

SUMMARY
The migration of mature dendritic cells (DC) into the draining lymph node (dLN) is thought to depend solely on the chemokine receptor CCR7. CD301b+ DCs migrate into the dLN after cutaneous allergen exposure and are required for T helper 2 (Th2) differentiation. We found that CD301b+ DCs poorly upregulated CCR7 expression after allergen exposure and required a second chemokine signal, mediated by CCR8 on CD301b+ DCs and its ligand CCL8, to exit the subcapsular sinus (SCS) and enter the LN parenchyma. CD169+SIGN-R1+ macrophages in interfollicular regions produced CCL8 after allergen exposure, which synergized with CCL21 in a Src kinase-dependent manner to promote CD301b+ DC migration. In CCR8-deficient mice, CD301b+ DCs remained in the SCS and were unable to enter the LN parenchyma, resulting in defective Th2 differentiation. We defined a CCR8-dependent stepwise mechanism of DC subset-specific migration, through which LN CD169+SIGN-R1+ macrophages control the polarization of the adaptive immune response.
Keywords: Dendritic cells, macrophages, migration, chemokines, Th2-differentiation, allergy
INTRODUCTION
Dendritic cells (DCs) play a central role in linking innate sensing with the activation and skewing of the adaptive immune response. Pattern recognition receptor (PRR) activation in DCs leads to a coordinated response known as DC maturation. DC maturation is classically characterized by a decrease in DC endocytosis, migration to the draining lymph nodes (dLN), an increase in major histocompatibility type II (MHC II) and costimulatory molecule expression, and the production of skewing cytokines involved in CD4+ T helper cell (Th) differentiation and activation. By reducing endocytosis and upregulating MHC II presentation, DC maturation creates a snapshot of the antigens encountered by the DC at the time of PRR activation. Once in the dLN, DCs display antigens as well as costimulatory molecules and skewing cytokines that are influenced by the inflammatory stimuli previously encountered. Thus, DCs can independently provide the precise costimulation and skewing cytokines required to promote the ideal adaptive immune response.
Although all conventional DCs share a robust capacity for antigen uptake and presentation, DC subsets show functional heterogeneity. Multiple subsets of DCs have been described that have different skewing effects on Th differentiation (Iwasaki and Medzhitov, 2015). For example, the heterogeneous CD103+ DC subsets have been shown to efficiently induce Th1 and Th17 differentiation (King et al., 2010; Lewis et al., 2011; Schlitzer et al., 2013). In contrast, the CD301b+PDL2+ DC population has been shown to be required for Th2 differentiation after cutaneous exposure to Th2-skewing stimuli (Gao et al., 2013; Kumamoto et al., 2013; Murakami et al., 2013). Given that Th differentiation is dependent on the DC subset, the migration of DC subsets must be differentially regulated in order to effectively skew the adaptive immune response. But how is this subset-specific migration controlled? One mechanism would be for DC subsets to express distinct PRRs, thereby allowing only certain subsets to be activated by any specific stimulus. However, although some subset-specific PRR expression has been described, most PRRs are widely expressed across DC subsets (Iwasaki and Medzhitov, 2015). Therefore, we hypothesized that subset-specific DC migration may be controlled by a distinct mechanism.
The processes governing DC positioning and migration are largely dependent on chemokines. Immature DCs express a wide variety of chemokine receptors on their surface, including CCR1, CCR2, CCR5, CCR6, CCR8, CXCR1, CXCR2, and CXCR4 (Dieu et al., 1998; Griffith et al., 2014; Sallusto et al., 1998). The reason for this broad expression remains unclear, but may be involved in positioning of DC precursors in peripheral tissues as well as migration of immature DCs to sites of inflammation. Once DCs are activated and undergo maturation, they upregulate CCR7 surface expression. Expression of CCR7 permits the maturing DCs to sense the CCR7 ligand CCL21 that is produced by the lymphatic endothelium (Worbs et al., 2017). This promotes their emigration from the peripheral tissues into the draining lymphatics where they travel via lymphatic flow into the LN sinuses, including the subcapsular sinus (SCS) (Tal et al., 2011; Weber et al., 2013). Once in the LN sinuses DCs again follow CCL21 gradients that in the SCS are established by the expression of the atypical chemokine scavenger receptor ACKR4 on the SCS ceiling (Ulvmar et al., 2014). After crossing the floor of the LN sinuses in the interfollicular regions (IFR), DCs continue to follow gradients of the CCR7 ligands, CCL19 and CCL21, to enter the T cell area where they can interact with naïve T cells (Braun et al., 2011). However, CCR7 may not be sufficient for proper DC positioning within the LN. In a Heligmosomoides polygyrus infection model, CXCR5 expression on DCs was shown to be necessary for their localization in interfollicular regions (IFR) of the dLN and induction of Tfh and Th2 responses (Leon et al., 2012). Thus, other chemokines may play a role in DC positioning and adaptive immune outcomes.
We hypothesized that alternative, non-CCR7 chemokine networks may be involved in subset-specific DC migration and entry into the dLN. We found that the Th2-skewing stimulus, papain, induced robust migration of CD301b+ DCs into the dLN, despite low surface levels of CCR7. Coinciding with CD301b+ DC entry, allergen immunization led to an upregulation in CCL8 expression from CD169+SIGN-R1+F4/80+ macrophages present within the IFR of dLN. CCL8 enhanced CCL21-mediated migration of CD301b+ dendritic cells in a Src-dependent manner and both CCR7 and CCR8 were required for the entry of CD301b+ DCs into the dLN after papain administration. CD301b+ DCs lacking CCR8 expression trafficked to the dLN but were retained in the SCS space, leading to a loss of Th2 differentiation in response to multiple Th2-skewing stimuli. These data reveal a chemokine-mediated mechanism by which LN macrophages control DC subset migration and the outcome of the adaptive immune response.
RESULTS
The migration of DC subsets is context dependent
To understand how the innate immune system might control DC subset migration, we first analyzed the migration of cutaneous DC subsets after immunization with ovalbumin protein (OVA) alone (Th0-immunization), OVA plus lipopolysaccharide (LPS) and poly I:C (Th1-immunization), or OVA plus papain (Th2-immunization). As others have shown, subcutaneous Th2-immunization promoted the specific migration of CD301b+ DCs into the dLN (Figures 1A–1C) (Kumamoto et al., 2013). Conversely, Th1-immunization specifically promoted the migration of CD103+ DCs into the dLN (Figures 1A–1C). Because female mice had a higher frequency of CD301b+ DCs in the dLN after both Th0- and Th2-immunization (Figure S1A), male mice were used for all DC migration experiments to control of baseline DC migration. As previously shown, CD301b+ DCs were absolutely required for Th2 differentiation after papain immunization (Figure 1D) (Kumamoto et al., 2013). Thus, allergen immunization specifically promoted the migration of CD301b+ DCs, which are required for Th2 differentiation.
Figure 1: CD301b+ DCs specifically migrate in response to Th2-skewing immunization.
