with NF-E2 biding to the locus control region of the β-globin gene enhancer (62). In addition to globin genes, NF-E2p45 regulates the expression of genes involved in multiple steps of heme biosynthesis in human erythroid cells, particularly the PBGD, UROD (63), and ferrochelatase genes (64), the exception is the rate-limiting enzyme ALAS2 (Figure 4A). NF-E2 KO zebrafish exhibit hypochromia in an early developmental stage (i.e., less hemoglobin staining is observed in the embryo) compared to wild-type animals, and a total loss of hemoglobin was observed in response to tert-butylhydroperoxide and diquat exposure, while wild-type fish shows only mild hypochromia. The authors argued that hypochromia in NF-E2 KO embryos may be related to decreased ALAS2 expression (48).
Nrf2 also regulates fetal γ-globin gene expression (65) and directly binds to the regulatory region of fetal (HBG1) and embryonic (HBE1) globin (66) genes. Furthermore, Nrf2 binds to the regulatory regions of the various heme biosynthesis genes, including ferrochelatase, although its regulatory role remains to be clarified (66). These results suggest roles for Nrf2 and NF-E2p45 in the expression of the β-globin and heme biosynthetic genes. Although transcriptional regulation does not occur in mature mammalian RBCs, CNC transcription factors may regulate heme and globin synthesis as well as antioxidant gene expression in the erythroblast stages, and contribute to the basal expression observed in mature RBCs (Figure 4B). From the evolutionary perspective, Nrf2 evolution from the ancestral transcription factors occurred around the period of the great oxidation event (67), which was roughly coincident with the period when the metazoan (i.e., predation evolved) and RBCs evolved. Thus, a tempting speculation is that Nrf2 may have evolved from the need for erythrocyte antioxidative function and its role has gradually diminished (i.e., retrogressed) after RBCs lost their nuclei after the branch to mammals. The possibilities remain to be clarified in the future.
Cross talk between Nrf2 and iron-sulfur cluster formation
Mitochondrial iron-sulfur cluster biogenesis is essential for a number of vital reactions, including respiration, the TCA cycle, and DNA synthesis, among others. Friedreich's ataxia (FRDA) is neuro- and cardio-degenerative disease caused by expansion of GAA repeats in intron 1 of the FXN gene and decreased expression of the gene product frataxin. Frataxin is a mitochondrial matrix protein that is indispensable for mitochondrial iron-sulfur cluster synthesis and heme synthesis in concert with iron-sulfur cluster assembly enzyme (ISCU) and ferrochelatase, respectively (68). Decreased frataxin function is thought to decrease iron utilization and cause iron overload in mitochondria. Indeed, iron accumulation has been observed in the brains of patients with FRDA (69), consistent with the observation that a conditional FXN knockout (FXN CKO) FRDA mouse model shows mitochondrial iron deposition distinct from the ferritin complex (70). FXN CKO mice exhibit elevated iron contents in tissues and cardiac hypertrophy with downregulation of the genes involved in iron-sulfur cluster biosynthesis (ISCU), heme synthesis (ALAD, UROS, and FECH) and iron transport (FPN1, FTH1, and FTL1), as well as upregulation of genes involved in iron uptake (TFR1 and Sec15l1) and mitochondrial import (Mfrn2) (71–73). An attenuation of Nrf2 activity in the FRDA model has been reported by several groups. The FXN CKO mice show decreased Nrf2 expression in the heart and a concomitant increase in Keap1 expression and GSK3β activation. GSK3β negatively regulates Nrf2 by inducing Nrf2 nuclear export and degradation through the phosphorylation of tyrosine kinase Fyn and Nrf2 itself, respectively (74). Nrf2 activation by oxidative stress is blocked in fibroblasts derived from patients with FRDA (75), as well as in motor neuron-like NSC34 cells expressing an FXN shRNA (76). The defective Nrf2 response in fibroblasts from patients with FRDA is rescued by treatment with Euk134, a cell-permeable catalase mimetic, indicating that chronic H2O2 generation causes this response (75). Furthermore, in the study by Shan et al., an FRDA mouse model hemizygous for mutant human FXN (YG8R mice) exhibits decreased Nrf2 activity and downregulation of its target genes in dorsal root ganglia (DRG) neurons and in FXN-knockdown cell line models using DRG (ND7/23 cells) and Schwann cells (T265 cells) (77). Interestingly, Sahdeo et al. also observed that dyclonine restores FXN expression by activating Nrf2, and Nrf2 directly binds to the evolutionarily conserved ARE in the upstream region of the FXN gene in lymphoblasts from patients with FRDA (78). Although the precise mechanisms of Nrf2 dysfunction have yet to be determined, these results indicate that impairments of Nrf2 function may participate in FRDA pathogenesis by altering the expression of antioxidant and iron metabolism/transport genes.
