

Abstract
In common with other self-cleaving RNAs, the lead-dependent ribozyme (leadzyme) undergoes dynamic fluctuations to a chemically activated conformation. We explored the connection between conformational dynamics and self-cleavage function in the leadzyme using a combination of NMR spin-relaxation analysis of ribose groups and conformational restriction via chemical modification. The functional studies were performed with a North-methanocarbacytidine modification that prevents fluctuations to C2′-endo conformations while maintaining an intact 2′-hydroxyl nucleophile. Spin-relaxation data demonstrate that the active-site Cyt-6 undergoes conformational exchange attributed to sampling of a minor C2′-endo state with an exchange lifetime on the order of microseconds to tens of microseconds. A conformationally restricted species in which the fluctuations to the minor species are interrupted shows a drastic decrease in self-cleavage activity. Taken together, these data indicate that dynamic sampling of a minor species at the active site of this ribozyme, and likely of related naturally occurring motifs, is strongly coupled to catalytic function. The combination of NMR dynamics analysis with functional probing via conformational restriction is a general methodology for dissecting dynamics-function relationships in RNA.
Keywords: conformational restriction, NMR dynamics, lead-dependent ribozyme, dynamics-function, sugar pucker
INTRODUCTION
A full understanding of the function of biological macromolecules requires a detailed understanding of their conformational dynamics to complement high-resolution structural data. This is particularly true in the case of RNA, for which rugged conformational landscapes are often limiting for function (Williamson 2000; Leulliot and Varani 2001; Perez-Canadillas and Varani 2001; Rambo and Doudna 2004; Al-Hashimi and Walter 2008; Walter 2009). Heteronuclear NMR has emerged as a powerful probe of the conformational dynamics of RNA on a variety of time scales (Bothe et al. 2011; Salmon et al. 2014), with a particular strength in the detection and characterization of low-population conformers, sampled on the microsecond to millisecond time scale, that may be functionally critical for RNA catalysis and recognition (Hoogstraten et al. 2000; Dethoff et al. 2012; Ravera et al. 2014; Zhao et al. 2014; Zhao and Zhang 2015; Furtig et al. 2016; Schnieders et al. 2017). Given a spectroscopic or biophysical analysis of a molecule's dynamics, true mechanistic insight can in principle be obtained by “dynamics-function” studies, in which a chosen dynamic mode is selectively quenched and the effect on catalysis, binding, or other functional output is assessed. The selective experimental quenching of dynamics, however, is significantly more challenging than the mutational ablation of an observed structural feature. As one effort toward dynamics-function studies in RNA, our group introduced the use of locked nucleic acid (LNA) modifications, which fix the ribose ring in a single sugar pucker conformer, thereby preventing the sampling of minor states with different ribose conformations (Julien et al. 2008).
Many of the small, self-cleaving class of catalytic RNA molecules, or ribozymes, rely on dynamics and conformational rearrangements to take up their chemically active forms (Scott et al. 1996; Wilson et al. 2007; Liu et al. 2014, 2017). The artificial lead-dependent ribozyme, or leadzyme (Fig. 1), which undergoes similar chemistry but with specificity for divalent lead as a cofactor (Pan and Uhlenbeck 1992; Pan et al. 1994), has been useful as a model system for structural and dynamic studies of this class of ribozymes by NMR (Legault and Pardi 1997; Hoogstraten et al. 1998, 2000; Legault et al. 1998; Westhof and Hermann 1999; Qi and Xia 2011; Hoogstraten and Sumita 2013).
FIGURE 1.
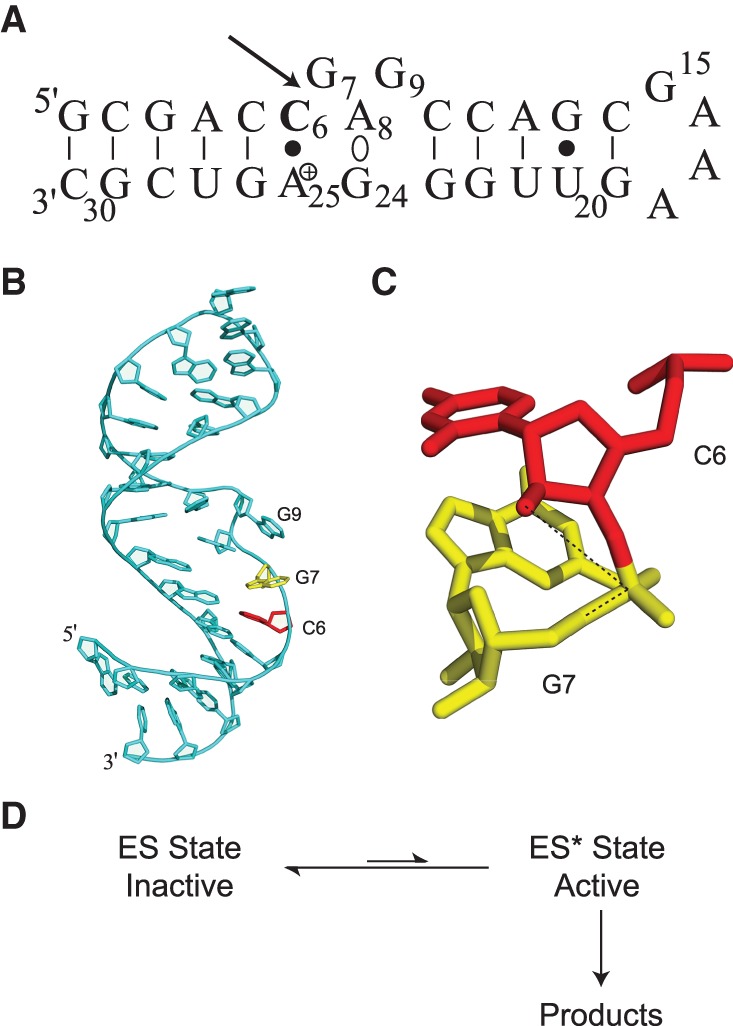
Sequence and three-dimensional structure of the lead-dependent ribozyme. (A) Leadzyme secondary structure, indicating the adenosine residue (Ade-25) with a shifted pKa of 6.5 as positively charged (Legault and Pardi 1997), the noncanonical dynamic A-G base-pairing interaction (Ade-8°Gua-24), and the self-cleavage site (arrow). (B) Overall structure of the leadzyme as observed in solution (Hoogstraten et al. 1998). Residues flanking the cleavage site are colored red (Cyt-6) and yellow (Gua-7). (C) Expanded view of Cyt-6 and Gua-7 (colored as in B), illustrating the lack of in-line alignment for the reactive groups (dotted line connecting nucleophilic O2′, reaction center phosphorus, and leaving group O5′). (D) Schematic reaction scheme for the leadzyme, illustrating the requirement for dynamic sampling of a rare (“invisible”) conformational state ES* for self-cleavage. Structure panels were prepared with PyMol (Delano 2002).
NMR spin-relaxation studies of conformational dynamics in catalytic RNA, including the leadzyme, have thus far been limited to the use of 13C resonances in the nucleotide base groups due to strong 13C–13C magnetic interactions in the backbone ribose ring. A general class of approaches to such interfering effects is provided by the introduction of 13C labels at specific sites within the nucleotide. Significant progress in atom-specific labeling has been made for DNA (Nelissen et al. 2016) and RNA (Hoogstraten and Johnson 2008; Nelissen et al. 2008; Le et al. 2015; Chen et al. 2016; Longhini et al. 2016a,b; LeBlanc et al. 2017). Our group has introduced a metabolically driven alternate-site labeling procedure in which nucleotides are biosynthetically produced by E. coli deficient in the pentose-phosphate pathway enzyme glucose-6-phosphate dehydrogenase (zwf genotype) grown with 2-13C-glycerol as the sole carbon source. Under these conditions, ribose-5-phosphate is synthesized only via transketolase and transaldolase operating on 12C–13C–12C isotopomers of three-carbon metabolites, resulting in nucleic acids with ribose groups 13C-labeled to high atom percent at C2′ and C4′ with no detectable 13C label at C1′, C3′, and C5′ sites (Johnson et al. 2006; Johnson and Hoogstraten 2008). With this labeling pattern, NMR spin-relaxation studies can be performed on 13C2′ and 13C4′ resonances in the complete absence of the one-bond JCC couplings that otherwise pose significant obstacles to data acquisition and interpretation. Comparisons of spin-relaxation data with uniformly labeled samples indicated significant errors introduced by 13C–13C interactions in the uniformly labeled molecules, emphasizing the importance of the selective labeling scheme (Johnson et al. 2006). We used this technology to perform a comprehensive spin-relaxation analysis of backbone ribose groups in the GCAA tetraloop (Johnson and Hoogstraten 2008). In the current work, we extend applications of alternate-site labeling techniques to catalytic RNA.
