

Abstract
RNA editing diversifies genomically encoded information to expand the complexity of the transcriptome. In ectothermic organisms, including Drosophila and Cephalopoda, where body temperature mirrors ambient temperature, decreases in environmental temperature lead to increases in A-to-I RNA editing and cause amino acid recoding events that are thought to be adaptive responses to temperature fluctuations. In contrast, endothermic mammals, including humans and mice, typically maintain a constant body temperature despite environmental changes. Here, A-to-I editing primarily targets repeat elements, rarely results in the recoding of amino acids, and plays a critical role in innate immune tolerance. Hibernating ground squirrels provide a unique opportunity to examine RNA editing in a heterothermic mammal whose body temperature varies over 30°C and can be maintained at 5°C for many days during torpor. We profiled the transcriptome in three brain regions at six physiological states to quantify RNA editing and determine whether cold-induced RNA editing modifies the transcriptome as a potential mechanism for neuroprotection at low temperature during hibernation. We identified 5165 A-to-I editing sites in 1205 genes with dynamically increased editing after prolonged cold exposure. The majority (99.6%) of the cold-increased editing sites are outside of previously annotated coding regions, 82.7% lie in SINE-derived repeats, and 12 sites are predicted to recode amino acids. Additionally, A-to-I editing frequencies increase with increasing cold-exposure, demonstrating that ADAR remains active during torpor. Our findings suggest that dynamic A-to-I editing at low body temperature may provide a neuroprotective mechanism to limit aberrant dsRNA accumulation during torpor in the mammalian hibernator.
Keywords: RNA-editing, hibernation, ADAR, temperature adaptation
INTRODUCTION
RNA editing promotes transcriptome diversity by expanding the coding capacity of the genome. The most common type of RNA editing is A-to-I editing, which is mediated by the adenosine deaminase that acts on RNA (ADAR) family of enzymes. ADARs recognize dsRNA structures of at least 23 base pairs (bp) and deaminate up to 50% or more of their adenosines (Bass and Weintraub 1988; Nishikura et al. 1991). Selective RNA editing impacts RNA structure and function in multiple ways including changing codons, altering splice site selection, and modifying regulation by miRNAs. A distinct mechanism targets long double-stranded RNA structures for hyperediting (Mannion et al. 2015; Walkley and Li 2017). Two broad roles for ADAR-mediated RNA editing: (i) altering codons and thus protein sequence and function, and (ii) restricting the accumulation of dsRNA structures, are particularly highlighted by their loss-of-function phenotypes in mammals (Walkley and Li 2017). The loss of ADARB1 in mice results in seizures and lethality 2–3 wk after birth due to loss of editing at a single recoding site in the glutamate receptor, GRIA2 (Brusa et al. 1995). In contrast, mice deficient for ADAR die at embryonic day 13.5 due to systemic activation of the innate immune response (Mannion et al. 2014). Consist with this, mutations in human ADAR result in the autoimmune disorder Aicardi–Goutieres syndrome, due to aberrant recognition of dsRNA and activation of innate immune signaling (Rice et al. 2012).
ADAR enzyme substrate specificity is sensitive to fluctuations in temperature because the stability of a dsRNA structure is influenced by temperature (Wan et al. 2012). As temperatures decrease, short dsRNA helices are stabilized, resulting in an increased number of potential ADAR substrates. In Drosophila and Cephlopod species, reduced environmental temperatures leads to decreased body temperature and increased RNA editing (Garrett and Rosenthal 2012; Savva et al. 2012; Buchumenski et al. 2017; Yablonovitch et al. 2017b). In flies, decreased temperatures result in increased editing at a recoding editing site in the ADAR mRNA itself, as well as increased editing at 55 additional protein coding (CDS) sites (Savva et al. 2012; Buchumenski et al. 2017). The absence of RNA editing results in perturbed acclimation to temperature changes, suggesting an adaptive function for temperature-sensitive RNA editing (Buchumenski et al. 2017). Similarly, octopus species that reside in colder temperatures have higher rates of RNA editing of potassium channels, which is an adaptation that alters Kv1 channel kinetics to improve function in the cold (Garrett and Rosenthal 2012). While temperature-sensitive A-to-I RNA editing has been described in these ectothermic animals, it is unknown if such temperature-sensitive RNA editing occurs in mammals, which typically maintain a constant high body temperature (Tb) despite environmental temperature fluctuations.
Hibernators provide a unique opportunity to assess whether temperature-dependent RNA-editing exists in mammals. Throughout winter, hibernating 13-lined ground squirrels cycle between extended periods of torpor at low Tb (>1 wk, ∼4°C), and short (<12 h) periods of arousal when Tb rapidly rises to the more typical mammalian temperature of ∼37°C (Fig. 1), despite constant, near freezing environmental temperatures.
FIGURE 1.
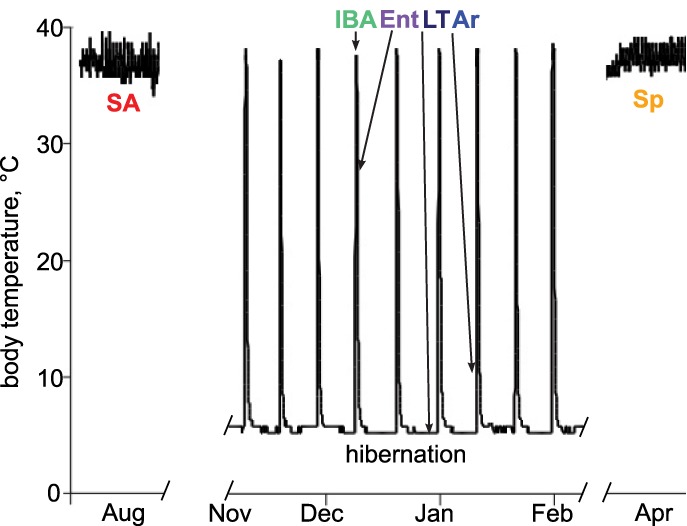
Thirteen-lined ground squirrels are heterothermic hibernators. Schematic of sampling time-points selected for RNA-seq analysis. Tb for sampled animals: InterBout Arousal (IBA) 34.1°C ± 2.9°C, Entrance (Ent) 25.4°C ± 1.8°C, Late Torpor (LT) 5.9°C ± 0.5°C, Arousing (Ar) 8.7°C ± 2.1°C. Tb was assumed to be 37°C for Summer Active (SA) and SpD (Spring Dark) animals due to observed animal activity.
In this study we exploit the natural temperature fluctuations of hibernation to determine, if similar to ectotherms, RNA editing is increased by low temperature in these seasonal heterotherms. Neural tissue from three brain regions was selected for analysis because ADAR activity is typically found at high levels in the mammalian brain, ADAR recoding events occur in neural enriched transcripts including ion-channels (Behm and Öhman 2016; Tan et al. 2017; Yablonovitch et al. 2017a), and ion channels and other brain transcripts have been shown to be adaptively edited to alter function across a range of temperatures in ectotherms (Palladino et al. 2000; Garrett and Rosenthal 2012; Yablonovitch et al. 2017b). Our data demonstrate that ADAR-mediated A-to-I RNA editing occurs at thousands of sites in the brain transcriptome of hibernating ground squirrels while they are torpid at low Tb.
RESULTS
Identification of RNA editing sites in squirrel brain tissues
Three brain regions, medulla, hypothalamus, and cerebrum, were selected for analysis of RNA editing due to their established roles in autonomic physiological processes, body temperature regulation and higher order cognitive processing, respectively. Furthermore, the medulla and hypothalamus remain relatively active compared to the cerebrum during the torpor phase of hibernation (Kilduff et al. 1990). A total of 90 paired-end, strand-specific RNA-seq libraries were generated from animals at six sampling points including two nonhibernating homeothermic stages: Summer Active (SA) and Spring Dark (SpD); and four of the heterothermic stages that characterize hibernation: Entrance (Ent), Late Torpor (LT), Arousing from torpor (Ar), and InterBout Arousal (IBA) (Figs. 1, 2A). These RNA-seq data were combined with data from testes and neonatal tissue samples to assemble a novel transcriptome that supplements the existing Ensembl annotations, which, because they are based largely on the automated recognition of protein sequence similarity, often lack complete mRNA annotations (e.g., missing 5′ and 3′ UTRs; see Materials and Methods). Over 86.1% ± 2.6% of the RNA-seq reads recovered from the brain samples were uniquely alignable, with 85.4% ± 1.2% of those uniquely mapped reads overlapping exon annotations defined in the novel transcriptome in contrast to only 53.4% ± 2.5% mapping to exons when annotated with the existing Ensembl transcriptome (Supplemental Fig. S1).