Concluding remarks
Since Nrf2 evolved after the branching of metazoans, it has played important roles in the defense mechanisms against oxidative stress and drug detoxification. Nrf2 restricts iron- or heme-mediated oxidative stress by regulating the expression of the FPN1, ferritin and HO-1 genes. Oxidative stress in the erythrocyte may be one of the most important selective forces driving the evolution of members of the CNC transcription factor family, including Nrf2. Currently, Nrf2 activators are being clinically tested in patients with malaria to combat heme-mediated oxidative stress. In the future, we will be able to treat oxidative stress-related diseases by employing Nrf2-mediated iron-detoxification pathways.
Author contributions
KI designed the structure of the review; SK, JM, TO, and KI wrote the manuscript.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Hiromi Yamazaki for critically reading and providing suggestions on the manuscript. This work was supported by a grant from MEXT/JSPS KAKENHI (Grant Number 26111010) and a Hirosaki University Institutional Research Grant.
References
- 1.Castresana J, Saraste M. Evolution of energetic metabolism: the respiration-early hypothesis. Trends Biochem Sci. (1995) 20:443–8. 10.1016/S0968-0004(00)89098-2 [DOI] [PubMed] [Google Scholar]
- 2.Russell MJ, Martin W. The rocky roots of the acetyl-CoA pathway. Trends Biochem Sci. (2004) 29:358–63. 10.1016/j.tibs.2004.05.007 [DOI] [PubMed] [Google Scholar]
- 3.Toyokuni S, Ito F, Yamashita K, Okazaki Y, Akatsuka S. Iron and thiol redox signaling in cancer: an exquisite balance to escape ferroptosis. Free Rad Biol Med. (2017) 108:610–26. 10.1016/j.freeradbiomed.2017.04.024 [DOI] [PubMed] [Google Scholar]
- 4.Itoh K, Mimura J, Yamamoto M. Discovery of the negative regulator of Nrf2, Keap1: a historical overview. Antioxid Redox Signal. (2010) 13:1665–78. 10.1089/ars.2010.3222 [DOI] [PubMed] [Google Scholar]
- 5.Sykiotis GP, Bohmann D. Stress-activated cap'n'collar transcription factors in aging and human disease. Sci Signal. (2010) 3:re3. 10.1126/scisignal.3112re3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moi P, Chan K, Asunis I, Cao A, Kan YW. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc Nat Acad Sci USA. (1994) 91:9926–30. 10.1073/pnas.91.21.9926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Itoh K, Igarashi K, Hayashi N, Nishizawa M, Yamamoto M. Cloning and characterization of a novel erythroid cell-derived CNC family transcription factor heterodimerizing with the small Maf family proteins. Mol Cell Biol. (1995) 15:4184–93. 10.1128/MCB.15.8.4184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shetlar MD, Hill HA. Reactions of hemoglobin with phenylhydrazine: a review of selected aspects. Environ Health Perspect. (1985) 64:265–81. 10.1289/ehp.8564265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shivdasani RA, Rosenblatt MF, Zucker-Franklin D, Jackson CW, Hunt P, Saris CJ, et al. Transcription factor NF-E2 is required for platelet formation independent of the actions of thrombopoietin/MGDF in megakaryocyte development. Cell (1995) 81:695–704. 10.1016/0092-8674(95)90531-6 [DOI] [PubMed] [Google Scholar]
- 10.Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. (1997) 236:313–22. 10.1006/bbrc.1997.6943 [DOI] [PubMed] [Google Scholar]