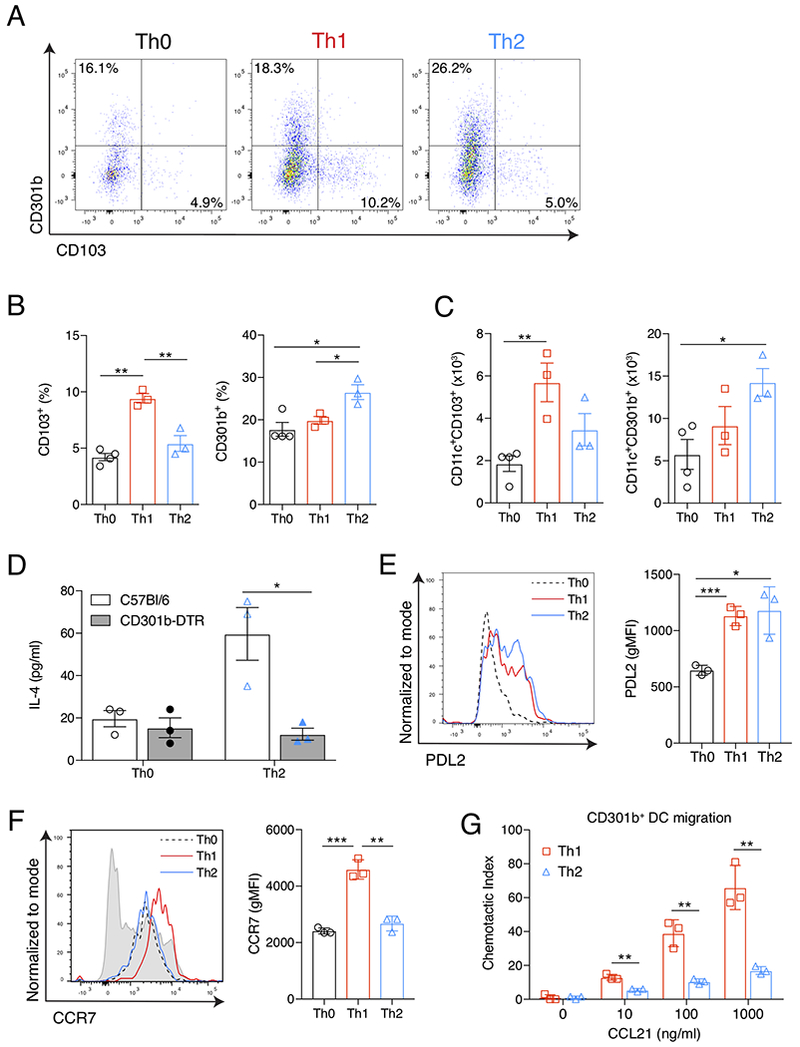
(A) Flow cytometric analysis of viable CD11c+ cells from the popliteal LNs of mice 24 hrs after footpad immunization with Th0- (OVA), Th1- (OVA, LPS & PolyI:C), or Th2-skewing (OVA & papain) immunizations. Numbers indicate percentage of CD11c+ cells in the indicated quadrant. (B) Quantification of percent CD103+ or CD301b+ cells out of total viable CD11c+ cells and (C) total number CD11c+CD103+ or CD11c+CD301b+ cells from the dLN as gated for in (A), 24 hrs after the indicated immunization. (D) ELISA of IL-4 in supernatant of LN cultures from WT (white bars) or CD301b-DTR mice (gray bars) after in vitro restimulation with OVA. All mice received DT and 2.5×106 CD4+ OTII cells prior to indicated immunization. (E) Surface expression of PDL2 and (F) CCR7 from viable CD11c+CD301b+ cells taken from the dLN 24 hrs after the indicated immunization and displayed compared to total CD11c+ cells from Th0-immunized mouse (gray shaded histogram). (G) Transwell chemotaxis to CCL21 of CD11c+CD301b+ cells sorted from the dLN 24 hrs after Th2-skewing immunization. Chemotactic index reflects the number of cells migrating through the Transwell at the indicated concentration over that of media alone. In all experiments, bars indicate mean values of individual data points presented. Error bars indicate SEM. Data are representative experiments from 3 independent experiments with 3-4 mice (A-F) or 3 replicates (G) each. * p<0.05, ** p<0.01, *** p<0.001, Student’s T test. See also Figure S1.
We next examined the phenotype of the CD301b+ DCs in the dLN 24 hours (hrs) after Th0-, Th1-, or Th2-immunizations. As expected, Th2-immunization led to an upregulation in PDL2 expression, an activation marker, on CD301b+ DCs (Figure 1E) (Gao et al., 2013; Kumamoto et al., 2013). This was not significantly different from that seen after a Th1-immunization, indicating that PDL2 upregulation is a common feature of CD301b+ DC activation by both allergens and PRRs (Figure 1E). Despite evidence of CD301b+ DC maturation and migration after Th2-immunization, CD301b+ DCs from the dLN of Th2-immunized mice did not upregulate CCR7 expression as compared to Th1-immunized mice (Figure 1F). There was a small increase in CCR7 expression on intracutaneous CD301b+ DCs after Th2-immunization compared to those after Th0-immunization, but these levels did not approach those seen on CD301b+ DCs after Th1-immunization (Figure S1B & C). This lower level of CCR7 had functional consequences; CD301b+ DCs from the dLN of Th2-immunized mice exhibited decreased chemotaxis toward CCL21 at all concentrations tested when compared to CD301b+ DCs isolated from the dLN of Th1-immunized mice (Figure 1G).
Th2-skewing immunization promotes a specific chemokine signature in the dLN
As Th2-immunization promoted robust CD301b+ DC migration without concurrently inducing optimal CCR7 expression, we hypothesized that additional allergen-induced chemokines may be involved in promoting CD301b+ DC migration from the periphery into the dLN. We focused on two potential sites of chemokine-mediated control: the point of DC entry into lymphatics (i.e., the skin) or the point of LN entry from the lymphatics. We focused our examination at 24 hrs after immunization, which corresponded with DC entry into the dLN (Sokol et al., 2008). In the skin, both Th1- and Th2-immunizations promoted the upregulation of the CCR2 chemokine ligands, Ccl2, Ccl7 and Ccl12 (Figure 2A). Th2-immunization induced the CCR8 ligand Ccl8, but this occurred past the window of DC emigration making it unlikely to be playing a role in CD301b+ DC migration (Figure 2A). Therefore, we turned our attention to the dLN. Within 24 hrs of footpad immunization, Th1-immunization led to upregulation of the CCR2 ligands, while Th2-immunization specifically promoted upregulation of Ccl8 (Figure 2B). Ccl8 upregulation was specific to Th2-immunization; Th1-immunization reduced the expression of both CCR8 ligands, Ccl1 and Ccl8 (Figure 2B).
Figure 2: Allergen immunization promotes a specific chemokine signature in the dLN.
