

Abstract
The analysis of illicit drugs in urban wastewater is the basis of wastewater‐based epidemiology (WBE), and has received much scientific attention because the concentrations measured can be used as a new non‐intrusive tool to provide evidence‐based and real‐time estimates of community‐wide drug consumption. Moreover, WBE allows monitoring patterns and spatial and temporal trends of drug use. Although information and expertise from other disciplines is required to refine and effectively apply WBE, analytical chemistry is the fundamental driver in this field. The use of advanced analytical techniques, commonly based on combined chromatography—mass spectrometry, is mandatory because the very low analyte concentration and the complexity of samples (raw wastewater) make quantification and identification/confirmation of illicit drug biomarkers (IDBs) troublesome. We review the most‐recent literature available (mostly from the last 5 years) on the determination of IDBs in wastewater with particular emphasis on the different analytical strategies applied. The predominance of liquid chromatography coupled to tandem mass spectrometry to quantify target IDBs and the essence to produce reliable and comparable results is illustrated. Accordingly, the importance to perform inter‐laboratory exercises and the need to analyze appropriate quality controls in each sample sequence is highlighted. Other crucial steps in WBE, such as sample collection and sample pre‐treatment, are briefly and carefully discussed. The article further focuses on the potential of high‐resolution mass spectrometry. Different approaches for target and non‐target analysis are discussed, and the interest to perform experiments under laboratory‐controlled conditions, as a complementary tool to investigate related compounds (e.g., minor metabolites and/or transformation products in wastewater) is treated. The article ends up with the trends and future perspectives in this field from the authors’ point of view. © 2016 by The Authors. Mass Spectrometry Reviews published by Wiley Periodicals, Inc. Mass Spec Rev 37:258–280, 2018
Keywords: mass spectrometry, drugs of abuse, urinary metabolites, wastewater‐based epidemiology
I. INTRODUCTION
Illicit drug use is a global problem with severe consequences, not only for people's health and welfare, but also as a clear threat to the stability and security of entire regions and economic and social development. Accordingly, there is a considerable financial cost related to illicit drug use, associated with drug use prevention, treatment of addicts, and on the fight against organized crime (Nutt et al., 2007; EMCDDA, 2012; UNODC, 2014). Policy makers need accurate and reliable information on the use of these substances in order to make evidence‐based decisions and to effectively allocate resources. The prevalence of illicit drug use has traditionally been estimated with direct subjective methods, like general population surveys and interviews, and indirect methods such as monitor drug‐related criminality, seizures, and hospital records (EMCDDA, 2015a). Despite considerable improvements with modern communication facilities, and the use of complementary methods such as targeted studies and statistical modeling, these survey methods mainly rely on the willingness of users to self‐report and to monitor actions. However, the social taboo related to illicit drug use might provoke non‐participation and false responses in such surveys that makes these methods vulnerable and potentially inaccurate. In addition, a common challenge with such methods is that they are time‐consuming, expensive, and complex (Banta‐Green & Field, 2011). Therefore, the development of new and complementary approaches is encouraged in order to obtain objective, low‐cost, fast, reliable, and comparable data.
The chemical analysis of illicit drug residues in untreated wastewater is as a valuable tool to complement existing approaches to monitor spatiotemporal patterns and trends of illicit drug use in large communities (Zuccato et al., 2008; Thomas et al., 2012; Ort et al., 2014c; EMCDDA, 2015b). This approach is termed wastewater‐based epidemiology (WBE), and relies on the principle that illicit drugs consumed by individuals are excreted, either unchanged or as a mixture of metabolites, into urban sewer networks. The quantitative measurement of these specific illicit drug biomarkers (IDBs) in wastewater samples reflects the drugs collectively excreted by users and enables data to be gathered on drug use by the community within the geographical boundary defined by the catchment area of a wastewater treatment plant (WWTP). Several crucial steps are involved in this approach (Fig. 1), which requires significant expertise from numerous research fields, and therefore, strong multi‐disciplinary collaboration (Ort et al., 2014a).
Figure 1.
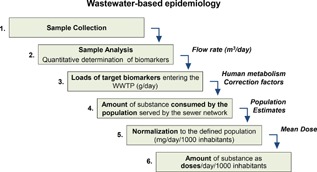
Main consecutive steps of the WBE approach and data required for each step (modified from Castiglioni et al. (2014)).
The WBE concept was initially proposed by Daughton in 2001 (Daughton, 2001) and first applied in 2005 through the estimation of cocaine use in Italy (Zuccato et al., 2005). Since then, WBE has expanded to include other illicit drugs, such as heroin, cannabis, and amphetamine‐like stimulants (van Nuijs et al., 2011; Castiglioni et al., 2014; EMCDDA, 2015b) and new psychoactive substances (NPS) (Reid, Derry, & Thomas, 2014; van Nuijs et al., 2014; Kinyua et al., 2015). Data have also been reported for WBE‐based analysis of alcohol (Reid et al., 2011; Mastroianni, Lopez de Alda, & Barcelo, 2014; Rodríguez‐Álvarez et al., 2014, 2015; Boogaerts et al., 2016), tobacco (Rodríguez‐Álvarez et al., 2014; Castiglioni et al., 2015; Tscharke, White, & Gerber, 2016), and counterfeit medicines (Venhuis et al., 2014).
An important step in the progression of WBE was accomplished with the establishment of a European‐wide network (Sewage analysis CORe group Europe [SCORE]), supported by the European Monitoring Centre for Drug and Drug Addiction (EMCDDA, 2015b). This network has since 2010 standardized the WBE approach, coordinated international studies (Thomas et al., 2012; Ort et al., 2014c), and conducted inter‐laboratory exercises for quality control purposes. The latter provided a means to estimate and critically assess the uncertainty related to the wastewater‐based estimates (Castiglioni et al., 2013). However, important WBE research has also been conducted in other continents such as Asia (Lai et al., 2013b; Khan et al., 2014; Li et al., 2014; Kim et al., 2015), Australia (Irvine et al., 2011; Prichard et al., 2012; Lai et al., 2013a; Tscharke et al., 2016, 2016), North America (Banta‐Green et al., 2009; Burgard et al., 2013; Subedi & Kannan, 2014), and Central and South America (Maldaner et al., 2012; Devault et al., 2014; Voloshenko‐Rossin et al., 2015; Bijlsma et al., 2016). Furthermore, a recently published review on neuropsychiatric pharmaceuticals and illicit drugs in WWTPs (Asimakopoulos & Kannan, 2016) is recommended for readers interested.
The increasing interest of WBE is clearly illustrated by the number of papers and citations on the topic (>200 papers in years 2005–2015, >5000 citations; ISI Web of Science), international conferences and workshops organized on this topic, and funding received from the European Commission (“http://score-cost.eu/”; “http://sewprof-itn.eu/”).
Chemical analysis of IDBs in wastewater plays an important key role within the WBE approach. Advanced analytical techniques and expertise is required to obtain accurate concentration data on IDBs in wastewater, because quantitative data are the basis of subsequent back‐calculations of IDB mass loads and drug use. Concentrations of IDBs in wastewater samples are generally around a factor 1000 lower than in human biological fluids (ng/L vs. ng/mL), which points out the challenge for quantitative analysis. Low analyte concentrations in combination with the complexity and unknown composition of the wastewater matrix might hamper not only the sensitive and accurate quantification but also a sound identification. Chromatography—mass spectrometry is the best‐suited approach to obtain the sensitivity, selectivity, and identification requirements in chemical analysis directed toward WBE.
Gas chromatography coupled to mass spectrometry (GC‐MS) (Mari et al., 2009; González‐Mariño et al., 2010) in general provides high levels of selectivity and sensitivity. However, derivatization of the target compounds is often necessary for most IDBs in order to make them compatible with GC. Consequently, sample treatment and measurement is generally laborious and time‐consuming. Liquid chromatography coupled to mass spectrometry (LC‐MS) is a more‐versatile technique that allows the determination of polar, low‐volatility, and/or thermolabile compounds, which most IDBs are, with less sample treatment and shorter chromatographic analysis times. Besides, the sample matrix (i.e., water) is completely compatible with this technique. LC—tandem mass spectrometry (LC‐MS/MS), for example, a triple quadrupole (QqQ) analyzer, is even more powerful, and has become the technique of choice for the quantitative determination of (known) IDBs in wastewater samples (van Nuijs et al., 2011).
Despite the predominance of LC‐MS/MS in WBE studies, the use of high‐resolution mass spectrometry (HRMS) has been recently explored and opens new perspectives in the analytical field. Its strong potential to screen and for identification purposes originates from the acquisition of accurate‐mass full‐spectrum data. LC‐HRMS is a powerful technique that allows the wide‐scope screening of many illicit drugs, metabolites and transformation products, as well as the investigation of NPS (Bijlsma et al., 2013b; Hernández et al., 2014; Ibáñez et al., 2014; Reid et al., 2014; Alechaga, Moyano, & Galceran, 2015; Bade et al., 2015c; Baz‐Lomba, Reid, & Thomas, 2016). There are several illustrative examples (as presented later in this manuscript) in the most‐recent literature that show that modern analytical chemistry is essential in order to increase our knowledge on trends in substance use in the general population.
In this article, an exhaustive review of the existing literature is not the goal. Rather, the objective is to present an overview of the approach by using the most‐recent literature available, with specific emphasis on the different analytical strategies applied for WBE. Most cited papers have been published within the last 5 years, except for some that have also been included because of their relevance in the development of the WBE approach. The predominance of LC‐MS/MS to quantify priority, well‐known, target IDBs and the essence to produce reliable and comparable results are illustrated. Accordingly, the importance to perform inter‐laboratory exercises and the need to analyze appropriate quality controls in each sample sequence is highlighted. Other crucial steps related to WBE, such as sample collection and sample pre‐treatment, are discussed. The article further focuses on novel analytical approaches, such as enantiomeric profiling, the different strategies for target and non‐target analysis with LC‐HRMS, and on the interest to perform in vivo or in vitro metabolism experiments and degradation laboratory experiments, as a useful tool to investigate metabolites and transformation products in wastewater. The article concludes with the trends and future perspectives in this discipline from the authors’ point of view.
II. TARGET AND NON‐TARGET APPROACHES
The terms target and non‐target analyses are widely employed in analytical chemistry. Other expressions, such as investigation of unknowns or suspect screening, are also frequently used, and illustrate the analytical challenges in complex fields, such as environmental analytical chemistry (Krauss, Singer, & Hollender, 2010; Schymanski et al., 2014b; Bletsou et al., 2015). With the implementation of LC‐HRMS in several analytical fields, the term post‐target analysis has been also employed, and illustrate the working mode in which full‐spectrum accurate‐mass techniques are applied (Hernández et al., 2005). Clarification of these terms is, however, necessary to fully understand the different strategies that can be applied to investigate the presence of IDBs in wastewater samples.
Target methodologies are commonly analyte‐dependent; that is, compound‐specific information is required before measurement. Based on this analyte‐specific information, highly sensitive and selective analytical methods can be developed, but other compounds that might be present in the samples will remain undetected. Target analysis is typically applied in methods based on LC and/or GC coupled to MS (Selected Ion Monitoring [SIM] mode) or tandem MS (Selected Reaction Monitoring [SRM] mode). A limited list of target compounds is included in the scope of the method, and only those previously selected ions/transitions are monitored. Reliable identification and quantification is commonly the main objective pursued in this type of analysis. This identification is achieved through acquisition of at least three ions in the SIM mode or two MS/MS transitions in the SRM mode, and evaluation of retention time (tR) and ion‐intensity ratios (SANTE/11945, 2015). The use of reference standards is compulsory with this methodology in order to optimize the mass spectrometric measurement parameters and for quantitative method validation.
This methodology can also be applied with HRMS instruments. However, thanks to the accurate‐mass full‐spectrum acquisition at good sensitivity, HRMS offers the possibility to investigate the presence of many other compounds, not only those initially targeted. Databases developed in‐house are commonly used to facilitate screening of a large number of compounds with HRMS. The information included in the database depends on the availability of a reference standard. When available, the information is very complete (i.e., tR, exact mass of the (de)protonated molecule and/or adducts, and the main fragment ions) to highly facilitate the analytical research. When the reference standard is not available, only limited information on the target analytes can be included in the database (e.g., molecular formula and exact mass, theoretical isotope distribution, predicted tR). Information reported in the literature on product ions can also be taken into account to facilitate compound identification. Even in this worst‐case situation (i.e., when no standard is available), accurate‐mass full‐spectrum data can be used for the tentative identification of the compounds in samples (Hernández et al., 2015a), a process where the interpretation and justification of the fragment ions observed is crucial. In any case, the fact that a search is directed toward a list of compounds implies a target approach, independent of the availability of reference standards. This target approach is commonly known as suspect screening (Krauss, Singer, & Hollender, 2010; Hug et al., 2014) whereas other authors use the term post‐target (Hernández et al., 2005; Ibáñez et al., 2008).
Target analysis based on HRMS commonly detects and identifies the compounds (i.e., qualitative analysis) in wastewater, because HRMS really takes advantage of its excellent performance for this type of application (Hernández et al., 2011a, 2014). However, recent studies have also been directed toward the quantitative analysis of IDBs in wastewater (González‐Mariño et al., 2012; Bijlsma et al., 2013b; Fedorova et al., 2013; van der Aa et al., 2013; Heuett et al., 2015), and a notable increase in the number of quantitative applications of HRMS that pursue a complete analysis (i.e., sensitive detection, reliable identification, accurate quantification) is expected in the near future.
A significant advantage of full‐spectrum acquisitions is that they also allow the investigation of any other compound, not only the list of selected contaminants, in a non‐target methodology. In contrast to the (post‐) target approach, a genuine non‐target analysis does not use any previous information on the compounds to be searched in the samples. Some authors use the term non‐target screening when they search for unknowns, which in a strict sense starts without any information on the compounds to be investigated. The term unknown does not necessarily mean that the compound discovered in the analysis is a new or an unreported compound. Following the elucidation processes, the unknown might turn out to be a known compound, which already has been reported in the literature, but unexpected or not specifically searched for in the samples. In a genuine non‐target analysis, there is no analyte selection a priori (neither before nor after MS acquisition). Under these circumstances, compound identification at trace levels in wastewater is a challenge, and commonly more than one elemental formula and several plausible structures are obtained for a given unknown detected in a sample (Ibáñez et al., 2008; Krauss, Singer, & Hollender, 2010; Schymanski et al., 2014a). More information on non‐target analysis applied in the environmental field can be found elsewhere (Hogenboom, van Leerdam, & de Voogt, 2009; Chiaia‐Hernandez et al., 2014; Hug et al., 2014; Schymanski et al., 2014b, 2015; van Leerdam et al., 2014).
Most IDBs are of (medium) high polarity; therefore, LC‐HRMS commonly uses TOF and Orbitrap analyzers. The absence of standardized mass spectral libraries in LC‐MS is an additional difficulty in non‐target analysis, opposite to GC‐MS with electron ionization (EI), where the availability of commercial libraries (e.g., NIST) offers the possibility to identify compounds by matching experimental and library spectra.
In order to have a more‐realistic and complete overview on the presence of organic contaminants in general, and on IDBs in particular, a combination of target (both with or without standards (i.e., suspect screening) and non‐target methodologies seems to be the most‐attractive approach (Hug et al., 2014). Furthermore, the combination of GC‐HRMS and LC‐HRMS (e.g., use the same QTOF instrument for both configurations) moves toward a more‐comprehensive screening of organic contaminants in the aquatic environment independently of their polarity and volatility (Hernández et al., 2015b).
III. SAMPLE COLLECTION APPROACHES AND SAMPLE PREPARATION
Measurements with high‐end instruments and sophisticated statistical analysis cannot compensate or reveal deficiencies in sample collection. Depending on the desired level of representativeness and accuracy, sample collection is as important as any subsequent steps. A sound understanding of the investigated environment is imperative and must be documented in detail. For most applications in WBE, the end user relies on 24‐hr composite influent (i.e., raw) wastewater samples collected routinely at the inlet of a WWTP. We briefly outline how such a sample is ideally collected (sampling) and how many samples are needed (monitoring) for specific applications.
A. Sampling
The number of consumers of a specific substance, pharmacokinetics, the population connected to a WWTP, and the hydraulic properties of the sewer system determine: (i) the number of toilet flushes that contain the substance of interest, and (ii) over which period an individual toilet flush expands when it passes the sampling location. These two factors determine the temporal variability on the scale of minutes, which in turn determines the required sampling frequency to collect a representative sample (i.e., how many samples must be collected and pooled over a certain period for analysis). Due to the potentially small number of relevant toilet flushes—at least for one of multiple substances analyzed in the collected sample—it is recommended that sampling intervals do not exceed 5–10 min at the influent of large WWTPs. Even shorter sampling intervals of 1–5 min are necessary for composite samples from effluents of individual premises. At the effluent of, for example, a prison, the toilet flushes extend over shorter periods and the absolute number of substance‐related flushes is typically much smaller than at the influent of a (large) WWTP, both leading to a substance pattern that fluctuates much more at the scale of a few minutes. This is also applicable to influents of very small WWTPs, where it may be beneficial to sample from the effluent of a primary treatment tank, which attenuates temporal fluctuations to some extent. However, please note that the latter also removes some particulate matter and that WWTP‐internal recirculation may influence mass loads there. Shorter sampling intervals are also required for the collection of representative samples that extend over periods <24 hr, for example, hourly composite samples to reliably assess diurnal variations. Please note, grab samples to determine diurnal variations are not suitable because of potential high short‐term variations. In addition, intra‐day variations in wastewater flows require samples to be collected in a flow‐ or volume‐weighted manner. More details and peculiarities for wastewater sampling in sewers, an overview of relevant literature, and a list of requirements for a series of typical applications is described elsewhere (De Keyser et al., 2010; Ort et al., 2010a,2010b; Ort, 2014; Coutu et al., 2016).