- 11.Yanagawa T, Itoh K, Uwayama J, Shibata Y, Yamaguchi A, Sano T, et al. Nrf2 deficiency causes tooth decolourization due to iron transport disorder in enamel organ. Genes Cells (2004) 9:641–51. 10.1111/j.1356-9597.2004.00753.x [DOI] [PubMed] [Google Scholar]
- 12.Kuroha T, Takahashi S, Komeno T, Itoh K, Nagasawa T, Yamamoto M. Ablation of Nrf2 function does not increase the erythroid or megakaryocytic cell lineage dysfunction caused by p45 NF-E2 gene disruption. J Biochem. (1998) 123:376–9. 10.1093/oxfordjournals.jbchem.a021947 [DOI] [PubMed] [Google Scholar]
- 13.Primiano T, Sutter TR, Kensler TW. Antioxidant-inducible genes. Adv Pharmacol. (1997) 38:293–328. 10.1016/S1054-3589(08)60989-8 [DOI] [PubMed] [Google Scholar]
- 14.Yamamoto M, Kensler TW, Motohashi H. The KEAP1-NRF2 system: a thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol Rev. (2018) 98:1169–203. 10.1152/physrev.00023.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim YC, Masutani H, Yamaguchi Y, Itoh K, Yamamoto M, Yodoi J. Hemin-induced activation of the thioredoxin gene by Nrf2. A differential regulation of the antioxidant responsive element by a switch of its binding factors. J Biol Chem. (2001) 276:18399–406. 10.1074/jbc.M100103200 [DOI] [PubMed] [Google Scholar]
- 16.Igarashi K, Sun J. The heme-Bach1 pathway in the regulation of oxidative stress response and erythroid differentiation. Antioxid Redox Signal. (2006) 8:107–18. 10.1089/ars.2006.8.107 [DOI] [PubMed] [Google Scholar]
- 17.Reichard JF, Motz GT, Puga A. Heme oxygenase-1 induction by NRF2 requires inactivation of the transcriptional repressor BACH1. Nucleic Acids Res. (2007) 35:7074–86. 10.1093/nar/gkm638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hintze KJ, Katoh Y, Igarashi K, Theil EC. Bach1 repression of ferritin and thioredoxin reductase1 is heme-sensitive in cells and in vitro and coordinates expression with heme oxygenase1, beta-globin, and NADP(H) quinone (oxido) reductase1. J Biol Chem. (2007) 282:34365–71. 10.1074/jbc.M700254200 [DOI] [PubMed] [Google Scholar]
- 19.Hentze MW, Kuhn LC. Molecular control of vertebrate iron metabolism: mRNA-based regulatory circuits operated by iron, nitric oxide, and oxidative stress. Proc Nat Acad Sci USA. (1996) 93:8175–82. 10.1073/pnas.93.16.8175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ganz T. Hepcidin and iron regulation, 10 years later. Blood (2011) 117:4425–33. 10.1182/blood-2011-01-258467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Drakesmith H, Nemeth E, Ganz T. Ironing out ferroportin. Cell Metab. (2015) 22:777–87. 10.1016/j.cmet.2015.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pietsch EC, Chan JY, Torti FM, Torti SV. Nrf2 mediates the induction of ferritin H in response to xenobiotics and cancer chemopreventive dithiolethiones. J Biol Chem. (2003) 278:2361–9. 10.1074/jbc.M210664200 [DOI] [PubMed] [Google Scholar]
- 23.Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell (2004) 117:285–97. 10.1016/S0092-8674(04)00343-5 [DOI] [PubMed] [Google Scholar]
- 24.Rouault TA, Klausner RD. Iron-sulfur clusters as biosensors of oxidants and iron. Trends Biochem Sci. (1996) 21:174–7. 10.1016/S0968-0004(96)10024-4 [DOI] [PubMed] [Google Scholar]