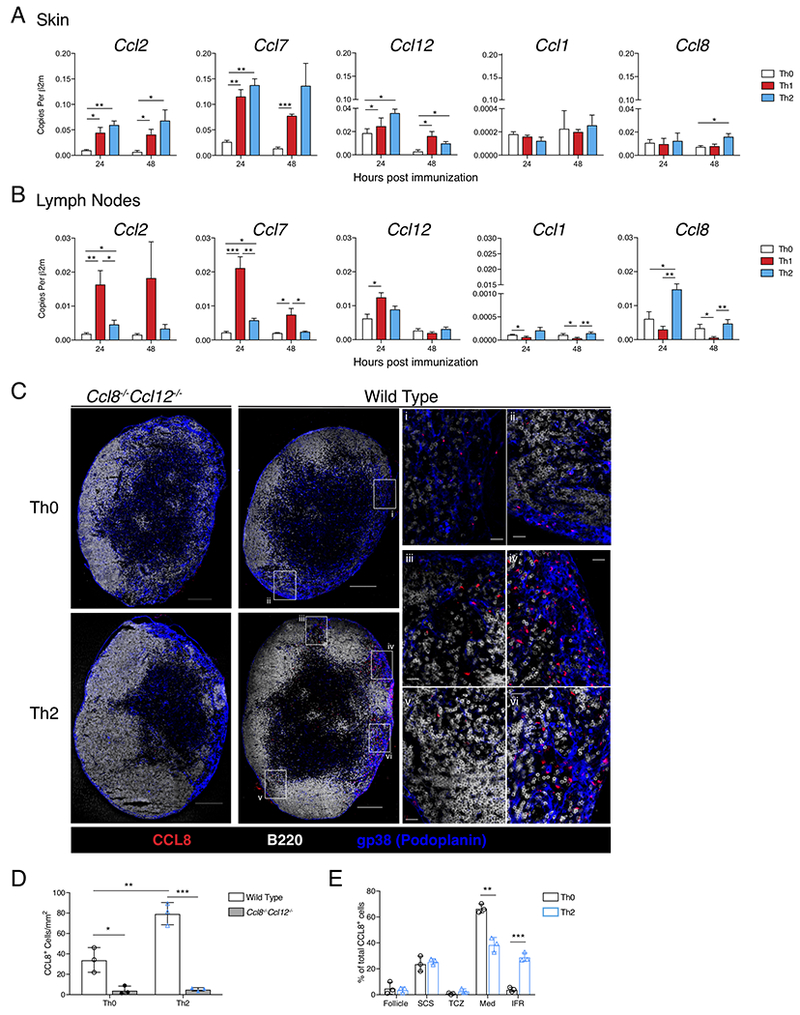
(A) QPCR analysis of footpad skin immunization site and (B) popliteal LN, 24 and 48 hrs after immunization with Th0-, Th1-, or Th2-skewing immunizations. Data is expressed as cDNA copies of indicated gene per copies of β2m. Each bar represents the mean of 3-6 mice from a representative of 4 independent experiments. Error bars indicate SEM. (C) CCL8 protein staining in whole LNs. Popliteal LNs of WT or Ccl8−/−Ccl12−/− mice were harvested 24 hrs after immunization. Confocal immunofluorescence of whole LNs shows CCL8 (red), B220 (white), and gp38 (blue), scale bar indicates 200μm. Insets show medullary (i) and medullary/follicular border (ii) in Th0-immunized WT mouse. In Th2-immunized WT mouse, insets show interfollicular regions (IFR) (iii, iv, and v) and medullary region (vi). Scale bar in insets indicates 20 μm. (D) Total CCL8+ cells per mm2 of LN section from WT and Ccl8−/−Ccl12−/− mice 24 hours after Th0 or Th2-immunization. (E) Percent of CCL8+ cells in each indicated region out of total CCL8+ cells per LN section in wild type Th0 or Th2-immunized mice: Follicle (B cell follicle), SCS (subcapsular sinus), TCZ (T cell zone), Med (medullary region), IFR. Data shown is a representative section from 3 mice used in each of 3 independent experiments. * p<0.05, ** p<0.01, *** p<0.001, Student’s T test. See also Figure S2.
After leaving the afferent lymphatics DCs enter the SCS space and the LN sinuses, which are lined by lymphatic endothelium and LN sinus macrophages, which must be traversed for DCs to enter the LN parenchyma (Girard et al., 2012; Qi et al., 2014). Using Ccl8−/−Ccl12−/− mice as a negative control, we examined the localization of CCL8 protein within dLN sections. In Th0-immunized wild type mice we detected CCL8 protein in two locations (Figure S2): adjacent to gp38+ lymphatic endothelium in the medullary region (Figure 2Ci) and within SCS regions, specifically where the SCS met the medullary region (Figure 2Cii). Concordant with the QPCR data, the number of discrete CCL8 expressing cells significantly increased after Th2-immunization in wild type mice but not in Ccl8−/−Ccl2−/− mice (Figure 2C & D). In addition to this absolute increase in CCL8+ cells, we found that Th2-immunization specifically promoted CCL8 protein expression within the IFR of the dLN (Figure 2Ciii, iv & v, Figure 2E). CCL8+ cells were still readily appreciated within the medullary region after Th2-immunization (Figure 2Cvi). However, given observations by others that IFR are the site of DC entry, we hypothesized that CCL8 produced in the IFR may play a role in the allergen-induced movement of CD301b+ DCs across LN sinuses and into the LN parenchyma (Braun et al., 2011; Woodruff et al., 2014).
CD301b+ DC entry into the dLN requires CCR8 signals
CCL8 and its functional human analogue CCL18 bind specifically to the CCR8 receptor (Islam et al., 2011; Islam et al., 2013). We hypothesized that CD301b+ DCs express CCR8 and that this chemokine system promotes CD301b+ DC trafficking into the dLN in response to Th2-skewing stimuli. Ccr8−/− mice showed decreased migration of mature, PDL2+CD301b+ DCs into the dLN at baseline (i.e., Th0-immunization) and after Th2-immunization (Figure 3A). This was not due to defective exit from or a decreased amount of CD301b+ DCs within the skin as there was no significant difference in the frequency of CD301b+ DCs in the skin of Ccr8−/− mice (Figure S3A). Perhaps due to the loss of CD301b+ DC migration, CD103+ DCs made a larger percentage of the total DC population in the dLN of Ccr8−/− mice, but were not present in significantly greater numbers (Figure 3B). In order to directly assess the migration of CD301b+ DCs from the skin to the dLN, we utilized Kaede transgenic mice, which express a photoconvertible protein (Kaede) that can be converted from green to red fluorescence (Kaedered) (Bromley et al., 2013; Tomura et al., 2008). As expected, we found that migration of CD301b+ DCs from the skin to the dLN (i.e., Kaedered cells) was completely dependent on CCR7 (Figure 3C). However, although some baseline migration of CD301b+ DCs was detected in Ccr8−/−Kaede+ mice, CCR8 was required for the allergen-induced DC emigration from the skin to the dLN (Figure 3D).
Figure 3: CCR8 is required for CD301b+ DC entry into the dLN.