FIGURE 2.

A-to-I RNA editing is widespread and increases at low temperature during torpor and arousing from torpor. (A) RNA-seq library information and RNA editing site identification approach; see Figure 1 for full description of physiological stages. (B) Proportion of variants identified by the GATK pipeline for each possible mismatch type that have significantly changed editing frequencies across hibernation sampling timepoints determined via GLM analysis (FDR < 0.01). (C) Heatmap depicting editing frequencies defined as the percentage of G containing reads over the reads with either A or G. Group 1 or Group 2 sites were defined by k-means clustering (k = 2 classes), and within each Group, editing sites and individual samples were ordered by hierarchical clustering. (D) The number of editing sites detected by the hyperediting approach in each brain region (panel) and hibernation state (color) normalized to the total number of uniquely mapped reads. P-values were determined via ANOVA. (E) Heatmap of editing frequencies for sites identified by the hyperediting approach that are significantly changing across hibernation states. Groups were defined with k-means clustering (k = 3 classes) and sites were ordered with hierarchical clustering. (F) Editing index for significant sites identified by the GATK or hyperediting approach across sampled hibernation states. The editing index is defined as the sum of all G containing reads for all significant sites over the sum of all reads at significant sites. Lines are fit using a loess method and shaded area indicates standard error estimates derived from the t-distribution.
To identify and characterize RNA editing sites, single nucleotide variants were found using the Genome Analysis ToolKit (GATK) best practices for calling variants from RNA-seq (Fig. 2; Supplemental Fig. S2; DePristo et al. 2011). Variants were called individually for each of the 90 samples, merged, filtered based on quality metrics, and assigned to a strand based on the strandedness of the paired end alignment orientation (Fig. 2; Supplemental Fig. S2A). In total, A-to-G was the most common variant detected, accounting for 26.3% of the single nucleotide polymorphisms (SNPs) identified. But a substantial proportion identified were non-A-to-G variants (Supplemental Fig. S2B). Because the animals used for this study were obtained from largely outbred populations they are expected to harbor genetic variation; the excess of transition substitutions (C-T, G-A, T-C, A-G) over transversions is consistent with this expected genetic diversity (Supplemental Fig. S2B). Despite the large number of polymorphisms recovered, however, we observed a 2.2-fold increase of A-to-G variants over the expected distribution derived from T-to-C variants, an enrichment not observed for other mismatch pairs (Supplemental Fig. S2C) and consistent with ADAR-mediated RNA editing activity.
There are 59 A-to-I editing sites that are conserved across mammals (Pinto et al. 2014). We recovered 51 of these 59 A-to-I RNA editing sites in the set of A-to-G variants identified prior to filtering to exclude sites with extremely high or low variant allele frequencies (Supplemental Table S1). These sites included amino acid recoding sites in the glutamate receptor GRIA2 (Gln560Arg in ground squirrel, ENSSTOG00000024582) and the serotonin receptor, HTR2C (Ile156Val, Ile156Met, Asn158Ser, Ile160Val, in ground squirrel, ENSSTOG00000028990) (Supplemental Fig. S3). In addition, we also recovered several mouse (n = 100) and human (n = 212) editing sites previously identified in a curated database of RNA editing sites (Supplemental Table S1; Ramaswami and Li 2014). Together, these observations demonstrate that, in addition to detecting polymorphic sites in the ground squirrel brain transcriptome, we also recover previously characterized RNA editing events in the set of A-to-G variants.
The 13-lined ground squirrel genome currently lacks high quality transcriptome and SNP annotations, complicating accurate assignment of variants as bona fide RNA-editing events using RNA-seq data. Commonly used methods for excluding polymorphisms from candidate RNA editing sites rely on SNP databases and therefore cannot be applied to remove SNPs from our candidate RNA-editing sites (Ramaswami et al. 2013). However, we reasoned that SNPs should be randomly distributed throughout our study population and would therefore not be enriched within samples derived from a specific hibernation state. We enumerated the number of reads with variant and reference alleles for each site and applied a statistical model to identify variants whose allele frequencies are significantly associated with a hibernation state. We applied a general linear model based ANOVA-like test (edgeR) to each set of the 12 possible mismatch types (i.e., A-to-G, A-to-C, T-to-G, etc.) and assessed changes in variant allele read counts while accounting for changes in the coverage at the site. From the original set of A-to-G variants (n = 179,295), we identified subsets of variants that are significantly associated with a hibernation state in the medulla (n = 990), hypothalamus (n = 3591), and cerebrum (n = 2962), respectively (FDR < 0.01). Moreover, these variants accounted for greater than 94% of the significant sites identified in each of these regions (Fig. 2B). The remaining minority population of variants (G-to-A, C-to-T, T-to-C, and A-to-T) were primarily transition mismatch variants that likely represent polymorphisms that passed our statistical cutoffs due to the large number of variants tested for significance. SNPs are distributed equally across both DNA strands, in contrast to RNA editing sites, which are specifically localized to the transcribed strand. The lack of enrichment for T-to-C variants in the set of hibernation state-dependent variants demonstrates that the significant A-to-G variant sites are not likely to be SNPs, but rather result from A-to-I RNA editing events mediated by the ADAR family of deaminases.
Widespread increases in A-to-I RNA editing during torpor
The editing frequencies of the sites that differed significantly by hibernation stage were clustered and visualized to ascertain their pattern of editing. Two clear patterns were evident after sites were classified into two groups by k-means clustering (Fig. 2C; Supplemental Fig. S4A). Group 1 comprises 82.9% of the editing sites and displays increased editing frequencies during late torpor and arousing from torpor where Tb at collection was 5.9°C ± 0.5°C and 8.7°C ± 2.1°C, respectively (Fig. 2F). Samples from animals in late torpor were at depressed Tb (<30°C) for >85% of the length of the previous torpor bout (7.7 ± 2.2 d) compared to samples from arousing hibernators which were at low Tb for the entire torpor bout (10.2 ± 2.0 d), and still had low Tb (<9°C) but were beginning to rewarm. The highest editing index is reached as the animals begin to arouse from torpor, indicating that editing frequencies are increased by both low temperature and the amount of time spent with Tb at low temperature.
In contrast to Group 1 sites, Group 2 sites were not enriched for any particular hibernation stage, suggesting that sites in this group are either editing sites that have variable editing frequencies independent of hibernation physiology or are contaminating SNPs that were not eliminated by our statistical approach. We also clustered the variant allele frequencies for the small number (n = 127) of significant non-A-to-G variants and found that these variants were not enriched for any particular hibernation stage, consistent with these minor sites likely being SNPs (Supplemental Fig. S5).
An independent statistical analysis was also performed to validate the suitability of the edgeR approach for detecting changes in editing frequencies. An ANOVA test was performed using the editing frequencies directly, in contrast to the edgeR approach which uses reference and variant allele read counts. Significant editing sites identified by ANOVA (FDR < 0.01) also demonstrated widespread cold-enriched editing, and a large proportion of the sites were also identified by the edgeR approach, albeit with less sensitivity (Supplemental Fig. S6). Additionally, we performed a two group comparison using edgeR of the low Tb states (Ar and LT) compared to the high Tb states (IBA, SpD, and SA) (Supplemental Fig. S7). This analysis also confirmed the large number of editing sites with increased editing frequency in the cold states (99.1% ± 0.25% of significant sites), with a small number of sites (0.82% ± 0.25%) displaying decreased editing frequencies in the cold states (Supplemental Table S2).
Hyperediting events are enriched in Late Torpor and Arousing stages
To corroborate the observed increases in RNA-editing during torpor we used a second approach to identify RNA editing sites. ADAR enzymes can edit adenosines in a dsRNA helix to a degree that generates hyperedited RNAs that are unable to be aligned to the genome after sequencing (Carmi et al. 2011). To identify these hyperediting events, we used a previously described approach that can reliably recover A-to-I editing with a low rate of false-positive SNP identification (Supplemental Fig. S8; Porath et al. 2014). This approach identifies hyperedited sites by converting all As in each unaligned read and the genome to G, prior to alignment. For successfully aligned reads, the original genome and read sequence are recovered and alignments with a large number of mismatches are retained as hyperedited (see Materials and Methods). This approach has also been used to identify RNA editing events in nonmodel organisms that lack curated databases of genetic variation (Liscovitch-Brauer et al. 2017; Porath et al. 2017b).