Ribose rings in nucleic acids generally are found in two conformations corresponding to two locations on the pseudorotation cycle (Altona and Sundaralingam 1972): C3′-endo or “North,” characteristic of A-form helices, and C2′-endo or “South,” characteristic of B-form helices (Fig. 2; Wijmenga and van Buuren 1998; Li and Szostak 2014; Wang and Woods 2016). Nonhelical nucleotides may be found in either conformer or in an equilibrium between the two, as can be analyzed by NMR J-coupling constants (Wijmenga and van Buuren 1998). In the leadzyme, the nucleotide immediately 5′ to the cleavage site, Cyt-6, has been determined to be C3′-endo by J-couplings and displays this conformation in both the NMR and crystallographic structures (Hoogstraten et al. 1998; Wedekind and McKay 1999, 2003). In addition, these ground-state structures show the reactive groups distant from the in-line conformation of nucleophile (2′-hydroxyl), reaction center (phosphorus), and leaving group (5′-oxygen) that is ideal for self-cleavage (Fig. 1). Together with other discrepancies between the biochemical and structural data in this system (Chartrand et al. 1997; Westhof and Hermann 1999; Hoogstraten and Sumita 2013), these data suggest that the active state of the leadzyme is a minor conformer that is distinct from those observed by NMR or crystallography and is sampled in dynamic fashion (Lemieux et al. 1998), with the reorientation of Cyt-6 from C3′-endo to C2′-endo a key part of the structural transition (Fig. 1D). The present work is designed as a specific test of this model.
FIGURE 2.
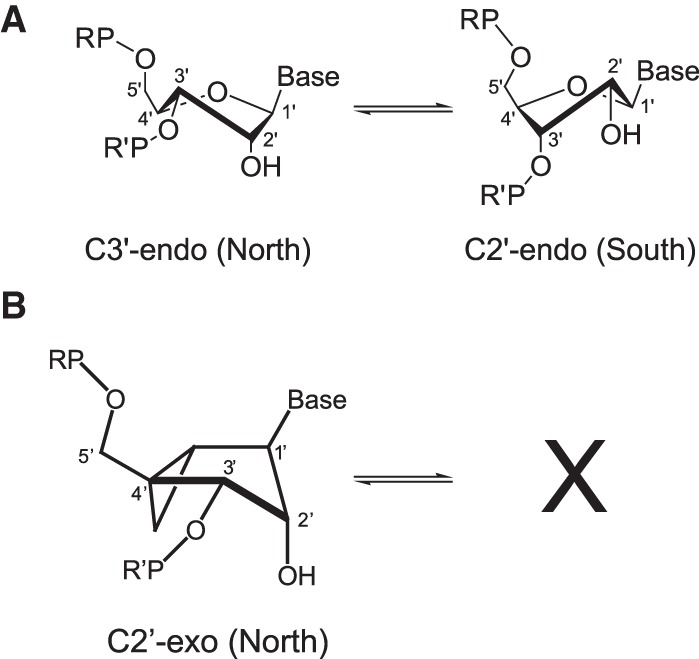
(A) Three-dimensional representations of the two primary pseudorotational conformations (sugar puckers) observed for ribose rings in nucleic acids. (B) Structure of the North-methanocarba nucleotide modification, which cannot take up South-like sugar pucker conformations, emphasizing the similar presentation of linkages and functional groups to unmodified C3′-endo nucleotides.
We previously applied LNA modification, which covalently fixes the nucleotide in the C3′-endo conformation and prevents fluctuations to C2′-endo, at several sites in the leadzyme internal loop (Gua-7, Gua-9, and Gua-24) as a probe of the importance of dynamics in activity (Julien et al. 2008). LNA, however, cannot be used at the critical Cyt-6 site, as the modification destroys the nucleophilic 2′-hydroxyl and thus abrogates self-cleavage completely. We have therefore opted to incorporate synthetic N-methanocarba (N-MC) nucleotides, which use a bicyclo[3.1.0]hexane pseudosugar structure to fix the sugar pucker in a C2′-exo (North) conformer without modification at the 2′ site (Fig. 2; Jeong et al. 1995; Choi et al. 2003). The deoxy versions of these nucleotides (Maier et al. 2004; Maderia et al. 2005; Wu et al. 2005) have been used to probe systems including DNA thermodynamics (Maderia et al. 2007) and thrombin-aptamer recognition (Saneyoshi et al. 2009), and an efficient synthesis of RNA oligonucleotides containing the 2′-hydroxyl variant of N-methanocarba nucleotides is available (Terrazas et al. 2011).
Work in several systems has demonstrated the compatibility of N-MC nucleotides with structural requirements for North sugar puckers, consistent with the display of functional groups in similar orientations to unmodified nucleotides (Fig. 2). An siRNA duplex containing a ribo-N-MC modification in the guide strand was taken up by the intracellular RNAi machinery and showed near-wild-type activity and target mRNA degradation (Terrazas et al. 2011). A crystal structure of a N-MC-dATP nucleotide bound at the active site of the model DNA polymerase Dpo4 superimposed nearly perfectly with an unmodified dATP bound at the same site, demonstrating selectivity of the polymerase for the North substrate conformation (Ketkar et al. 2012a), and no protein contacts to the cyclopropane ring were observed in either Dpo4 or human DNA polymerase iota (Ketkar et al. 2012b). Both N-MC and S-MC nucleotides have been shown to reproduce the preferences of the corresponding unmodified nucleotides for rotamers at the sugar-base glycosidic bond (torsion angle χ) and ring-adjacent C3′–C4′ bond (γ) (Marquez et al. 1997, 2004). Finally, inhibition of HIV reverse transcriptase by N-MC-AZT was kinetically indistinguishable from that mediated by wild-type AZT (Marquez et al. 1998). In short, N-methanocarba nucleotides are good structural and functional mimics of unmodified nucleotides in their North conformation. When substituted at Cyt-6 of the leadzyme, N-methanocarbacytidine (N-MCC) would be expected to reproduce the observed ground-state structure, and thus to be functionally active in the absence of sugar pucker dynamics coupled to self-cleavage.
In this work, we have combined NMR spin relaxation analyses of ribose pucker dynamics at cytosine residues in the leadzyme, enabled by the metabolically driven site-specific stable isotope labeling scheme (Hoogstraten and Johnson 2008), with functional probing via N-MCC substitution at the active site Cyt-6. We find that, nearly uniquely among cytosine residues in the molecule, Cyt-6 shows dynamic sampling of a minor conformer with substantial chemical shift changes at 13C2′ and 13C4′ and an exchange lifetime on the order of 10 µsec. N-MCC substitution reveals that conformational restriction to North conformations at this site results in a drastic loss of self-cleavage activity. Taken together, these results indicate that dynamic sampling of a low-population “invisible” state of the active-site ribose is an important factor in the catalytic cycle of the leadzyme, suggesting that reorientation to a C2′-endo sugar pucker at Cyt-6 is characteristic of the active state of the molecule (ES* in Fig. 1). Given the similarity of the leadzyme chemical mechanism to naturally occurring self-cleaving ribozymes, similar factors may very well be at play in those biologically relevant species as well (Cochrane and Strobel 2008). The combination of NMR ribose spin relaxation with conformational restriction via chemical modification is a generally useful technology for probing the functional importance of ribose pucker dynamics in RNA.
RESULTS
Specific labeling of RNA
The use of standard uniformly 13C-labeled samples of RNA poses serious obstacles to NMR spin relaxation analysis along the RNA backbone. Large one-bond 13C–13C scalar and dipolar couplings within the ribose ring significantly interfere with the acquisition and interpretation of relaxation data, which generally relies on the approximation that the behavior of the heteronucleus (13C, in this case) is dominated by interactions with the directly bonded 1H atom. Subtypes of valid data may be acquired in some circumstances in DNA (Shajani and Varani 2008) and RNA (Clay et al. 2017), but the most general solution is to acquire data under conditions in which 13C nuclei have only 12C neighbors along the carbon chain. Our group's metabolically driven labeling scheme using glucose-6-phosphate dehydrogenase (zwf) mutants of E. coli and 2-13C-glycerol as a carbon source achieves this aim via high atom percent 13C labeling at C2′ and C4′ sites (2′,4′-13C2) and undetectable 13C elsewhere in the ribose (Johnson et al. 2006; Hoogstraten and Johnson 2008; Johnson and Hoogstraten 2008). In addition, in RNA oligonucleotides of more than a few residues, substantial overlap in the C2′/H2′ and C4′/H4′ regions of two-dimensional NMR spectra greatly reduces the number of signals that can be analyzed. In this work, we enabled ribose spin-relaxation studies in the 30-nucleotide leadzyme construct via in vitro transcription in which cytidine nucleotides are labeled in 2′,4′-13C2 fashion as described above and the remaining nucleotides are present at natural isotopic abundance.
Figure 3A shows the ribose-sugar region of a non-constant-time 1H-13C HSQC NMR spectrum of the resulting sample, demonstrating signals arising from 13C2′ and 13C4′ labels along with the complete absence of 13C signals in the regions containing C1′, C3′, and C5′ signals from this molecule (Legault et al. 1998). Slices along the 13C axis through cross-peaks in this spectrum display singlets rather than the triplets which arise from the ∼40 Hz one-bond 13C–13C couplings in uniformly labeled samples, indicating the efficiency of the alternate-site scheme (Fig. 3B). A comprehensive analysis of the isotopomers present in this labeling scheme has been published previously and is consistent with the complete absence of one-bond 13C–13C interactions (Johnson et al. 2006). Expansions of the analyzed signals (Fig. 3C,D) demonstrate the presence of all expected peaks (Legault et al. 1998), albeit with substantial overlap for H4′/C4′ signals in helical regions that prevented analysis for these signals for five of the eight cytosine residues. A number of minor signals present in this spectrum that were not consistently observed are indicated with asterisks and attributed to a small component of misfolded ribozyme. Assignments of all analyzed signals to the proper resonances of the correctly folded species were confirmed based on a 3D 13C HSQC-NOESY spectrum (not shown). Based on the results in Figure 3, spin-relaxation experiments on this sample can be taken as rigorously free of contributions from 1JCC interactions. In addition, the restriction of labeling to cytosine residues removes 22 of the 30 signals from each atom type. As a result, all of the H2′–C2′ and three of the eight H4′–C4′ peaks were sufficiently resolved for relaxation analysis.