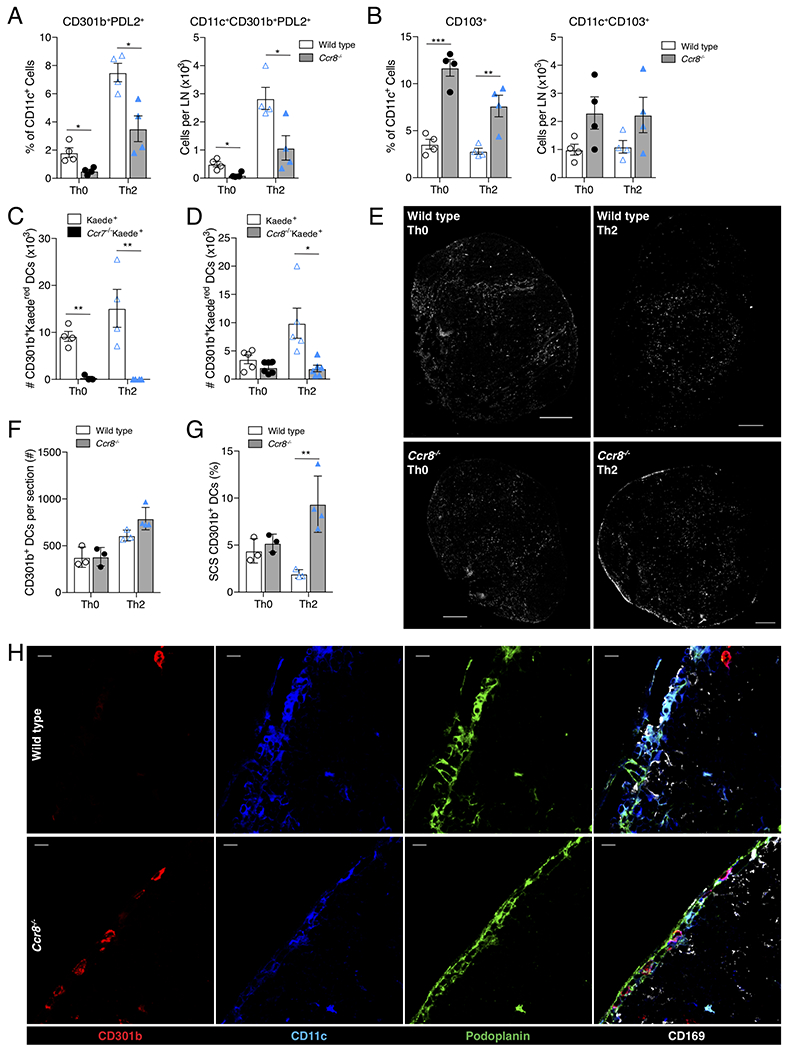
(A) Quantification of CD301b+PDL2+ DCs and (B) CD103+ DCs in the dLN of Th0 or Th2 immunized WT (clear bars) or Ccr8−/− (gray bars) mice as determined by flow cytometry. Percentages reflect percent of CD301b+PDL2+ or CD103+ out of total CD11c+ cells, numbers reflect total cells per popliteal LN. (C & D) Flow cytometry of inguinal LNs was performed 24 hrs after immunization of photoconverted flank skin of Kaede+ transgenic mice. (C) Total CD11c+CD301b+Kaedered (i.e., photoconverted) cells per inguinal LN are shown in comparison to Ccr7−/−Kaede+ and (D) Ccr8−/−Kaede+ groups. (E) CD301b staining (white) of whole popliteal LNs of WT or Ccr8−/− mice 24 hrs after Th0 or Th2 immunization. Scale bar indicates 200μm. (F) Quantification of total number CD11c+CD301b+ cells per entire LN section or (G) percent of total CD11c+CD301b+ within the SCS (identified by podoplanin and CD169 immunohistochemical staining) 24 hrs after immunization. (H) CD301b+ DCs within the SCS space of Ccr8−/− mice, but not WT mice, 24 hrs after Th2 immunization. Panels show single stains of CD301b (red), CD11c (blue), podoplanin (green), and overlay of those stains and CD169 (white). Scale bars indicate 10μm. Images (E & H) are one representative of 2-3 mice per condition and 4 independent experiments. Bar graphs represent mean value of individual mouse data points (3-6 mice per condition per experiment) taken from a representative of 3 independent experiments (A, B, D, F, G, H), or 2 independent experiments (C). Error bars represent SEM. * p<0.05, ** p<0.01, *** p<0.001, Student’s T test. See also Figure S3.
Our data suggested that CD301b+ DCs specifically required CCR8 for entry into the dLN after Th2-immunization. However, by immunofluorescence of the dLN after Th2-immunization, we found no significant difference in the total number of CD301b+ DCs between WT and Ccr8−/− mice (Figure 3E & 3F). Instead, whereas CD301b+ DCs generally localized to the T-B interface of the dLN in WT mice after a Th2-immunization, in Th2-immunized Ccr8−/− mice CD301b+ DCs appeared to be excluded from entering the dLN and were retained at the periphery of the LN (Figure 3E). Closer inspection after Th2-immunization revealed an accumulation of CD301b+ DCs in the SCS space of Ccr8−/− mice (Figure 3G & 3H). Because standard LN digestion protocols do not adequately release SCS stroma, these SCS CD301b+ DCs were unintentionally excluded in flow cytometry experiments leading to the decrease in CD301b+ DC numbers by flow cytometry, but not immunofluorescence (Fletcher et al., 2011; Gray et al., 2012). We were able to confirm this using enhanced digestion methods designed to release LN stromal cells (Fletcher et al., 2011). Although standard digestion methods showed a decreased percentage of Kaedered skin emigrant CD301b+ DCs in Ccr8−/− mice (Figure 3D), no such defect was seen after enhanced digestion, indicating that enhanced digestion released CD301b+ DCs from the SCS (Figure S3B). Taken together, these data indicated that while CCR7 was essential for CD301b+ DC entry into the draining lymphatics, CCR8 played an important role in promoting their migration across the floor of the LN sinus and into the dLN following a Th2-immunization.
CD169+SIGN-R1+ macrophages produce Ccl8, which is required for CD301b+ DC entry
Our initial CCL8 staining showed it to be closely associated with the lymphatic endothelium of the dLN, but we were unable to detect CCL8 protein or RNA in Lyve-1+ or gp38+ lymphatic endothelium (data not shown). We next hypothesized that LN sinus macrophages that are found in association with lymphatic endothelium, were the source of CCL8. LN sinus macrophages include CD169+SIGN-R1−F4/80− SCS macrophages as well as F4/80+ medullary macrophages, which have been divided into the CD169−F4/80+ medullary cord macrophages and CD169+SIGN-R1+F4/80+ medullary sinus macrophages (Gray and Cyster, 2012). In accordance with their name, medullary cord macrophages are present in the LN medullary region and SCS macrophages make up the floor of the SCS overlying B cell follicles. However, the CD169+SIGN-R1+ medullary sinus macrophages can be found along lymphatic endothelium of the medullary region as well as the IFR (Gray and Cyster, 2012). CD169+ macrophages in the lamina propria have been shown to produce CCL8 in murine colitis models (Asano et al., 2015). Similarly, we found that CCL8 protein could be detected within CD169+F4/80+ macrophages after Th2-skewing immunization and that these cells could be visualized in apposition with CD301b+ DCs (Figure 4A and Figure S4A). Based on localization in serial sections, CCL8+CD169+ cells also expressed SIGN-R1, consistent with the F4/80+CD169+SIGN-R1+ macrophage population (Figure S4B). CD169+SIGN-R1− SCS macrophages were specifically localized in the SCS floor adjacent to B cell follicles (Figure 4B, SCS), while CD169+SIGN-R1+ macrophages were present in the medullary region (Figure 4B, med) as well as in the IFR (Figure 4B, IFR). To confirm that CD169+SIGN-R1+ macrophages were the source of Ccl8, we utilized enhanced enzymatic digestion techniques to free LN resident macrophages for further analysis (Fletcher et al., 2011). Using SIGN-R1 positive selection by magnetic beads, we found that Ccl8 and Siglec1 (CD169) were both enriched in the SIGN-R1+ population (Figure 4C). We next performed flow cytometric sorting of SIGN-R1+ cells, which identified the CD169+SIGN-R1+CD11c+CD11b+ macrophages as the source of Ccl8 (Figure 4D). This enrichment of Ccl8 expression by QPCR was confirmed using an alternative sorting scheme to identify the CCL8-producing macrophages based on expression of CD11c+CD103+CD11bhighCD169+F4/80+ (Figure S5A & B). Finally, to confirm our cell sorting data, we depleted CD169+SIGN-R1+F4/80+ macrophages using clodronate-mediated depletion (Figure S5A). Clodronate treatment completely abolished Siglec1 and Ccl8 expression within the dLN (Figure 4E). Together, these data indicated that CD169+SIGN-R1+ macrophages, located in the IFR and LN medullary area, were the source of CCL8 in the dLN.
Figure 4: CCL8 is produced in the dLN by CD169+SIGN-R1+ LN macrophages that interact with CD301b+ DCs.