B. Monitoring
Ideally, one would analyze 365 daily composite samples per year. Due to limited resources, this amount of samples is usually not feasible. Different (research/epidemiological) questions require different monitoring designs, and obviously more samples provide higher accuracy. Random day‐to‐day variability, systematic weekly cycles, and seasonal fluctuations influence baseline variation. Together with the desired level of sensitivity and significance, the baseline variation determines the required number and distribution of samples. To date, two monitoring design studies have investigated different approaches that aim to answer different questions. They are based on a few available long‐term time series; that is, data sets with observations over more than 28 consecutive days. In brief, it was found that the baseline variation of almost all investigated drug residues in five different catchments (population sizes that range from approx. 7000 to 1.3 million people) did not exceed 80% (as coefficient of variation), not accounting for any temporal correlation (EMCDDA, 2016). Based on these data, Ort et al. (2014b) proposed that 56 stratified random samples (10 working days and 4 weekend days per quarter) are suitable to estimate an annual mean with an accuracy of 10%. Humphries et al. (2016) evaluated more informed approaches, which rely on knowledge about weekly cycles. In the presence of strong weekly cycles (e.g., cocaine or MDMA), it is efficient to distribute monitoring days systematically by considering peak, mid, and through usage days. The identification of weekly cycles requires an intensive, monitoring period. In return, the accuracy, particularly of intra‐annual trends, improves the informed routine monitoring compared to an uninformed scheme with the same reduced number of samples. In the context to assess effects of interventions, the baseline variation, the magnitude of the effect to be shown, and the confidence level determine the number of required samples.
C. Collection of (Meta)Information
An example of a questionnaire to collect (meta)information relevant to evaluate the appropriateness of sampling and to facilitate the interpretation of results is can be found (Ort et al., 2014c). This paper encompasses catchment properties (e.g., hydraulic residence time, population size, and how it was estimated), as well as details on sample collection and handling.
D. Stability of IDs Biomarkers in the Samples Under Storage Conditions
It is often not possible to immediately analyse samples for the presence of IDBs. It is, therefore, necessary to store wastewater samples until analysis under conditions that avoid transformation of the analytes, because biotransformation during storage would lead to false interpretations of WBE data, even if analytical methods are accurate.
For most IDBs, experiments have been performed to evaluate stability for different storage conditions, including various temperatures, pH values, and preservation agent additions, and for different time frames (reviewed in detail by McCall et al., (2016)). Storage of wastewater samples at −20°C ensures stability of most IDBs for at least 3 weeks. For some IDBs (i.e., the amphetamine‐type stimulants and 11‐nor‐9‐carboxy‐delta‐9‐tetrahydrocannabinol [THC‐COOH]), experiments have revealed that they are even stable at −20°C for more than 15 weeks (McCall et al., 2016). Stability is ensured for only several days at 4°C, whereas other IDBs (e.g., cocaine) are within several hours transformed at this temperature (McCall et al., 2016).
Acidification of the samples increases stability of the majority of IDBs, but significantly enhances biotransformation of THC‐COOH. Because multi‐analyte determinations are often applied in WBE studies, it is, therefore, not advisable to acidify the samples before storage. The effect of the addition of preservation agents such as sodium metabisulfite (Na2S2O5) has also been studied, and revealed higher stability for cocaine and its metabolite benzoylecgonine (McCall et al., 2016). However, for the other IDBs, more experiments need to be performed in order to evaluate the efficacy to add preservation agents to wastewater samples for storage purposes.
E. Sample Treatment
In order to remove solid particles from the wastewater samples, two strategies can be followed: (i) a filtration step with membrane of glass fiber filters with pore sizes as low as 0.1 μm, or (ii) a centrifugation step. In addition, because concentrations of most IDBs in wastewater are in the ng/L–μg/L range, a preconcentration step is generally required prior to analysis in order to achieve the necessary quantification limits. However, some modern analytical instruments allow direct injection of filtered or centrifuged wastewater due to the high sensitivity provided (Chiaia, Banta‐green, & Field, 2008; Berset, Brenneisen, & Mathieu, 2010; Bisceglia et al., 2010; Lai et al., 2011; Boix et al., 2015).
A sample‐preparation step is commonly needed not only to preconcentrate analytes, but also to remove matrix components that might interfere with the analytical measurement (e.g., ionization suppression/enhancement processes in LC–MS) of IDBs.
By far, the most‐used procedure reported in the literature is off‐line solid‐phase extraction (SPE) with sample volumes between 50 and 1000 mL (van Nuijs et al., 2011). Popular SPE sorbents used for a reproducible extraction of IDBs from wastewater are based on a polymeric backbone with reversed‐phase or cation‐exchange properties. Some methods demonstrated that off‐line SPE sample preparation could also be incorporated in a fully automated on‐line SPE application that is directly linked to the LC‐MS analysis (Postigo, Lopez de Alda, & Barceló, 2008; Fedorova et al., 2013; Heuett et al., 2015).
Some alternative ways of sample preparation can be found in the literature. González‐Mariño et al. (2009) applied commercially available molecular imprinted polymers (MIPs) to extract and concentrate amphetamine‐type stimulants from wastewater, and reported better performance of the MIPs in terms of selectivity, sensitivity, accuracy, and precision compared to off‐line SPE. The main drawbacks of this approach were no possibility for multi‐class analysis, and higher analysis time and cost. The usefulness of solid‐phase micro extraction (SPME) for sample preparation to analyze amphetamine‐type stimulants or THC‐COOH in wastewater has been demonstrated (Racamonde et al., 2012, 2013). SPME is highly compatible with GC‐MS, and shows good performance; however, the main drawback was the limited applicability to determinate multi‐class compounds, which is desired for WBE purposes.
IV. APPLICATIONS OF LOW‐RESOLUTION MASS SPECTROMETRY
Low‐resolution MS systems, with either ion trap or QqQ analyzers, are the most widely applied for the quantitative determination of IDBs in wastewater. These MS systems typically operate in the MS/MS mode, where one or more product ions are monitored by selecting appropriate precursor‐to‐product ion transitions (i.e., SRM). Even with this approach, there are some probabilities that other compounds, not related to the analyte, can share the same transition. Therefore, at least two transitions is required to be monitored, and the presence of a compound is considered to be confirmed if both transitions produce a chromatographic peak at the same retention time, that corresponds to that of the injection of a reference standard. Identity confirmation is of the utmost importance because it gives the required confidence to the reported results and reduces the likelihood of reporting false positives. The confirmation of identity is especially relevant when analyzing highly contaminated and complex matrices such as influent wastewater samples. For confirmation, not only the acquisition of several transitions is needed, but also the compliance of the ion ratio between reference standards and samples. This important aspect is discussed in more detail in section VII “Relevant analytical parameters and quality control.”
Although ion‐trap analyzers have been used in this field (Bones, Thomas, & Paull, 2007; Gheorghe et al., 2008; Postigo, Lopez de Alda, & Barceló, 2008; Martínez‐Bueno et al., 2011), LC‐MS/MS with QqQ has become the most‐popular technique due to its excellent performance in terms of robustness, dynamic range, sensitivity, and selectivity. These characteristics, together with the compatibility of LC‐MS/MS with aqueous samples and mostly polar analytes targeted in WBE studies, allow one to notably simplify sample treatment. LC‐MS/MS with QqQ can nowadays be considered as the workhorse in analytical laboratories that deal with WBE. This fact has also been illustrated in monitoring studies of illicit drugs in wastewater, where the majority of laboratories applied this technique (Castiglioni et al., 2008; Thomas et al., 2012; Ort et al., 2014c).
Some of the early studies, used only one transition for quantification (Chiaia, Banta‐green, & Field, 2008; Gheorghe et al., 2008; Bartelt‐Hunt et al., 2009; Metcalfe et al., 2010). However, it is nowadays widely accepted that confirmation of analyte identity in LC‐MS/MS‐based methodologies requires a minimum of two transitions (European Commission, 2002). Yet, the acquisition of more transitions per compound would obviously give more confidence to the confirmation process, and is feasible with the latest analytical instruments with faster acquisition times (Boleda, Galceran, & Ventura, 2007; Bijlsma et al., 2009, 2014a). The limited fragmentation in ESI, frequently with low‐abundance secondary and tertiary transitions, can limit the absolute number of useful MRM transitions for identification in some particular cases. Nevertheless, with the excellent sensitivity of the new instruments, the acquisition of, at least, two transitions is commonly not a problem. Typically the most‐abundant transition is selected to favor quantification at low concentrations (quantifier, Q), and the other ones are acquired for confirmation (qualifier, q). At this point, it is also important to understand the fragmentation pattern of the analytes under experimental conditions to allow selection of analyte‐specific fragments in order to minimize potential interferences of the matrix and/or background noise. Hence, it might occur that the most‐sensitive transition presents more matrix interference and/or background noise, and might therefore not be the most appropriate for quantification. As an example, THC‐COOH, a metabolite of the active ingredient found in cannabis, was measured in the positive electrospray ionization (ESI) mode. In this mode, the transition m/z 345 > 327 is occasionally selected for quantification (Table 1). However, this transition corresponds to a non‐specific loss of water, which might be more prone to interferences when analyzing complex matrices, such as wastewater (Pozo et al., 2006). This fact is demonstrated in Figure 2, where three different transitions were acquired to measure THC‐COOH in influent wastewater. The transitions 345 > 327 and 345 > 299, corresponding to losses of H2O and HCOOH, respectively, presented higher noise than the less‐abundant, but more‐selective, transition 345 > 193. These matrix interferences has also been observed in the negative mode; when a non‐specific loss of CO2 (343 > 299) was measured.
Table 1.
SRM transitions most often used with LC‐QqQ‐MS instruments to determine IDBs in wastewater during the monitoring campaign 2015 of SCORE (“http://score-cost.eu/”)
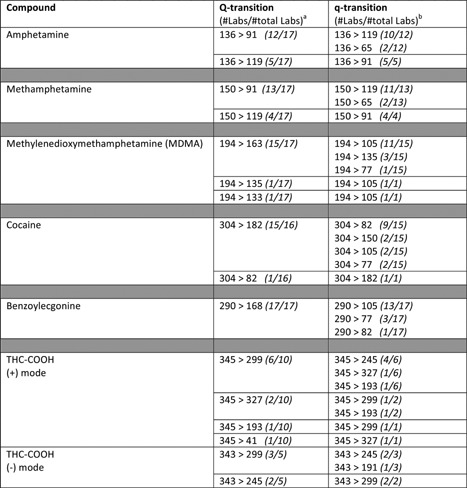
Note: More‐detailed information regarding the analytical procedures can be found elsewhere: (Castiglioni et al., 2006; Postigo, Lopez de Alda, & Barceló, 2008; van Nuijs et al., 2009; Berset et al., 2010; Karolak et al., 2010; Lai et al., 2011; Fedorova et al., 2013; Senta et al., 2013; Andrés‐Costa et al., 2014; Bijlsma et al., 2014a; Borova et al., 2014; Devault et al., 2014; Kankaanpää et al., 2014; Tscharke et al., 2015; Castrignanò, Lubben, & Kasprzyk‐Hordern, 2016). It is noteworthy that not all analytical methodologies used during the monitoring campaign were published.
aNumber of laboratories that selected the SRM transition (Q) for quantification/the total of laboratories that used LRMS and determine the IBD.
bNumber of laboratories that selected the SRM transition (q) for confirmation/the total of laboratories that selected the same Q‐transition (see alsoa).
Figure 2.
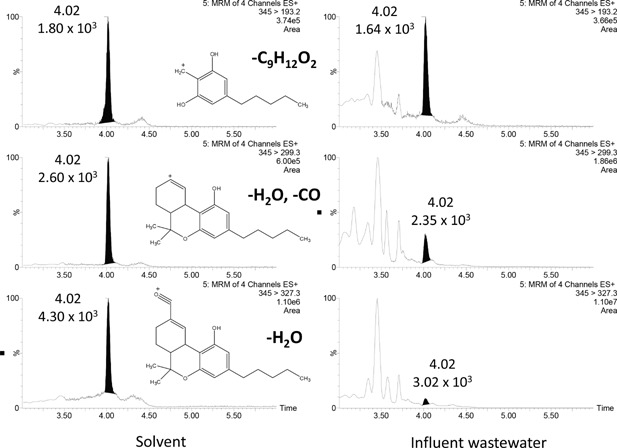
Selectivity of THC‐COOH transitions.
Table 1 shows the quantification (Q) and confirmation (q) transitions used during the monitoring campaign 2015 coordinated by SCORE to measure the most widely studied IDBs in influent wastewater in WBE research. In general, the Q transition is, in most studies, the same with few exceptions. However, more differences are observed in the qualifier transition used for confirmation.
The Q‐transitions selected for cocaine and its metabolite benzoylecgonine (BE) are in practically all cases 304 > 182 and 290 > 168, respectively. Both correspond to the neutral loss of benzoic acid, specific for cocaine and its metabolites (Castiglioni et al., 2008; Bijlsma et al., 2011). In relation to the q‐transition, some differences were observed, but 304 > 82 and 290 > 105 were most frequently selected. The Q‐ and q‐transitions for amphetamine are in most cases 136 > 91 and 136 > 119, and for methamphetamine, 150 > 91 and 150 > 119, respectively. More variation occurs in the selection of transitions for MDMA, although most laboratories use 194 > 163 for quantification.
THC‐COOH has been measured in the negative‐ and positive‐ESI modes. Obviously, different transitions/ions are selected in each case. The determination of THC‐COOH is more problematic than other drugs investigated. The use of different ionization modes, the poorer sensitivity for this compound, and the matrix interferences that affect LC–MS/MS analysis make its determination more troublesome (Vazquez‐Roig, Blasco, & Picó, 2013; Bijlsma et al., 2014b; Ort et al., 2014c). Moreover, other non‐instrumental factors, such as possible sorption to solids (Harman, Reid, & Thomas, 2011), in‐sewer and in‐sample stability (McCall et al., 2016), might also play an important role in the difficulty to get satisfactory results for this compound. Although some difficulties might be related to its different physico‐chemical properties (lower polarity) compared with other illicit drugs and metabolites, an unambiguous explanation has not yet been found. The problems associated to the determination of THC‐COOH have been corroborated with the results of inter‐laboratory exercises, where data for this compound indicate that recoveries of spiked amounts of THC‐COOH to wastewater invariably are low; those data suggest a systematic underestimation of the true concentrations of THC‐COOH in this type of matrix.
The examples, shown above, illustrate that the selection of appropriate transitions is not a banal aspect, and it requires a detailed study before, considering not only the abundance of the ions (used as common criterion), but also specificity and associated issues such as background noise.
V. APPLICATIONS OF HIGH‐RESOLUTION MASS SPECTROMETRY
HRMS is a powerful technique with many different applications in the investigation of IDBs in wastewater, from the screening of large number of compounds, the elucidation of unknowns, the identification of new metabolites and degradation/transformation products (TPs), to quantification of target analytes at low concentrations. Several reviews have been published on the use of HRMS to determine of licit and illicit drugs in environmental analysis, and are recommended for those researchers interested in this field (Wong & MacLeod, 2009; Petrovic et al., 2010; Farré et al., 2012; Hernández et al., 2012, 2014; Vazquez‐Roig, Blasco, & Picó, 2013; Kaufmann, 2014).
A. Wide‐Scope Screening
HRMS allows the efficient screening of a large variety of compounds, including IDBs, in wastewater. Its potential comes from the acquisition of accurate‐mass full‐spectrum data (Kaufmann et al., 2010). These data allow one to screen of compounds in a post‐target way without the need to pre‐select the analytes for method development, as stated in previous sections. Furthermore, the presence of compounds initially not considered, such as new substances and metabolites/TPs, can be also investigated from data acquired in a retrospective way without the need for additional analysis (Hernández et al., 2011a; Bijlsma et al., 2013b). This ability is advantageous, because in some occasions, samples might already have been discarded or the analytes are degraded, so additional sample injections might not be possible. In this way, the screening can be further widened by reprocessing raw data without the need to perform new analysis.