- 25.Marro S, Chiabrando D, Messana E, Stolte J, Turco E, Tolosano E, et al. Heme controls ferroportin1 (FPN1) transcription involving Bach1, Nrf2 and a MARE/ARE sequence motif at position−7007 of the FPN1 promoter. Haematologica (2010) 95:1261–8. 10.3324/haematol.2009.020123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Theurl I, Hilgendorf I, Nairz M, Tymoszuk P, Haschka D, Asshoff M, et al. On-demand erythrocyte disposal and iron recycling requires transient macrophages in the liver. Nat Med. (2016) 22:945–51. 10.1038/nm.4146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu XB, Nguyen NB, Marquess KD, Yang F, Haile DJ. Regulation of hepcidin and ferroportin expression by lipopolysaccharide in splenic macrophages. Blood Cells Mol Dis. (2005) 35:47–56. 10.1016/j.bcmd.2005.04.006 [DOI] [PubMed] [Google Scholar]
- 28.Nemeth E, Rivera S, Gabayan V, Keller C, Taudorf S, Pedersen BK, et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest. (2004) 113:1271–6. 10.1172/JCI200420945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weiss G, Goodnough LT. Anemia of chronic disease. N Engl J Med. (2005) 352:1011–23. 10.1056/NEJMra041809 [DOI] [PubMed] [Google Scholar]
- 30.Wang L, Johnson EE, Shi HN, Walker WA, Wessling-Resnick M, Cherayil BJ. Attenuated inflammatory responses in hemochromatosis reveal a role for iron in the regulation of macrophage cytokine translation. J Immunol. (2008) 181:2723–31. 10.4049/jimmunol.181.4.2723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang L, Harrington L, Trebicka E, Shi HN, Kagan JC, Hong CC, et al. Selective modulation of TLR4-activated inflammatory responses by altered iron homeostasis in mice. J Clin Invest. (2009) 119:3322–8. 10.1172/JCI39939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Habib A, Polavarapu R, Karmali V, Guo L, Van Dam R, Cheng Q, et al. Hepcidin-ferroportin axis controls toll-like receptor 4 dependent macrophage inflammatory responses in human atherosclerotic plaques. Atherosclerosis (2015) 241:692–700. 10.1016/j.atherosclerosis.2015.06.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harada N, Kanayama M, Maruyama A, Yoshida A, Tazumi K, Hosoya T, et al. Nrf2 regulates ferroportin 1-mediated iron efflux and counteracts lipopolysaccharide-induced ferroportin 1 mRNA suppression in macrophages. Arch Biochem Biophys. (2011) 508:101–9. 10.1016/j.abb.2011.02.001 [DOI] [PubMed] [Google Scholar]
- 34.Nairz M, Schleicher U, Schroll A, Sonnweber T, Theurl I, Ludwiczek S, et al. Nitric oxide-mediated regulation of ferroportin-1 controls macrophage iron homeostasis and immune function in Salmonella infection. J Exp Med. (2013) 210:855–73. 10.1084/jem.20121946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gomes P, Fleming Outeiro T, Cavadas C. Emerging role of sirtuin 2 in the regulation of mammalian metabolism. Trends Pharmacol Sci. (2015) 36:756–68. 10.1016/j.tips.2015.08.001 [DOI] [PubMed] [Google Scholar]
- 36.Yang X, Park SH, Chang HC, Shapiro JS, Vassilopoulos A, Sawicki KT, et al. Sirtuin 2 regulates cellular iron homeostasis via deacetylation of transcription factor NRF2. J Clin Invest. (2017) 127:1505–16. 10.1172/JCI88574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Telen MJ. Beyond hydroxyurea: new and old drugs in the pipeline for sickle cell disease. Blood (2016) 127:810–9. 10.1182/blood-2015-09-618553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Belcher JD, Chen C, Nguyen J, Milbauer L, Abdulla F, Alayash AI, et al. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood (2014) 123:377–90. 