To assess the specificity of this pipeline for recovering A-to-I editing events, we independently identified hyperedited sites for all possible mismatch types. The majority (93.7%) of hyperedited sites identified were A-to-G mismatches (Supplemental Fig. S9A), with the remaining sites comprising T-to-C and other transitions, with very few transversions. Thousands of unique A-to-G hyperediting sites were identified (n = 85,737); the number of these sites increased strongly during torpor and as animals aroused from torpor in all three brain regions (2.35-, 2.52-, and 2.21-fold increase in Ar compared to SA in the cerebrum, hypothalamus, and medulla, respectively), consistent with the increased editing frequency identified using the GATK + edgeR approach (Fig. 2D; Supplemental Fig. S9B). In contrast to the low Tb LT and Ar stages, the number of hyperediting sites did not fluctuate across the homeothermic stages (SpD, SA) or warm heterothermic stages (IBA, Ent) (Fig. 2D; Supplemental Fig. S9C,D).
We next computed the editing frequency for the hyperedited sites using only reads aligned to the unmodified genome (i.e., without changing A-to-G) and tested these sites for significant changes across hibernation states, in the same manner as the GATK + edgeR approach. We recovered 1634 significant hyperediting sites (FDR < 0.01) (Fig. 2E) and used k-means clustering to define three groups of hyperedited sites. Three groups were chosen for the hyperedited sites as three groups more clearly separated cold-enriched editing sites from non-cold-enriched sites (Fig. 2F; Supplemental Fig. S4B,C). The need for three groups is in part because the hyperedited sites are more enriched for editing sites and depleted for SNPs compared to the GATK approach (Supplemental Fig. S9A), and therefore do not segregate as easily into two classes of sites (Supplemental Fig. S4C). The sites in Groups 3 and 4 displayed similar increased editing frequencies when animals were in late torpor or arousing from torpor, whereas Group 5 sites were largely invariant across hibernation states, similar to the Group 2 sites identified by the GATK + edgeR pipeline (Fig. 2F).
The Group 3 and 4 editing sites identified by the hyperediting approach and the Group 1 sites identified by the GATK pipeline were designated as cold-enriched editing sites (Fig. 3A). The sites identified by both the GATK and the hyperediting pipeline that were not called significant in the ANOVA-like edgeR analysis were designated as a set of constitutively edited sites with editing frequency independent of temperature or hibernation physiology (Fig. 3B,C). We required these nonsignificant constitutively edited sites to be identified by both pipelines to reduce the likelihood of misclassifying a SNP as an editing site.
FIGURE 3.

Novel editing sites have an ADAR sequence motif and are enriched in regions with dsRNA character. (A) Euler diagram depicting the number of significant editing sites detected by the GATK or hyperediting pipeline. Cold-enriched sites were defined as the union of the two sets of significant sites. (B) Euler diagram illustrating the set of constitutively edited sites defined as the nonsignificant editing sites (FDR > 0.5) identified by both the GATK and the hyperediting pipeline. Only sites identified by both methods were selected to reduce the likelihood of misclassifying a SNP as an editing site. (C) Editing index for cold-enriched and constitutively edited sites. (D) Heatmap depicting editing sites conserved in either mouse or human, ordered by hierarchical clustering. Predicted locations of these sites are based on annotations in the RADAR database. (E) Sequence logo of sequences surrounding all cold-enriched or constitutive editing sites (±5 nt) or editing sites identified in mouse in the RADAR database. A set of randomly positioned sites was also generated by randomly selecting an A nucleotide position from transcripts containing editing sites. (F) Quantification of dsRNA character of each editing site via blastn by aligning the reverse complement of ±100 nt regions surrounding editing sites, to a ±2 kb region surrounding the editing site or to the end of the transcript. Sites with an e-value <0.1 with alignment over the editing site are considered paired dsRNA. As a negative control, the nonreverse complement, denoted as (−), of each ±100 nt region was also quantified and the number of second best alignments are shown.
Between brain regions there were differences in the number of cold-enriched editing sites identified by both the GATK and hyperediting pipeline (Supplemental Fig. S10A). Editing sites that were not identified in all three brain regions exhibited lower read coverage (Supplemental Fig. S10B) and sites in the medulla had the lowest read coverage, mirroring the lower number of sites found in the medulla compared to other regions. We attempted to identify editing sites that displayed strong evidence for region specific RNA editing, however when individual sites were examined, they displayed nonsignificant trends toward cold-enriched RNA editing, and in aggregate had an increased editing index at both LT and Ar stages, similar to the significant sites (Supplemental Fig. S10C,D). These observations suggest that the differences in the number of cold-enriched RNA editing sites between regions are in part explained by technical differences in detection limits and within sampling group variability between replicates.
Conservation of mRNA editing sites
We compared the set of common significantly edited sites (n = 5165) identified by the hyperediting or the GATK pipeline (Fig. 3A) to previously characterized mouse or human editing sites collected in the RADAR database of editing sites (Ramaswami and Li 2014). Twenty-seven cold-enriched sites were previously identified in humans, and four identified in mouse, with 2 of the 31 sites being identified in both the mouse and human sets. This set of 29 sites includes intronic editing sites in the glutamate receptors; GRIA2, GRIA3, and GRIK2, UTR editing sites in the RNA binding proteins PUM2 and DGCR8, and the recoding site in the DNA glycosylase NEIL1 (Lys242Arg) (Supplemental Table S1; Yeo et al. 2010). The editing frequencies for these 29 conserved sites were clustered, and additionally demonstrated the same pattern of enriched editing frequencies in late torpor and arousing animals as observed when clustering all of the significant editing sites (Fig. 3D).
Sequence and structural features of cold-enriched mRNA editing sites
We next examined the sequence and structural context of the cold-enriched sites and the set of editing sites whose editing frequencies did not vary across samples. Both cold-enriched and constitutively edited sites have a sequence preference for depletion of guanosine 5′ of the edited adenosine and enrichment for guanosine 3′ of the site, a pattern not observed for randomly selected adenosines (Fig. 3E). This sequence preference is similar to the sequence context preferred for ADAR enzymes for editing and to the sequence preferences for previously identified RNA editing sites in mouse and humans in the RADAR database (Eggington et al. 2011).
ADAR deaminates adenosines in dsRNA structures of at least 23 bp in length that form by pairing with editing complementary sequences (ECS) (Nishikura et al. 1991). Identifying ECS sequences is challenging as they can be located proximal to the editing site, in distal regions such as an intronic ECS that base-pairs with an exon, or mediated through intermolecular interactions (Bass and Weintraub 1988; Reenan 2005; Ramaswami et al. 2015). To identify potential ECS sequences in our ground squirrel data, a 201 nt region surrounding each editing site was reverse complemented and aligned pairwise to a larger region (4001 nt) surrounding the editing site. Alignments matching the larger region represent sequences capable of base-pairing with the editing site and were enumerated for cold-enriched, constitutive, and a set of shuffled control sites (Fig. 3F; Supplemental Fig. S11). A large percentage of cold-enriched (25.2%) and constitutive (38.0.%) editing sites had identifiable ECS sequences, in contrast to nonreverse complemented negative controls (9.6% and 16.4% for cold-enriched and constitutive, respectively) or shuffled control sites (0.2% and 0.1%). Taken together these results demonstrate that the cold-enriched and constitutive editing sites both have sequence and structural features consistent with known A-to-I editing sites.
mRNA editing events are enriched in SINE element repeats
A-to-I editing events in mammals are predominantly located in SINE-element derived tandem inverted repeats (Porath et al. 2017a). A much smaller proportion are located in exonic sequences, with very few cases of editing sites that recode amino acids, in contrast to Drosophila or Cephalopods, where recoding events are more common (Liscovitch-Brauer et al. 2017). We therefore next assessed the genomic distribution of the ground squirrel brain editing sites. The cold-enriched editing sites are primarily localized in retained intron sequences, similar to the distribution of RNA editing sites in human brain (Fig. 4A and Materials and Methods) (Hwang et al. 2016). Constitutively edited sites have a similar distribution to the cold-enriched sites, indicating that the temperature-sensitive RNA-editing sites are not preferentially localized to exonic or coding sequences (Fig. 4B). A total of 90.1% of the constitutive and 82.5% of the cold-enriched editing sites reside in SINE repeat regions, with the majority being STRID repeat elements (Fig. 4C,D). In the squirrel lineage, the STRID SINE repeats are derived from tRNA, similar to ID-elements in mice and analogous to 7SL-RNA derived Alu elements in primates (Churakov et al. 2010; Vassetzky and Kramerov 2013). These results demonstrate that the temperature-sensitive editing sites are not preferentially localized to coding or exonic regions, and are primarily repeat-derived.