(A) Confocal immunofluorescence of whole popliteal LN (top) and area of interest (bottom) 24 hrs after Th2-skewing immunization in WT or Ccl8−/−Ccl12−/− mice as indicated. Panels show single stains of CCL8 (white), CD169 (blue), CD301b (red) and overlay. Scale bars in top panels represent 200μm, scale bars in bottom panels represent 10μm. Images are representative of 3 mice in each of 3 independent experiments. (B) Confocal immunofluorescence of whole popliteal LN with insets showing CD169+SIGN-R1+ macrophage populations in medullary regions (med) and IFR (IFR), as well as CD169+SIGN-R1− SCS macrophages in the SCS (SCS). CD169 (red), SIGN-R1 (white), gp38 (green), and B220 (blue). Scale bar in whole LN represents 200 μm, those in insets represent 10 μm. (C) QPCR analysis of total cells (Pre) or MACS positively selected SIGN-R1+ or SIGN-R1− cells from popliteal LNs. (D) Gating scheme to identify macrophage populations from total viable lymphocytes obtained 24 hours after Th2-immunization from dLN, and QPCR analysis of the indicated populations showing Ccl8 expression. Colored histograms correspond to colored gates. (E) QPCR from total LNs of control liposome- or clodronate-treated mice 24 hrs after Th2-mediated immunization. Data points are from individual mice (4-6) in one representative experiment out of three independent trials. (F) Decreased percentage and number of CD301b+ DCs in Ccl8−/−Ccl12−/− mice after Th2-skewing immunization, based on flow cytometry of viable CD11c+CD301b+ cells. Data points are from dLNs of individual mice (3-4) from one of three independent trials. Cells in (C) and (D) were obtained using enhanced enzymatic digestion, while those in (F) were obtained using routine enzymatic digestion. QPCR data presented as copies of indicated transcript over Gapdh. Bar graphs represent mean value of indicated data points, error bars represent SEM. * p<0.05, ** p<0.01, *** p<0.001, Student’s T test. See also Figure S4 and S5.
CCR8 has two ligands in the mouse: CCL1 and CCL8 (Griffith et al., 2014). CCL1 has been shown to be produced by lymphatic endothelium on the floor of the SCS and to promote entry of monocyte-derived DCs into the dLN (Qu et al., 2004). We found that Th2-skewing immunization did not promote Ccl1 production in the dLN (Figure 2B). However, to ensure that the defect in CD301b+ DC migration was due to CCL8, and not CCL1, we examined CD301b+ DC migration in Ccl8−/− mice. Due to the genes’ proximity, Ccl8−/− mice are only currently available as Ccl8−/−Ccl12−/− dual knock-out mice (Tsou et al., 2007). Consistent with our data from Ccr8−/− mice using standard enzymatic digestion, we found that Ccl8−/−Ccl12−/− mice exhibited defective CD301b+ DC migration by flow cytometry in both total frequency and number after Th2-skewing immunization (Figure 4F). Therefore, we conclude that CCL8 production from CD169+SIGN-R1+ macrophages is required for CD301b+ DC entry into the dLN after Th2-skewing immunization.
Analysis of Ccl1-Ccl8 dual reporter transgenic mice confirms Ccl8 production by CD169+SIGN-R1+ macrophages
In order to verify our CCL8 expression data, we generated a transgenic mouse that Reports the Expression of the CCR8 ligands (REC8). To allow for detection of Ccl1 and Ccl8 expression, we inserted mCherry at the start codon of Ccl1 and eGFP at the start codon of Ccl8 in a bacterial artificial chromosome that contained the Ccl1 and Ccl8 genes (Figure 5A). Consistent with RNA data from dLNs (Figure 2B), Ccl1-mCherry expression was unaffected while Ccl8-eGFP expression was induced by Th2-immunization (Figure 5B). CCL1 has been described to be produced by CD3+ T cells as well as lymphatic endothelium (Miller and Krangel, 1992; Qu et al., 2004). We were unable to detect Ccl1-mCherry expression in lymphatic endothelial cells, blood endothelial cells or fibroblastic reticular cells of the dLN (Figure S5C). Instead, we found that Ccl1-mCherry+ cells were uniformly CD3+, while Ccl8-eGFP+ cells were CD11b+ (Figure 5C). Accurate Ccl1 and Ccl8 reporting by REC8 mice was confirmed by enrichment of Ccl1 and Ccl8 RNA within the mCherry+ and eGFP+ populations, respectively (Figure 5D). The expression of CD11b on Ccl8-eGFP+ cells suggested that Ccl8-eGFP marked CD169+SIGN-R1+ macrophages. Confirming this, CD169+SIGN-R1+ macrophages exhibited Ccl8-eGFP expression, which was induced by Th2-immunization, but not Ccl1-mCherry expression. (Figure 5E). These data indicate Th2-immunization specifically induces Ccl8 expression by CD169+SIGN-R1+ macrophages.
Figure 5: CCR8 ligands are differentially expressed in the dLN.
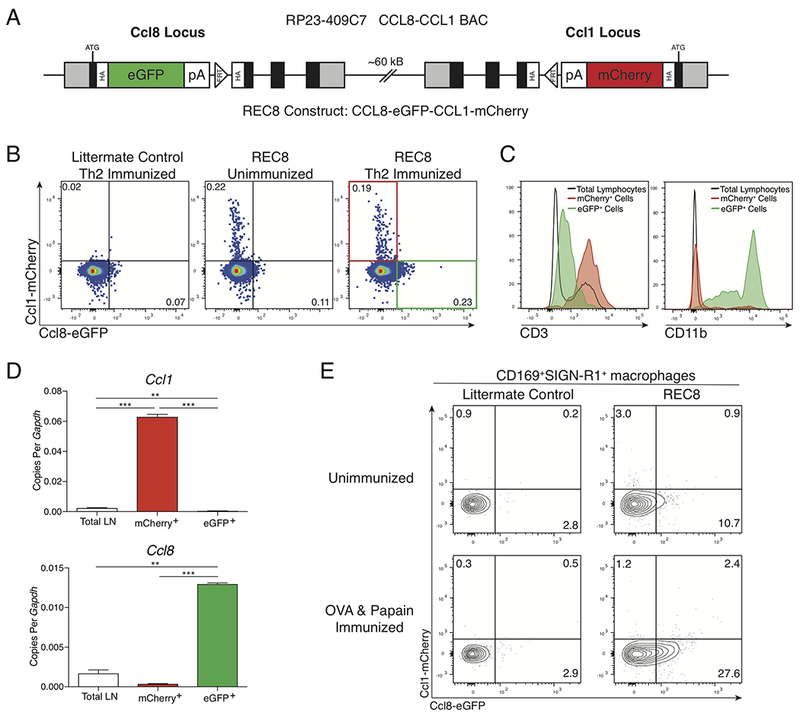
(A) Schematic of REC8 transgene construct indicating insertion of eGFP into the Ccl8 locus and mCherry into the Ccl1 locus of the RP23-409C7 BAC. Gray box, non-coding exons; black box, endogenous coding exons; green box, eGFP ORF; red box, mCherry ORF; clear box, HA tags or SV40 poly(A); clear triangle, Flippase Recognition Target (FRT) site. (B) Expression of mCherry and eGFP in total live dLN cells in unimmunized REC8 mice or in Th2-immunized REC8 mice or littermate control WT mice. Numbers are percent of total cells in indicated gates. (C) Expression of CD3 and CD11b in total live dLN cells (black), mCherry+ cells (red), or eGFP+ cells (green) gated as in (B), isolated from REC8 mice 24 hours after Th2-skewing immunization. (D) QPCR analysis of Ccl1 or Ccl8 expression in total LN (clear), or FACS sorted mCherry+ cells (red bar) and eGFP+ cells (green bar), gated as in (B) from dLNs harvested from REC8 mice 24 hours after Th2-skewing immunization. (E) Expression of mCherry and eGFP on live, CD11b+CD169+SIGN-R1+ cells harvested from dLN of unimmunized WT and REC8 mice and 24 hours after Th2-skewing immunization. Bar graphs represent mean value of replicates (2 data points per bar). Data shown are representative of three independent experiments, except (D) which is representative of two independent experiments. Error bars represent SEM. ** p<0.01, *** p<0.001, Student’s T test. See also Figure S5.