10.1182/blood-2013-04-495887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Keleku-Lukwete N, Suzuki M, Otsuki A, Tsuchida K, Katayama S, Hayashi M, et al. Amelioration of inflammation and tissue damage in sickle cell model mice by Nrf2 activation. Proc Nat Acad Sci USA. (2015) 112:12169–74. 10.1073/pnas.1509158112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu LC, Sun CW, Ryan TM, Pawlik KM, Ren J, Townes TM. Correction of sickle cell disease by homologous recombination in embryonic stem cells. Blood (2006) 108:1183–8. 10.1182/blood-2006-02-004812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Taguchi K, Maher JM, Suzuki T, Kawatani Y, Motohashi H, Yamamoto M. Genetic analysis of cytoprotective functions supported by graded expression of Keap1. Mol Cell Biol. (2010) 30:3016–26. 10.1128/MCB.01591-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bauer DE, Kamran SC, Orkin SH. Reawakening fetal hemoglobin: prospects for new therapies for the beta-globin disorders. Blood (2012) 120:2945–53. 10.1182/blood-2012-06-292078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krishnamoorthy S, Pace B, Gupta D, Sturtevant S, Li B, Makala L, et al. Dimethyl fumarate increases fetal hemoglobin, provides heme detoxification, and corrects anemia in sickle cell disease. JCI Insight (2017) 2:96409. 10.1172/jci.insight.96409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Umbreit J. Methemoglobin–it's not just blue: a concise review. Am J Hematol. (2007) 82:134–44. 10.1002/ajh.20738 [DOI] [PubMed] [Google Scholar]
- 45.Rifkind JM, Nagababu E, Ramasamy S, Ravi LB. Hemoglobin redox reactions and oxidative stress. Redox Rep. (2003) 8:234–7. 10.1179/135100003225002817 [DOI] [PubMed] [Google Scholar]
- 46.Kuhn V, Diederich L, Keller TCS, IV, Kramer CM, Luckstadt W, Panknin C, et al. Red blood cell function and dysfunction: redox regulation, nitric oxide metabolism, anemia. Antioxid Redox Signal. (2017) 26:718–42. 10.1089/ars.2016.6954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang DL, Wu J, Shah BN, Greutelaers KC, Ghosh MC, Ollivierre H, et al. Erythrocytic ferroportin reduces intracellular iron accumulation, hemolysis, and malaria risk. Science (2018) 359:1520–3. 10.1126/science.aal2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Williams LM, Lago BA, McArthur AG, Raphenya AR, Pray N, Saleem N, et al. The transcription factor, nuclear factor, erythroid 2 (Nfe2), is a regulator of the oxidative stress response during Danio rerio development. Aquatic Toxicol. (2016) 180:141–54. 10.1016/j.aquatox.2016.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cappellini MD, Fiorelli G. Glucose-6-phosphate dehydrogenase deficiency. Lancet (2008) 371:64–74. 10.1016/S0140-6736(08)60073-2 [DOI] [PubMed] [Google Scholar]
- 50.Chu CS, Bancone G, Nosten F, White NJ, Luzzatto L. Primaquine-induced haemolysis in females heterozygous for G6PD deficiency. Malaria J. (2018) 17:101. 10.1186/s12936-018-2248-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reilly TP, Woster PM, Svensson CK. Methemoglobin formation by hydroxylamine metabolites of sulfamethoxazole and dapsone: implications for differences in adverse drug reactions. J Pharmacol Exp Therapeut. (1999) 288:951–9. [PubMed] [Google Scholar]
- 52.Chan JY, Kwong M, Lo M, Emerson R, Kuypers FA. Reduced oxidative-stress response in red blood cells from p45NFE2-deficient mice. Blood (2001) 97:2151–8. 10.1182/blood.V97.7.2151 [DOI] [PubMed] [Google Scholar]