The most‐used HRMS analyzers are undoubtedly TOF and Orbitrap. Both instruments can be efficiently coupled with LC, although TOF MS has the advantage of easy coupling with ultra‐high performance liquid chromatography (UHPLC). On the contrary, restrictions due to the lower scan speed can limit the applicability of coupling an Orbitrap with UHPLC (Hernández et al., 2014). As a consequence of the intrinsic characteristics of these analyzers, the use of LC‐HRMS enables screening for a large number of IDBs at satisfactory sensitivity within one analysis. Obviously, the restrictions derived from the chromatographic and ionization processes and from sample pre‐treatment have to be taken into account in a search for contaminants. TOF analyzers have been widely used to screen licit and illicit drugs, and their potential has been well‐documented (Hernández et al., 2011a, 2014). Mass resolution typically ranges from 20,000 up to recently 80,000 FWHM, whereas mass accuracy <2 ppm and quantitative linear ranges start to become usual. Following the invention by Makarov (Hardman & Makarov, 2003; Hu et al., 2005; Makarov et al., 2006), the Orbitrap has gained popularity in the investigation of emerging contaminants (de Voogt et al., 2011; Fedorova et al., 2013). An Orbitrap possesses high mass resolution (>100,000 FWHM), high mass accuracy (<5 ppm) and acceptable dynamic range (5 · 103). The newest instruments can reach up to 450,000 FWHM at m/z 200 and sub‐ppm mass accuracy. However, the main drawback is its scanning speed, which is inverse to mass resolution. Thus, a compromise between achievable resolution and adequate chromatography must be found (Kellmann et al., 2009; Makarov & Scigelova, 2010).
Hybrid configurations increase the potential of these analyzers for screening purposes. The most‐common are Q‐TOF and LIT‐Orbitrap, although other possibilities exist, such as IT‐TOF and Q‐Orbitrap. These hybrid instruments provide relevant structural information by obtaining accurate‐mass product‐ion spectra after MS/MS experiments. Information obtained with MS/MS is highly useful to confirm potential positives revealed by, for example, HRMS or QqQ analysis, and to elucidate structures of unknowns or suspect compounds. However, the pre‐selection of the precursor ion is required for MS/MS product‐ion generation, and, therefore, a second injection is in principle needed. In order to overcome these limitations, product‐ion spectra can be collected with data‐dependent acquisition (DDA). In this mode, the first scan usually works as the survey scan, where data are processed “on‐the‐fly” to search for potential compounds of interest based on predefined selection criteria; for example, intensity threshold or a suspect inclusion list. If the selection criteria are met or the included ion is observed, then a second MS/MS scan (data‐dependent) is performed. The major advantage of this approach is the collection of “clean” structural information in just one injection. The main limitations are the intensity threshold itself, as well as the size of the inclusion list (number of suspects searched); both can negatively affect the achievable duty cycle. Hence, a decrease in the number of data points (i.e., the number of scans), affects the detectability of chromatographic peaks.
Advantageously, most current instruments also allow the acquisition of full‐scan spectra at different collision energies in just one injection. As a function of the instrument and/or manufacturer, this operation mode for the QTOF analyzer is known as MSE (Castro‐Perez et al., 2002; Plumb et al., 2006; Díaz et al., 2011; Hernández et al., 2011a) in the case of Waters, broadband collision‐induced dissociation (bbCID) (Dasenaki et al., 2015) in the case of Bruker, or all‐ions MS/MS (Kinyua et al., 2015) in the case of Agilent. Likewise for Q‐Orbitrap instruments from Thermo, this type of acquisition is also possible, and known as All Ion Fragmentation (AIF) (Coscollà et al., 2014; Berendsen et al., 2015), or variable Data‐Independent Analysis (vDIA) (Zomer & Mol, 2015). These approaches are possible thanks to the availability of collision gas inside the collision cell of the hybrid instruments. With the application of low energy in the collision cell, fragmentation is minimized, and the information obtained corresponds normally to the parent molecule ((de)protonated and/or adducts in some cases). At high collision energy, fragmentation of the molecule is favored. In addition, the high‐energy function provides not only fragmentation spectra similar to MS/MS experiments, but also the isotopic pattern of the fragments, and it conserves adduct and/or dimer information as the quadrupole works as an ion guide. In this way, (de)protonated molecule and fragment ion data collection are both enabled in a single acquisition without the need to select the precursor ion; therefore, not affecting negatively the duty cycle.
One of the main difficulties in wide‐scope screening is to ensure that the method can detect and identify all compounds included in the target list. Reference standards are obviously required for a final confirmation of the identity, but also needed to perform method validation. Qualitative validation of the screening is a key aspect, but a laborious and time‐consuming task. The objective is to ensure that the method detects a given compound at an established minimum concentration; therefore, the screening detection limit (SDL) is the main parameter evaluated. To this aim, water samples spiked at different levels need to be tested to establish the SDL as the lowest analyte concentration tested that can be detected (wiht the most abundant ion; that is, normally the (de)protonated molecule). In absence of guidelines in the environmental field, the approach used in other fields, such as pesticide residue analysis (SANCO, 2013) or doping control analysis (Pozo et al., 2007) can be adopted. To accept the empirical value of SDL, it is necessary to have at least 95% of positive detections in the spiked samples tested. Another key parameter is the limit of identification; that is, the lowest concentration tested for which the compound can be detected (only one ion) and identified (at least two accurate‐mass ions, with acceptable mass errors). For identification, other parameters have to be also considered, such as retention time and ion‐ratios (see below).
Qualitative validation is normally performed with selected compounds from the target list that are taken as a model, due to the extreme difficulties to validate the method for the huge number of compounds that might be included in the target list. Once the methodology is validated, the screening is applied to sample analysis. The occurrence of false positives is drastically minimized if strict criteria are applied for identification on the basis of the accurate‐mass data; however, one cannot ignore the possibility of false negatives for those compounds that were not previously validated.
Although the qualitative potential of HRMS is evident, quantitative applications have been more limited until now, mainly because HRMS analyzers typically show lower sensitivity and narrower dynamic range than QqQ instruments that operate in SRM mode. Thus, most research until now has been focused on identification and elucidation purposes. However, Orbitrap and the latest TOF instruments show improved performance to prompt their use also for quantification of IDBs in wastewater (González‐Mariño et al., 2012; Bijlsma et al., 2013b).
The strategy used for HRMS screening strongly depends on the availability of reference standards, as described previously. Nevertheless, when dealing with thousands of compounds, it is almost impossible to have all reference standards in the laboratory. One of the main benefits with HRMS is that reference standards are not strictly required in a first step of the process, because a tentative identification of suspect compounds can be made on the basis of the obtained information. Obviously, reference standards highly facilitate the analytical task in the screening, and are required for ultimate and unambiguous confirmation (Fig. 3 ); however, they may be acquired only in a final stage when solid well‐founded evidence exists on the presence of the compound in the sample. In this way, laboratories do not need to acquire all reference standards before analysis, with the subsequent problems of availability (e.g., TPs), costs, and expiry dates (Ibáñez et al., 2014).
Figure 3.

(Post)‐target screening strategy.
In the absence of reference standards, HRMS data might be sufficient for a tentative identification (Fig. 3). An example, identification of 2‐ethylidene‐1,5‐dimethyl‐3,3‐diphenylpyrrolidine (EDDP), a metabolite of methadone, in wastewater by LC‐QTOF MS, is shown in Figure 4. By combining the information obtained in the low‐energy (LE) function on the protonated molecule and in the high‐energy (HE) function on the fragment ions, tentative identification of this metabolite was feasible without any reference standard. The use of UHPLC also facilitated the assignment of the chromatographic peaks that corresponded to this compound (note that some ions present in the HE spectra did not correspond to EDDP; marked as x in the figure).
Figure 4.
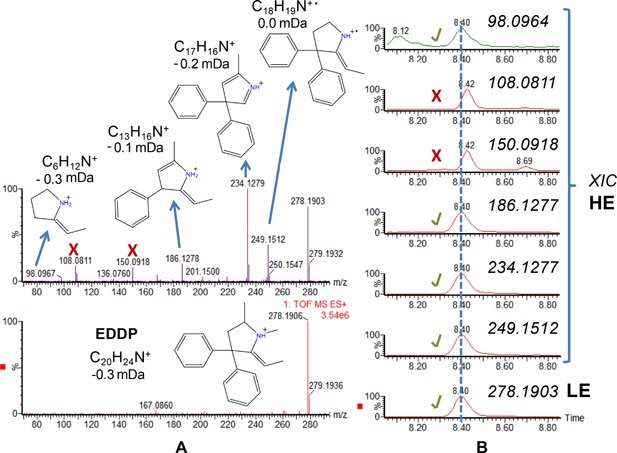
Tentative identification of EDDP, a methadone metabolite. (A) LE mass spectrum (bottom) HE mass spectrum (top). (B) XICs of the protonated molecule and several fragment ions (X indicates that the fragment ion is not related to EDDP).
Successful identification depends on the quality of the information provided (e.g., more than one accurate‐mass is required, low mass errors give more confidence to the process) and on the knowledge of mass spectrometry fragmentation rules to properly justify the fragment ions observed. Previous data reported in the literature on fragment ions (in nominal and accurate mass), which can also be available in databases such as MassBank (Horai et al., 2010; “http://www.massbank.jp/”), are useful to the analyst. Another interesting tool is retention‐time prediction, which helps to discard potential false positives and to focus the elucidation process on only those peaks that fit the predicted tR. These tR predictors are based on quantitative structure‐retention relationships (QSRRs) that vary from very simple, which incorporate a single descriptor (Kern et al., 2009; Bade et al., 2015b), to more complex, which use a large number of descriptors (Miller et al., 2013; Bade et al., 2015a; Gago‐Ferrero et al., 2015; Munro et al., 2015; Barron and McEneff, 2016); however, all of these predictors are based on a single column, commonly a reversed phase (C18) column. A direct‐mapping technique (PredRet) able to predict retention times across different chromatographic systems has also recently been developed (Stanstrup, Neumann, & Vrhovšek, 2015), but is limited to compounds having to be within the PredRet database. Although this area is rapidly advancing, the principal limitation is the use of a single column for most predictions, with further optimization needed for a truly transferable prediction technique.
One of the issues that still remains without broad consensus is the criteria applied for confident identification/confirmation. It is clear that a combination of parameters is required for this aim, including retention time and MS data. Accurate‐mass measurements give more confidence for a reliable identification than nominal mass data, and this factor is recognized in several guidelines, which, for example, give more identification points to HRMS ions than LRMS ions (European Commission, 2002). In addition, the ion intensity ratio is commonly used as a key parameter in the identification process. It is widely accepted that at least two accurate‐mass ions are required for a confident identification with HRMS. However, two main issues need to be considered: (i) what is the acceptable mass error? (ii) what is the maximum deviation acceptable in the ion ratio? Moreover, another parameter helpful in the process is the isotopic distribution, especially when abundant isotope ions such as chlorine, sulphur, or bromine are present. Several situations might occur that lead to different degrees of confidence in identification. For example, Schymanski et al. (2014a) propose up to 5 levels of confidence in a non‐target analysis. These levels range from only exact mass to unequivocal molecular formula, and then tentative candidate(s) followed by probable structure to a fully confirmed structure with a reference standard.
Figure 5 summarizes the key parameters in the detection and identification of a compound with HRMS. As shown in this figure, different scenarios might occur as a function on the information provided, and on the availability of reference standard (Bade et al., 2015c; Hernández et al., 2015a; Nácher‐Mestre et al., 2016).
Figure 5.
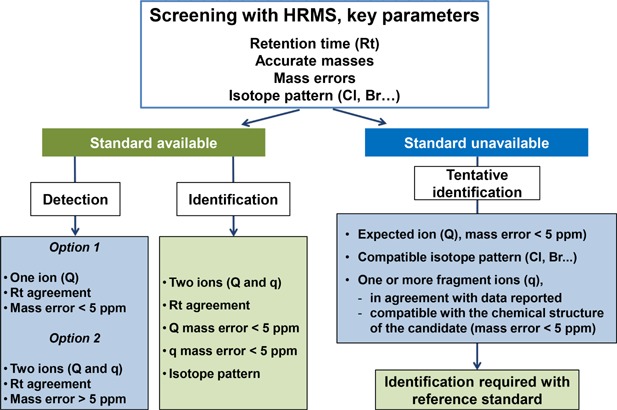
Detection and identification criteria in screening of illicit drugs with HRMS (modified from Nácher‐Mestre et al. (2016)).
When a reference standard is available, compounds can be detected or identified. Detection is considered satisfactory when the most‐abundant ion (Q), commonly the (de)protonated molecule, is found at the expected tR (±0.1 min,) and mass error <5 ppm (SANTE/11945, 2015). Another likely situation for detection is to find two representative ions (i.e., the most‐abundant ion (Q) and a fragment/adduct ions(q) at the expected tR), but with mass errors between 5 and 20 ppm. The latter situation seems to occur when the signal intensity is low (favored at low analyte concentrations). In that case, an additional effort is recommended to investigate more accurate‐mass ions and/or repeat sample injection. Identification is based on the presence of at least two representative ions (Q, q) at the expected tR with mass errors <5 ppm. Additionally, q/Q ratios should fit with those for reference standards within tolerance limits (SANTE/11945, 2015). Identification under these conditions is highly reliable and can be considered as the ideal situation.
When the reference standard is not available, a tentative identification can be made when an expected ion with mass error <5 ppm is observed, together with its characteristic isotopic pattern. Subsequently, the fragment ions should be evaluated by comparing the data with, for example, data reported in the literature or justified by the accurate‐mass fragments taking into account the structure of the molecule. However, for structure confirmation, injection of the reference standard is eventually required.
B. Non‐Target Analysis
Full‐spectrum accurate‐mass acquisition provided with HRMS also opens the possibility to investigate non‐target compounds in water (Ibáñez et al., 2005; Krauss, Singer, & Hollender, 2010; Hernández et al., 2011b; Díaz et al., 2012; Hug et al., 2014; Schymanski et al., 2015). A true “unbiased” non‐target screening, without any a priori information on the compounds to be detected, is an analytical challenge. This process needs expertise, and is complex and time‐consuming. The main difficulties associated with this process when applied to environmental or wastewater samples come from: (i) the complexity and unknown composition of the sample that is investigated, (ii) the presence of many peaks in the total ion chromatogram with the most‐abundant corresponding commonly to compounds other than the analytes, and (iii) the low analyte concentrations. Thus, the main problem is to prioritize the most “relevant” chromatographic peaks in the sample, because the majority will not be associated to drugs, in order to focus the subsequent elucidation process on those compounds. From the HRMS information, a complex process has to be applied that establishes the empirical formula of the unknown compound, searches chemical databases for potential candidates, and finally assigns the chemical structure of the discovered compound.
In a true non‐target analysis, the maximum number of compounds from very different physico‐chemical characteristics should be investigated. Therefore, a combination of GC‐HRMS and LC‐HRMS seems to be the most‐appropriate way to achieve this aim. Due to the complementarity of these two techniques this approach can be seen as the most‐comprehensive to advance toward the desired “universal” screening (Hernández et al., 2015b). Obviously, some “difficult” compounds would not likely be included in a wide‐scope screening. For example, very polar/ionic analytes would require specific chromatographic separation (e.g., HILIC). Thus, a combination of C18 and HILIC chromatographic columns would render a wider scope for LC‐amenable compounds. It must be taken also into account that universality of the screening should not refer only to the techniques of measurement but also to the sample treatment. From this point of view, a generic extraction, or even better, direct analysis of samples into the MS system, would be the best option to avoid compound losses during sample manipulation.
Because no pre‐selection of analytes is made in non‐target analysis, the compounds discovered in water samples might belong to the illicit drugs group, but also to any other family of organic contaminants or their metabolites. The fact that most IDBs are of medium to high polarity makes LC‐HRMS the most‐attractive approach for their potential identification. However, the absence of standardized mass spectral libraries in LC‐MS is an additional difficulty in non‐target analysis. Advances in the creation of mass spectra libraries for LC‐MS/MS analysis will be of help in the near future, but at the moment the analyst does not count on the aid of standardized libraries to facilitate a non‐target analysis.
An intermediate situation between target and true non‐target analysis is the application of “biased” non‐target approaches, where, for example, the formation of “unknown” metabolites/TPs from a given parent compound is investigated with “in vitro” or “in vivo” experiments, degradation laboratory experiments, or in‐silico models (Reid et al., 2014). Hence, the investigation is focused on chemically related compounds (e.g., share a common fragment, moiety, or mass defect) or on compounds that have specific atoms in their structure that give a distinctive isotopic signature (e.g., Cl, Br, S). Here, the number of chemically meaningful structures, which can be assigned to an unknown peak, is limited to structures that show a close relationship with the parent compound (Krauss, Singer, & Hollender, 2010). This issue will briefly be treated in the following section.
C. Investigation of Metabolites and Transformation Products
The investigation of metabolites and TPs of illicit drugs in water samples is a current topic of research (Bletsou et al., 2015). WBE is based on the analysis of key biomarkers of drugs. These IDBs can be the parent compound and/or the major urinary metabolite(s). For the most known and widely consumed drugs, information on human metabolism is already available, and has allowed establishment of benzoylecgonine as the main metabolite and IDB of cocaine, THC‐COOH as main metabolite and IDB of cannabis, or that methamphetamine, amphetamine, and MDMA are mainly excreted as unchanged compounds. However, this information is usually scarce for NPS and, therefore, the main biomarker of use is generally not well‐established.