CCR8 promotes CCR7-mediated migration of CD301b+ DCs
Our data suggest that while both CCR7 and CCR8 are necessary for CD301b+ DC migration, CCR8 plays a specific role in promoting CD301b+ DC migration across the SCS and into the LN parenchyma. Ccr8 mRNA has been detected in dermal DC populations, but the detection of CCR8 protein has been hampered by the lack of suitable reagents (Jakubzick et al., 2006; Qu et al., 2004; Sallusto et al., 1998; Yabe et al., 2015). Flow cytometric staining using commercially available anti-mouse CCR8 antibodies consistently showed staining of DCs from Ccr8−/− mice (data not shown). Therefore, to determine the expression of CCR8 on CD301b+ DCs we utilized a fluorescently-labeled ligand of CCR8, human CCL1 (hCCL1). hCCL1 binds to the murine CCR8 receptor, inducing calcium flux and chemotaxis in transfected cell lines (Islam et al., 2011). Using this reagent to fluorescently “label” CCR8, we found that compared to Ccr8−/− mice, CD11c+CD11b+CD301b− DCs (CD11c+ DCs) did not express detectable CCR8, but that CD11c+CD11b+CD301b+ DCs (i.e., CD301b+ DCs) from the dLNs of Th0-immunized mice expressed detectable CCR8 that increased after Th2-immunization (Figure 6A and S6A). We found no significant difference in CCR7 expression between WT and Ccr8−/− DCs after Th0- or Th2-immunization (Figure 6B). As expected, ex vivo CCL21 stimulation of CD301b+ DCs promoted CCR7 internalization, but this was unaffected by co-stimulation with CCL8 (Figure S6B). Likewise, CCL21 stimulation had no effect on CCR8 expression (Figure S6B). These data suggest that CCR8 is expressed by CD301b+ DCs, and that CCR8 expression does not affect CCR7 expression.
Figure 6: CCL8 synergizes with CCL21 to specifically promote CD301b+ DC migration.
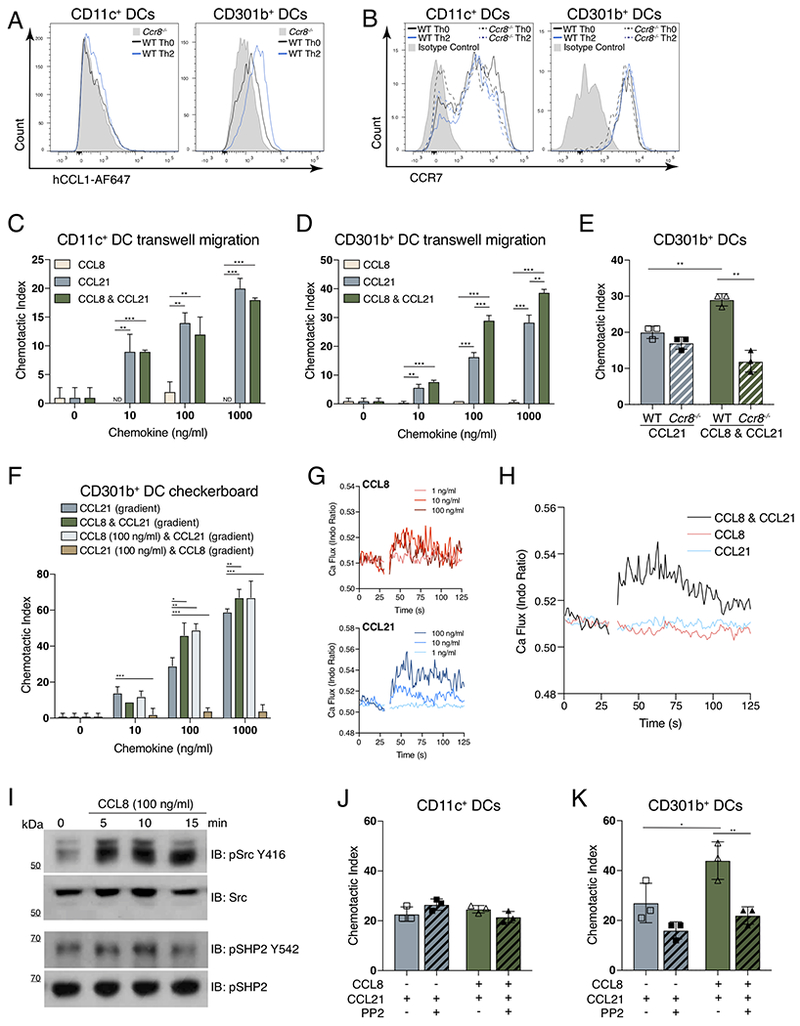
(A) Flow cytometric expression hCCL1-AF647 binding to CD11c+CD11b+CD301b− (CD11c+ DCs) or CD11c+CD11b+CD301b+ (CD301b+ DCs) cells obtained from dLNs of WT mice after the Th0 (black histogram) or Th2-skewing (blue histogram) immunization or from Ccr8−/− mice after Th0 immunization (gray shaded histogram). (B) Flow cytometric cell surface expression of CCR7 on CD11c+ DCs or CD301b+ DCs from the dLN after Th0-immunization in WT (black solid) or Ccr8−/− (black dotted) mice, or after Th2-immunization in WT (blue solid) or Ccr8−/− (blue dotted) mice. Isotype control from WT Th0-immunized mice is shown (gray shaded histogram). (C) Transwell chemotaxis of CD11c+ DCs or (D) CD301b+ DCs to indicated concentrations of CCL8, CCL21 or CCL8 & CCL21. (E) Transwell chemotaxis of CD301b+ DCs isolated from WT or Ccr8−/− mice to 100 ng/ml of indicated chemokines. (F) Checkerboard Transwell migration assay of CD301b+ DCs in response to CCL21 gradient alone (blue), CCL8 & CCL21 combined gradient (green), fixed CCL8 concentration and CCL21 gradient (light blue), and fixed CCL21 concentration and CCL8 gradient (brown). (G) Analysis of calcium flux in CD301b+ DCs, gap indicates addition of CCL8 or CCL21 at the indicated concentration. (H) Calcium flux in CD301b+ DCs, gap indicates addition of 1 ng/ml of CCL8 & CCL21 (black), CCL8 (pink), and CCL21 (blue). (I) Western blot of Src and SHP2 phosphorylation in serum-starved 4DE4 cells stably transfected with human CCR8. (J) Transwell chemotaxis of CD11c+ DCs or (K) CD301b+ DCs in response to 100 ng/ml of the indicated chemokines with or without 10μM PP2 (Src inhibitor). In all panels, cells were harvested from dLNs 24 hrs after the indicated immunization. (A, B, G & H) are representative graphs from four independent experiments, 3 mice per group. Cells were obtained by pooling dLNs from 30-50 mice (C-F, J, & K) or 12 mice (G-H) 24 hrs after Th2-immunization and using fluorescence activated cell sorting to obtain CD11c+ DCs or CD301b+ DCs. Bar graphs represent mean value of replicates (2-3 data points per bar), and are one representative trial of four independent experiments. Error bars represent SEM. * p<0.05, ** p<0.01, *** p<0.001, Student’s T test. See also Figure S6.