- 53.Suzuki T, Kelly VP, Motohashi H, Nakajima O, Takahashi S, Nishimura S, et al. Deletion of the selenocysteine tRNA gene in macrophages and liver results in compensatory gene induction of cytoprotective enzymes by Nrf2. J Biol Chem. (2008) 283:2021–30. 10.1074/jbc.M708352200 [DOI] [PubMed] [Google Scholar]
- 54.Kawatani Y, Suzuki T, Shimizu R, Kelly VP, Yamamoto M. Nrf2 and selenoproteins are essential for maintaining oxidative homeostasis in erythrocytes and protecting against hemolytic anemia. Blood (2011) 117:986–96. 10.1182/blood-2010-05-285817 [DOI] [PubMed] [Google Scholar]
- 55.Diederich L, Suvorava T, Sansone R, Keller TCS, IV, Barbarino F, Sutton TR, et al. On the effects of reactive oxygen species and nitric oxide on red blood cell deformability. Front Physiol. (2018) 9:332. 10.3389/fphys.2018.00332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li C, Stocker R. Heme oxygenase and iron: from bacteria to humans. Redox Rep. (2009) 14:95–101. 10.1179/135100009X392584 [DOI] [PubMed] [Google Scholar]
- 57.Vinogradov SN, Hoogewijs D, Bailly X, Mizuguchi K, Dewilde S, Moens L, et al. A model of globin evolution. Gene (2007) 398:132–42. 10.1016/j.gene.2007.02.041 [DOI] [PubMed] [Google Scholar]
- 58.Freitas TA, Saito JA, Hou S, Alam M. Globin-coupled sensors, protoglobins, and the last universal common ancestor. J Inorgan Biochem. (2005) 99:23–33. 10.1016/j.jinorgbio.2004.10.024 [DOI] [PubMed] [Google Scholar]
- 59.Gasiorek JJ, Nouhi Z, Blank V. Abnormal differentiation of erythroid precursors in p45 NF-E2(–/–) mice. Exp Hematol. (2012) 40:393–400. 10.1016/j.exphem.2012.01.007 [DOI] [PubMed] [Google Scholar]
- 60.Gasiorek JJ, Blank V. Regulation and function of the NFE2 transcription factor in hematopoietic and non-hematopoietic cells. Cell Mol Life Sci. (2015) 72:2323–35. 10.1007/s00018-015-1866-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lu SJ, Rowan S, Bani MR, Ben-David Y. Retroviral integration within the Fli-2 locus results in inactivation of the erythroid transcription factor NF-E2 in Friend erythroleukemias: evidence that NF-E2 is essential for globin expression. Proc Nat Acad Sci USA. (1994) 91:8398–402. 10.1073/pnas.91.18.8398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sawado T, Igarashi K, Groudine M. Activation of beta-major globin gene transcription is associated with recruitment of NF-E2 to the beta-globin LCR and gene promoter. Proc Nat Acad Sci USA. (2001) 98:10226–31. 10.1073/pnas.181344198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rheinemann L, Seeger TS, Wehrle J, Pahl HL. NFE2 regulates transcription of multiple enzymes in the heme biosynthesis pathway. Haematologica (2014) 99:e208–10. 10.3324/haematol.2014.106393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tugores A, Magness ST, Brenner DA. A single promoter directs both housekeeping and erythroid preferential expression of the human ferrochelatase gene. J Biol Chem. (1994) 269:30789–97. [PubMed] [Google Scholar]