Although WBE is solely based on the measurement of appropriate metabolites that result from human excretion (commonly the major and most‐stable ones), there is also a concern about the presence of many other metabolites and TPs of illicit and licit drugs in the aquatic environment. Especially, possible long‐term (chronic) effects on organisms and effects of combined exposure to multiple compounds is of concern (van der Aa et al., 2013). The detection and identification of these compounds is a challenge for environmental analytical chemists, and different approaches can be followed to this aim. In the case of known metabolites and TPs, already reported in the literature, an inclusion list of target analytes can be made. From an analytical point of view, they can be treated similarly to their parent compound with the above‐mentioned target methodologies. Furthermore, retrospective analysis is also feasible by reviewing the acquired MS data. For example, metabolites of drugs have been retrospectively investigated in wastewater samples previously analyzed for parent compounds only. In this way, several metabolites were tentatively identified without the need of additional analysis, and illustrate the potential of HRMS in this field (Hernández et al., 2011a; Bijlsma et al., 2013b).
Regarding unknown metabolites and TPs, the use of common fragmentation pathways between the parent compound and metabolites/TPs might discover new/unexpected compounds. In this strategy, a common behavior in their fragmentation is assumed. The presence of additional chromatographic peaks at the accurate masses of the fragments might reveal the presence of analyte‐related compounds. The accurate‐mass spectra and appropriate study of the fragmentation might finally allow the (tentative) identification of metabolites/TPs (Thurman et al., 2005; García‐Reyes, Molina‐Díaz, & Fernández‐Alba, 2007; Hernández et al., 2011b). This approach can be extended based not only on the fragmentation pathway of the parent compound, but also on that of metabolites/TPs detected in samples (Hernández et al., 2009).
A simpler approach is prediction of possible metabolites or TPs with computational (in silico) prediction tools (Reid et al., 2014; Kirchmair et al., 2015). Many different methodologies to predict metabolites or sites of metabolism have been reported recently. The metabolic fate of a molecule depends on its chemical reactivity toward metabolic processes that can occur, as well as on its interactions (affinity and binding orientation) with the biotransformation enzymes involved. The prediction system should, therefore, properly be selected after consideration of the organism or the system where metabolites/TPs are formed. Commercially available and freely accessible programs have been applied in this prediction step. Kirchmair et al. (2015), reviewed 10 different software for predicting metabolites, but only two are without costs and an additional one is solely available for academia (Kirchmair et al., 2015). This free‐availability is probably the main reason that the University of Minnesota Pathway Prediction System (UM‐PPS: (“http://eawag-bbd.ethz.ch/”) is one of the most‐common prediction tools in suspect metabolite/TPs screening (Kern et al., 2009).
Prediction of metabolites/TPs is followed by HRMS analysis; the exact mass for each of the predicted compound is extracted from the chromatogram and checked by a comparison with control samples. The plausibility of the chromatographic tR, isotopic pattern, and ionization efficiency are used as further filters to narrow down the number of candidate peaks. The structures of suspected compounds are tentatively identified based on the observed fragmentation pattern.
A suitable strategy to investigate metabolites and TPs makes use of “in vitro” or “in vivo” metabolism experiments, and of laboratory or field‐degradation experiments under controlled conditions, which can identify known and unknown metabolites or TPs of selected drugs, respectively. In the recent literature, several examples can be found that deal with the investigation of drug metabolites with in vitro or in vivo experiments (Pozo et al., 2014; Takayama et al., 2014; Holm et al., 2015; Meyer et al., 2015; Ibáñez et al., 2016). These experiments are very useful and allow the discovery of metabolites, which might be expected and detected in wastewater, and are, therefore, potential target IDBs in future WBE‐based studies. The discovery of metabolites is of particular relevance for new drugs; that is, NPS whose metabolism is not well known. Other papers study the degradation of illicit drugs, after spiked samples have been subjected to processes, such as hydrolysis, photodegradation, chlorination, biodegradation, or any other process of interest (Postigo et al., 2011; Bijlsma et al., 2013a; Boix et al., 2013, 2014). In all cases, HRMS plays a key role in the tentative identification of the compounds formed. Furthermore, the use of specialized software is critical for a rapid and efficient comparison between the full scan data set from untreated and treated samples, because manual inspection of TIC chromatogram to look for visible peaks can easily fail in complex matrices with low analyte concentrations.
VI. CHIRAL ANALYSIS
The determination of specific metabolic excretion products of illicit drugs in wastewater is not always possible, and makes difficult the differentiation between consumption of drugs and direct disposal of unused drugs. In such situations, chiral analysis can be applied because most illicit drugs are chiral, and are subject to stereoselective human metabolism (Emke et al., 2014; Evans & Kasprzyk‐Hordern, 2014).
MDMA (3,4‐methylenedioxymethamphetamine) and MDA (3,4‐methylenedioxyamphetamine) profiling in wastewater is an excellent example of the importance of chiral enantioselective analysis to distinguish between consumption and direct disposal of unused MDMA and MDA. Both drugs have one asymmetric carbon center and therefore they can exist in the form of two enantiomeric pairs, which differ both quantitatively and qualitatively in pharmacological activity: S(+)‐enantiomers are more amphetamine‐like stimulants and R(−)‐enantiomers are more hallucinogenic (Kasprzyk‐Hordern & Baker, 2012a). Both MDMA and MDA have no medical usage and are synthesized and abused in racemic forms. Their human metabolism is stereoselective and leads to the enrichment of excreted drugs with their R(−)‐enantiomers. However, if the presence of MDA in urine is due to MDMA abuse and not direct MDA use, an enrichment of MDA with S(+)‐enantiomer takes place (Moore et al., 1996). It is, therefore, expected that after consumption, both drugs will be present in urine and wastewater enriched with R(−)‐enantiomers. Indeed, Kasprzyk‐Hordern and Baker (2012a) reported, in a first study of this kind, that MDMA was enriched with the R(−)‐enantiomer due to preferential metabolism of S(+)‐MDMA in humans. Furthermore, the identified MDA was enriched with S(+)‐enantiomer, to suggest that its presence might be associated with MDMA consumption and its subsequent metabolism into S(+)‐MDA and not intentional MDA use (if the latter were true, MDA in wastewater would be enriched with R(−)‐enantiomer).
Surprisingly, in several instances, during Europe‐wide monitoring undertaken by the SCORE group in 2011–13 (Thomas et al., 2012; Emke et al., 2014; Ort et al., 2014c), unexpectedly high loads of MDMA were observed. For example, in 2011, aberrantly high mass loads of MDMA were observed in the wastewater of Utrecht in the Netherlands. These loads highly deviated from the results observed in the previous monitoring campaign in 2010 (Bijlsma et al., 2012). Enantiomeric profiling as shown in Figure 6 revealed that MDMA was racemic (enantiomeric fraction [EF] = 0.54), which indicated its direct disposal in the sewage system and further explains high loads of MDMA quantified in Utrecht wastewater during the sampling week in 2011 (average load was 20‐times higher than in 2010). In contrast, the samples from 2010 (green line in Fig. 6) showed an average EF of 0.65 that corresponded to excretion profiles in urine after consumption of MDMA (Emke et al., 2014). This direct disposal could be the result of a police raid into an illegal production facility that took place 2 days before the monitoring started (Emke et al., 2014). The police estimated that 30 kg of raw MDMA or tablets had been disposed under the pressure of the police raid.
Figure 6.
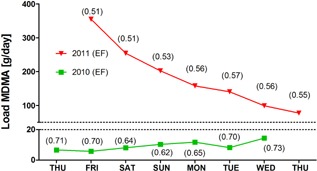
MDMA loads during two separate weeks sampled in 2010 and 2011 in the sewage treatment plant of Utrecht, the Netherlands, and their corresponding enantiomeric fractions (EF) (Emke et al., 2014).
Enantiomeric profiling of wastewater represents a powerful tool that allow to determine if mass loads of studied drugs actually originated from consumption, disposal of unused drugs, or production waste.
Cellobiohydrolase (CBH) is the most widely used stationary phase for enantiomeric profiling of amphetamines with chiral liquid chromatography coupled with tandem mass spectrometry. This cellulase enzyme is immobilized onto 5 μm silica beads with an isoelectric point (pI) of 3.9. It contains multiple chiral centres and mechanisms for ionic, hydrophobic, and hydrogen bonding. It has been successfully applied with an isocratic mobile phase of H2O with 10% 2‐propanol and 1 mM NH4OH, to amphetamine‐like compounds (Figs. 6 and 7) (Bagnall et al., 2012; Kasprzyk‐Hordern & Baker, 2012a,2012b; Emke et al., 2014). There are several factors that have to be taken into account to achieve satisfactory chiral recognition on CBH. These are primarily temperature, pH and mobile‐phase composition. Mobile‐phase pH plays a key role in chiral recognition because it influences ionization of both analytes and CBH. Due to the isoelectric point of 3.9, CBH is negatively charged at pH>pI, so increasing the mobile phase pH will facilitate ionic interactions with positively charged analyte (e.g., amphetamine or MDMA). This interaction will facilitate longer retention times and higher enantioselectivity. Hydrophobic interactions and hydrogen bonding can be influenced with mobile phases that contain different nature and percentage of organic modifiers and ionic strength. Less than 20% of organic modifier is allowed to avoid denaturation of this enzymatic chiral selector. Lower percentages of organic modifiers lead to higher retention in the case of amphetamine‐like compounds. Isopropanol, methanol, or acetonitrile are usually used as organic modifiers. They are characterized by different elution strengths; for example, methanol has a lower elution strength than isopropranol. Other factors, which should be considered are (Camacho‐Muñoz et al., 2016): (i) correct sampling and sample‐preparation protocols that do not introduce stereoselectivity (e.g., microbial metabolic degradation of analytes might lead to incorrect estimation of enantiomeric fractions, or use of charged eluting agents during SPE (e.g., methanol modified with ammonium hydroxide) might lead to loss of chiral recognition in the CBH column), and (ii) elimination of matrix effects (via robust sample preparation approaches and use of deuterated or 13C‐labeled internal standards).
Figure 7.
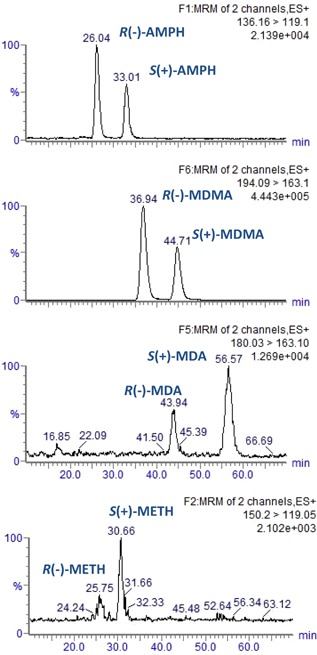
Mass chromatograms show chiral drugs: amphetamine (AMPH), MDMA, MDA and methamphetamine (METH) in wastewater obtained with CBH column and HPLC‐QqQ MS (modified from Kasprzyk‐Hordern & Baker (2012b)).
Enantiomeric profiling has been used in WBE as a complementary tool alongside non‐chiral multi‐residue methods that use reversed phase (C18) stationary materials. This additional analysis required an ad hoc sample preparation, which meant a higher quantity of sample, and a more time‐consuming and less‐cost effective analysis. A recently developed multi‐residue method combined chiral recognition capability of the CBH‐based stationary materials with multi‐residue separation potential of the C18‐based materials. The methodology enabled detection and quantification of all targeted (chiral and non‐chiral) human biomarkers in wastewater along with satisfactory enantiomeric separations of 18 analytes and a unique single sample‐preparation step (Castrignanò, Lubben, & Kasprzyk‐Hordern, 2016).
VII. RELEVANT ANALYTICAL PARAMETERS AND QUALITY CONTROL
The use of advanced analytical techniques and the expertise of the analyst are essential to obtain accurate quantitative data for IDBs in wastewater samples. In addition, appropriate measures for quality control are required to obtain reliable data.
Obviously, prior to its application, the analytical methodology needs to be fully validated for all analytes in terms of linearity, trueness/accuracy (evaluated by means of recovery experiments) and precision (as repeatability RSD), selectivity/specificity, and limits of detection (LOD) and quantification (LOQ). One of the main drawbacks in this field is the lack of guidelines specifically directed toward analysis of IDBs in wastewater. In absence of such guidelines, recommendations in other fields, such as pesticide residue analysis (SANCO, 2013; SANTE/11945, 2015), residues in products of animal origin (European Commission, 2002) bioanalytical methods (EMA, 2012), or clear water act programs (EPA, 2007) can be used as guidelines.
Commonly, a minimum of 5 replicates are required to check the accuracy and precision at the targeted LOQ, and at least one other higher level, for example, 10 times the targeted LOQ. A quantitative analytical method should be demonstrated at the initial validation, but also later with sample batches to perform quality controls (QCs) that provide acceptable recovery at each spiking level and for each analyte. Acceptable mean recoveries for IDBs in wastewater are typically in the range 70–120%, with an associated repeatability RSD ≤ 20%, (e.g., as established for pesticides in SANCO/12571/2013 (SANCO, 2013)).
As previously described in this review, LC‐MS systems are most widely applied in WBE studies. Yet, matrix effects are one of the main problems associated with the correct determination of IDBs with these techniques. Matrix effects result from the competition between matrix co‐eluting components and analytes in the ionization process (Trufelli et al., 2011). This competition affects quantification of the analytes and must, therefore, be removed, minimized, or corrected for. Quantification with matrix‐matched calibration is suitable and is frequently used in some research fields, where a representative blank matrix can be easily obtained. However, the variability of the chemical composition of wastewater and the common presence of some compounds in the samples used as “blank” poses difficulties to use this approach in WBE. Instead, the standard additions method might be used, but it would imply many more injections in the LC‐MS system, apart from the need to adjust the additions accordingly to the analyte concentration in the sample for appropriate application of this methodology. Furthermore, the high level of some compounds makes it problematic to maintain linearity in the calibration curve. Hence, the use of isotope‐labeled internal standards (ILIS) is the most common way to correct for matrix effects, but also for potential errors associated with sample manipulation and storage if the ILIS is added to the sample just after sample collection (i.e., as surrogate). When available, the use of a labeled analyte is recommended to ensure a satisfactory correction of matrix effects. However, it should be noted that, during method validation, there is an absolute need to thoroughly evaluate if the labeled IS accurately corrects for matrix effects.
The estimation of limits of detection (LODs) and limits of quantification (LOQs) is usually performed based on a signal‐to‐noise (s/n) ratio of 1:3 and 1:10, respectively. More‐realistic LOQs can be estimated at s/n 10 for the quantitation transition, but also at s/n 3 for the qualification transition to ensure not only the quantification of the compound but also its reliable identification (Bijlsma et al., 2014a). In Orbitrap data processing, noise is filtered out. Therefore, the common s/n approach to derive LOQ and LOD does not apply. Instead, LOQs in Orbitrap MS are determined based on the lowest concentration of an ILIS in pure water that produces an appreciable signal that meets the identification criteria (de Voogt et al., 2011; Bijlsma et al., 2013b). The matrix effect is calculated from the ratio of the responses of the ILIS in the matrix and in pure water and used to calculate the actual LOQ in that matrix. Some guidelines, like SANCO, defined the method LOQ as the lowest spiking level of the validation that meets the method performance acceptability—that definition is a stricter criterion that can also be applied in WBE. Anyway, an estimation of LODs and LOQs in wastewater is complicated, because notable variations in chemical composition between samples occur. Representative matrix‐matched standards cannot be easily prepared, due to the presence of analytes in most influent samples, and because of the variations in sample composition from one sample to the other. This fact makes problematic a rational comparison of these parameters among different published methodologies. An estimation of LODs/LOQs from standards in solvent at least could make a fair comparison between instrumental performances, although not analytical methods. An efficient and realistic approach to estimate these parameters in a homogeneous way is from inter‐laboratory exercises where all participants analyze the same samples with their own analytical methodology.
Confirmation of positives is an essential aspect in the analysis. To this aim, guidelines such as that from the European Commission (European Commission, 2002) can be adopted, where the confirmation of positive findings is based on the collection of identification points (IPs). The number of IPs earned depends on the mass analyzer used. Thus, for QqQ low‐resolution instruments used in tandem MS, a minimum of two transitions should be monitored for a safe positive finding, whereas in high‐resolution instruments at least two ions must be monitored. In addition, the accomplishment of the ion ratio between recorded transitions and a retention time within the maximum tolerances allowed are required.
Not only quantification, but also confirmation of the identity could be affected by matrix interferences. As stated before, two transitions (Q, q) are normally acquired in LC‐MS/MS analysis. One of the most‐controversial issues is the accomplishment of the ion ratio between these transitions (q/Q ratio) required for confirmation. Ion ratios in samples might be affected by matrix components; for example, when at least one of the two selected SRM transitions is shared with the matrix, which might lead to unexpected ion ratios with deviations that exceed the maximum allowed. This situation is more prone to happen when the specificity of the neutral losses involved in the selected transitions is not evaluated and common losses, such as the loss of H2O or CO2, are implicated. Non‐specific transitions are a weakness not yet tackled with the current guidelines and should be considered in this research field (Delatour, Mottier, & Gremaud, 2007). In this situation, the acquisition of all available transitions is recommended to facilitate confirmation of the positives by testing the additional transitions acquired. In cases of non‐compliance, the sample might be reported as positive, but include a comment on the non‐compliance of the ratio, and report the actual q/Q ratio deviation obtained. Additional work would be necessary to test if any interfering compound actually affected the q/Q ratio.