FIGURE 4.

Editing sites are predominantly located in SINE elements in intronic sequences. (A) Genomic distribution of cold-enriched editing sites. Annotations were derived using both the Ensembl 85 annotation and the novel transcriptome built in this study. (B) Distribution of constitutively edited sites. (C) Number of cold-enriched editing sites within repeat classes and repeat families defined by repeatMasker annotations. (D) Repeat classification for constitutively edited sites.
Functional impact of editing
We next assessed the potential functional impacts of the cold-enriched editing on specific mRNAs. We used snpEFF to annotate the predicted effects of A-to-G substitution based on gene annotations from Ensembl. The majority of sites reside in either intron or intergenic regions, and are not predicted to overlap with splice acceptor or donor sequences (Fig. 5A). Just 13 sites are predicted to be either moderately or highly deleterious, 12 of these are recoding events and one is in a splice acceptor site (Fig. 5B; Supplemental Table S3). Additionally, eight CDS editing sites are silent, and do not recode an amino acid.
FIGURE 5.

Predicted functional consequences of editing. (A) Summary of predicted impacts of cold-enriched editing events as defined by SnpEff using Ensembl annotations. (B) Heatmap depicting RNA editing frequencies for editing sites in CDS regions or predicted to be deleterious by SNPeff analysis (Moderate or High impact). Sites without read coverage are colored gray. Columns are ordered by sample group and rows are ordered by hierarchical clustering. (C) Heatmap depicting cold-enriched sites with significant seasonally altered editing frequencies (FDR < 0.01). Comparisons were made between heterothermic warm samples (Ent and IBA) and homeothermic warm samples (SA and SpD). Rows and columns are clustered by hierarchical clustering.
Examining the editing frequencies for the predicted deleterious sites revealed that the recoding sites in PLEKHM2, SYTL5 and MCF2L2 were additionally edited during IBA and Ent stages, suggesting that the editing of these sites is seasonally regulated (Fig. 5B). We therefore examined the cold-enriched editing sites to additionally classify sites with seasonally altered editing frequencies. Seasonally regulated editing sites were identified by comparing the warm heterothermic stages IBA (34.1°C ± 2.9°C) and Ent (25.4°C ± 1.8°C) to warm homeothermic stages SpD (37°C) and SA (37°C). From this analysis we identified 102 sites with significantly increased editing frequencies in IBA and Ent compared to SpD and SA (FDR < 0.01) (Fig. 5C), demonstrating that a small proportion of the cold-enriched sites (1.9%) are also edited in a seasonally dependent manner.
We selected 10 of the putative RNA-editing sites for validation in a subset of the ground squirrels by dideoxy sequencing of the corresponding liver genomic gDNA (Supplemental Figs. S12–S14). The selected sites included nine of the temperature-sensitive sites, seven predicted to be deleterious and two intronic sites, and the universally edited Gln560Arg site in GRIA2. Additionally a Group 2 site identified by the GATK pipeline that was predicted to be a SNP rather than an editing site was selected as a positive control for detecting homozygous vs. heterozygous SNPs via this method. cDNA and gDNA sequencing demonstrated that none of the predicted RNA editing events were actually SNPs, and additionally validated the predicted SNP in DKK3 mRNA (Supplemental Fig. S14). Validated recoding events include the splice-acceptor site alteration in ZCCHC8 (ENSSTOG00000001683), a component of the Nuclear EXosome Targeting complex (NEXT) and a recoding event in Z3CH18 (Thr377Ala, ENSSTOG00000022431) a scaffolding protein that links the NEXT complex to the Cap Binding Complex to promote exonucleolytic cleavage of snRNA and replication dependent histone mRNAs (Meola et al. 2016). We also verified the recoding site in EIF3A (Arg378Gly, ENSSTOG00000002512), the largest subunit of EIF3 which is required for EIF3 assembly and translation initiation (Wagner et al. 2014). In addition, editing caused the amino acid replacement at the previously described Lys242Arg site in NEIL1 (ENSSTOG00000005781), a DNA glycosylase whose substrate specificity is altered by the arginine substitution (Yeo et al. 2010), and a recoding event in one subunit of gamma-aminobutyric acid receptor, GABRA4 (Arg307Gly, ENSSTOG00000001951), which is a ligand-gated chloride channel for the main inhibitory neurotransmitter in mammalian brains.
The splice-acceptor editing site in the ZCCHC8 mRNA occurs in a retained intron. We therefore investigated if cold-enriched RNA editing at this site modulated the frequency of intron retention. The editing frequency at the splice-acceptor site increases up to 21.0% in late torpor and 53.3% during arousal compared to a near absence of editing observed in the other states (Supplemental Fig. S15). However, the relative proportion of the retained intron did not increase in the cold animals (LT and Ar), suggesting that editing of this mRNA occurs post-transcriptionally and therefore does not impact its splicing.
mRNA editing frequencies increase during torpor
Our observation that editing frequencies increased in the LT and Ar animals compared to the IBA and Ent animals could be the result of either RNA editing during entrance into torpor as Tb falls below 23°C, or of editing throughout the 1–2 wk torpor period while Tb is maintained at 5°C–6°C. To distinguish between these possibilities, cerebrum samples were taken at an earlier stage of torpor, when Tb is <30°C for 1.2 ± 0.3 d. Editing frequencies for intronic sites in FBXW7 and CDH9 were determined by dideoxy DNA sequencing of cDNA from five early torpor animals for comparison to five LT animals. These sites were chosen because they had nearly undetectable editing in the IBA and Ent animals, but had greatly increased A-to-G substitutions during LT and Ar, providing the greatest dynamic range with which to assess the change in editing frequency across the torpor bout (Fig. 6A). Because the editing frequencies in early torpor were higher than those observed at Ent, some editing has already occurred in the first 1–2 d with Tb < 23°C, but the editing frequencies at both sites were significantly lower during early torpor than late torpor, indicating that editing occurred across the torpor bout despite the continuously low Tb. The increased editing frequency during torpor and arousal from torpor is unlikely to be explained by changes in ADAR abundance, as we observe only a 19.5% ± 6.3% increase in ADAR mRNA in LT compared to IBA (Supplemental Fig. S16). This small increase is most likely explained by a relatively increased stability of ADAR mRNA compared to the mRNA pool (Grabek et al. 2015), and is unlikely to affect the ADAR protein pool because initiation of translation, and thus translation of any newly edited mRNAs, is blocked at the low Tb of torpor (van Breukelen and Martin 2001). Increased editing frequencies across the torpor bout could in principle be the result of either de novo editing by ADARs, or alternatively, selective stabilization of mRNAs edited prior to ET. To distinguish between these possibilities we examined the normalized read counts containing unedited (A) or edited (G) nucleotides across hibernation stages. During LT and Ar the normalized abundance of edited transcripts increased while the abundance of unedited transcripts decreased proportionally (Fig. 6B; Supplemental Fig. S17). Given the absence of transcription at the low Tb of torpor (van Breukelen and Martin 2002), these findings favor a model in which de novo ADAR activity throughout the period of low Tb results in increasing editing frequencies.
FIGURE 6.

A-to-I editing frequencies increase during torpor. (A) Editing frequencies for intronic sites in FBXW7 and CHD9 determined by Sanger sequencing from cerebrum samples (n = 5) at early torpor (ET) or late torpor (LT). Editing frequencies determined by RNA-seq are also shown for comparison. (B) For all cold-enriched sites the average normalized abundance (counts per million) for reads containing reference (A) or edited (G) nucleotides is shown.