FIGURE 3.

(A) Two-dimensional non-constant-time HSQC spectrum for the ribose resonances of the 2′–4′-13C2-Cyt labeled leadzyme. Boxes indicate the regions in which the five types of ribose 1H-13C cross peaks arising from cytosine residues in this molecule would be expected (Legault et al. 1998). Predicted C5′ chemical shifts are folded from the ∼65 to 66 ppm region due to the restricted sweep width of the spectrum. (Inset) Schematic of the alternate-site labeling pattern with 13C-labeled sites in red. (B) Section along the 13C axis for a representative H2′–C2′ peak demonstrating the lack of multiplet structures that would arise from any one-bond 13C–13C interactions present. (C,D) Expansions of the 2′ and 4′ regions from A with the analyzed peaks labeled. “ov” indicates overlapped helical 4′ resonances not analyzed in this work, daggers indicate signals arising from 3′-end heterogeneity, and asterisks indicate inconsistently observed peaks that are attributed to a minor population of misfolded RNA (see text).
Ribose dynamics at the cleavage site
The rate of decay of transverse NMR magnetization, commonly denoted R2 when measured under spin-echo (Carr–Purcell–Meiboom–Gill, CPMG) conditions and R1ρ when measured under spin-lock conditions, is sensitive to conformational changes on microsecond to millisecond timescales that cause a change in chemical shift (Δω) of the nucleus according to
| (1) |
| (2) |
where R0 represents the relaxation rate in the absence of exchange contributions. In the case of exchange between two states a and b with different chemical shifts, the exchange contribution to R1ρ data is given in the fast-exchange limit by
| (3) |
where pa and pb are the populations of the interconverting states, kex is the sum of the forward and reverse rate constants for the interconversion, and ωeff is the effective strength of the spin-locking field, obtained as the vector sum of the resonance offset and the applied field strength ω1 = γB1/2π. Exchange lifetimes τex are reported as the reciprocal of kex. Thus, exchange processes give rise to increased R2 values compared to those observed in molecular regions for which exchange on these time scales is not present. For ribose rings in nucleic acids, interconversions between C3′-endo and C2′-endo conformers (Fig. 2) give rise to substantial changes in 13C chemical shifts, especially at the C2′, C3′, and C4′ sites (Ebrahimi et al. 2001; Rossi and Harbison 2001). , in contrast, depends only on the chemical structure at the carbon site (especially the near-constant C-H bond length), the overall tumbling of the molecule, and the presence of sub-nanosecond local dynamics, which will tend to decrease the relaxation rate. Thus, in stable helical regions, near-constant R1ρ values for a given carbon type are generally observed (Johnson and Hoogstraten 2008). Increased values of R1ρ in nonhelical regions can be taken as arising from microsecond-to-millisecond conformational exchange (Palmer 2004).
We acquired R1ρ measurements of C2′ and C4′ nuclei to detect and analyze the sampling of minor conformers for cytidine nucleotides in the leadzyme. As an example of the data obtained, Figure 4 shows decay curves for transverse relaxation for two internal-loop resonances (Cyt-6 C2′ and C4′) compared to a typical helical resonance (Cyt-28 C2′) at a spin-lock power ωeff of 3000 Hz. The loop resonances display a significant increase in decay rate well outside the error in the measurements, consistent with the presence of an exchange contribution to relaxation. Figure 5 collects R1ρ values for all observed resonances at a single ω1 value of 2500 Hz. With the exception of Cyt-10 C4′, discussed below, resonances from Watson–Crick helical regions indeed show near-constant values across the molecule, consistent with the absence of microsecond-scale internal dynamics in helical regions. For the active-site residue Cyt-6, in contrast, both C2′ and C4′ resonances show substantially increased R1ρ values compared to helical residues, indicating strong Rex contributions from sampling of a minor conformer. This residue is found by both NMR (Hoogstraten et al. 1998) and crystallographic (Wedekind and McKay 1999, 2003) structural analysis to take up an A-form structure extending the flanking Watson–Crick helix, and is fully C3′-endo according to NMR J-coupling constants (Legault et al. 1998). Chemical shifts at 13C2′ and 13C4′ are expected to be strongly dependent on changes in sugar pucker, with a particularly dramatic shift possible if the glycosidic angle simultaneously shifts from anti to syn; in contrast, simple changes in the exocyclic γ torsion lead to relatively minor chemical shift changes at these two sites (Ebrahimi et al. 2001; Rossi and Harbison 2001). Therefore, the most straightforward interpretation of exchange dynamics at these sites is the sampling of a low-population invisible state with the Cyt-6 ribose in the C2′-endo conformation, although other more complicated motional models cannot be ruled out.
FIGURE 4.
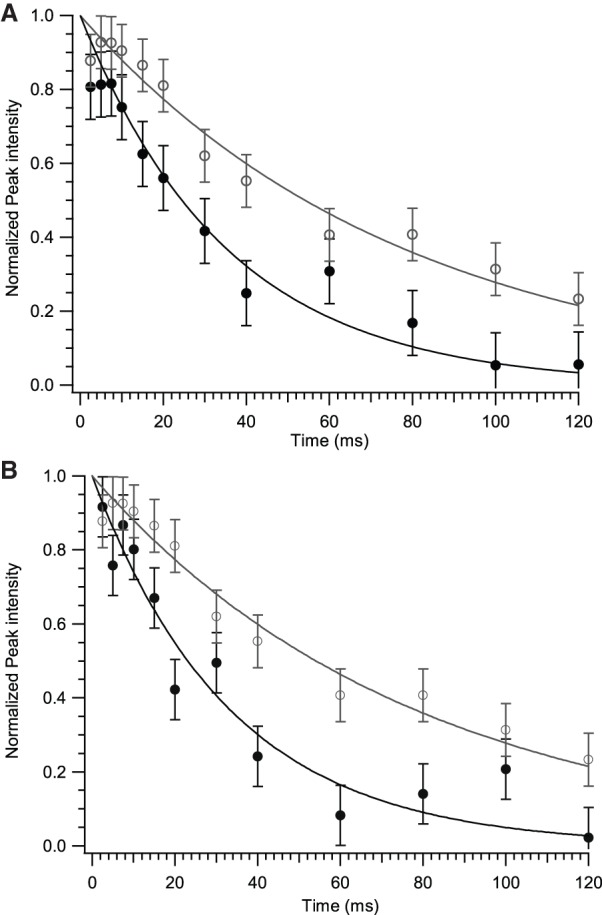
Illustrative R1ρ transverse relaxation curves at γB1/2π = 3.0 KHz for internal-loop peaks. (A) Cyt-6 C2′ (solid); (B) Cyt-6 C4′ (solid). Open symbols in both panels are the helical residue Cyt-28 C2′ for comparison. Curves were normalized such that extrapolated intensity at t = 0 was unity. Error bars were derived from threefold replicas of a single time point.
FIGURE 5.
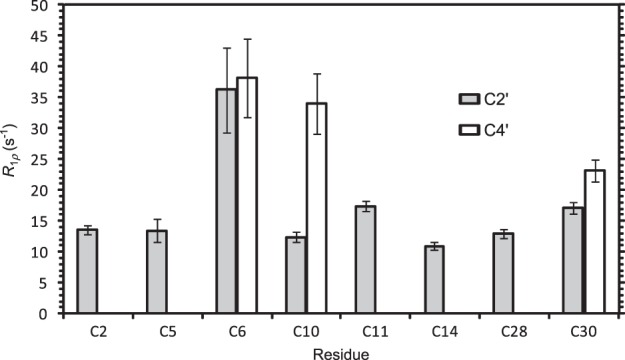
Transverse relaxation rates for the 2′,4′-13C2-cytidine-leadzyme. Values correspond to R1ρ data at γB1/2π = 3.0 KHz. Error bars are as reported by the fit software for a single-exponential decay.