During a recent inter‐laboratory study undertaken by the SCORE group, one of the questions raised related to the significant variations in q/Q ratios reported by the different participants (Castiglioni et al., 2013), even with the same SRM transitions. As an example, for cocaine, with 304 > 182 (Q) and 304 > 82 (q), ion ratios from different laboratories ranged from 0.12 to 0.38, and seemed to notably depend on the instrument used and on the parameters optimized in each laboratory (e.g., cone and collision voltages). In addition, some variations from batch to batch were also observed. Nevertheless, these variations were not relevant to confirm identity, because it is recommended that q/Q ratios are measured and evaluated for accomplishment by every laboratory within each sample batch with the standards included in the sequence of sample analysis.
Apart from the validation of the method in each laboratory before its routine use, the use of internal quality controls (QCs) is of primary importance to track possible daily method variations. In theory, an internal QC is a final check of the correct execution of all the processes that are included in the analytical protocol. Due to the difficulties to obtain blank wastewater samples, those with the lowest concentrations expected are preferred to prepare spiked samples used as internal QCs. This means that samples from locations with low drug use and/or collected in the mid‐week, where the consumption of illicit drugs is expected to be lower than in the weekends, would fit better to prepare the QCs. Despite this fact that low concentrations of IDBs are expected, samples selected should be analyzed to accurately know the potential analyte concentration in these “blank” samples, in order to be subtracted from the QC prepared. Although there is no guideline in this field, QCs individual recoveries from 60 to 140% might be acceptable in wastewater analysis, as suggested in the SANCO guidelines (SANCO, 2013). Other guidelines recommend an acceptable range of 80–120% at the LOQ level (EMA, 2012). When QC recoveries are out of this interval, the sample sequence of analysis should be repeated. If the problem persists, then appropriate measures should be taken to ensure the accuracy of the method.
The implementation of quality assurance practices to analytical chemistry is recognized as a prerequisite to produce data with known metrological qualities. Regardless of the target analyte or sample type, quality assurance and quality control are the cornerstones to analytical data validation. In addition to daily internal performance verifications via QC materials, a good quality assurance plan should include regular external performance evaluations for an independent assessment of analytical proficiency. The goal of inter‐laboratory studies is to demonstrate that participation in this exercise leads to improved quality of analytical results. The results are of crucial interest for laboratories because these provide clear information of their measurement capabilities. It has to be pointed out that participation is either voluntary or forced by external requirements (e.g., legal, accreditation, control bodies). Inter‐laboratory exercises involve comparison of participants’ results on a set of spiked water samples. Care needs to be taken, to ensure concentrations are equal in all aliquots, especially when particulate matter should also be analyzed. This also holds true when subsampling from composite samples (Capel and Larson, 1996). The separate results are compared with the median of the entire group to determine the accuracy of the method used. By using a set of two samples spiked at different levels with the same compound a so called Youden pair is created. Visualization of these results in a Youden plot show within‐ and between‐laboratory variability. Also individual constant errors become visible, and random errors and systematical errors can be recognized.
Several inter‐laboratory studies have been organized by the SCORE group on selected IDBs regularly monitored in WBE. In the period 2011–2015 five consecutive sets of interlaboratory exercises have been organized, initially with methanolic solutions of analytes that gradually moved to more‐complicated matrices such as extracts and genuine wastewater samples. These were the first inter‐laboratory exercises organized to determine IDBs in wastewater. The results of this stepwise inter‐laboratory testing and the improvements made during this 5‐year period can help to optimize the analytical procedures of participating laboratories and aid in a reliable interpretation of WBE data. To this end, concentrations of IDBs in wastewater provided by laboratories that did not perform well in the interlaboratory exercises were not taken into account; however, together with experts in the field, solutions for the analytical deviations are sought.
VIII. GENERAL SUMMARY AND PERSPECTIVES
The investigation of IDBs in wastewater is a subject of current interest in analytical chemistry. The complexity of the matrix under investigation, the low analyte concentrations commonly found, and the need to detect and quantify not only the parent compounds but also the main metabolites of drugs, make this task an analytical challenge. In addition, human metabolism is not always well known, and the target biomarkers might be unknown. In this field, liquid chromatography combined with low‐ and high‐resolution MS is an indispensable tool. Different strategies can be applied to investigate IDBs in wastewater (discussed in this review) from quantification of target compounds to the detection and (tentative) identification of unknown metabolites and TPs.
Quantification of IDBs in wastewater is a requisite in WBE in order to estimate drug use in populations. However, the presence of drug residues in wastewater also has other implications. For example, there is a possible environmental impact when compounds are not completely removed by WWTPs. Indeed, low removal rates for certain illicit drugs, such as MDMA, ketamine, and methadone, have been observed (Bijlsma et al., 2012). Thus low removal rates implies that drug residues can be present in effluent wastewater, and finally reach the aquatic environment. Therefore, the determination of illicit drugs and metabolites in effluent wastewater and surface water is also of interest, as well as the investigation of potential TPs that can be formed in the environment. Here, concentration levels are much lower than in influent wastewater and, therefore, excellent sensitivity is required for the analytical methods.
In this field, under continuous development, several trends and needs can be highlighted:
There is a need to increase the knowledge of metabolites that might be present in the aquatic environment. Especially for NPS, there is a lack of metabolism studies, and target compounds are still not fully identified. Here, the use of HRMS is highly promising due to its potential to detect and identify drug‐related compounds to make use of different approaches, such as the “common fragmentation pathway,” which is very useful to find parent‐chemically related compounds. Advantageously, a retrospective analysis can be made at any time from HRMS data to facilitate the search of compounds of interest (e.g., new discovered metabolites) in samples previously analyzed. Furthermore, from the perspective of environmental chemistry, it is of interest to extend this research to TPs, which in many occasions are still unknown.
The development of wide‐scope screening will surely be one of the key features of HRMS in the next few years. The use of long lists of selected compounds is essential to facilitate target screening with or without (suspect screening) reference standards. To maintain the “universality” of this approach, generic sample treatments would be required in order to avoid potential losses of analytes during sample handling. Some problematic compounds, mainly those present in ionic form, will always require specific/individual methods, because they cannot be included in multi‐residue/multi‐class methodologies, due to their special physico‐chemical characteristics.
In line with the previous point, it is expected to widen future screening to include NPS. However, it will not be easy to obtain satisfactory results in this case because of two main limitations: (i) very low concentrations expected in the samples as a consequence of the limited use of these compounds in comparison with “conventional” drugs, such as cocaine, cannabis, or amphetamines; (ii) continuous appearance of new NPS with changes in the chemical structure that complicate their detection and identification. Related to the last point, HRMS will be a powerful tool to identify NPS and unknown metabolites.
The combination of non‐biased and biased non‐target analysis with HRMS will be one of the key research fields in the near future. Hopefully, this combined approach will allow one to discover new compounds of interest, such as unknown or non‐searched metabolites, TPs and/or new drugs. Their low concentrations in most cases will be an extra difficulty to obtain satisfactory results.
LC‐MS/MS with triple quadrupole mass analyzers will still be the reference technique to quantify selected IDBs in target methodologies, due to its excellent sensitivity and selectivity. However, new instruments with improved performances will widen the number of compounds included in the method up to several hundreds. The incomparable sensitivity reached with this instrumentation will obtain data on the presence of NPS in wastewaters, with expected concentrations at the pg/L level.
A common protocol of action should be used in WBE studies to produce homogeneous and comparable data at different sites and provide the most reliable estimates of drug use. Uncertainty factors associated with the different steps involved in WBE (sampling, chemical analysis, stability of drug biomarkers in sewage, back‐calculation of drug use, estimation of population size) have to be considered in order to minimize the uncertainty of the entire procedure (Castiglioni et al., 2013). A best practice protocol has been recently proposed and adopted by Europe‐wide studies. Three phases of the approach are included: sampling and sample handling, storage treatment during sampling, chemical analysis‐quality control (EMCDDA, 2016). Currently, the most‐urgent needs for future research are: (i) improve the quality of chemical analyses by following specific quality requirements; (ii) improve the knowledge on stability of IDBs in‐sewer; (iii) plan additional pharmacokinetic studies to produce reliable human excretion profiles for IDBs; (iv) explore novel possibilities to estimate the population size in a catchment.
The improved characteristics of HRMS instruments, mainly sensitivity and linear dynamic range, will make this technique attractive for quantitative analysis as well. The search for the desired “All‐in‐One” method and instrument will still continue in the coming years, because combination of all desired features in just one method/instrument is an exciting issue: qualitative and quantitative analysis, with possibilities for structural elucidation of unknowns. Future developments of HRMS will surely be related to scan‐speed improvements for Orbitrap, which for example will allow more efficient combination to UHPLC, or GC, whereas improvements in TOF MS analyzers will increase the mass‐resolving power. Advances will include development of tribrid analyzers that incorporate new possibilities such as Ion Mobility Spectrometry. Improvements in accurate‐mass full‐acquisition data processing with more‐powerful and user‐friendly software are also expected. In the near future, we will see a growth of HRMS applications, not only in WBE, but also in other fields, such as environmental research, food‐safety, toxicology and doping control analysis.
It will be necessary to harmonize the criteria for reliable identification/confirmation of compounds detected in samples. Relevant parameters, as the number and specificity of ions required, maximum ion‐ratio deviations allowed, mass errors accepted are needed to facilitate comparison of data and to avoid false positives or negatives due to the use of dissimilar criteria among different authors.
The limits of detection and/or quantification of the methods is a requisite to perform realistic comparison of data in WBE studies. The non‐detection might be translated to figures that depend on the LOD; that is, the method sensitivity. The comparison of drug consumption among populations makes use in some occasions of very low concentration data for less‐consumed drugs; this comparison might be distorted with unrealistic LOD/LOQ data. There is a need to harmonize criteria for their estimation in WBE.
Further development of chiral analytical methods and wider application of enantiomeric profiling of wastewater are expected in the coming years. This expectation is because enantiomeric profiling of illicit/abused drugs in wastewater represents a powerful tool to allow one to differentiate between consumption and disposal of unused drugs or production waste in WBE. Advances are needed in the design of new, more‐robust stationary materials to separate multi‐residue mixtures of chiral drugs within a shorter method time.
A wastewater validation guideline, based on the existing guidelines, is desired including key aspects related to quality control that must be carefully considered in WBE. Internal and external quality controls are both needed for a reliable analytical methodology. The organization of interlaboratory exercises are imperative from two perspectives: (i) to produce reliable WBE data, and (ii) to improve analytical performance of participating laboratories.
The ultimate goal of WBE is to provide results in real‐time. This data can be achieved with biosensors. Biosensors have already been applied in WBE to screen PSA, prostate cancer biomarker (Yang et al., 2015c), DNA (Yang et al., 2015a), and cocaine (Yang et al., 2016). There are, however, several issues that need to be addressed; mainly the relatively low sensitivity of biosensors and most probably low selectivity. Due to fast advancements in the field, wide application of biosensors in WBE is envisaged (Yang et al., 2015b).
The potential of WBE will be expanded to other aspects of public health. Sewage contains a hidden wealth of highly complex chemical information about biological processes that occur in the human body (Daughton, 2011). Specific biomarkers could serve as indicators of exposure, stress, vulnerability to disease, acquired disease, or health. Biomarkers include endogenous compounds produced in response to stress or indicative of health, adducts of endogenous chemicals and xenobiotics, or metabolites of detoxification or intoxication processes from xenobiotic exposure (Daughton, 2012). Theoretically, such biomarkers could serve as collective measures of community‐wide health or disease, and could provide a means to conduct epidemiology in near‐real time. It has, therefore, the capability to serve as an early‐warning system in pandemics (Ort et al., 2014a). Quantitative analysis of “health biomarkers” in sewage might allow one to monitor changes over time; for example, in response to specific campaigns, identification of trends and, inter‐community comparisons in relation to their health, diet, lifestyle, and environment (Thomas & Reid, 2011).
It is essential to widen the knowledge on selection of the most‐appropriate IDB in WBE and to take into account specific requirements that a proper biomarker should fulfil in order to ensure the reliability of the back‐calculated estimates. The main requirements for an IDB are: (i) excretion in consistent amounts in urine; (ii) detectable in urban wastewater; (iii) stable in wastewater; (iv) human excretion as unique source. These characteristics are essential to perform a reliable quantitative analysis of the substance under investigation and should be, therefore, carefully considered in future studies to select new IDBs.
IX. ABBREVIATIONS
- CBH
cellobiohydrolase
- EDDP
2‐ethylidene‐1,5‐dimethyl‐3,3‐diphenylpyrrolidine
- EF
enantiomeric fraction
- EI
electron ionization
- EMCDDA
European Monitoring Centre for Drugs and Drug Addiction
- ESI
electrospray ionization
- GC‐MS
gas chromatography coupled to mass spectrometry
- HE
high collision energy
- HRMS
high‐resolution mass spectrometry
- IDB
illicit drug biomarker
- ILIS
isotope‐labeled internal standards
- IP
identification point
- LC‐MS
liquid chromatography coupled to mass spectrometry
- LC‐MS/MS
liquid chromatography coupled to tandem mass spectrometry
- LE
low collision energy
- LOD
limits of detection
- LOQ
limits of quantification
- LRMS
low‐resolution mass spectrometry
- MDA
3,4‐methylenedioxyamphetamine
- MDMA
3,4‐methylenedioxymethamphetamine
- NPS
new psychoactive substances
- QC
quality control
- QqQ
triple quadrupole analyzer
- QSRRs
quantitative structure‐retention relationships
- SCORE
sewage analysis CORe group Europe
- SDL
screening detection limit
- SIM
selected ion monitoring
- s/n
signal‐to‐noise
- SRM
selected reaction monitoring
- TIC
total ion chromatogram
- TPs
transformation products
- tR
retention time
- UHPLC
ultra‐high performance liquid chromatography
- WBE
wastewater‐based epidemiology
- WWTP
wastewater treatment plant
ACKNOWLEDGMENTS
Financial support by the European Union's Program for research, technological development, and demonstration SEWPROF (project no. 317205) and the COST‐action SCORE (Action no. ES 1307) are gratefully acknowledged. Dr. Félix Hernández acknowledges the Spanish Ministry of Economy and Competitiveness for financial support in the field of illicit drug research (Ref CTQ2015‐65603‐P). Dr. Alexander van Nuijs acknowledges the Research Foundation Flanders (FWO) for his post‐doctoral fellowship. Dr. Lubertus Bijlsma acknowledges NPS‐Euronet (HOME/2014/JDRUG/AG/DRUG/7086), co‐funded by the European Union, for his post‐doctoral fellowship. This publication reflects the views only of the authors, and the European Commission cannot be held responsible for any use that might be made of the information contained therein.