We next performed in vitro chemotaxis assays to determine what role CCR8 and its ligand, CCL8, played in CD301b+ DC migration. CCL8 had no effect on migration of the CCR8 negative, CD11c+ DCs (Figure 6C). Despite expression of CCR8, CD301b+ DCs showed no migration in response to CCL8 alone (Figure 6D). However, the combination of CCL8 and the CCR7 ligand CCL21 were synergistic in CD301b+ DCs, leading to an almost doubling of migration at the physiologic 100 ng/ml concentration of these chemokines (Figure 6D). This synergism was also seen with the other CCR8 ligand, CCL1 (Figure S6C). Using in vitro assays with μM chemokine concentrations, human CCL1, CCL2, CCL7, CCL8 and CCL11 have been shown to synergize with CCL21 by the formation of heteromeric chemokine complexes that promote the migration of mature human DCs (Paoletti et al., 2005). Therefore, we examined whether this synergistic effect on CCR7-mediated CD301b+ DC migration was similarly indiscriminate, but found no effect of murine CCL2, CCL7, and CCL11 on CD301b+ DC migration in the presence or absence of CCL21 (Figure S6D-F). Indeed, the synergism in migration induced by murine CCL8 and CCL21 was dependent on CCR8 expression in CD301b+ DCs, as it was absent in Ccr8−/− CD301b+ DCs. (Figure 6E). To determine whether a CCL8 gradient was necessary for its synergistic effect, we performed a checkerboard chemotaxis assay in which CD301b+ DCs harvested from the dLN of Th2-immunized mice were exposed to combinations of CCL8 and CCL21 in either gradients or fixed concentrations. While CCL21-mediated migration required a CCL21 gradient, the synergistic effect of CCL8 on CCL21-induced migration of CD301b+ DCs was independent of the presence of a CCL8 gradient (Figure 6F). These data suggest that the production of CCL8 is an innate control mechanism to specifically promote the migration of Th2-skewing CD301b+ DCs by altering their responsiveness to CCL21 gradients.
Chemokine receptors are G protein-coupled receptors (GPCRs) that classically signal through Gαi to promote cell migration. We hypothesized that CCL8 augmented CCL21-mediated migration in CD301b+ DCs by enhancing GPCR-mediated signaling. To study this, we measured calcium (Ca2+) flux in CD301b+ DCs isolated from the dLN of Th2-immunized mice. We were able to detect Ca2+ flux in CD301b+ DCs exposed to either CCL8 or CCL21 at 10 ng/ml and 100 ng/ml concentrations (Figure 6G). Although exposure to either CCL8 or CCL21 at 1 ng/ml did not induce Ca2+ flux, the combination of CCL8 and CCL21 at a 1 ng/ml did induce Ca2+ flux (Figure 6H). This indicated that perhaps CCL8 increases the sensitivity of CD301b+ DCs to low levels of CCL21 as we found no augmentation in Ca2+ flux at higher chemokine concentrations (Figure S6G). In addition to inducing calcium signaling, we hypothesized that CCL8 might activate other signaling pathways that contribute to cell migration. It was recently reported that prostaglandin E2 augments CCL21-induced human DC migration by increasing signaling via Src kinase and the tyrosine phosphatase SHP2 (Hauser et al., 2016). Using a human CCR8 transfected cell line (Islam et al., 2011), we found that murine CCL8 stimulation induced the phosphorylation of Src and SHP2 within 5 min and 10 min, respectively (Figure 6I). Therefore, we hypothesized that CCL8 may similarly promote signaling through the Src and SHP2 pathway to enhance CCL21-mediated migration in CD301b+ DCs. Chemotaxis of CD11c+ DCs to CCL21 was unaffected by the presence of the Src inhibitor, PP2 (Figure 6J). However, in CD301b+ DCs, Src inhibition abolished the synergistic effect of CCL8 on CCL21-mediated migration (Figure 6K). Thus, CCL8 directly promotes Src-SHP2 signaling and augments CCL21-mediated migration in CD301b+ DCs in a Src-dependent manner.
CCR8 mediated entry of CD301b+ DCs into the dLN is required for Th2 differentiation
Our data suggest that the production of CCL8 by CD169+SIGN-R1+ macrophages promotes the migration of CCR7loCD301b+ DCs by inducing a Src-dependent signaling pathway. This led us to hypothesize that the production of CCL8 by CD169+SIGN-R1+ macrophages was a mechanism by which the innate immune system controlled the polarization of the adaptive immune response. Therefore, we examined whether CCR8 deficiency led to alterations in Th development by examining the immune response in Ccr8−/− mice that had previously received WT naïve OVA-specific CD4+ OTII cells. In response to the Th2-immunization OVA & papain, we found a significant defect in Th2 differentiation based on IL-4 production during in vitro restimulation, but found no defect in IFNγ production on restimulation after Th1-skewing immunization (Figure 7A). To assess whether this defect in Th2 differentiation was limited to papain immunization, we next examined CD4+ T cell differentiation after immunization with the alternative Th2-skewing agent, OVA/Alum, or infection with Nippostrongylus brasiliensis (N. brasiliensis). Regardless of the Th2-skewing immunization, we found a significant defect in Th2 differentiation based on IL-4 and IL-13 production from transferred OVA-specific CD4+ OTII cells in Ccr8−/− mice (Figure 7B, 7C, and S7). These data indicated that Th2 differentiation in response to cutaneous immune stimuli requires CCR8.
Figure 7: CCR8 on CD301b+ DCs is required for allergen-induced Th2 differentiation.
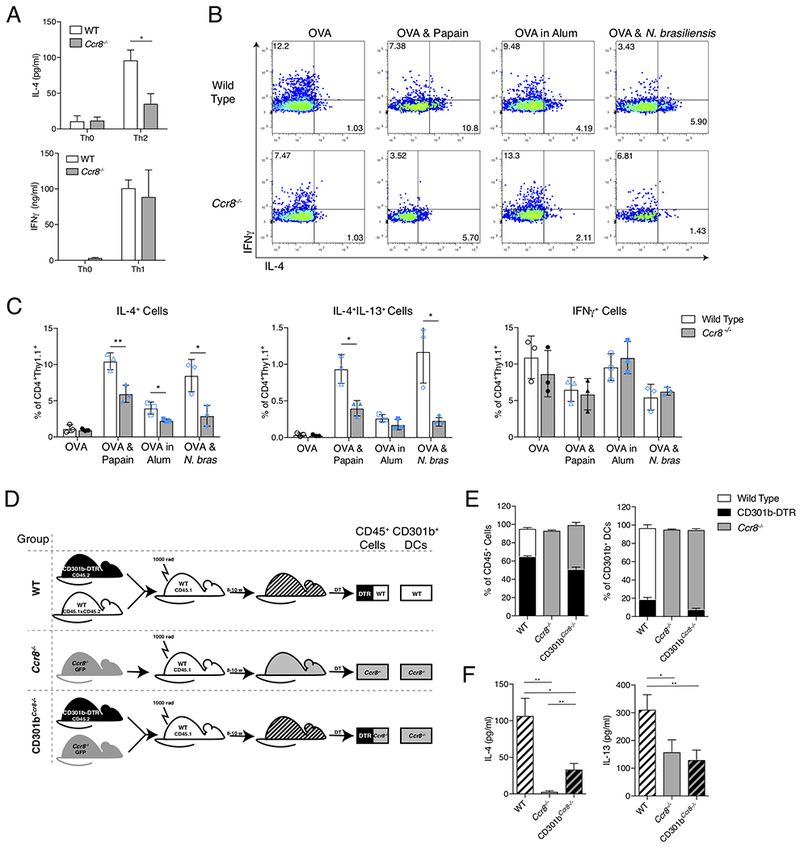
(A) IL-4 and IFNγ concentrations in supernatants following in vitro restimulation of total dLN cells from WT or Ccr8−/− mice after Th0 or Th2-skewing immunizations (IL-4) or Th0 or Th1-skewing immunizations (IFNγ). Mice received 2.5×105 CD4+ OTII cells prior to immunization, dLNs were harvested 5 days after immunization for in vitro restimulation with OVA, and supernatants were obtained 4 days later. (B) Intracellular IL-4 and IFNγ staining of CD4+Thy1.1+ viable cells 4 days after OVA, OVA & Papain, or OVA in Alum immunization or 5 days after OVA & N. brasiliensis infection. Mice received 2.5-5×105 naïve CD4+Thy1.1+ OTII cells prior to immunization Representative plots are shown with percentage of cells in quadrants indicated. (C) Quantification of IL-4+, IL4+IL-13+ or IFNγ+ staining of CD4+Thy1.1+ cells after immunization or infection as in (B). (D) Generation of BM chimeras. (E) Chimerism by flow cytometric analysis of total viable CD45+ cells and CD11c+CD301b+ DCs from popliteal LNs 3 days after last DT injection. (F) IL-4 and IL-13 concentrations in supernatants from in vitro restimulation of total LN cells from indicated BM chimeras. Chimeric mice received 2.5×105 CD4+ OTII cells and DT prior to Th2-skewing immunization. dLNs were harvested 5 days after immunization and cultured with OVA for 4 days prior to supernatant harvest. Data shown is representative of 3 independent experiments (A-C) or 5 independent experiments (D & E), with 3-5 mice per condition in each experiment. Bar graphs represent mean value, error bars represent SEM. * p<0.05, ** p<0.01, *** p<0.001, Student’s T test. See also Figure S7.