DISCUSSION
Our results demonstrate that widespread RNA editing occurs in three brain regions of a mammalian hibernator during torpor, when body temperature remains near freezing for multiple days. Temperature-sensitive RNA editing has been observed previously in Drosophila species exposed to acute temperature changes (Buchumenski et al. 2017; Yablonovitch et al. 2017b), and in Cephalopods, where ocean temperatures are negatively correlated with editing frequencies in a potassium channel mRNA across multiple species (Garrett and Rosenthal 2012). In both cases, increased A-to-I editing contributes to temperature adaptation, and is associated with increased editing in coding regions. In this study we find that ADAR-mediated RNA editing is also greatly increased in hibernating ground squirrels during torpor, when the animal's temperature hovers near freezing for several days. The degree of editing at most of these sites increases with increasing time spent cold and then returns to baseline within 3 h of temperature restoration to 37°C during spontaneous arousal (Figs. 2, 6), although some (1.9%) edited sites persist throughout the torpor-arousal cycle (Fig. 5B,C). Only 20 of the hibernation edited sites with either pattern lie within coding regions, and just 12 of these would recode the corresponding protein. Instead, the vast majority of the edited sites reside in SINE-family interspersed repeats, as is typical of nonhibernating, homeothermic mammals (Porath et al. 2017a).
It is clear from our data that the dominant effect of the greatly enhanced RNA editing during the cold phase of hibernation is to target sequences that are likely to form dsRNA rather than to diversify the proteome for cold adaptation. Nevertheless, it remains formally plausible that one or more of the small set (n = 12) of edited sites predicted to recode amino acids (Fig. 5B) could improve the corresponding protein's function in the cold, as documented for the Kv1 potassium channel in the octopus (Garrett and Rosenthal 2012), and thus be adaptive for hibernation. Interestingly, five of the 12 recoding events occur in edited sites that appear to be enhanced seasonally rather than strictly by temperature (Fig. 5B), specifically at two sites in PLEKHM2 and SYTL5, and one site in MCF2L2. These may be particularly important to help neurons function in the cold, because the level of editing remains consistently elevated throughout the torpor-arousal cycle, including during interbout arousals when edited transcripts can be actively translated into protein (Frerichs et al. 1998). Conversely, the seven remaining potential recoding events are less likely to substantially affect their corresponding protein pools because translational initiation is arrested at temperatures below 18°C (van Breukelen and Martin 2001) and the proportion of transcript that is edited quickly declines as Tb recovers to 37°C during IBA (Figs. 2, 6), when translation resumes and the bulk of protein synthesis during hibernation occurs. For these transcripts, recoded proteins synthesized from cold-edited mRNAs are expected to reach their greatest concentrations just as euthermic Tb is restored rather than during the time of greatest need for their cold-adapted function, i.e., as Tb declines during entrance into torpor and throughout multiple days at low Tb in torpor. These kinetics taken together with the low editing frequencies for recoded sites (<40%) suggest that only a small fraction of the proteins made from the cold edited transcripts would be recoded.
Because transcription is largely suppressed during the near-freezing temperatures of torpor (van Breukelen and Martin 2002), ADAR enzymes must be active post-transcriptionally for edited sites to accumulate during hibernation (Fig. 6). Recent work suggests that the bulk of ADAR mediated RNA editing (>93%) occurs cotranscriptionally in cultured human cells (Hsiao et al. 2018) and Drosophila (Rodriguez et al. 2012). Unlike the torpid hibernators, these cells were transcriptionally active; thus, even in the presence of active transcription at least some ADAR editing is apparently post-transcriptional. It is also noteworthy that RNA-editing was first described in transcriptionally quiescent Xenopus oocytes (Bass 2002) and can also occur in RNA viruses in the absence of transcription (Taylor et al. 2005), hence transcription is not absolutely required for RNA editing by ADAR.
The data reveal modest differences in the extent of hyperediting and number of significant RNA editing sites discovered in each brain region. The cerebrum had the lowest number of hyperedited sites, followed by the medulla and the hypothalamus (Fig. 2D). This pattern mirrors both the number of detectable transcripts in each region (17,575 ± 156, 17,894 ± 244, and 18,089 ± 115, in the cerebrum, medulla, and hypothalamus, respectively) and the mRNA abundance of the ADAR isoform responsible for hyperediting dsRNA (Supplemental Fig. S16; Mannion et al. 2014; Liddicoat et al. 2015). Despite this difference in the overall number of sites, the fold increase in hyperediting is similar among the three brain regions. Taken together these findings suggest that the observed differences reflect differences in baseline RNA editing levels rather than regional differences in cold-enriched RNA editing. For editing sites that were called significant using the edgeR approach, there were also differences in the number of sites identified in each region. The medulla had the lowest number of sites recovered, which correlated with the read depth at these sites (Supplemental Fig. S10B). Upon further examination we find little evidence for regional differences in RNA editing, with most of the observed differences explained by read coverage, and variability among each sample in a hibernation group (Supplemental Fig. S10C,D).
ADAR activity during torpor provides a mechanism to destabilize dsRNA structures that form at lower temperatures, and by restricting the accumulation of dsRNA, RNA editing could prevent inappropriate activation of innate immune sensors upon rewarming as the animals arouse from torpor (Liddicoat et al. 2015). ADAR activity during torpor could also promote the nuclear retention of subsets of transcripts in via P54NRB, which recognizes inosine containing RNAs and sequesters them within the nucleus (Zhang and Carmichael 2001). The majority of cold-enriched editing that we observed in the hibernators occurs in polyadenylated transcripts with retained introns (Fig. 4A), which are unlikely to produce a functional protein product. Nuclear retention of such transcripts could be used to prevent wasteful translation of inappropriately processed mRNAs generated during temperature transitions in the torpor-arousal cycle.
Hibernators are unique among mammals in their ability to allow body temperature to fall to near freezing and remain there for days to weeks at a time with no evidence of irreversible cellular damage or loss of organismal function on arousal (Dave et al. 2012). Given that homeothermy is an evolutionarily recent invention found in birds and mammals, the ability to maintain cellular function and integrity while cold in hibernation is likely a retained ancestral trait (Lovegrove et al. 2014). To date there is largely evidence against (Villanueva-Cañas et al. 2014) but little evidence for (Matos-Cruz et al. 2017) genetically encoded cold-adaptation of proteins that could support function at low temperature during hibernation. mRNA editing has the potential to cause adaptive changes in proteins that leave no signature in the genome; this mechanism is used for temperature adaptation of proteins in ectotherms (Garrett and Rosenthal 2012; Savva et al. 2012; Buchumenski et al. 2017). Here we demonstrate for the first time rampant, temperature-dependent RNA editing during hibernation, with most sites falling outside of protein coding regions. While we cannot rule out that the few protein recoding events observed in this study are adaptive for improved function in the cold, the bulk of the editing, as typical of homeothermic mammals studied previously, is directed toward interspersed repeats that engage in dsRNA formation. Blocking and marking regions of dsRNA at low temperature represses activation of the innate immune response (O'Connell et al. 2015). Suppression of inflammation during hibernation has been demonstrated in multiple tissues (for review, see Bouma et al. 2010) and is likely to be adaptive.
MATERIALS AND METHODS
Tissue collection
Tissues were collected at precise timepoints based on Tb (Fig. 1) from five ground squirrels in each of six distinct physiological stages. Animals from two homeothermic and four heterothermic (hibernation) phases were used (Fig. 1). Under deep isoflurane anesthesia, animals were euthanized via exsanguination and perfused with ice cold saline. The brain was extracted from the skull, and then the brainstem and cerebellum were removed. The medulla and hypothalamus were dissected; for this study, these two regions and the remaining telencephalon plus diencephalon (excluding the hypothalamus, henceforth referred to as cerebrum) were snap frozen in liquid nitrogen and stored at −80°C until RNA extraction.
RNA-seq
Either the entire frozen tissue (medulla, hypothalamus), or 100 mg of tissue pulverized under liquid nitrogen with a mortar and pestle (cerebrum) were homogenized in ice cold TRIzol Reagent (Invitrogen) on ice using a polytron (Brinkman), and then purified using the Direct-zol RNA MiniPrep Kit (Zymo Research), following the manufacturer's recommendations. RNA was assessed for quantity (NanoDrop, Thermo Scientific) and quality (RIN > 8; Bioanalyzer, Agilent Technologies) and then 1 µg was submitted to the University of Colorado Denver Genomics and Microarray Core for library preparation and sequencing. Strand-specific RNA-seq libraries were constructed using the TruSeq Stranded mRNA Library Prep Kit (Illumina) which selects mRNAs using oligodT; 126 or 151 nt paired-end sequence reads were collected for each sample after multiplexing via Illumina HiSeq 2500 (cerebrum, 10 samples/lane) or 4000 (hypothalamus and medulla, 15 samples/lane), respectively. Samples (replicates 1–5 from each physiological state) were assigned to a sequencing lane by multiplexing all of the first replicates from each state, followed by the second replicates etc., to avoid batch effects and assure that roughly equivalent numbers of individual libraries representing each state were spread among the sequencing lanes. Sequencing library statistics are provided in Supplemental Table S4.