Relaxation-dispersion analysis of exchange
According to Equation 3, the measurement of transverse relaxation rates as the spin-lock power ω1 is parametrically varied allows the determination of the motional parameters pa pb (Δω)2 (often expressed as Φex) and kex. If independent estimates of the state populations are available, the chemical shift difference between them (Δω) can be calculated directly from Φex; otherwise, a lower bound can be placed on Δω by considering the limiting case pa = pb = 0.5 (Δωmin2 = Φex/0.25). Observation of such relaxation-dispersion effects also provides a more unambiguous demonstration of the presence of conformational exchange than data at a single value of ω1. Therefore, we obtained R1ρ data for our 13C2-Cyt sample at ω1 values varying from 1.5 to 6.0 KHz. All measured relaxation rates are collected in the Supplemental Information, Supplemental Table S1. Figure 6 shows such relaxation-dispersion data for the two active-site resonances, Cyt-6 C2′ and Cyt-6 C4′, in comparison to a typical helical resonance, Cyt-28 C2′. In data of this type, for exchange processes on the scale of a few tens of microseconds or more, a 6 kHz applied field is sufficient to nearly or completely suppress Rex, thus leading R1ρ to converge to the asymptotic value at the upper end of such dispersion plots according to Equation 3 (Johnson and Hoogstraten 2008). In the data in Figure 6, only weak variation of dispersion effects with ω1 is observed, and the exchange lifetimes cannot be determined quantitatively. We have simulated relaxation-dispersion curves based on a number of combinations of parameters in an attempt to determine the limits of dynamic properties consistent with the present data (see Supplemental Material). We found that τex values of less than ∼2 µsec would require unrealistically large chemical-shift changes to reproduce the observed magnitude of Rex (Supplemental Material, Supplemental Fig. S1A–C), whereas lifetimes greater than ∼30 to 40 µsec are inconsistent with the observed weak ω1 dependence (Supplemental Fig. S1D). The exchange lifetime can therefore be qualitatively taken to be on the order of 10 µsec. Using an exchange value of 10.0 µsec yields a Δωmin value of roughly 3.0 ppm, consistent with the expected magnitude of chemical-shift changes of these atoms upon C3′-endo to C2′-endo conversion (Ebrahimi et al. 2001; Rossi and Harbison 2001). For a 5% population of the minor state, the data is consistent with chemical shift changes of ∼7 ppm, corresponding to shifts expected for a concerted shift from C3′-endo/anti to C2′-endo/syn. Although the lack of strong variation of relaxation rate with ω1 requires caution in quantitative interpretation, especially given possible variations in , the analyses in Figure 6 and Supplemental Figure S1 unambiguously demonstrate a dynamic process at this ribose with an exchange lifetime on the order of 10 µsec. With dispersion effects on comparable lifetimes observed at both 13C sites with the ribose, the most parsimonious explanation of these data is the sampling of a minor C2′-endo state at this ribose. More precise quantitation of the exchange lifetimes would require extension of the dispersion curve above ω1 = 6 kHz, a regime often difficult to access due to excessive power deposition in the NMR probe. Recent work with the most advanced current cryoprobes allows the extension of this limit in favorable cases (Ban et al. 2017; Reddy et al. 2018).
FIGURE 6.
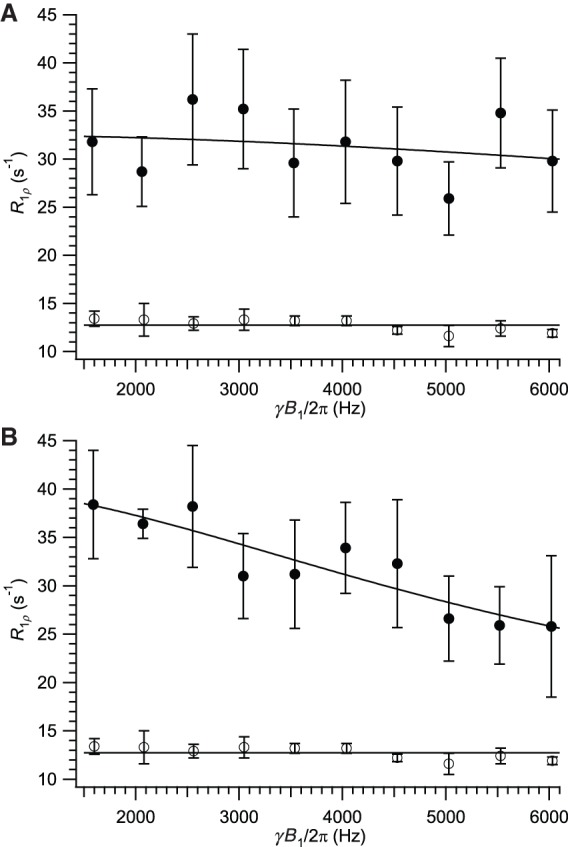
Relaxation-dispersion curves. (A) Cyt-6 C2′ (solid); (B) Cyt-6 C4′ (solid). Open symbols in both panels are the helical residue Cyt-28 C2′ for comparison. Fits to the upper curves are to Equations 2 and 3 with R1ρ fixed to 12.7 sec−1 based on the mean value for helical Cyt-6 C2′. Fits in which was allowed to vary resulted in extremely poor parameter determination. Fit parameters: Cyt-6 C2′, kex = (1.0 ± 0.8) × 105 sec−1, Φex = (2.0 ± 1.3) × 106, Δωmin = (3.0 ± 1.0) ppm; Cyt-6 C4′, kex = (3.6 ± 0.5) × 104 sec−1, Φex = (9.9 ± 0.7) × 105, Δωmin = (2.1 ± 0.1) ppm.
Activity of a conformationally restricted leadzyme variant
We have demonstrated the use of LNA modifications, which covalently restrict the corresponding nucleotide to C3′-endo conformations using a methylene bridge between C4′ and the 2′-hydroxyl oxygen, as a functional probe of ribose dynamics in systems including leadzyme catalysis (Julien et al. 2008) and hairpin ribozyme docking (A McClain, N White, and CG Hoogstraten, unpubl.). LNA, however, destroys the RNA 2′-hydroxyl nucleophile, and would therefore be incompatible with self-cleavage if used at the Cyt-6 active site. We therefore adopted the north-locked methanocarbacytidine (N-MCC) nucleotide modification (Fig. 2), which restricts the nucleotide to an envelope C2′-exo conformation, leaving the 2′-hydroxyl group in an axial disposition similar to a ribose in a C3′-endo conformation (Fig. 2; Jeong et al. 1995; Choi et al. 2003; Terrazas et al. 2011). North-methanocarba nucleotides have been demonstrated to be excellent structural and functional mimics for unmodified nucleotides in the C3′-endo conformation in a number of contexts (Marquez et al. 1998; Terrazas et al. 2011; Ketkar et al. 2012a,b) and to reproduce the preferences of unmodified nucleotides for glycosidic and exocyclic torsion angles (Marquez et al. 1997, 2004). When incorporated at Cyt-6 in the leadzyme, this modification is thus expected to only minimally perturb the ground-state structure, in which the ribose is stably C3′-endo (Hoogstraten et al. 1998; Legault et al. 1998; Wedekind and McKay 2003). The fluctuations to a minor C2′-endo conformation implied by the above spin-relaxation data, however, are completely abolished.
The self-cleavage activity of the leadzyme as a function of added [Pb2+] was determined by quantitating the conversion of fluorescently labeled full-length RNA to cleavage product upon incubation with varying concentrations of Pb(II) on a denaturing polyacrylamide gel. Figure 7 shows a comparison of the self-cleavage kinetics of wild-type and N-MCC-6 leadzyme constructs at pH 7.0. A maximum cleavage rate of roughly 1 min−1 for the wild-type sequence is consistent with previous observations at this pH (Pan et al. 1994). The decay of cleavage rates at elevated [Pb2+] is characteristic of the system and is ascribed to the formation of polyhydroxy-Pb(II) clusters in solution (Pan et al. 1994; Yajima et al. 2007; Julien et al. 2008). We observe a dramatic decrease in the rate of self-cleavage upon N-MCC modification, with kobs values at the optimum 25 µM Pb2+ decreasing from (1.2 ± 0.1) min−1 to (0.013 ± 0.007) min−1. Thus, the specific quenching of dynamics at the Cyt-6 ribose group has drastically reduced the self-cleavage activity of the leadzyme.
FIGURE 7.
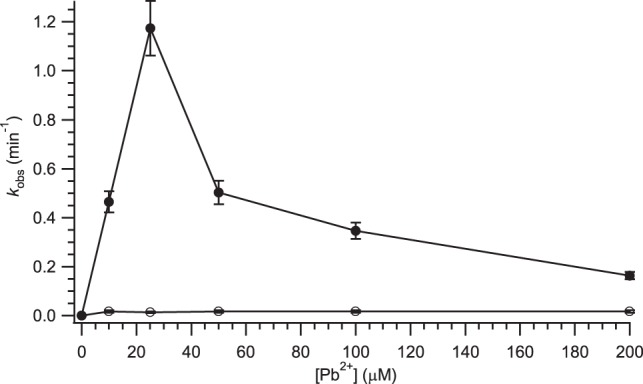
Self-cleavage rate constants for wild-type (solid symbols) and N-MCC-6 modified (open symbols) leadzyme sequences. For the wild-type sequence, biexponential decay was observed due to incomplete folding, and the faster of the two resulting rate constants is plotted (cf. Materials and Methods). Relative errors (σn−1) were averaged across a given data set.
Taken together, the sampling of a minor conformer detected by spin relaxation and the loss of activity upon restriction to a region of the pseudorotational cycle consistent with the ground state suggest that repuckering of this nucleotide from C3′-endo to C2′-endo is directly coupled to catalytic function, i.e., lies along the reaction coordinate. The effects of selective conformational restriction indeed strengthen our identification of the exchange dynamics observed by NMR with a C3′-endo/C2′-endo equilibrium at the active-site ribose.