- 65.Macari ER, Lowrey CH. Induction of human fetal hemoglobin via the NRF2 antioxidant response signaling pathway. Blood (2011) 117:5987–97. 10.1182/blood-2010-10-314096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Campbell MR, Karaca M, Adamski KN, Chorley BN, Wang X, Bell DA. Novel hematopoietic target genes in the NRF2-mediated transcriptional pathway. Oxid Med Cell Longevity (2013) 2013:120305. 10.1155/2013/120305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gacesa R, Dunlap WC, Barlow DJ, Laskowski RA, Long PF. Rising levels of atmospheric oxygen and evolution of Nrf2. Sci Rep. (2016) 6:27740. 10.1038/srep27740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lane DJ, Merlot AM, Huang ML, Bae DH, Jansson PJ, Sahni S, et al. Cellular iron uptake, trafficking and metabolism: key molecules and mechanisms and their roles in disease. Biochim Biophys Acta (2015) 1853:1130–44. 10.1016/j.bbamcr.2015.01.021 [DOI] [PubMed] [Google Scholar]
- 69.Waldvogel D, van Gelderen P, Hallett M. Increased iron in the dentate nucleus of patients with Friedrich's ataxia. Ann Neurol. (1999) 46:123–5. [DOI] [PubMed] [Google Scholar]
- 70.Puccio H, Simon D, Cossee M, Criqui-Filipe P, Tiziano F, Melki J, et al. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat Genet. (2001) 27:181–6. 10.1038/84818 [DOI] [PubMed] [Google Scholar]
- 71.Whitnall M, Suryo Rahmanto Y, Sutak R, Xu X, Becker EM, Mikhael MR, et al. The MCK mouse heart model of Friedreich's ataxia: alterations in iron-regulated proteins and cardiac hypertrophy are limited by iron chelation. Proc Nat Acad Sci USA. (2008) 105:9757–62. 10.1073/pnas.0804261105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huang ML, Becker EM, Whitnall M, Suryo Rahmanto Y, Ponka P, Richardson DR. Elucidation of the mechanism of mitochondrial iron loading in Friedreich's ataxia by analysis of a mouse mutant. Proc Nat Acad Sci USA. (2009) 106:16381–6. 10.1073/pnas.0906784106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Whitnall M, Suryo Rahmanto Y, Huang ML, Saletta F, Lok HC, Gutierrez L, et al. Identification of nonferritin mitochondrial iron deposits in a mouse model of Friedreich ataxia. Proc Nat Acad Sci USA. (2012) 109:20590–5. 10.1073/pnas.1215349109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Anzovino A, Chiang S, Brown BE, Hawkins CL, Richardson DR, Huang ML. Molecular alterations in a mouse cardiac model of Friedreich ataxia: an impaired Nrf2 response mediated via upregulation of Keap1 and activation of the Gsk3beta axis. Am J Pathol. (2017) 187:2858–75. 10.1016/j.ajpath.2017.08.021 [DOI] [PubMed] [Google Scholar]
- 75.Paupe V, Dassa EP, Goncalves S, Auchere F, Lonn M, Holmgren A, et al. Impaired nuclear Nrf2 translocation undermines the oxidative stress response in Friedreich ataxia. PLoS ONE (2009) 4:e4253. 10.1371/journal.pone.0004253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.D'Oria V, Petrini S, Travaglini L, Priori C, Piermarini E, Petrillo S, et al. Frataxin deficiency leads to reduced expression and impaired translocation of NF-E2-related factor (Nrf2) in cultured motor neurons. Int J Mol Sci. (2013) 14:7853–65. 10.3390/ijms14047853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shan Y, Schoenfeld RA, Hayashi G, Napoli E, Akiyama T, Iodi Carstens M, et al. Frataxin deficiency leads to defects in expression of antioxidants and Nrf2 expression in dorsal root ganglia of the Friedreich's ataxia YG8R mouse model. Antioxid Redox Signal. (2013) 19:1481–93. 10.1089/ars.2012.4537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sahdeo S, Scott BD, McMackin MZ, Jasoliya M, Brown B, Wulff H, et al. Dyclonine rescues frataxin deficiency in animal models and buccal cells of patients with Friedreich's ataxia. Hum Mol Genet. (2014) 23:6848–62. 10.1093/hmg/ddu408 [DOI] [PMC free article] [PubMed] [Google Scholar]