Transcriptome assembly
A custom transcriptome assembly was built using all 90 brain, plus three neonatal and two testes libraries. The neonatal libraries were constructed from three regions of a P1 neonate, head, thorax and abdomen. The testes libraries were prepared from two aliquots of RNA isolated from one adult male testes during spring recrudescence following hibernation. The 95 RNA-seq libraries were aligned first to the 13-lined ground squirrel mitochondrial DNA sequence (Hampton et al. 2011) and then the remaining reads were aligned to SpeTri2.0 (Ensembl release 88, retaining contigs ≥10,000 nt) with HISAT2 (v 2.0.4, Kim et al. 2015). BAM files were filtered to remove reads mapping to more than one genomic location (bamtools, v 2.4.0) and duplicates (Picard Tools, v 1.83), and the remaining reads were used for guided transcriptome reconstruction using StringTie (v. 1.3.3b, with options --rf, -j 3, -c 3) (Kim et al. 2015; Pertea et al. 2015). Transcriptomes were then merged to generate a single assembly using TACO (v. 0.7.2, with option --filter-min-expr st to 5.0) (Niknafs et al. 2016). The reconstructed transcriptome was used to define splice junctions for STAR mapping and annotating editing sites.
RNA editing detection with the GATK pipeline
Candidate RNA editing sites were identified by following the GATK pipeline for variant calling from RNA-seq data as described in https://gatkforums.broadinstitute.org/gatk/discussion/4067/best-practices-for-variant-discovery-in-rnaseq. RNA-seq libraries were trimmed with cutadapt (v.1.8.3) (Martin 2011) (cerebrum) or Illuminas bcl2fastq (medulla and hypothalamus) to remove Illumina truseq adapters, then aligned to the SpeTri2.0 (Ensembl 85) genome assembly supplemented with the transcript assembly annotations built as described above. Alignment was performed using STAR (v.2.5.1b with default parameters) in two-pass mode as recommended by GATK (Dobin et al. 2013). Duplicate alignments were marked with MarkDuplicates from Picard (v.2.7.0), and read alignments over splice junctions were split into independent alignments using SplitNTrim from GATK (v3.5-0-ge91472d). Variants were called using HaplotypeCaller (-stand_call_conf 20.0, -stand_emit_conf 20.0), filtered to remove sites with Qual by Depth (QD) < 2.0 or Fisher Strand Score (FS) > 30. Variants were then merged and used as input for base recalibration. Variant calling was then rerun with the updated base recalibration table. This process was repeated twice to establish proper base recalibration as recommended by GATK for samples without a known set of SNPs. Final variant calls were then merged and filtered to remove sites with Depth (DP) < 20. The strandedness of the variant was determined by calculating the mean strand bias (positive or negative stranded reads / total number of reads) determined based on the read alignment strand and mate (R1 or R2), and averaged over all the libraries. Variants with >0.80 positive or negative strand bias were assigned to a strand, whereas others were excluded as ambiguous. Reference and variant alleles were counted at each site using only reads with unique alignments, MAPQ > 10, base quality scores ≥20, not marked as duplicate, secondary, QC failed, or mate-non-mapped alignments. Additionally, reads with variant sites located in the first 6 nt were also excluded to avoid biases from random hexamer priming. Variants with extreme variant allele frequencies were then filtered to remove any variants that were not present at least >5% variant allele or <95% frequencies in any of the 90 libraries to enrich for variants with dynamic allele frequencies across samples.
Hyperediting detection
Hyperedited regions were identified following the methods described in a published method and were implemented with custom Python scripts (Porath et al. 2014). Briefly, reads that did not align after two-pass STAR mapping were subjected to filtering to exclude low-quality reads and reads with highly repetitive sequences, as previously described. Following filtering, A nucleotides were converted to Gs in forward stranded reads (from paired mate R2) and mapped to a genome also with As changed to Gs to capture sense alignments, or a genome with Ts changed to Cs to capture antisense alignments. Similarly in reads from paired mate R1 (reverse stranded) Ts were changed to Cs and aligned to genomes with either T to C or A to G changes. Alignments were performed using BWA (v0.7.10) with no gaps and only two allowed mismatches (Li and Durbin 2009). Successful alignments were then processed to identify mismatches between the original read and genome sequences. Alignments with high quality mismatches passing a stringent set of filters to reduce misalignment artifacts were retained as hyperedited reads as previously described (Porath et al. 2014). The described procedure was repeated for each of the 12 possible mismatch types to determine the specificity of this approach for A-to-G mismatches. Hyperedited regions were defined in each read as the region spanning the first and last mismatch. Hyperedited clusters were defined by merging all overlapping regions or those located within 20 nt. Clusters and sites supported by fewer than two reads were discarded. Editing frequencies were computed for hyperedited sites by using reads aligned during STAR two-pass mapping to the unmodified genome with reference and variant alleles counted as described for the GATK pipeline.
Differential editing analysis
Reference allele counts at each candidate editing site were normalized using library sizes derived from the total number of exonic alignments in each library and scaled with TMM normalization using the R package, edgeR (v3.18.1). Normalization factors were then propagated to the variant allele counts. A GLM model was next constructed, ∼0 + Animal + Hibernation Stage:Allele, whereby Animal = animal id, Hibernation Stage = IBA, Ent, Lt, Ar, Spd, or SA, and Allele = A or G. P-values were obtained using glmLRT, with contrasts set to test for sites with variable G counts across sampled hibernation stages while controlling for changes in A counts within each animal (McCarthy et al. 2012). Sites with FDR < 0.01 were considered significant. This procedure was independently applied for all possible pairwise allelic combinations (i.e., A-to-T, A-to-G, A-to-C, T-to-A, T-to-G, etc.). Pairwise comparisons were also tested between cold (Lt and Ar) and warm samples (Spd, SA, and IBA), and between winter warm (IBA and Ent) and summer samples (SA and Spd). An independent statistical approach was also used, which used an ANOVA to test for differences in editing frequencies between any sampled state. P-values were corrected for multiple hypothesis testing using the Benjamini–Hochberg approach, and FDR < 0.01 was considered significant. Editing frequencies were visualized using the R package ComplexHeatmap (v1.14.0) (Gu et al. 2016). K-means clustering was performed on editing frequencies mean-centered and scaled across all samples using the flexclust package (1.3–4) with the kmeans++ initialization function (Leisch 2006). Sites without sufficient read coverage to compute editing frequencies were set to zero for k-means classification. Individual samples and editing site ordering in the displayed heatmaps were determined by hierarchical clustering of euclidean distances with the complete linkage method.
RNA editing site annotation and functional effect prediction using Snpeff
The genomic region that each RNA editing site overlapped was annotated using a reference that combined annotations from Ensembl 85 and the novel transcriptome built in this study. An editing site was annotated to a novel mRNA exon if the editing site resided within an exon from a novel transcript of a protein coding gene that did not overlap an Ensembl annotated CDS exon. An editing site was annotated to a novel ncRNA exon if the editing site resided within an exon from a novel transcript of a gene that was not annotated as a protein coding gene by Ensembl. Editing sites were annotated as intronic if there was no overlap with an exon, but resided within the gene boundaries. Additionally, editing sites were also annotated as intronic if the overlapping exon has evidence of being a retained intron misannotated as an exon in the novel transcriptome, a common event in brain tissues (Braunschweig et al. 2014). Specifically, if a novel exon overlapped two or more CDS exons, the exon was considered a retained intron, and editing sites residing in these regions were classified as intronic. The potential functional impacts of the cold-enriched editing sites were annotated with SnpEff (v.4.3b) using Ensembl 85 annotations (Cingolani et al. 2012). The novel transcriptome was not used for predicting the functional impacts of the editing events due to the lack of CDS annotations in the novel transcriptome.