DISCUSSION
This work combines detailed NMR spin-relaxation analysis of the sampling of a minor state of the ribose ring at the active site of the lead-dependent ribozyme with the use of a conformationally restricted nucleotide to test the activity of a variant in which that sampling is prevented, providing an unambiguous correlation between conformational dynamics and function. Due to the limited range of spin-lock powers available, the quantitative precision of the motional analysis is limited, but the overall conclusion that a minor state corresponding to a change in 13C chemical shift of several ppm of both the C2′ and C4′ resonances at Cyt-6 is sampled on approximately the 10-microsecond time scale is unambiguous.
The mechanism of self-cleavage of the in vitro selected leadzyme, nucleophilic attack of a 2′-hydroxyl on the adjoining phosphate with departure of the 5′-oxygen, is common to the prominent Mg2+-activated family of self-cleaving ribozymes that includes the paradigmatic hammerhead, hairpin, and hepatitis delta motifs (Esteban et al. 1997; Fedor and Williamson 2005; Cochrane and Strobel 2008) as well as several more recently discovered species (Roth et al. 2014; Harris et al. 2015; Li et al. 2015; Weinberg et al. 2015). The proper alignment of these groups for facile “in-line” attack is a substantial contributor to the catalytic power of these motifs (Soukup and Breaker 1999; Emilsson et al. 2003). Based on structural and computational data in several systems, the coupling of active-site alignment with the assumption of a C2′-endo conformation at the active site, either via static structural stabilization or dynamic sampling, may be a common feature of these motifs (Cochrane and Strobel 2008; Lee et al. 2009, 2016; Heldenbrand et al. 2014; Gaines and York 2016). For example, the corresponding nucleotide in the hairpin ribozyme is C3′-endo in an NMR structure of the inactive loop A in isolation (Cai and Tinoco 1996), but converts to C2′-endo upon formation of the active state via tertiary interface formation with loop B (Rupert and Ferré-D'Amaré 2001). In the case of the leadzyme, a binding partner to conformationally capture the active state is not available, and self-cleavage instead proceeds via dynamic sampling of a minor, catalytically proficient state. The combination of NMR spin-relaxation and related techniques and functional probing via conformational restriction is a powerful tool to map out such functionally relevant states that are invisible to standard techniques of structure determination.
Interestingly, the data in Figure 5 indicate that an exchange process also perturbs the chemical shift of the Cyt-10 C4′ resonance. This nucleus falls in the ribose ring of a Watson–Crick base pair adjoining the internal loop, for which repuckering dynamics are not necessarily expected, and comparable dispersion effects are not observed for the C2′ resonance of this nucleotide. This apparent discrepancy may be resolved by reference to the adjoining Gua-9 residue, whose base is extruded from the helix via a sharp bend of the backbone, forms no stable hydrogen bonds in solution, and is poorly ordered in the solution structure (Hoogstraten et al. 1998). A LNA substitution at this site resulted in a dramatic (up to 20-fold) gain-of-function phenotype, strongly indicating functionally relevant dynamics at Gua-9 (Julien et al. 2008). A reorientation of the backbone to a minor conformer at this site could put the base of either residue Gua-9 or Ade-8 in position to supply a strong ring-current chemical shift to the Cyt-10 C4′ nucleus. Thus, a strong Δω effect and resulting Rex contribution at this site could plausibly arise from movements of these bases relative to a static Cyt-10 ribose moiety, and we tentatively ascribe the enhanced relaxation at Cyt-10 C4′ (Fig. 5) to this indirect exchange effect.
The bicyclic N-MCC nucleotide modification was designed to restrict the nucleotide to the C2′-exo conformation (approximately 342° on the pseudorotation cycle), closely resembling the C3′-endo or “North” conformation (0° to 36°) characteristic of A-form helices (cf. Fig. 2; Altona and Sundaralingam 1972). Substantial experimental evidence for both ribo- and deoxyribo-MC nucleotides in a wide variety of contexts supports the assumption that these modifications do not perturb oligonucleotide structure beyond the intended restriction to a particular region of the pseudorotation cycle (Marquez et al. 1997, 1998, 2004; Maderia et al. 2007; Terrazas et al. 2011; Ketkar et al. 2012a,b). In a 1.8 Å crystal structure of two north-methanocarba-thymidine (N-MCT) nucleotides incorporated into a self-complementary DNA dodecamer, however, an unexpected left-handed stem–loop formed, with the N-dMCT residues located within the loop and taking up pseudorotation angles of 48°–52° (Pallan et al. 2012). The structural context of these nucleotides varies from the present study in important ways, including the lack of the 2′-hydroxyl group on both the methanocarba and native nucleotides and the presence of the unusual left-handed structural motif. This study thus serves as an important reminder that the structural effects of nucleotide modifications do depend to some extent on context. In the present study, the N-MCC nucleotide is incorporated at a site where the native structure stabilizes a C3′-endo pseudorotation, and we therefore believe that our interpretation of the modification as stabilizing the observed ground-state conformation of the ribozyme while preventing the formation of a minor conformer with Cyt-6 in the C2′-endo state is fully justified.
Several existing lines of evidence support the model that the most-populated structure of the leadzyme, as observed by NMR and crystallography, does not correspond to the active form of the motif, and that fluctuations to a minor active conformer (ES* in Fig. 1) are necessary for self-cleavage. This evidence includes a severe mismatch between the high-resolution structures and the extant biochemical data in the system (Pan et al. 1994; Chartrand et al. 1997), as well as the failure of the active-site groups to adopt a catalytically relevant “in-line attack” conformation in the ground-state solution structure. In addition, two modifications that destabilize the observed structure, an 8-bromo-guanosine substitution at position 24 that tends to favor syn glycosidic conformation and an LNA modification at position 9 that disrupts the native C2′-endo state, have been found to substantially increase the self-cleavage rate (Yajima et al. 2007; Julien et al. 2008). In the simplified two-state model of Figure 1, these modifications are taken as perturbing the conformational equilibrium in favor of the active ES* state, whereas the N-MCC-6 species analyzed herein has the opposite effect of opposing the formation of ES*. In practice, the ES and ES* states presumably correspond to two families of conformers that are distinguished by their relative abilities to approach the chemical transition state without further thermally activated conformational transitions. The data in this paper suggest that the sampling of the Cyt-6 C2′-endo sugar pucker corresponds to a transition from the ES to the ES* state, rather than from one conformer to another within the ES ensemble.
In cases where NMR-detected exchange lifetimes at multiple sites quantitatively coincide, parsimony strongly supports a single concerted exchange process affecting all of the affected sites, as has been seen in RNase A (Cole and Loria 2002; Kovrigin et al. 2003) and the GCAA tetraloop (Johnson and Hoogstraten 2008). In the present work, the precision of exchange lifetime determination is insufficient to judge whether those observed at multiple sites are in quantitative agreement, although they can be said to fall in the same order of magnitude. The simplest explanation for our data is the existence of a minor conformer in which Cyt-6 is C2′-endo and the chemical shifts of both Cyt-6 C2′ and C4′ are modulated accordingly, but other more complicated motional models, including indirect effects from reorientation of the base of Gua-7, cannot be ruled out. Interestingly, an earlier relaxation-dispersion study of side-chain 13C resonances in the leadzyme found multiple processes near the active site, including an ∼40 µs lifetime perturbation of Ade-25 C2 that was attributed to base-pair opening dynamics exposing the protonated adenine ring to solvent exchange (Hoogstraten et al. 2000). In addition, variants in which the ground-state A25:C6 base pair is substituted with more stable base pairs including the isosteric G:U are inactive (Legault et al. 1998), whereas activity is maintained if that base pair is weakened by modifications including an abasic structure at residue 25 (Chartrand et al. 1997). Based on these data, we have inferred that the A25:C6 base pair is broken, likely by base flipping at Cyt-6, as part of the transition from ES to the active conformation ES* (Hoogstraten et al. 2000). Because relaxation data do not directly provide correlations between observations at various sites, we cannot rigorously conclude that the observed base dynamics are coupled to the Cyt-6 sugar pucker dynamics analyzed here. Nevertheless, it is tempting to speculate that the opening of the protonated A25:C6 base pair releases structural constraints on the backbone and thereby permits the adoption of the C2′-endo pucker that the current results imply is central to the formation of the catalytically relevant conformation. From this perspective, it is interesting that a base flipping process consisting of a concerted conformational shift from C3′-endo/anti to C2′-endo/syn can result in chemical shift changes of up to 9–10 ppm (Ebrahimi et al. 2001; Rossi and Harbison 2001). Shift changes of this magnitude could yield the Rex effects seen in Figures 5 and 6 even for relatively low (5% or less) populations of the minor activated state, consistent with the results of LNA probing data (Julien et al. 2008).