REFERENCES
- Alechaga É, Moyano E, Galceran MT. 2015. Wide‐range screening of psychoactive substances by FIA‐HRMS: Identification strategies. Anal Bioanal Chem 407:4567–4580. [DOI] [PubMed] [Google Scholar]
- Andrés‐Costa MJ, Rubio‐López N, Morales Suárez‐Varela M, Pico Y. 2014. Occurrence and removal of drugs of abuse in Wastewater Treatment Plants of Valencia (Spain). Environ Pollut 194:152–162. [DOI] [PubMed] [Google Scholar]
- Asimakopoulos A, Kannan K. 2016. Neuropsychiatric pharmaceuticals and illicit drugs in wastewater treatment plants: A review. Environ Chem 13:541–576. [Google Scholar]
- Bade R, Bijlsma L, Miller TH, Barron LP, Sancho JV, Hernández F. 2015a. Suspect screening of large numbers of emerging contaminants in environmental waters using artificial neural networks for chromatographic retention time prediction and high resolution mass spectrometry data analysis. Sci Total Environ 538:934–941. [DOI] [PubMed] [Google Scholar]
- Bade R, Bijlsma L, Sancho JV, Hernández F. 2015b. Critical evaluation of a simple retention time predictor based on LogKow as a complementary tool in the identification of emerging contaminants in water. Talanta 139:143–149. [DOI] [PubMed] [Google Scholar]
- Bade R, Rousis NI, Bijlsma L, Gracia‐Lor E, Castiglioni S, Sancho JV, Hernandez F. 2015c. Screening of pharmaceuticals and illicit drugs in wastewater and surface waters of Spain and Italy by high resolution mass spectrometry using UHPLC‐QTOF MS and LC‐LTQ‐Orbitrap MS. Anal Bioanal Chem 407:8979–8988. [DOI] [PubMed] [Google Scholar]
- Bagnall JP, Evans SE, Wort MT, Lubben AT, Kasprzyk‐Hordern B. 2012. Using chiral liquid chromatography quadrupole time‐of‐flight mass spectrometry for the analysis of pharmaceuticals and illicit drugs in surface and wastewater at the enantiomeric level. J Chromatogr A 1249:115–129. [DOI] [PubMed] [Google Scholar]
- Banta‐Green C, Field J. 2011. City‐wide drug testing using municipal wastewater. Significance 8:70–74. [Google Scholar]
- Banta‐Green CJ, Field JA, Chiaia AC, Sudakin DL, Power L, De Montigny L. 2009. The spatial epidemiology of cocaine, methamphetamine and 3,4‐methylenedioxymethamphetamine (MDMA) use: A demonstration using a population measure of community drug load derived from municipal wastewater. Addiction 104:1874–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barron LP, McEneff GL. 2016. Gradient liquid chromatographic retention time prediction for suspect screening applications: A critical assessment of a generalised artificial neural network‐based approach across 10 multi‐residue reversed‐phase analytical methods. Talanta 147:261–270. [DOI] [PubMed] [Google Scholar]
- Bartelt‐Hunt SL, Snow DD, Damon T, Shockley J, Hoagland K. 2009. The occurrence of illicit and therapeutic pharmaceuticals in wastewater effluent and surface waters in Nebraska. Environ Pollut 157:786–791. [DOI] [PubMed] [Google Scholar]
- Baz‐Lomba JA, Reid MJ, Thomas KV. 2016. Target and suspect screening of psychoactive substances in sewage‐based samples by UHPLC‐QTOF. Anal Chim Acta 914:81–90. [DOI] [PubMed] [Google Scholar]
- Berendsen BJA, Wegh RS, Meijer T, Nielen MWF. 2015. The assessment of selectivity in different quadrupole‐Orbitrap mass spectrometry acquisition modes. J Am Soc Mass Spectrom 26:337–346. [DOI] [PubMed] [Google Scholar]
- Berset J‐D, Brenneisen R, Mathieu C. 2010. Analysis of llicit and illicit drugs in waste, surface and lake water samples using large volume direct injection high performance liquid chromatography‐electrospray tandem mass spectrometry (HPLC‐MS/MS). Chemosphere 81:859–866. [DOI] [PubMed] [Google Scholar]
- Bijlsma L, Sancho JV, Pitarch E, Ibáñez M, Hernández F. 2009. Simultaneous ultra‐high‐pressure liquid chromatography‐tandem mass spectrometry determination of amphetamine and amphetamine‐like stimulants, cocaine and its metabolites, and a cannabis metabolite in surface water and urban wastewater. J Chromatogr A 1216:3078–3089. [DOI] [PubMed] [Google Scholar]
- Bijlsma L, Sancho JV, Hernández F, Niessen WMA. 2011. Fragmentation pathways of drugs of abuse and their metabolites based on QTOF MS/MS and MS E accurate‐mass spectra. J Mass Spectrom 46:865–875. [DOI] [PubMed] [Google Scholar]
- Bijlsma L, Emke E, Hernández F, De Voogt P. 2012. Investigation of drugs of abuse and relevant metabolites in Dutch sewage water by liquid chromatography coupled to high resolution mass spectrometry. Chemosphere 89:1399–1406. [DOI] [PubMed] [Google Scholar]
- Bijlsma L, Boix C, Niessen WMA, Ibáñez M, Sancho JV, Hernández F. 2013a. Investigation of degradation products of cocaine and benzoylecgonine in the aquatic environment. Sci Total Environ 443:200–208. [DOI] [PubMed] [Google Scholar]
- Bijlsma L, Emke E, Hernández F, De Voogt P. 2013b. Performance of the linear ion trap Orbitrap mass analyzer for qualitative and quantitative analysis of drugs of abuse and relevant metabolites in sewage water. Anal Chim Acta 768:102–110. [DOI] [PubMed] [Google Scholar]
- Bijlsma L, Beltrán E, Boix C, Sancho JV, Hernández F. 2014a. Improvements in analytical methodology for the determination of frequently consumed illicit drugs in urban wastewater. Anal Bioanal Chem 406:4261–4272. [DOI] [PubMed] [Google Scholar]
- Bijlsma L, Serrano R, Ferrer C, Tormos I, Hernández F. 2014b. Occurrence and behavior of illicit drugs and metabolites in sewage water from the Spanish Mediterranean coast (Valencia region). Sci Total Environ 487:703–709. [DOI] [PubMed] [Google Scholar]
- Bijlsma L, Botero‐Coy AM, Rincón RJ, Peñuela GA, Hernández F. 2016. Estimation of illicit drug use in the main cities of Colombia by means of urban wastewater analysis. Sci Total Environ 565:984–993. [DOI] [PubMed] [Google Scholar]
- Bisceglia KJ, Roberts AL, Schantz MM, Lippa KA. 2010. Quantification of drugs of abuse in municipal wastewater via SPE and direct injection liquid chromatography mass spectrometry. Anal Bioanal Chem 398:2701–2712. [DOI] [PubMed] [Google Scholar]
- Bletsou AA, Jeon J, Hollender J, Archontaki E, Thomaidis NS. 2015. Targeted and non‐targeted liquid chromatography‐mass spectrometric workflows for identification of transformation products of emerging pollutants in the aquatic environment. TrAC Trends Anal Chem 66:32–44. [Google Scholar]
- Boix C, Ibáñez M, Sancho JV, Niessen WMA, Hernández F. 2013. Investigating the presence of omeprazole in waters by liquid chromatography coupled to low and high resolution mass spectrometry: Degradation experiments. J Mass Spectrom 48:1091–1100. [DOI] [PubMed] [Google Scholar]
- Boix C, Ibáñez M, Bijlsma L, Sancho JV, Hernández F. 2014. Investigation of cannabis biomarkers and transformation products in waters by liquid chromatography coupled to time of flight and triple quadrupole mass spectrometry. Chemosphere 99:64–71. [DOI] [PubMed] [Google Scholar]
- Boix C, Ibáñez M, Sancho JV, Rambla J, Aranda JL, Ballester S, Hernández F. 2015. Fast determination of 40 drugs in water using large volume direct injection liquid chromatography‐tandem mass spectrometry. Talanta 131:719–727. [DOI] [PubMed] [Google Scholar]
- Boleda MR, Galceran MT, Ventura F. 2007. Trace determination of cannabinoids and opiates in wastewater and surface waters by ultra‐performance liquid chromatography‐tandem mass spectrometry. J Chromatogr A 1175:38–48. [DOI] [PubMed] [Google Scholar]
- Bones J, Thomas KV, Paull B. 2007. Using environmental analytical data to estimate levels of community consumption of illicit drugs and abused pharmaceuticals. J Environ Monit 9:701–707. [DOI] [PubMed] [Google Scholar]
- Boogaerts T, Covaci A, Kinyua J, Neels H, van Nuijs ALN. 2016. Spatial and temporal trends in alcohol consumption in Belgian cities: A wastewater‐based approach. Drug Alcohol Depend 160:170–176. [DOI] [PubMed] [Google Scholar]
- Borova VL, Maragou NC, Gago‐Ferrero P, Pistos C, Thomaidis NS. 2014. Highly sensitive determination of 68 psychoactive pharmaceuticals, illicit drugs, and related human metabolites in wastewater by liquid chromatography‐tandem mass spectrometry. Anal Bioanal Chem 406:4273–4285. [DOI] [PubMed] [Google Scholar]
- Burgard DA, Fuller R, Becker B, Ferrell R, Dinglasan‐Panlilio M. 2013. Potential trends in Attention Deficit Hyperactivity Disorder (ADHD) drug use on a college campus: Wastewater analysis of amphetamine and ritalinic acid. Sci Total Environ 450:242–249. [DOI] [PubMed] [Google Scholar]
- Camacho‐Muñoz D, Petrie B, Castrignanò E, Kasprzyk‐Hordern B. 2016. Enantiomeric profiling of chiral pharmacologically active compounds in the environment with the usage of chiral liquid chromatography coupled with tandem mass spectrometry. Curr Anal Chem 12:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capel PD, Larson SJ. 1996. Evaluation of selected information on splitting devices for water samples. US Geol Surv Water‐Resources Investig Rep 95‐4141:1–103. [Google Scholar]
- Castiglioni S, Zuccato E, Crisci E, Chiabrando C, Fanelli R, Bagnati R. 2006. Identification and measurement of illicit drugs and their metabolites in urban wastewater by liquid chromatography‐tandem mass spectrometry. Anal Chem 78:8421–8429. [DOI] [PubMed] [Google Scholar]
- Castiglioni S, Zuccato E, Chiabrando C, Fanelli R, Bagnati R. 2008. Mass spectrometric analysis of illicit drugs in wastewater and surface water. Mass Spectrom Rev 27:378–394. [DOI] [PubMed] [Google Scholar]
- Castiglioni S, Bijlsma L, Covaci A, Emke E, Hernández F, Reid M, Ort C, Thomas KV, Van Nuijs ALN, De Voogt P, Zuccato E. 2013. Evaluation of uncertainties associated with the determination of community drug use through the measurement of sewage drug biomarkers. Environ Sci Technol 47:1452–1460. [DOI] [PubMed] [Google Scholar]
- Castiglioni S, Thomas KV, Kasprzyk‐Hordern B, Vandam L, Griffiths P. 2014. Testing wastewater to detect illicit drugs: State of the art, potential and research needs. Sci Total Environ 487:613–620. [DOI] [PubMed] [Google Scholar]
- Castiglioni S, Senta I, Borsotti A, Davoli E, Zuccato E. 2015. A novel approach for monitoring tobacco use in local communities by wastewater analysis. Tob Control 24:38–42. [DOI] [PubMed] [Google Scholar]
- Castrignanò E, Lubben A, Kasprzyk‐Hordern B. 2016. Enantiomeric profiling of chiral drug biomarkers in wastewater with the usage of chiral liquid chromatography coupled with tandem mass spectrometry. J Chromatogr A 1438:84–99. [DOI] [PubMed] [Google Scholar]
- Castro‐Perez J, Hoyes J, Major H, Preece S. 2002. Advances in MS‐based approaches for drug and metabolism studies. Chromatographia 55:S59. [Google Scholar]
- Chiaia AC, Banta‐green C, Field J. 2008. Eliminating solid phase extraction with large‐Volume injection LC/MS/MS: Analysis of illicit and legal drugs and human urine indicators in US wastewaters. Environ Sci Technol I:8841–8848. [DOI] [PubMed] [Google Scholar]
- Chiaia‐Hernandez AC, Schymanski EL, Kumar P, Singer HP, Hollender J. 2014. Suspect and nontarget screening approaches to identify organic contaminant records in lake sediments. Anal Bioanal Chem 406:7323–7335. [DOI] [PubMed] [Google Scholar]
- Coscollà C, León N, Pastor A, Yusà V. 2014. Combined target and post‐run target strategy for a comprehensive analysis of pesticides in ambient air using liquid chromatography‐Orbitrap high resolution mass spectrometry. J Chromatogr A 1368:132–142. [DOI] [PubMed] [Google Scholar]
- Coutu S, Pouchon T, Queloz P, Vernaz N. 2016. Integrated stochastic modeling of pharmaceuticals in sewage networks. Stoch Environ Res Risk Assess 30:1087–1097. [Google Scholar]
- de Voogt P, Emke E, Helmus R, Panteliadis P, van Leerdam JA. 2011. Determination of illicit drugs in the water cycle by LC‐Orbitrap MS In: Castiglioni S, Zuccato E, Fanelli R, editors. Illicit drugs in the environment: Occurrence, analysis, and fate using mass spectrometry. New York, NY: Wiley; pp. 85–114. [Google Scholar]
- Díaz R, Ibáñez M, Sancho JV, Hernández F. 2011. Building an empirical mass spectra library for screening of organic pollutants by ultra‐high‐pressure liquid chromatography/hybrid quadrupole time‐of‐flight mass spectrometry. Rapid Commun Mass Spectrom 25:355–369. [DOI] [PubMed] [Google Scholar]
- Díaz R, Ibáñez M, Sancho JV, Hernández F. 2012. Target and non‐target screening strategies for organic contaminants, residues and illicit substances in food, environmental and human biological samples by UHPLC‐QTOF‐MS. Anal Methods 4:196–209. [Google Scholar]
- Dasenaki ME, Bletsou AA, Koulis GA, Thomaidis NS. 2015. Qualitative multiresidue screening method for 143 veterinary drugs and pharmaceuticals in milk and fish tissue using liquid chromatography quadrupole‐time‐of‐flight mass spectrometry. J Agric Food Chem 63:4493–4508. [DOI] [PubMed] [Google Scholar]
- Daughton CG, Daughton CG. 2001. Pharmaceuticals and personal care products in the environment, scientific and regulatory issues. In: Daughton CG, Jones‐Lepp TL, editors. Washington: American Chemical Society; pp. 348–364. [Google Scholar]
- Daughton CG. 2011. Illicit drugs: Contaminants in the environment and utility in forensic epidemiology. Rev Environ Contam Toxicol 210:59–110. [DOI] [PubMed] [Google Scholar]
- Daughton CG. 2012. Using biomarkers in sewage to monitor community‐wide human health: Isoprostanes as conceptual prototype. Sci Total Environ 424:16–38. [DOI] [PubMed] [Google Scholar]
- De Keyser W, Gevaert V, Verdonck F, De Baets B, Benedetti L. 2010. An emission time series generator for pollutant release modelling in urban areas. Environ Model Softw 25:554–561. [Google Scholar]
- Delatour T, Mottier P, Gremaud E. 2007. Limits of suspicion, recognition and confirmation as concepts that account for the confirmation transitions at the detection limit for quantification by liquid chromatography‐tandem mass spectrometry. J Chromatogr A 1169:103–110. [DOI] [PubMed] [Google Scholar]
- Devault DA, Néfau T, Pascaline H, Karolak S, Levi Y. 2014. First evaluation of illicit and licit drug consumption based on wastewater analysis in Fort de France urban area (Martinique, Caribbean), a transit area for drug smuggling. Sci Total Environ 490:970–978. [DOI] [PubMed] [Google Scholar]
- EMA. 2012. Guideline on bioanalytical method validation, European Medicines Agency, Committee for Medicinal Products for Human Use. doi: EMEA/CHMP/EWP/192217/2009
- EMCDDA. 2012. MCDDA publishes 2012 report on the state of the drugs problem in Europe. Annual report. doi: 10.2810/64775 [DOI] [PubMed]
- EMCDDA. 2015a. The EMCDDA's five key epidemiological indicators [WWW Document]. URL http://www.emcdda.europa.eu/themes/key-indicators
- EMCDDA. 2015b. Wastewater analysis and drugs: A European multi‐city study.
- EMCDDA. 2016. Assessing illicit drugs in wastwater In: Castiglioni S, Vandam L, Griffiths P, editors. Assessing illicit drugs in wastewater: Advances in wastewater‐Based drug epidemiology, EMCDDA insights 22. Luxembourg: Publications Office of the European Union; pp. 1–82. [Google Scholar]
- Emke E, Evans S, Kasprzyk‐Hordern B, de Voogt P. 2014. Enantiomer profiling of high loads of amphetamine and MDMA in communal sewage: A Dutch perspective. Sci Total Environ 487:666–672. [DOI] [PubMed] [Google Scholar]
- EPA. 2007. Method 1694: Pharmaceuticals and personal care products in water, soil, sediment, and biosolids by HPLC/MS/MS, Environmental Protection Agency: Method 1694.
- European Commission E. 2002. Decision 2002/657/EC, implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results.