RNA editing site validation
PCR primers (Supplemental Table S5) were designed to amplify gDNA or cDNA surrounding candidate editing sites. Six cold-enriched editing sites were validated by both gDNA and cDNA sequencing of cold (Ar) and warm (SpD) animal samples from either liver for gDNA or brain tissue for cDNA, (ZCCHC8, EIF3A, ZC3H18, AMIGO2, FBXW7, CHD9), in addition to the positive control site in GRIA2 (Supplemental Fig. S14). The negative control DKK3 site (Supplemental Fig. S14) and three cold-enriched editing sites were validated by gDNA sequencing from liver gDNAs alone (GABRA4, NEIL1, and ZNF483) (Data not shown). PCR products were subjected to dideoxy sequencing. Peak heights from chromatograms were determined with the ThermoFisher QC app tool (https://apps.thermofisher.com/apps/dashboard/#/).
Differential gene expression and splicing analysis
Read counts were computed using featureCounts from the subread package (v1.4.4) using the custom transcriptome annotations (Liao et al. 2014). Lowly expressed genes were filtered if there were not at least two counts per million in at least four samples. Normalized FPKM values were calculated using edgeR. Differentially expressed genes were identified using an ANOVA-like test for any variation across hibernation state. Relative exon usage values were computed using DEXSeq (v.1.16.10).
Editing site conservation
Genome coordinates for human (hg19) and mouse (mm9) editing sites were downloaded from the RADAR database (Ramaswami and Li 2014). These coordinates were converted to mm10 coordinates using UCSC liftover chain files and the liftOver tool. Squirrel RNA editing sites were converted to mm10 coordinates using the UCSC liftover chain and liftOver tool, and shared editing sites were identified using the R package valr (v0.3.1) (Riemondy et al. 2017). Editing sites conserved in mammals were taken from the Supplemental Data of Pinto et al. (2014) and compared to the squirrel editing sites as described above.
Sequence motifs
Sequences 5 nt upstream and downstream from each editing site were extracted and sequence logos were computed with weblogo (v3.5.0) (Crooks et al. 2004). A set of negative control shuffled editing sites were constructed for the cold-enriched sites by randomly selecting an adenosine nucleotide in the same transcript as each editing site. Random sites were drawn from exonic sequences if the editing site was exonic, otherwise the sites were drawn from pre-mRNA transcript coordinates.
RNA structure predictions
Editing sites were classified as falling within dsRNA using an approach as previously described (Li et al. 2009). Briefly, the reverse complement of a 201 nt region centered on the editing site was aligned to a larger surrounding region (4001 nt) to identify sequence capable of base-pairing to the editing site. blastn (v2.2.29, -strand “plus” -word_size 7 -evalue “0.1”) was used in the two sequence alignment mode. Alignments were filtered to keep alignments with e-value <0.01 and alignment regions >20 nt overlapping the editing site. Negative controls were generated by aligning the region surrounding the editing site to the same stranded larger region and selecting the second best alignment using the aforementioned criteria. Additional details and examples of regions identified as dsRNA are provided in Supplemental Figure S11. Example dsRNA regions were folded using RNAcofold and visualized using RNAplot from the ViennaRNA package (Lorenz et al. 2011).
DATA DEPOSITION
The RNA-seq raw data, transcriptome assembly, and processed data have been deposited at GEO under accession number GSE106947. The RNA editing pipelines were implemented as a snakemake pipeline (Köster and Rahmann 2012), and custom scripts for hyperediting detection were written in Python and C++. Statistical procedures, data-processing, and visualizations were implemented in R. The pipeline, scripts, and a link to a UCSC Genome Browser trackhub with the RNA-seq data can be found at https://github.com/rnabioco/rnaedits.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge support from the RNA Bioscience Initiative at the University of Colorado School of Medicine (K.A.R., A.E.G., E.W., J.R.H., and S.L.M.) and the National Institutes of Health (R21 NS088315 to S.L.M. and J.R.H.). We thank K. Diener at the University of Colorado Cancer Center Genomics Resource for expert assistance with DNA library construction and sequencing (supported by NIH grant P30 CA046934) and E. Epperson for brain dissection (supported by NIH grant R01 HL089049 to S.L.M.).
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.066522.118.
REFERENCES
- Bass BL. 2002. RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem 71: 817–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass BL, Weintraub H. 1988. An unwinding activity that covalently modifies its double-stranded RNA substrate. Cell 55: 1089–1098. [DOI] [PubMed] [Google Scholar]
- Behm M, Öhman M. 2016. RNA editing: a contributor to neuronal dynamics in the mammalian brain. Trends Genet 32: 165–175. [DOI] [PubMed] [Google Scholar]
- Bouma HR, Carey HV, Kroese FGM. 2010. Hibernation: the immune system at rest? J Leukoc Biol 88: 619–624. [DOI] [PubMed] [Google Scholar]
- Braunschweig U, Barbosa-Morais NL, Pan Q, Nachman EN, Alipanahi B, Gonatopoulos-Pournatzis T, Frey B, Irimia M, Blencowe BJ. 2014. Widespread intron retention in mammals functionally tunes transcriptomes. Genome Res 24: 1774–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brusa R, Zimmermann F, Koh D-S, Feldmeyer D, Gass P, Seeburg PH, Sprengel R. 1995. Early-onset epilepsy and postnatal lethality associated with an editing-deficient GluR-B allele in mice. Science 270: 1677–1680. [DOI] [PubMed] [Google Scholar]
- Buchumenski I, Bartok O, Ashwal-Fluss R, Pandey V, Porath HT, Levanon EY, Kadener S. 2017. Dynamic hyper-editing underlies temperature adaptation in Drosophila. PLoS Genet 13: e1006931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmi S, Borukhov I, Levanon EY. 2011. Identification of widespread ultra-edited human RNAs. PLoS Genet 7: e1002317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churakov G, Sadasivuni MK, Rosenbloom KR, Huchon D, Brosius J, Schmitz J. 2010. Rodent evolution: back to the root. Mol Biol Evol 27: 1315–1326. [DOI] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. 2012. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6: 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia J-M, Brenner SE. 2004. WebLogo: a sequence logo generator. Genome Res 14: 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dave KR, Christian SL, Perez-Pinzon MA, Drew KL. 2012. Neuroprotection: lessons from hibernators. Comp Biochem Physiol B Biochem Mol Biol 162: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43: 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggington JM, Greene T, Bass BL. 2011. Predicting sites of ADAR editing in double-stranded RNA. Nat Commun 2: 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frerichs KU, Smith CB, Brenner M, DeGracia DJ, Krause GS, Marrone L, Dever TE, Hallenbeck JM. 1998. Suppression of protein synthesis in brain during hibernation involves inhibition of protein initiation and elongation. Proc Natl Acad Sci 95: 14511–14516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett S, Rosenthal JJC. 2012. RNA editing underlies temperature adaptation in K+ channels from polar octopuses. Science 335: 848–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabek KR, Behn CD, Barsh GS, Hesselberth JR, Martin SL. 2015. Enhanced stability and polyadenylation of select mRNAs support rapid thermogenesis in the brown fat of a hibernator. Elife 4: e04517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Eils R, Schlesner M. 2016. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32: 2847–2849. [DOI] [PubMed] [Google Scholar]
- Hampton M, Melvin RG, Kendall AH, Kirkpatrick BR, Peterson N, Andrews MT. 2011. Deep sequencing the transcriptome reveals seasonal adaptive mechanisms in a hibernating mammal. PLoS One 6: e27021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao Y-HE, Bahn JH, Yang Y, Lin X, Tran S, Yang E-W, Quinones-Valdez G, Xiao X. 2018. RNA editing in nascent RNA affects pre-mRNA splicing. Genome Res 28: 812–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang T, Park C-K, Leung AKL, Gao Y, Hyde TM, Kleinman JE, Rajpurohit A, Tao R, Shin JH, Weinberger DR. 2016. Dynamic regulation of RNA editing in human brain development and disease. Nat Neurosci 19: 1093–1099. [DOI] [PubMed] [Google Scholar]
- Kilduff TS, Miller JD, Radeke CM, Sharp FR, Heller HC. 1990. 14C-2-deoxyglucose uptake in the ground squirrel brain during entrance to and arousal from hibernation. J Neurosci 10: 2463–2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, Salzberg SL. 2015. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12: 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köster J, Rahmann S. 2012. Snakemake—a scalable bioinformatics workflow engine. Bioinformatics 28: 2520–2522. [DOI] [PubMed] [Google Scholar]