MATERIALS AND METHODS
NMR sample preparation
2′-4′-13C2 nucleotides were synthesized in E. coli in metabolically driven fashion using minor modifications of published procedures (Johnson et al. 2006; Hoogstraten and Johnson 2008; Johnson and Hoogstraten 2008). A kanamycin-resistant zwf strain of E. coli (JW1841-1; Coli Genetic Stock Center, Yale) was used to combine the glucose-6-phosphate dehydrogenase null (zwf) genotype with antibiotic selection. Selective 13C labeling of ribonucleotide ribose groups at the 2′ and 4′ sites was confirmed using NMR spectroscopy of monomeric nucleotides. Purified 2′–4′-13C2-CTP was incorporated into an in vitro transcription reaction from a synthetic DNA template to yield a 30 nt leadzyme construct (Fig. 1) which has been extensively studied by NMR (Legault and Pardi 1997; Hoogstraten et al. 1998, 2000; Legault et al. 1998). RNA was purified under native conditions using a preparative Sephadex-75 column (McKenna et al. 2007). Final buffer conditions were 0.6 mM RNA, 10 mM sodium phosphate, 100 mM sodium chloride, 200 µM EDTA, and trace DSS at pH 5.5. The sample was repeatedly lyophilized in 99.8% D2O, suspended in 255 µL of 99.96% D2O to a final RNA concentration of 0.6 mM, and placed in a reduced-volume NMR tube (Shigemi).
NMR spectroscopy and data analysis
Peak assignments for the sample were obtained via comparison with published values (Legault et al. 1998) and verified with a three-dimensional NOESY-HSQC (not shown). Spin-relaxation data were acquired on a Varian UNITY INOVA 600 MHz (13C at 150 MHz) spectrometer at 25°C using standard pulse sequences provided by the manufacturer (VNMR/Biopack) (Lukavsky and Puglisi 2001). Transverse relaxation curves were acquired at ten spin-lock power settings with γB1/2π varying from 1.5 to 6.0 kHz. Twelve time points varying up to 120 msec for R1ρ and 13 time points between 10 and 1200 msec for R1 were acquired in random order, with one time point repeated three times for error analysis. At some powers, additional data was acquired at an array of shorter delays for analysis of unusually fast-relaxing peaks.
Data analysis
NMR spectra were processed using NMRPipe 8.9 (Delaglio et al. 1995) and then exported into Sparky 3.115 for relaxation analysis. Peak heights derived from Sparky were fit to a two-parameter single-exponential decay in IgorPro 6.0 (WaveMetrics) to determine the reported relaxation rates. Errors were as reported by the fitting software. The fast-relaxing C2′ peak of cytidine-6 was partially overlapped with a slower-relaxing peak, complicating analysis. Therefore, a more sophisticated data fitting procedure within NMRPipe was used for this resonance. Spectra were transformed using a Lorentz-Gauss window function, compiled into a pseudo-3D data matrix, and the overlapping peaks were simultaneously fit via modeling as a 3D product of two 1D Gaussian line shapes and a time evolution profile. The resulting evolution curves were fit to exponential relaxation with Monte Carlo estimation of parameter error. Application of this procedure to selected nonoverlapped peaks produced results identical within error to regular exponential fitting from peak height data (not shown). R1ρ values were corrected for offset effects using
where θ = tan−1 (ω1/Ω) and Ω is the peak offset from the 13C carrier frequency (Palmer 2004; Palmer and Massi 2006). Preliminary CPMG spectra showed no evidence for additional dispersion effects on the millisecond time scale (not shown) and detailed analysis of this type was therefore not pursued. For the exchange rates observed here, the exchange lifetimes are substantially below the millisecond-scale reciprocal chemical shift changes. Thus, neither CPMG nor R1ρ data can separately determine chemical shift changes and state populations, and the simple expressions in Equations 1–3 are appropriate (Palmer 2004).
Ribozyme cleavage assays
Leadzyme RNA samples used for kinetic studies were derived via solid-phase synthesis incorporating a 5′-fluorescein tag. The following sequence was used: 5′-Fl- GCGACC6GAGCCAGCGAAAGUUGGGAGUCGC-3′.
For C6-North samples, the indicated cytidine at position 6 was replaced with a cytidine carrying a bicyclo[3.1.0]hexane (North-methanocarba-cytidine, N-MCC) pseudosugar modification. The wild-type sequence was chemically synthesized by the W.M. Keck Foundation Biotechnology Resource laboratory at Yale University. The RNA was deprotected using tetrabutylammonium fluoride (TBAF) solution in tetrahydrofuran (THF) for 6 h at room temperature. The bicyclo[3.1.0]hexane cytidine phosphoramidite was synthesized as previously described (Terrazas et al. 2011) and incorporated into the leadzyme sequence with TBAF deprotection by Dr. Montserrat Terrazas at the Barcelona Institute of Science and Technology. Deprotected RNAs were desalted on Sephadex G25 (Sigma-Aldrich) and then exchanged repeatedly against RNase-free double deionized water followed by 15 mM MOPS pH 7.0 using Amicon Ultra 15 centrifugal filtration units. After exchange was completed, RNAs were analyzed for purity on a 20% denaturing polyacrylamide gel.
RNA concentrations were determined with a single-stranded extinction coefficient (ε) of 2.95 × 105 M−1cm−1 for both wild-type and N-MCC-6 modified sequences. RNAs were renatured via heating at 85°C for 2 min then allowed to cool slowly to room temperature. Samples were equilibrated at the assay temperature of 27°C for 15 min, the reaction was initiated by addition of freshly prepared Pb(OAc)2, and data was collected for 3 h. Typical 50 µL cleavage reactions contained 1 µM RNA, 15 mM MOPS pH 7.0, and various concentrations of Pb(OAc)2 (10, 25, 50, 100, 200, and 300 µM). For each time point, a 5 µL aliquot was removed and quenched via addition of 5 µL of a 9 M urea/50 mM EDTA mixture followed by freezing on dry ice. Reactions were analyzed on denaturing 20% polyacrylamide gels with visualization on a Bio-Rad Fluor-S MAX multiImager with excitation at 494 nm, measurement of emission at 518 nm, and band quantification using Quantity One software (BioRad). Intensities for substrate and 5′ cleavage product were corrected for background and reported as percent of substrate remaining (substrate/[substrate + product]). Time points representing the average of at least two runs were fit to single and double exponential functions using the software Igor Pro 5.04B. For the wild-type species, a standard F-test of additional terms (Bevington and Robinson 2003) yielded a statistically significant improvement in fit according to using a double exponential in most cases, presumably indicating the presence of a misfolded, slowly reacting fraction (typically 35%–40%). The faster of the two rate constants is plotted in Figure 7. For the N-MCC modified leadzyme, no reproducible multiexponential behavior was observed at any [Pb2+], and the reported kobs values therefore derive from a single-exponential fit.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
The authors are grateful to Dr. Frank Delaglio (National Institute of Standards and Technology) for assistance with data processing and Professor Montserrat Terrazas (Institute for Research in Biomedicine [IRB]-Barcelona) for synthesis of the N-MCC-containing oligonucleotide. This work was funded in part by Center for Cancer Research, National Cancer Institute, National Institutes of Health (V.E.M.) and by a National Science Foundation-BIO award (MCB-1715816) to C.G.H.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.067579.118.