- Evans SE, Kasprzyk‐Hordern B. 2014. Applications of chiral chromatography coupled with mass spectrometry in the analysis of chiral pharmaceuticals in the environment. Trends Environ Anal Chem 1:e34–e51. [Google Scholar]
- Farré M, Kantiani L, Petrovic M, Pérez S, Barceló D. 2012. Achievements and future trends in the analysis of emerging organic contaminants in environmental samples by mass spectrometry and bioanalytical techniques. J Chromatogr A 1259:86–99. [DOI] [PubMed] [Google Scholar]
- Fedorova G, Randak T, Lindberg RH, Grabic R. 2013. Comparison of the quantitative performance of a Q‐Exactive high‐resolution mass spectrometer with that of a triple quadrupole tandem mass spectrometer for the analysis of illicit drugs in wastewater. Rapid Commun Mass Spectrom 27:1751–1762. [DOI] [PubMed] [Google Scholar]
- Gago‐Ferrero P, Schymanski EL, Bletsou AA, Aalizadeh R, Hollender J, Thomaidis NS. 2015. Extended suspect and non‐Target strategies to characterize emerging polar organic contaminants in raw wastewater with LC‐HRMS/MS. Environ Sci Technol 49:12333–12341. [DOI] [PubMed] [Google Scholar]
- García‐Reyes JF, Molina‐Díaz A, Fernández‐Alba AR. 2007. Identification of pesticide transformation products in food by liquid chromatography/time‐of‐flight mass spectrometry via “fragmentation‐degradation” relationships. Anal Chem 79:307–321. [DOI] [PubMed] [Google Scholar]
- Gheorghe A, van Nuijs A, Pecceu B, Bervoets L, Jorens PG, Blust R, Neels H, Covaci A. 2008. Analysis of cocaine and its principal metabolites in waste and surface water using solid‐phase extraction and liquid chromatography‐ion trap tandem mass spectrometry. Anal Bioanal Chem 391:1309–1319. [DOI] [PubMed] [Google Scholar]
- González‐Mariño I, Quintana JB, Rodríguez I, Rodil R, González‐Peñas J, Cela R. 2009. Comparison of molecularly imprinted, mixed‐mode and hydrophilic balance sorbents performance in the solid‐phase extraction of amphetamine drugs from wastewater samples for liquid chromatography‐tandem mass spectrometry determination. J Chromatogr A 1216:8435–8441. [DOI] [PubMed] [Google Scholar]
- González‐Mariño I, Quintana JB, Rodríguez I, Cela R. 2010. Determination of drugs of abuse in water by solid‐phase extraction, derivatisation and gas chromatography‐ion trap‐tandem mass spectrometry. J Chromatogr A 1217:1748–1760. [DOI] [PubMed] [Google Scholar]
- González‐Mariño I, Quintana JB, Rodríguez I, González‐Díez M, Cela R. 2012. Screening and selective quantification of illicit drugs in wastewater by mixed‐mode solid‐phase extraction and quadrupole‐time‐of‐flight liquid chromatography‐mass spectrometry. Anal Chem 84:1708–1717. [DOI] [PubMed] [Google Scholar]
- Hardman M, Makarov A. 2003. Interfacing the orbitrap mass analyzer to an electrospray ion source. Anal. Chem. 75:1699–1705. [DOI] [PubMed] [Google Scholar]
- Harman C, Reid M, Thomas KV. 2011. In situ calibration of a passive sampling device for selected illicit drugs and their metabolites in wastewater, and subsequent year‐long assessment of community drug usage. Environ Sci Technol 45:5676–5682. [DOI] [PubMed] [Google Scholar]
- Hernández F, Pozo ÓJ, Sancho JV, López FJ, Marín JM, Ibáñez M. 2005. Strategies for quantification and confirmation of multi‐class polar pesticides and transformation products in water by LC‐MS2 using triple quadrupole and hybrid quadrupole time‐of‐flight analyzers. TrAC Trends Anal Chem 24:596–612. [Google Scholar]
- Hernández F, Grimalt S, Pozo ÓJ, Sancho JV. 2009. Use of ultra‐high‐pressure liquid chromatography‐quadrupole time‐of‐flight MS to discover the presence of pesticide metabolites in food samples. J Sep Sci 32:2245–2261. [DOI] [PubMed] [Google Scholar]
- Hernández F, Bijlsma L, Sancho JV, Díaz R, Ibáñez M. 2011a. Rapid wide‐scope screening of drugs of abuse, prescription drugs with potential for abuse and their metabolites in influent and effluent urban wastewater by ultrahigh pressure liquid chromatography‐quadrupole‐time‐of‐flight‐mass spectrometry. Anal Chim Acta 684:96–106. [DOI] [PubMed] [Google Scholar]
- Hernández F, Sancho JV, Ibáñez M, Portolés T. 2011b. Time‐of‐Flight and quadrupole time‐of‐flight mass spectrometry for identifying unknown contaminants and degradation products in the environment. Encycl Anal Chem doi: 10.1002/9780470027318.a9233 [DOI] [Google Scholar]
- Hernández F, Sancho JV, Ibáñez M, Abad E, Portolés T, Mattioli L. 2012. Current use of high‐resolution mass spectrometry in the environmental sciences. Anal Bioanal Chem 403:1251–1264. [DOI] [PubMed] [Google Scholar]
- Hernández F, Ibáñez M, Bade R, Bijlsma L, Sancho JV. 2014. Investigation of pharmaceuticals and illicit drugs in waters by liquid chromatography‐high‐resolution mass spectrometry. TrAC Trends Anal Chem 63:140–157. [Google Scholar]
- Hernández F, Ibañez M, Botero‐Coy AM, Bade R, Bustos‐Lopez MC, Rincon J, Moncayo A, Bijlsma L. 2015a. LC‐QTOF MS screening of more than 1,000 licit and illicit drugs and their metabolites in wastewater and surface waters from the area of Bogotá, Colombia. Anal Bioanal Chem 407:6405–6416. [DOI] [PubMed] [Google Scholar]
- Hernández F, Ibáñez M, Portolés T, Cervera MI, Sancho JV, López FJ. 2015b. Advancing towards universal screening for organic pollutants in waters. J Hazard Mater 282:86–95. [DOI] [PubMed] [Google Scholar]
- Heuett NV, Ramirez CE, Fernandez A, Gardinali PR. 2015. Analysis of drugs of abuse by online SPE‐LC high resolution mass spectrometry: Communal assessment of consumption. Sci Total Environ 511:319–330. [DOI] [PubMed] [Google Scholar]
- Hogenboom AC, van Leerdam JA, de Voogt P. 2009. Accurate mass screening and identification of emerging contaminants in environmental samples by liquid chromatography‐hybrid linear ion trap Orbitrap mass spectrometry. J Chromatogr A 1216:510–519. [DOI] [PubMed] [Google Scholar]
- Holm NB, Pedersen AJ, Dalsgaard PW, Linnet K. 2015. Metabolites of 5F‐AKB‐48, a synthetic cannabinoid receptor agonist, identified in human urine and liver microsomal preparations using liquid chromatography high‐resolution mass spectrometry. Drug Test Anal 7:199–206. [DOI] [PubMed] [Google Scholar]
- Horai H, Arita M, Kanaya S, Nihei Y, Ikeda T, Suwa K, Ojima Y, Tanaka K, Tanaka S, Aoshima K, Oda Y, Kakazu Y, Kusano M, Tohge T, Matsuda F, Sawada Y, Hirai MY, Nakanishi H, Ikeda K, Akimoto N, Maoka T, Takahashi H, Ara T, Sakurai N, Suzuki H, Shibata D, Neumann S, Iida T, Tanaka K, Funatsu K, Matsuura F, Soga T, Taguchi R, Saito K, Nishioka T. 2010. MassBank: A public repository for sharing mass spectral data for life sciences. J Mass Spectrom 45:703–714. [DOI] [PubMed] [Google Scholar]
- Hu Q, Noll RJ, Li H, Makarov A, Hardman M, Cooks RG. 2005. The orbitrap: A new mass spectrometer. J Mass Spectrom 40:430–443. [DOI] [PubMed] [Google Scholar]
- Hug C, Ulrich N, Schulze T, Brack W, Krauss M. 2014. Identification of novel micropollutants in wastewater by a combination of suspect and nontarget screening. Environ Pollut 184:25–32. [DOI] [PubMed] [Google Scholar]
- Humphries MA, Bruno R, Lai FY, Thai PK, Holland BR, O'Brien JW, Ort C, Mueller JF. 2016. Evaluation of monitoring schemes for wastewater‐based epidemiology to identify drug use trends using cocaine, methamphetamine, MDMA and methadone. Environ Sci Technol 50:4760–4768. [DOI] [PubMed] [Google Scholar]
- Ibáñez M, Sancho JV, Pozo ÓJ, Niessen WMA, Hernández F. 2005. Use of quadrupole time‐of‐flight mass spectrometry in the elucidation of unknown compounds present in environmental water. Rapid Commun Mass Spectrom 19:169–178. [DOI] [PubMed] [Google Scholar]
- Ibáñez M, Sancho JV, Hernández F, McMillan D, Rao R. 2008. Rapid non‐target screening of organic pollutants in water by ultraperformance liquid chromatography coupled to time‐of‐light mass spectrometry. TrAC Trends Anal Chem 27:481–489. [Google Scholar]
- Ibáñez M, Sancho JV, Bijlsma L, Van Nuijs ALN, Covaci A, Hernández F. 2014. Comprehensive analytical strategies based on high‐resolution time‐of‐flight mass spectrometry to identify new psychoactive substances. TrAC Trends Ana Chem 57:107–117. [Google Scholar]
- Ibáñez M, Pozo AJ, Sancho JV, Orengo T, Haro G, Hernandez F. 2016. Analytical strategy to investigate 3,4‐methylenedioxypyrovalerone (MDPV) metabolites in consumers’ urine by high‐resolution mass spectrometry. Anal Bioanal Chem 408:151–164. [DOI] [PubMed] [Google Scholar]
- Irvine RJ, Kostakis C, Felgate PD, Jaehne EJ, Chen C, White JM. 2011. Population drug use in Australia: A wastewater analysis. Forensic Sci Int 210:69–73. [DOI] [PubMed] [Google Scholar]
- Kankaanpää A, Ariniemi K, Heinonen M, Kuoppasalmi K, Gunnar T. 2014. Use of illicit stimulant drugs in Finland: A wastewater study in ten major cities. Sci Total Environ 487:696–702. [DOI] [PubMed] [Google Scholar]
- Karolak S, Nefau T, Bailly E, Solgadi A, Levi Y. 2010. Estimation of illicit drugs consumption by wastewater analysis in Paris area (France). Forensic Sci Int 200:153–160. [DOI] [PubMed] [Google Scholar]
- Kasprzyk‐Hordern B, Baker DR. 2012a. Estimation of community‐wide drugs use via stereoselective profiling of sewage. Sci. Total Environ. 423:142–150. [DOI] [PubMed] [Google Scholar]
- Kasprzyk‐Hordern B, Baker DR. 2012b. Enantiomeric profiling of chiral drugs in wastewater and receiving waters. Environ Sci Technol 46:1681–1691. [DOI] [PubMed] [Google Scholar]
- Kaufmann A, Butcher P, Maden K, Walker S, Widmer M. 2010. Comprehensive comparison of liquid chromatography selectivity as provided by two types of liquid chromatography detectors (high resolution mass spectrometry and tandem mass spectrometry): “Where is the crossover point?”. Anal Chim Acta 673:60–72. [DOI] [PubMed] [Google Scholar]
- Kaufmann A. 2014. Combining UHPLC and high‐resolution MS: A viable approach for the analysis of complex samples? TrAC Trends Anal Chem 63:113–128. [Google Scholar]
- Kellmann M, Muenster H, Zomer P, Mol H. 2009. Full scan MS in comprehensive qualitative and quantitative residue analysis in food and feed matrices: How much resolving power is required? J Am Soc Mass Spectrom 20:1464–1476. [Google Scholar]
- Kern S, Fenner K, Singer HP, Schwarzenbach RP, Hollender J. 2009. Identification of transformation products of organic contaminants in natural waters by computer‐aided prediction and high‐resolution mass spectrometry. Environ Sci Technol 43:7039–7046. [DOI] [PubMed] [Google Scholar]
- Khan U, van Nuijs ALN, Li J, Maho W, Du P, Li K, Hou L, Zhang J, Meng X, Li X, Covaci A. 2014. Application of a sewage‐based approach to assess the use of ten illicit drugs in four Chinese megacities. Sci Total Environ 487:710–721. [DOI] [PubMed] [Google Scholar]
- Kim KY, Lai FY, Kim H‐Y, Thai PK, Mueller JF, Oh J‐E. 2015. The first application of wastewater‐based drug epidemiology in five South Korean cities. Sci Total Environ 524‐525:440–446. [DOI] [PubMed] [Google Scholar]
- Kinyua J, Covaci A, Maho W, McCall A‐K, Neels H, van Nuijs ALN. 2015. Sewage‐based epidemiology in monitoring the use of new psychoactive substances: Validation and application of an analytical method using LC‐MS/MS. Drug Test Anal 7:812–818. [DOI] [PubMed] [Google Scholar]
- Kirchmair J, Göller AH, Lang D, Kunze J, Testa B, Wilson ID, Glen RC, Schneider G. 2015. Predicting drug metabolism: Experiment and/or computation? Nat Rev Drug Discov 14:387–404. [DOI] [PubMed] [Google Scholar]
- Krauss M, Singer H, Hollender J. 2010. LC‐high resolution MS in environmental analysis: From target screening to the identification of unknowns. Anal Bioanal Chem 397:943–951. [DOI] [PubMed] [Google Scholar]
- Lai FY, Ort C, Gartner C, Carter S, Prichard J, Kirkbride P, Bruno R, Hall W, Eaglesham G, Mueller JF. 2011. Refining the estimation of illicit drug consumptions from wastewater analysis: Co‐analysis of prescription pharmaceuticals and uncertainty assessment. Water Res 45:4437–4448. [DOI] [PubMed] [Google Scholar]
- Lai FY, Bruno R, Hall W, Gartner C, Ort C, Kirkbride P, Prichard J, Thai PK, Carter S, Mueller JF. 2013a. Profiles of illicit drug use during annual key holiday and control periods in Australia: Wastewater analysis in an urban, a semi‐rural and a vacation area. Addiction 108:556–565. [DOI] [PubMed] [Google Scholar]
- Lai FY, Bruno R, Leung HW, Thai PK, Ort C, Carter S, Thompson K, Lam PKS, Mueller JF. 2013b. Estimating daily and diurnal variations of illicit drug use in Hong Kong: A pilot study of using wastewater analysis in an Asian metropolitan city. Forensic Sci Int 233:126–132. [DOI] [PubMed] [Google Scholar]
- Li J, Hou L, Du P, Yang J, Li K, Xu Z, Wang C, Zhang H, Li X. 2014. Estimation of amphetamine and methamphetamine uses in Beijing through sewage‐based analysis. Sci Total Environ 490:724–732. [DOI] [PubMed] [Google Scholar]
- Makarov A, Scigelova M. 2010. Coupling liquid chromatography to Orbitrap mass spectrometry. J Chromatogr A 1217:3938–3945. [DOI] [PubMed] [Google Scholar]
- Makarov A, Denisov E, Kholomeev A, Balschun W, Lange O, Strupat K, Horning S. 2006. Performance evaluation of a hybrid linear ion trap/orbitrap mass spectrometer. Anal Chem 78:2113–2120. [DOI] [PubMed] [Google Scholar]
- Maldaner AO, Schmidt LL, Locatelli MAF, Jardim WF, Sodré FF, Almeida FV, Pereira CEB, Silva CM. 2012. Estimating cocaine consumption in the brazilian federal district (FD) by sewage analysis. J Braz Chem Soc 23:861–867. [Google Scholar]
- Mari F, Politi L, Biggeri A, Accetta G, Trignano C, Di Padua M, Bertol E. 2009. Cocaine and heroin in waste water plants: A 1‐year study in the city of Florence, Italy. Forensic Sci Int 189:88–92. [DOI] [PubMed] [Google Scholar]
- Martínez‐Bueno MJ, Uclés S, Hernando MD, Fernández‐Alba AR. 2011. Development of a solvent‐free method for the simultaneous identification/quantification of drugs of abuse and their metabolites in environmental water by LC‐MS/MS. Talanta 85:157–166. [DOI] [PubMed] [Google Scholar]
- Mastroianni N, Lopez de Alda M, Barcelo D. 2014. Analysis of ethyl sulfate in raw wastewater for estimation of alcohol consumption and its correlation with drugs of abuse in the city of Barcelona. J Chromatogr A 1360:93–99. [DOI] [PubMed] [Google Scholar]
- McCall A‐K, Bade R, Kinyua J, Lai FY, Thai PK, Covaci A, Bijlsma L, van Nuijs ALN, Ort C. 2016. Critical review on the stability of illicit drugs in sewers and wastewater samples. Water Res 88:933–947. [DOI] [PubMed] [Google Scholar]
- Metcalfe C, Tindale K, Li H, Rodayan A, Yargeau V. 2010. Illicit drugs in Canadian municipal wastewater and estimates of community drug use. Environ Pollut 158:3179–3185. [DOI] [PubMed] [Google Scholar]
- Meyer MR, Holderbaum A, Kavanagh P, Maurer HH. 2015. Low resolution and high resolution MS for studies on the metabolism and toxicological detection of the new psychoactive substance methoxypiperamide (MeOP). J Mass Spectrom. 50:1163–1174. [DOI] [PubMed] [Google Scholar]
- Miller TH, Musenga A, Cowan DA, Barron LP. 2013. Prediction of chromatographic retention time in high‐resolution anti‐doping screening data using artificial neural networks. Anal Chem 85:10330–10337. [DOI] [PubMed] [Google Scholar]
- Moore KA, Mozayani A, Fierro MF, Poklis A. 1996. Distribution of 3,4‐methylenedioxymethamphetamine (MDMA) and 3,4‐methylenedioxyamphetamine (MDA) stereoisomers in a fatal poisoning. Forensic Sci Int 83:111–119. [DOI] [PubMed] [Google Scholar]