- Leisch F. 2006. A toolbox for K-centroids cluster analysis. Comput Stat Data Anal 51: 526–544. [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JB, Levanon EY, Yoon J-K, Aach J, Xie B, LeProust E, Zhang K, Gao Y, Church GM. 2009. Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science 324: 1210–1213. [DOI] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. 2014. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930. [DOI] [PubMed] [Google Scholar]
- Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, Li JB, Seeburg PH, Walkley CR. 2015. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 349: 1115–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liscovitch-Brauer N, Alon S, Porath HT, Elstein B, Unger R, Ziv T, Admon A, Levanon EY, Rosenthal JJC, Eisenberg E. 2017. Trade-off between transcriptome plasticity and genome evolution in cephalopods. Cell 169: 191–202.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz R, Bernhart SH, Höner Zu Siederdissen C, Tafer H, Flamm C, Stadler PF, Hofacker IL. 2011. ViennaRNA Package 2.0. Algorithms Mol Biol 6: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovegrove BG, Lobban KD, Levesque DL. 2014. Mammal survival at the Cretaceous–Palaeogene boundary: metabolic homeostasis in prolonged tropical hibernation in tenrecs. Proc Biol Sci 281: 20141304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannion NM, Greenwood SM, Young R, Cox S, Brindle J, Read D, Nellåker C, Vesely C, Ponting CP, McLaughlin PJ, et al. 2014. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep 9: 1482–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannion N, Arieti F, Gallo A, Keegan LP, O'Connell MA. 2015. New insights into the biological role of mammalian ADARs; the RNA editing proteins. Biomolecules 5: 2338–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17: 10–12. [Google Scholar]
- Matos-Cruz V, Schneider ER, Mastrotto M, Merriman DK, Bagriantsev SN, Gracheva EO. 2017. Molecular prerequisites for diminished cold sensitivity in ground squirrels and hamsters. Cell Rep 21: 3329–3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy DJ, Chen Y, Smyth GK. 2012. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res 40: 4288–4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meola N, Domanski M, Karadoulama E, Chen Y, Gentil C, Pultz D, Vitting-Seerup K, Lykke-Andersen S, Andersen JS, Sandelin A, et al. 2016. Identification of a nuclear exosome decay pathway for processed transcripts. Mol Cell 64: 520–533. [DOI] [PubMed] [Google Scholar]
- Niknafs YS, Pandian B, Iyer HK, Chinnaiyan AM, Iyer MK. 2016. TACO produces robust multisample transcriptome assemblies from RNA-seq. Nat Methods 14: 68–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikura K, Yoo C, Kim U, Murray JM, Estes PA, Cash FE, Liebhaber SA. 1991. Substrate specificity of the dsRNA unwinding/modifying activity. EMBO J 10: 3523–3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell MA, Mannion NM, Keegan LP. 2015. The epitranscriptome and innate immunity. PLoS Genet 11: e1005687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palladino MJ, Keegan LP, O'Connell MA, Reenan RA. 2000. A-to-I pre-mRNA editing in Drosophila is primarily involved in adult nervous system function and integrity. Cell 102: 437–449. [DOI] [PubMed] [Google Scholar]
- Pertea M, Pertea GM, Antonescu CM, Chang T-C, Mendell JT, Salzberg SL. 2015. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 33: 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto Y, Cohen HY, Levanon EY. 2014. Mammalian conserved ADAR targets comprise only a small fragment of the human editosome. Genome Biol 15: R5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porath HT, Carmi S, Levanon EY. 2014. A genome-wide map of hyper-edited RNA reveals numerous new sites. Nat Commun 5: 4726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porath HT, Knisbacher BA, Eisenberg E, Levanon EY. 2017a. Massive A-to-I RNA editing is common across the Metazoa and correlates with dsRNA abundance. Genome Biol 18: 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porath HT, Schaffer AA, Kaniewska P, Alon S, Eisenberg E, Rosenthal J, Levanon EY, Levy O. 2017b. A-to-I RNA editing in the earliest-diverging eumetazoan phyla. Mol Biol Evol 34: 1890–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswami G, Li JB. 2014. RADAR: a rigorously annotated database of A-to-I RNA editing. Nucleic Acids Res 42: D109–D113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswami G, Zhang R, Piskol R, Keegan LP, Deng P, O'Connell MA, Li JB. 2013. Identifying RNA editing sites using RNA sequencing data alone. Nat Methods 10: 128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswami G, Deng P, Zhang R, Anna Carbone M, Mackay TFC, Li JB. 2015. Genetic mapping uncovers cis-regulatory landscape of RNA editing. Nat Commun 6: 8194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reenan RA. 2005. Molecular determinants and guided evolution of species-specific RNA editing. Nature 434: 409–413. [DOI] [PubMed] [Google Scholar]
- Rice GI, Kasher PR, Forte GMA, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA, et al. 2012. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat Genet 44: 1243–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riemondy KA, Sheridan RM, Gillen A, Yu Y, Bennett CG, Hesselberth JR. 2017. valr: reproducible genome interval analysis in R. F1000Res 6: 1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez J, Menet JS, Rosbash M. 2012. Nascent-seq indicates widespread cotranscriptional RNA editing in Drosophila. Mol Cell 47: 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savva YA, Jepson JEC, Sahin A, Sugden AU, Dorsky JS, Alpert L, Lawrence C, Reenan RA. 2012. Auto-regulatory RNA editing fine-tunes mRNA re-coding and complex behaviour in Drosophila. Nat Commun 3: 790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan MH, Li Q, Shanmugam R, Piskol R, Kohler J, Young AN, Liu KI, Zhang R, Ramaswami G, Ariyoshi K, et al. 2017. Dynamic landscape and regulation of RNA editing in mammals. Nature 550: 249–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor DR, Puig M, Darnell MER, Mihalik K, Feinstone SM. 2005. New antiviral pathway that mediates hepatitis C virus replicon interferon sensitivity through ADAR1. J Virol 79: 6291–6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Breukelen F, Martin SL. 2001. Translational initiation is uncoupled from elongation at 18°C during mammalian hibernation. Am J Physiol Regul Integr Comp Physiol 281: R1374–R1379. [DOI] [PubMed] [Google Scholar]
- van Breukelen F, Martin SL. 2002. Reversible depression of transcription during hibernation. J Comp Physiol B 172: 355–361. [DOI] [PubMed] [Google Scholar]
- Vassetzky NS, Kramerov DA. 2013. SINEBase: a database and tool for SINE analysis. Nucleic Acids Res 41: D83–D89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva-Cañas JL, Faherty SL, Yoder AD, Albà MM. 2014. Comparative genomics of mammalian hibernators using gene networks. Integr Comp Biol 54: 452–462. [DOI] [PubMed] [Google Scholar]
- Wagner S, Herrmannová A, Malík R, Peclinovská L, Valášek LS. 2014. Functional and biochemical characterization of human eukaryotic translation initiation factor 3 in living cells. Mol Cell Biol 34: 3041–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkley CR, Li JB. 2017. Rewriting the transcriptome: adenosine-to-inosine RNA editing by ADARs. Genome Biol 18: 205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y, Qu K, Ouyang Z, Kertesz M, Li J, Tibshirani R, Makino DL, Nutter RC, Segal E, Chang HY. 2012. Genome-wide measurement of RNA folding energies. Mol Cell 48: 169–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yablonovitch AL, Deng P, Jacobson D, Li JB. 2017a. The evolution and adaptation of A-to-I RNA editing. PLoS Genet 13: e1007064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yablonovitch AL, Fu J, Li K, Mahato S, Kang L, Rashkovetsky E, Korol AB, Tang H, Michalak P, Zelhof AC, et al. 2017b. Regulation of gene expression and RNA editing in Drosophila adapting to divergent microclimates. Nat Commun 8: 1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo J, Goodman RA, Schirle NT, David SS, Beal PA. 2010. RNA editing changes the lesion specificity for the DNA repair enzyme NEIL1. Proc Natl Acad Sci 107: 20715–20719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Carmichael GG. 2001. The fate of dsRNA in the nucleus: a p54nrb-containing complex mediates the nuclear retention of promiscuously A-to-I edited RNAs. Cell 106: 465–475. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.