REFERENCES
- Al-Hashimi HM, Walter NG. 2008. RNA dynamics: it is about time. Curr Opin Struct Biol 18: 321–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altona C, Sundaralingam M. 1972. Conformational analysis of the sugar ring in nucleosides and nucleotides. A new description using the concept of pseudorotation. J Am Chem Soc 94: 8205–8212. [DOI] [PubMed] [Google Scholar]
- Ban D, Smith CA, de Groot BL, Griesinger C, Lee D. 2017. Recent advances in measuring the kinetics of biomolecules by NMR relaxation dispersion spectroscopy. Arch Biochem Biophys 628: 81–91. [DOI] [PubMed] [Google Scholar]
- Bevington PR, Robinson DK. 2003. Data reduction and error analysis for the physical sciences, 3rd ed McGraw-Hill, New York. [Google Scholar]
- Bothe JR, Nikolova EN, Eichhorn CD, Chugh J, Hansen AL, Al-Hashimi HM. 2011. Characterizing RNA dynamics at atomic resolution using solution-state NMR spectroscopy. Nat Methods 8: 919–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z, Tinoco I Jr. 1996. Solution structure of loop A from the hairpin ribozyme from tobacco ringspot virus satellite. Biochemistry 35: 6026–6036. [DOI] [PubMed] [Google Scholar]
- Chartrand P, Usman N, Cedergren R. 1997. Effect of structural modifications on the activity of the leadzyme. Biochemistry 36: 3145–3150. [DOI] [PubMed] [Google Scholar]
- Chen B, LeBlanc R, Dayie TK. 2016. SAM-II riboswitch samples at least two conformations in solution in the absence of ligand: implications for recognition. Ang Chem Int Ed 55: 2724–2727. [DOI] [PubMed] [Google Scholar]
- Choi Y, Moon HR, Yoshimura Y, Marquez VE. 2003. Recent advances in the synthesis of conformationally locked nucleosides and their success in probing the critical question of conformational preferences by their biological targets. Nucleosides Nucleotides Nucleic Acids 22: 547–557. [DOI] [PubMed] [Google Scholar]
- Clay MC, Ganser LR, Merriman DK, Al-Hashimi HM. 2017. Resolving sugar puckers in RNA excited states exposes slow modes of repuckering dynamics. Nucleic Acids Res 45: e134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochrane JC, Strobel SA. 2008. Catalytic strategies of self-cleaving ribozymes. Acc Chem Res 41: 1027–1035. [DOI] [PubMed] [Google Scholar]
- Cole R, Loria JP. 2002. Evidence for flexibility in the function of ribonuclease A. Biochemistry 41: 6072–6081. [DOI] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. 1995. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6: 277–293. [DOI] [PubMed] [Google Scholar]
- Delano WL. 2002. The PyMOL molecular graphics system. DeLano Scientific, Palo Alto, CA. [Google Scholar]
- Dethoff EA, Petzold K, Chugh J, Casiano-Negroni A, Al-Hashimi HM. 2012. Visualizing transient low-populated structures of RNA. Nature 491: 724–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahimi M, Rossi P, Rogers C, Harbison GS. 2001. Dependence of 13C NMR chemical shifts on conformations of RNA nucleosides and nucleotides. J Magn Reson 150: 1–9. [DOI] [PubMed] [Google Scholar]
- Emilsson GM, Nakamura S, Roth A, Breaker RR. 2003. Ribozyme speed limits. RNA 9: 907–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteban JA, Banerjee AR, Burke JM. 1997. Kinetic mechanism of the hairpin ribozyme. J Biol Chem 272: 13629–13639. [DOI] [PubMed] [Google Scholar]
- Fedor MJ, Williamson JR. 2005. The catalytic diversity of RNAs. Nat Rev Mol Cell Biol 6: 399–412. [DOI] [PubMed] [Google Scholar]
- Furtig B, Schnieders R, Richter C, Zetzsche H, Keyhani S, Helmling C, Kovacs H, Schwalbe H. 2016. Direct 13C-detected NMR experiments for mapping and characterization of hydrogen bonds in RNA. J Biomol NMR 64: 207–221. [DOI] [PubMed] [Google Scholar]
- Gaines CS, York DM. 2016. Ribozyme catalysis with a twist: active state of the twister ribozyme in solution predicted from molecular simulation. J Am Chem Soc 138: 3058–3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KA, Lunse CE, Li S, Brewer KI, Breaker RR. 2015. Biochemical analysis of pistol self-cleaving ribozymes. RNA 21: 1852–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldenbrand H, Janowski PA, Giambasu G, Giese TJ, Wedekind JE, York DM. 2014. Evidence for the role of active site residues in the hairpin ribozyme from molecular simulations along the reaction path. J Am Chem Soc 136: 7789–7792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogstraten CG, Johnson JE Jr. 2008. Metabolic labeling: taking advantage of bacterial pathways to prepare spectroscopically useful isotope patterns in proteins and nucleic acids. Concepts Magn Reson A 32: 34–55. [Google Scholar]
- Hoogstraten CG, Sumita M. 2013. Lead and RNA. In Encyclopedia of metalloproteins (ed. Uversky V, Kretsinger RH, Permyakov EA), pp. 1166–1173. Springer, New York. [Google Scholar]
- Hoogstraten CG, Legault P, Pardi A. 1998. NMR solution structure of the lead-dependent ribozyme: evidence for dynamics in RNA catalysis. J Mol Biol 284: 337–350. [DOI] [PubMed] [Google Scholar]
- Hoogstraten CG, Wank JR, Pardi A. 2000. Active site dynamics in the lead-dependent ribozyme. Biochemistry 39: 9951–9958. [DOI] [PubMed] [Google Scholar]
- Jeong LS, Marquez VE, Yuan CS, Borchardt RT. 1995. 4′,1′a-methanocarbocyclic adenosine analogues as potential inhibitors of S-adenosylhomocysteine hydrolase. Heterocycles 41: 2651–2656. [Google Scholar]
- Johnson JE Jr, Hoogstraten CG. 2008. Extensive backbone dynamics in the GCAA RNA tetraloop analyzed using 13C NMR spin relaxation and specific isotope labeling. J Am Chem Soc 130: 16757–16769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JE Jr, Julien KR, Hoogstraten CG. 2006. Alternate-site isotopic labeling of ribonucleotides for NMR studies of ribose conformational dynamics in RNA. J Biomol NMR 35: 261–274. [DOI] [PubMed] [Google Scholar]
- Julien KR, Sumita M, Chen P-H, Laird-Offringa IA, Hoogstraten CG. 2008. Conformationally restricted nucleotides as a probe of structure-function relationships in RNA. RNA 14: 1632–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketkar A, Zafar MK, Banerjee S, Marquez VE, Egli M, Eoff RL. 2012a. Differential furanose selection in the active sites of archaeal DNA polymerases probed by fixed-conformation nucleotide analogues. Biochemistry 51: 9234–9244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketkar A, Zafar MK, Banerjee S, Marquez VE, Egli M, Eoff RL. 2012b. A nucleotide-analogue-induced gain of function corrects the error-prone nature of human DNA polymerase iota. J Am Chem Soc 134: 10698–10705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovrigin EL, Cole R, Loria JP. 2003. Temperature dependence of the backbone dynamics of ribonuclease A in the ground state and bound to the inhibitor 5′-phosphothymidine (3′–5′)pyrophosphate adenosine 3′-phosphate. Biochemistry 42: 5279–5291. [DOI] [PubMed] [Google Scholar]
- Le MT, Brown RE, Simon AE, Dayie TK. 2015. In vivo, large-scale preparation of uniformly 15N- and site-specifically 13C-labeled homogeneous, recombinant RNA for NMR studies. Methods Enzymol 565: 495–535. [DOI] [PubMed] [Google Scholar]
- LeBlanc RM, Longhini AP, Le Grice SFJ, Johnson BA, Dayie TK. 2017. Combining asymmetric 13C-labeling and isotopic filter/edit NOESY: a novel strategy for rapid and logical RNA resonance assignment. Nucleic Acids Res 45: e146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TS, Giambasu GM, Sosa CP, Martick M, Scott WG, York DM. 2009. Threshold occupancy and specific cation binding modes in the hammerhead ribozyme active site are required for active conformation. J Mol Biol 388: 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T-S, Radak BK, Harris ME, York DM. 2016. A two-metal-ion-mediated conformational switching pathway for HDV ribozyme activation. ACS Catal 6: 1853–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legault P, Pardi A. 1997. Unusual dynamics and pKa shift at the active site of a lead-dependent ribozyme. J Am Chem Soc 119: 6621–6628. [Google Scholar]
- Legault P, Hoogstraten CG, Metlitzky E, Pardi A. 1998. Order, dynamics, and metal binding in the lead-dependent ribozyme. J Mol Biol 284: 325–335. [DOI] [PubMed] [Google Scholar]
- Lemieux S, Chartrand P, Cedergren R, Major F. 1998. Modeling active RNA structures using the intersection of conformational space: application to the lead-activated ribozyme. RNA 4: 739–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leulliot N, Varani G. 2001. Current topics in RNA-protein recognition: control of specificity and biological function through induced fit and conformational capture. Biochemistry 40: 7947–7956. [DOI] [PubMed] [Google Scholar]
- Li L, Szostak JW. 2014. The free energy landscape of pseudorotation in 3′–5′ and 2′–5′ linked nucleic acids. J Am Chem Soc 136: 2858–2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Lunse CE, Harris KA, Breaker RR. 2015. Biochemical analysis of hatchet self-cleaving ribozymes. RNA 21: 1845–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wilson TJ, McPhee SA, Lilley DM. 2014. Crystal structure and mechanistic investigation of the twister ribozyme. Nat Chem Biol 10: 739–744. [DOI] [PubMed] [Google Scholar]