- Munro K, Miller TH, Martins CPB, Edge AM, Cowan DA, Barron LP. 2015. Artificial neural network modelling of pharmaceutical residue retention times in wastewater extracts using gradient liquid chromatography‐high resolution mass spectrometry data. J Chromatogr A 1396:34–44. [DOI] [PubMed] [Google Scholar]
- Nácher‐Mestre J, Ibáñez M, Serrano R, Boix C, Bijlsma L, Lunestad BT, Hannisdal R, Alm M, Hernández F, Berntssen MHG. 2016. Investigation of pharmaceuticals in processed animal by‐products by liquid chromatography coupled to high‐resolution mass spectrometry. Chemosphere 154:231–239. [DOI] [PubMed] [Google Scholar]
- Nutt D, King LA, Saulsbury W, Blakemore C. 2007. Development of a rational scale to assess the harm of drugs of potential misuse. Lancet (London, England) 369:1047–1053. [DOI] [PubMed] [Google Scholar]
- Ort C, Lawrence MG, Reungoat J, Mueller JF. 2010a. Sampling for PPCPs in wastewater systems: Comparison of different sampling modes and optimization strategies. Environ Sci Technol 44:6289–6296. [DOI] [PubMed] [Google Scholar]
- Ort C, Lawrence MG, Rieckermann J, Joss A. 2010b. Sampling for pharmaceuticals and personal care products (PPCPs) and illicit drugs in wastewater systems: Are your conclusions valid? A critical review. Environ Sci Technol 44:6024–6035. [DOI] [PubMed] [Google Scholar]
- Ort C, Banta‐Green C, Bijlsma L, Castiglioni S, Emke E, Gartner C, Kasprzyk‐Hordern B, Reid MJ, Rieckermann J, van Nuijs ALN. 2014a. Sewage‐based epidemiology requires a truly transdisciplinary approach. GAIA 23:266–268. [Google Scholar]
- Ort C, Eppler JM, Scheidegger A, Rieckermann J, Kinzig M, Sörgel F. 2014b. Challenges of surveying wastewater drug loads of small populations and generalizable aspects on optimizing monitoring design. Addiction 109:472–481. [DOI] [PubMed] [Google Scholar]
- Ort C, van Nuijs ALN, Berset JD, Bijlsma L, Castiglioni S, Covaci A, de Voogt P, Emke E, Fatta‐Kassinos D, Griffiths P, Hernández F, González‐Mariño I, Grabic R, Kasprzyk‐Hordern B, Mastroianni N, Meierjohann A, Nefau T, Östman M, Pico Y, Racamonde I, Reid M, Slobodnik J, Terzic S, Thomaidis N, Thomas KV. 2014c. Spatial differences and temporal changes in illicit drug use in Europe quantified by wastewater analysis. Addiction 109:1338–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ort C. 2014. Quality assurance/quality control in wastewater sampling In: Zhang C, Mueller JF, Mortimer M, editors. Quality assurance & quality control of environmental field samples. London, UK: Future Science. [Google Scholar]
- Petrovic M, Farré M, Lopez de Alda M, Perez S, Postigo C, Köck M, Radjenovic J, Gros M, Barcelo D. 2010. Recent trends in the liquid chromatography‐mass spectrometry analysis of organic contaminants in environmental samples. J Chromatogr A 1217:4004–4017. [DOI] [PubMed] [Google Scholar]
- Plumb RS, Johnson KA, Rainville P, Smith BW, Wilson ID, Castro‐Perez JM, Nicholson JK. 2006. UPLC/MS E; a new approach for generating molecular fragment information for biomarker structure elucidation. Rapid Commun. mass Spectrom. 20:1989–1994. [DOI] [PubMed] [Google Scholar]
- Postigo C, Lopez de Alda M, Barceló D. 2008. Fully automated determination in the low nanogram per liter level of different classes of drugs of abuse in sewage water by on‐line solid‐phase extraction‐liquid chromatography‐electrospray‐tandem mass spectrometry. Anal Chem 80:3123–3134. [DOI] [PubMed] [Google Scholar]
- Postigo C, Sirtori C, Oller I, Malato S, Maldonado MI, López de Alda M, Barceló D. 2011. Solar transformation and photocatalytic treatment of cocaine in water: Kinetics, characterization of major intermediate products and toxicity evaluation. Appl Catal B Environ 104:37–48. [DOI] [PubMed] [Google Scholar]
- Pozo OJ, Sancho JV, Ibáñez M, Hernández F, Niessen WMA. 2006. Confirmation of organic micropollutants detected in environmental samples by liquid chromatography tandem mass spectrometry: Achievements and pitfalls. TrAC Trends Anal Chem 25:1030–1042. [Google Scholar]
- Pozo OJ, Van Eenoo P, Deventer K, Delbeke FT. 2007. Development and validation of a qualitative screening method for the detection of exogenous anabolic steroids in urine by liquid chromatography‐tandem mass spectrometry. Anal Bioanal Chem 389:1209–1224. [DOI] [PubMed] [Google Scholar]
- Pozo OJ, Ibáñez M, Sancho JV, Lahoz‐Beneytez J, Farre M, Papaseit E, de la Torre R, Hernández F. 2014. Mass spectrometric evaluation of mephedrone in vivo human metabolism: Identification of phase I and phase II metabolites, including a novel succinyl conjugate. Drug Metab Dispos 43:248–257. [DOI] [PubMed] [Google Scholar]
- Prichard J, Lai FY, Kirkbride P, Bruno R, Ort C, Carter S, Hall W, Gartner C, Thai PK, Mueller JF. 2012. Measuring drug use patterns in Queensland through wastewater analysis. Trends Issues Crime Crim Justice 1–8. [Google Scholar]
- Racamonde I, Villaverde‐de‐Sáa E, Rodil R, Quintana JB, Cela R. 2012. Determination of Δ9‐tetrahydrocannabinol and 11‐nor‐9‐carboxy‐Δ9‐tetrahydrocannabinol in water samples by solid‐phase microextraction with on‐fiber derivatization and gas chromatography‐mass spectrometry. J Chromatogr A 1245:167–174. [DOI] [PubMed] [Google Scholar]
- Racamonde I, Rodil R, Quintana JB, Cela R. 2013. In‐sample derivatization‐solid‐phase microextraction of amphetamines and ecstasy related stimulants from water and urine. Anal Chim Acta 770:75–84. [DOI] [PubMed] [Google Scholar]
- Reid MJ, Langford KH, Mørland J, Thomas KV. 2011. Analysis and interpretation of specific ethanol metabolites, ethyl sulfate, and ethyl glucuronide in sewage effluent for the quantitative measurement of regional alcohol consumption. Alcohol Clin Exp Res 35:1593–1599. [DOI] [PubMed] [Google Scholar]
- Reid MJ, Baz‐Lomba JA, Ryu Y, Thomas KV. 2014. Using biomarkers in wastewater to monitor community drug use: A conceptual approach for dealing with new psychoactive substances. Sci. Total Environ. 487:651–658. [DOI] [PubMed] [Google Scholar]
- Reid MJ, Derry L, Thomas KV. 2014. Analysis of new classes of recreational drugs in sewage: Synthetic cannabinoids and amphetamine‐like substances. Drug Test Anal 6:72–79. [DOI] [PubMed] [Google Scholar]
- Rodríguez‐Álvarez T, Rodil R, Cela R, Quintana JB. 2014. Ion‐pair reversed‐phase liquid chromatography‐quadrupole‐time‐of‐flight and triple‐quadrupole‐mass spectrometry determination of ethyl sulfate in wastewater for alcohol consumption tracing. J Chromatogr A 1328:35–42. [DOI] [PubMed] [Google Scholar]
- Rodríguez‐Álvarez T, Racamonde I, González‐Mariño I, Borsotti A, Rodil R, Rodríguez I, Zuccato E, Quintana JB, Castiglioni S. 2015. Alcohol and cocaine co‐consumption in two European cities assessed by wastewater analysis. Sci Total Environ 536:91–98. [DOI] [PubMed] [Google Scholar]
- Rodríguez‐Álvarez T, Rodil R, Rico M, Cela R, Quintana JB. 2014. Assessment of local tobacco consumption by liquid Chromatography−Tandem mass spectrometry sewage analysis of nicotine and its metabolites, cotinine and trans‐3′‐hydroxycotinine, after enzymatic deconjugation. Anal Chemsitry 86:10274–10281. [DOI] [PubMed] [Google Scholar]
- SANCO. 2013. Guidance document on analytical quality control and validation procedures for pesticide residues analysis in food and feed.
- SANTE/11945. 2015. Guidance document on analytical quality control and method validation procedures for pesticide residues analysis in food and feed.
- Schymanski EL, Jeon J, Gulde R, Fenner K, Ru M, Singer HP, Hollender J. 2014a. Identifying small molecules via high resolution mass spectrometry: Communicating confidence. Environ Sci Technol 48:2097–2098. [DOI] [PubMed] [Google Scholar]
- Schymanski EL, Singer HP, Longrée P, Loos M, Ruff M, Stravs MA, Ripollés Vidal C, Hollender J. 2014b. Strategies to characterize polar organic contamination in wastewater: Exploring the capability of high resolution mass spectrometry. Environ Sci Technol 48:1811–1818. [DOI] [PubMed] [Google Scholar]
- Schymanski EL, Singer HP, Slobodnik J, Ipolyi IM, Oswald P, Krauss M, Schulze T, Haglund P, Letzel T, Grosse S, Thomaidis NS, Bletsou A, Zwiener C, Ibáñez M, Portolés T, de Boer R, Reid MJ, Onghena M, Kunkel U, Schulz W, Guillon A, Noyon N, Leroy G, Bados P, Bogialli S, Stipaničev D, Rostkowski P, Hollender J. 2015. Non‐target screening with high‐resolution mass spectrometry: Critical review using a collaborative trial on water analysis. Anal Bioanal Chem 407:6237–6255. [DOI] [PubMed] [Google Scholar]
- Senta I, Krizman I, Ahel M, Terzic S. 2013. Integrated procedure for multiresidue analysis of dissolved and particulate drugs in municipal wastewater by liquid chromatography‐tandem mass spectrometry. Anal Bioanal Chem 405:3255–3268. [DOI] [PubMed] [Google Scholar]
- Stanstrup J, Neumann S, Vrhovšek U. 2015. PredRet: Prediction of retention time by direct mapping between multiple chromatographic systems. Anal Chem 87:9421–9428. [DOI] [PubMed] [Google Scholar]
- Subedi B, Kannan K. 2014. Mass loading and removal of select illicit drugs in two wastewater treatment plants in New York State and estimation of illicit drug usage in communities through wastewater analysis. Environ Sci Technol 48:6661–6670. [DOI] [PubMed] [Google Scholar]
- Takayama T, Suzuki M, Todoroki K, Inoue K, Min JZ, Kikura‐Hanajiri R, Goda Y, Toyo'oka T. 2014. UPLC/ESI‐MS/MS‐based determination of metabolism of several new illicit drugs, ADB‐FUBINACA, AB‐FUBINACA, AB‐PINACA, QUPIC, 5F‐QUPIC and α‐PVT, by human liver microsome. Biomed Chromatogr 28:831–838. [DOI] [PubMed] [Google Scholar]
- Thomas KV, Reid MJ. 2011. What else can the analysis of sewage for urinary biomarkers reveal about communities? Environ Sci Technol 45:7611–7612. [DOI] [PubMed] [Google Scholar]
- Thomas KV, Bijlsma L, Castiglioni S, Covaci A, Emke E, Grabic R, Hernández F, Karolak S, Kasprzyk‐Hordern B, Lindberg RH, Lopez de Alda M, Meierjohann A, Ort C, Pico Y, Quintana JB, Reid M, Rieckermann J, Terzic S, van Nuijs ALN, de Voogt P. 2012. Comparing illicit drug use in 19 European cities through sewage analysis. Sci Total Environ 432:432–439. [DOI] [PubMed] [Google Scholar]
- Thurman EM, Ferrer I, Zweigenbaum JA, García‐Reyes JF, Woodman M, Fernández‐Alba AR. 2005. Discovering metabolites of post‐harvest fungicides in citrus with liquid chromatography/time‐of‐flight mass spectrometry and ion trap tandem mass spectrometry. J Chromatogr A 1082:71–80. [DOI] [PubMed] [Google Scholar]
- Trufelli H, Palma P, Famiglini G, Cappiello A. 2011. An overview of matrix effects in liqiud chromatography‐mass spectrometry. Mass Spectrom Rev 30:491–509. [DOI] [PubMed] [Google Scholar]
- Tscharke B, Chen C, Gerber JP, White JM. 2015. Trends in stimulant use in Australia: A comparison of wastewater analysis and population surveys. Sci Total Environ 536:331–337. [DOI] [PubMed] [Google Scholar]
- Tscharke BJ, White JM, Gerber JP. 2016. Estimates of tobacco use by wastewater analysis of anabasine and anatabine. Drug Test Anal 8:702–707. [DOI] [PubMed] [Google Scholar]
- Tscharke B, Chen C, Gerber JP, White JM. 2016. Temporal trends in drug use in Adelaide, South Australia by wastewater analysis. Sci Total Environ 565:384–391. [DOI] [PubMed] [Google Scholar]
- UNODC. 2014. World Drug Report 2014, Trends in Organized Crime. doi: [DOI]
- van der Aa M, Bijlsma L, Emke E, Dijkman E, van Nuijs ALN, van de Ven B, Hernández F, Versteegh A, de Voogt P. 2013. Risk assessment for drugs of abuse in the Dutch watercycle. Water Res 47:1848–1857. [DOI] [PubMed] [Google Scholar]
- van Leerdam JA, Vervoort J, Stroomberg G, de Voogt P. 2014. Identification of unknown microcontaminants in dutch river water by liquid chromatography‐high resolution mass spectrometry and nuclear magnetic resonance spectroscopy. Environ Sci Technol 48:12791–12799. [DOI] [PubMed] [Google Scholar]
- van Nuijs ALN, Tarcomnicu I, Bervoets L, Blust R, Jorens PG, Neels H, Covaci A. 2009. Analysis of drugs of abuse in wastewater by hydrophilic interaction liquid chromatography‐tandem mass spectrometry. Anal Bioanal Chem 395:819–828. [DOI] [PubMed] [Google Scholar]
- van Nuijs ALN, Castiglioni S, Tarcomnicu I, Postigo C, de Alda ML, Neels H, Zuccato E, Barcelo D, Covaci A. 2011. Illicit drug consumption estimations derived from wastewater analysis: A critical review. Sci Total Environ 409:3564–3577. [DOI] [PubMed] [Google Scholar]
- van Nuijs ALN, Gheorghe A, Jorens PG, Maudens K, Neels H, Covaci A. 2014. Optimization, validation, and the application of liquid chromatography‐tandem mass spectrometry for the analysis of new drugs of abuse in wastewater. Drug Test Anal 6:861–867. [DOI] [PubMed] [Google Scholar]
- Vazquez‐Roig P, Blasco C, Picó Y. 2013. Advances in the analysis of legal and illegal drugs in the aquatic environment. TrAC Trends Anal Chem 50:65–77. [Google Scholar]
- Venhuis BJ, de Voogt P, Emke E, Causanilles A, Keizers PHJ. 2014. Success of rogue online pharmacies: Sewage study of sildenafil in the Netherlands. BMJ 349:g4317. [DOI] [PubMed] [Google Scholar]
- Voloshenko‐Rossin A, Gasser G, Cohen K, Gun J, Cumbal‐Flores L, Parra‐Morales W, Sarabia F, Ojeda F, Lev O. 2015. Emerging pollutants in the Esmeraldas watershed in Ecuador: Discharge and attenuation of emerging organic pollutants along the San Pedro–Guayllabamba–Esmeraldas rivers. Environ Sci Process Impacts 17:41–53. [DOI] [PubMed] [Google Scholar]
- Wong CS, MacLeod SL. 2009. JEM spotlight: Recent advances in analysis of pharmaceuticals in the aquatic environment. J Environ Monit 11:923–936. [DOI] [PubMed] [Google Scholar]
- Yang Z, Anglès d'Auriac M, Goggins S, Kasprzyk‐Hordern B, Thomas KV, Frost CG, Estrela P. 2015a. A novel DNA biosensor using a ferrocenyl intercalator applied to the potential detection of human population biomarkers in wastewater. Environ Sci Technol 49:5609–5617. [DOI] [PubMed] [Google Scholar]
- Yang Z, Kasprzyk‐Hordern B, Frost CG, Estrela P, Thomas KV. 2015b. Community sewage sensors for monitoring public health. Environ Sci Technol 49:5845–5846. [DOI] [PubMed] [Google Scholar]
- Yang Z, Kasprzyk‐Hordern B, Goggins S, Frost CG, Estrela P. 2015c. A novel immobilization strategy for electrochemical detection of cancer biomarkers: DNA‐directed immobilization of aptamer sensors for sensitive detection of prostate specific antigens. Analyst 140:2628–2633. [DOI] [PubMed] [Google Scholar]
- Yang Z, Castrignanò E, Estrela P, Frost CG, Kasprzyk‐Hordern B. 2016. Community sewage sensors towards evaluation of drug use trends: Detection of cocaine in wastewater with DNA‐Directed immobilization aptamer sensors. Sci Rep 6:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zomer P, Mol JGJ. 2015. Simultaneous quantitative determination, identification and qualitative screening of pesticides in fruits and vegetables using LC‐Q‐Orbitrap™‐MS. Food Addit Contam Part A Chem Anal Control Expo Risk Assess 32:1628–1636. [DOI] [PubMed] [Google Scholar]
- Zuccato E, Chiabrando C, Castiglioni S, Calamari D, Bagnati R, Schiarea S, Fanelli R. 2005. Cocaine in surface waters: A new evidence‐based tool to monitor community drug abuse. Environ Heal A Glob Access Sci Source 4:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccato E, Chiabrando C, Castiglioni S, Bagnati R, Fanelli R. 2008. Estimating community drug abuse by wastewater analysis. Environ Health Perspect 116:1027–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]