- Liu Y, Wilson TJ, Lilley DMJ. 2017. The structure of a nucleolytic ribozyme that employs a catalytic metal ion. Nat Chem Biol 13: 508–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longhini AP, LeBlanc RM, Becette O, Salguero C, Wunderlich CH, Johnson BA, D'Souza VM, Kreutz C, Dayie TK. 2016a. Chemo-enzymatic synthesis of site-specific isotopically labeled nucleotides for use in NMR resonance assignment, dynamics and structural characterizations. Nucleic Acids Res 44: e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longhini AP, LeBlanc RM, Dayie TK. 2016b. Chemo-enzymatic labeling for rapid assignment of RNA molecules. Methods 103: 11–17. [DOI] [PubMed] [Google Scholar]
- Lukavsky PJ, Puglisi JD. 2001. RNAPack: an integrated NMR approach to RNA structure determination. Methods 25: 316–332. [DOI] [PubMed] [Google Scholar]
- Maderia M, Wu J, Bax A, Shenoy S, O'Keefe B, Marquez VE, Barchi JJ Jr. 2005. Engineering DNA topology with locked nucleosides: a structural study. Nucleosides Nucleotides Nucleic Acids 24: 687–690. [DOI] [PubMed] [Google Scholar]
- Maderia M, Shenoy S, Van QN, Marquez VE, Barchi JJ. 2007. Biophysical studies of DNA modified with conformationally constrained nucleotides: comparison of 2′-exo (north) and 3′-exo (south) ‘locked’ templates. Nucleic Acids Res 35: 1978–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier MA, Choi Y, Gaus H, Barchi JJ Jr, Marquez VE, Manoharan M. 2004. Synthesis and characterization of oligonucleotides containing conformationally constrained bicyclo[3.1.0]hexane pseudosugar analogs. Nucleic Acids Res 32: 3642–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquez VE, Ezzitouni A, Siddiqui MA, Russ P, Ikeda H, George C. 1997. Conformational analysis of nucleosides constructed on a bicyclo[3.1.0]hexane template. Structure-antiviral activity analysis for the northern and southern hemispheres of the pseudorotational cycle. Nucleosides Nucleotides 16: 1431–1434. [Google Scholar]
- Marquez VE, Ezzitouni A, Russ P, Siddiqui MA, Ford H, Feldman RJ, Mitsuya H, George C, Barchi JJ. 1998. HIV-1 reverse transcriptase can discriminate between two conformationally locked carbocyclic AZT triphosphate analogues. J Am Chem Soc 120: 2780–2789. [Google Scholar]
- Marquez VE, Ben-Kasus T, Barchi JJ, Green KM, Nicklaus MC, Agbaria R. 2004. Experimental and structural evidence that herpes 1 kinase and cellular DNA polymerase(s) discriminate on the basis of sugar pucker. J Am Chem Soc 126: 543–549. [DOI] [PubMed] [Google Scholar]
- McKenna SA, Kim I, Puglisi EV, Lindhout DA, Aitken CE, Marshall RA, Puglisi JD. 2007. Purification and characterization of transcribed RNAs using gel filtration chromatography. Nat Protoc 2: 3270–3277. [DOI] [PubMed] [Google Scholar]
- Nelissen FH, van Gammeren AJ, Tessari M, Girard FC, Heus HA, Wijmenga SS. 2008. Multiple segmental and selective isotope labeling of large RNA for NMR structural studies. Nucleic Acids Res 36: e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelissen FHT, Tessari M, Wijmenga SS, Heus HA. 2016. Stable isotope labeling methods for DNA. Prog Nucl Magn Reson Spectrosc 96: 89–108. [DOI] [PubMed] [Google Scholar]
- Pallan PS, Marquez VE, Egli M. 2012. The conformationally constrained N-methanocarba-dT analogue adopts an unexpected C4′-exo sugar pucker in the structure of a DNA hairpin. Biochemistry 51: 2639–2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer AG III. 2004. NMR characterization of the dynamics of biomacromolecules. Chem Rev 104: 3623–3640. [DOI] [PubMed] [Google Scholar]
- Palmer AG III, Massi F. 2006. Characterization of the dynamics of biomacromolecules using rotating-frame spin relaxation NMR spectroscopy. Chem Rev 106: 1700–1719. [DOI] [PubMed] [Google Scholar]
- Pan T, Uhlenbeck OC. 1992. A small metalloribozyme with a two-step mechanism. Nature 358: 560–563. [DOI] [PubMed] [Google Scholar]
- Pan T, Dichtl B, Uhlenbeck OC. 1994. Properties of an in vitro selected Pb2+ cleavage motif. Biochemistry 33: 9561–9565. [DOI] [PubMed] [Google Scholar]
- Perez-Canadillas JM, Varani G. 2001. Recent advances in RNA-protein recognition. Curr Opin Struct Biol 11: 53–58. [DOI] [PubMed] [Google Scholar]
- Qi X, Xia T. 2011. Structure, dynamics, and mechanism of the lead-dependent ribozyme. Biomol Concepts 2: 305–314. [DOI] [PubMed] [Google Scholar]
- Rambo RP, Doudna JA. 2004. Assembly of an active group II intron-maturase complex by protein dimerization. Biochemistry 43: 6486–6497. [DOI] [PubMed] [Google Scholar]
- Ravera E, Salmon L, Fragai M, Parigi G, Al-Hashimi H, Luchinat C. 2014. Insights into domain-domain motions in proteins and RNA from solution NMR. Acc Chem Res 47: 3118–3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy J, Pratihar S, Ban D, Frischkorn S, Becker S, Griesinger C, Lee D. 2018. Simultaneous determination of fast and slow dynamics in molecules using extreme CPMG relaxation dispersion experiments. J Biomol NMR 70: 1–9. [DOI] [PubMed] [Google Scholar]
- Rossi P, Harbison GS. 2001. Calculation of 13C chemical shifts in RNA nucleosides: structure-13C chemical shift relationships. J Magn Reson 151: 1–8. [DOI] [PubMed] [Google Scholar]
- Roth A, Weinberg Z, Chen AG, Kim PB, Ames TD, Breaker RR. 2014. A widespread self-cleaving ribozyme class is revealed by bioinformatics. Nat Chem Biol 10: 56–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupert PB, Ferré-D'Amaré AR. 2001. Crystal structure of a hairpin ribozyme-inhibitor complex with implications for catalysis. Nature 410: 780–786. [DOI] [PubMed] [Google Scholar]
- Salmon L, Yang S, Al-Hashimi HM. 2014. Advances in the determination of nucleic acid conformational ensembles. Annu Rev Phys Chem 65: 293–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saneyoshi H, Mazzini S, Avino A, Portella G, Gonzalez C, Orozco M, Marquez VE, Eritja R. 2009. Conformationally rigid nucleoside probes help understand the role of sugar pucker and nucleobase orientation in the thrombin-binding aptamer. Nucleic Acids Res 37: 5589–5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnieders R, Richter C, Warhaut S, de Jesus V, Keyhani S, Duchardt-Ferner E, Keller H, Wohnert J, Kuhn LT, Breeze AL, et al. 2017. Evaluation of 15N-detected H-N correlation experiments on increasingly large RNAs. J Biomol NMR 69: 31–44. [DOI] [PubMed] [Google Scholar]
- Scott WG, Murray JB, Arnold JRP, Stoddard BL, Klug A. 1996. Capturing the structure of a catalytic RNA intermediate: the hammerhead ribozyme. Science 274: 2065–2069. [DOI] [PubMed] [Google Scholar]
- Shajani Z, Varani G. 2008. 13C Relaxation studies of the DNA target sequence for HhaI methyltransferase reveal unique motional properties. Biochemistry 47: 7617–7625. [DOI] [PubMed] [Google Scholar]
- Soukup GA, Breaker RR. 1999. Relationship between internucleotide linkage geometry and the stability of RNA. RNA 5: 1308–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrazas M, Avino A, Siddiqui MA, Marquez VE, Eritja R. 2011. A direct, efficient method for the preparation of siRNAs containing ribo-like North bicyclo[3.1.0]hexane pseudosugars. Org Lett 13: 2888–2891. [DOI] [PubMed] [Google Scholar]
- Walter NG. 2009. The blessing and curse of RNA dynamics: past, present, and future. Methods 49: 85–86. [DOI] [PubMed] [Google Scholar]
- Wang X, Woods RJ. 2016. Insights into furanose solution conformations: beyond the two-state model. J Biomol NMR 64: 291–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedekind JE, McKay DB. 1999. Crystal structure of a lead-dependent ribozyme revealing metal binding sites relevant to catalysis. Nat Struct Biol 6: 261–268. [DOI] [PubMed] [Google Scholar]
- Wedekind JE, McKay DB. 2003. Crystal structure of the leadzyme at 1.8 Å resolution: metal ion binding and the implications for catalytic mechanism and allo site ion regulation. Biochemistry 42: 9554–9563. [DOI] [PubMed] [Google Scholar]
- Weinberg Z, Kim PB, Chen TH, Li S, Harris KA, Lunse CE, Breaker RR. 2015. New classes of self-cleaving ribozymes revealed by comparative genomics analysis. Nat Chem Biol 11: 606–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westhof E, Hermann T. 1999. Leadzyme RNA catalysis. Nat Struct Biol 6: 208–209. [DOI] [PubMed] [Google Scholar]
- Wijmenga SS, van Buuren BNM. 1998. The use of NMR methods for conformational studies of nucleic acids. Prog Nucl Magn Reson Spectrosc 32: 287–387. [Google Scholar]
- Williamson JR. 2000. Induced fit in RNA-protein recognition. Nat Struct Biol 7: 834–837. [DOI] [PubMed] [Google Scholar]
- Wilson TJ, Nahas M, Araki L, Harusawa S, Ha T, Lilley DM. 2007. RNA folding and the origins of catalytic activity in the hairpin ribozyme. Blood Cells Mol Dis 38: 8–14. [DOI] [PubMed] [Google Scholar]
- Wu Z, Maderia M, Barchi JJ Jr, Marquez VE, Bax A. 2005. Changes in DNA bending induced by restricting nucleotide ring pucker studied by weak alignment NMR spectroscopy. Proc Natl Acad Sci 102: 24–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yajima R, Proctor DJ, Kierzek R, Kierzek E, Bevilacqua PC. 2007. A conformationally restricted guanosine analog reveals the catalytic relevance of three structures of an RNA enzyme. Chem Biol 14: 23–30. [DOI] [PubMed] [Google Scholar]
- Zhao B, Zhang Q. 2015. Measuring residual dipolar couplings in excited conformational states of nucleic acids by CEST NMR spectroscopy. J Am Chem Soc 137: 13480–13483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Hansen AL, Zhang Q. 2014. Characterizing slow chemical exchange in nucleic acids by carbon CEST and low spin-lock field R1ρ NMR spectroscopy. J Am Chem Soc 136: 20–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.