To determine whether this phenotype seen in Ccr8−/− mice was the result of CCR8 deficiency specifically on the CD301b+ DC subset, we produced mixed bone marrow (BM) chimeras utilizing WT, CD301b-DTR, and Ccr8−/− mice. Utilizing the scheme illustrated in Figure 7D, we produced three groups of mice: WT, Ccr8−/−, and CD301bCcr8−/−. WT mice were mixed chimeras of WT and CD301b-DTR cells, allowing us to control for any effect caused by depletion of CD301b+ DCs derived from CD301b-DTR precursors, while Ccr8−/− mice were standard BM chimeras into WT mice (Figure 7E). CD301bCcr8−/− mice were mixed chimeras of CD301b-DTR and Ccr8−/− BM cells, allowing us to examine mice in which the overall immune compartment was mixed, but the CD301b+ DC compartment was derived from Ccr8−/− mice after DT-mediated depletion (Figure 7E). As expected, Ccr8−/− BM chimeras showed a defect in Th2 differentiation as measured by IL-4 and IL-13 production on in vitro restimulation of previously transferred OVA-specific CD4+ T cells (Figure 7F). CD301bCcr8−/− mice, in which all CD301b+ DCs were CCR8-deficient but the rest of the immune cells were a mixture of CCR8-sufficient and CCR8-deficient cells, also showed a defect in Th2 differentiation (Figure 7F). Restricting complete CCR8 deficiency to the CD301b+ DC population led to a significant loss of IL-4 production that was intermediate to that seen between the WT and Ccr8−/− groups, and a loss of IL-13 production that was indistinguishable from the level in Ccr8−/− chimeras. Since both Th2 and Tfh cells produce IL-4, but only Th2 cells produce IL-13, we believe these data indicate that CCR8 expression on CD301b+ DCs is required for their migration into the dLN after allergen exposure, and is necessary for subsequent Th2 differentiation. However, the intermediate level of IL-4 production seen in CD301bCcr8−/− chimeras compared with the Ccr8−/− chimeras may indicate that another, non-CD301b+ DC involved in Tfh development, requires CCR8 for entry into the dLN and subsequent Tfh differentiation (Kumamoto et al., 2016).
DISCUSSION
A fundamental paradigm in immunity is the role of DCs in bridging the innate and adaptive immune responses. DCs are positioned throughout the periphery where they are poised for early detection of pathogens. Pathogens induce DC maturation through activation of their PRRs, which leads to CCR7 upregulation and migration into the dLN. Within the dLN, DCs can interact with naïve T cells to activate the adaptive immune response by providing antigen presentation, costimulation and skewing signals. However, this paradigm does not address the evidence that specific DC subsets play distinct roles in T cell activation. Dermal IRF4+CD301b+PDL2+ DCs and lung KLF4+ DCs migrate to dLNs after allergen exposure and have been shown to play a necessary role in Th2 differentiation, while IRF4+KLF4− DCs play a specific role in the differentiation of Th17 cells (Gao et al., 2013; Kumamoto et al., 2013; Tussiwand et al., 2015; Williams et al., 2013). Given that DC subset migration is stimulus specific and that DC subsets induce different adaptive immune outputs, we were interested in defining the mechanisms that promote this specificity in DC migration. To do this, we utilized subcutaneous papain immunization, which promotes Th2-differentiation via the migration of CD301b+ dermal DCs into the dLN. Despite robust migration to the dLN after Th2-immunization, CD301b+ DCs did not upregulate CCR7 compared to after Th1-immunization, leading us to hypothesize that additional mechanisms might promote CD301b+ DC migration in vivo.
Chemokines are key regulators controlling the migration and positioning of immune cells. CCR7 and its ligands CCL19 and CCL21 control the migration of mature DCs from the periphery to dLNs (Worbs et al., 2017). Within the dLN, chemokines promote the positioning of DCs, T cells and accessory cells within specific niches where they allow for optimal T cell activation and effector differentiation (Griffith et al., 2014; Lian and Luster, 2015). There is evidence that CCR8 and its ligand CCL1 may play a role in the migration of monocyte-derived DCs from the skin and conventional DCs from the lung to the dLN (Jakubzick et al., 2006; Qu et al., 2004). However, we were unable to detect any induction of Ccl1 transcript or reporter expression in the dLN in response to Th2-immunization. Instead, Th2-immunization was specifically associated with upregulation of the other CCR8 ligand, Ccl8, in the dLN that temporally correlated with DC migration. CCR8 has been associated with allergic inflammation and is present on Th2 cells, immature DCs and monocytes. CCR8 has also been shown to play a potential role in the skewing of the adaptive immune response, although the results have been mixed. Ccr8−/− mice show attenuated effector type-2 immune responses to Schistosoma mansoni soluble egg antigen, OVA, and cockroach antigen-induced allergic airway inflammation, as well as allergic skin inflammation in a model of atopic dermatitis (Chensue et al., 2001; Islam et al., 2011). However, these defects were thought to be the result of defective CCR8 expression by Th2 cells. Arguing in favor of a role for CCR8 expression on DCs, in a model of Th1- and Th17-mediated contact hypersensitivity, Ccr8−/− mice were shown to have increased numbers of CD11c+ cells in the dLN associated with increased Th1 and Th17 differentiation (Yabe et al., 2015). At the same time, others have not been able to find a role for CCR8 in Th2-mediated inflammation (Chung et al., 2003; Goya et al., 2003). The cause for this discrepancy is unknown, but our data suggest it may be due to the dependence of different models on CD301b+ dermal DCs. We found that disparate Th2-skewing stimuli, including papain, alum and N. brasiliensis infection, all require CCR8 for CD301b+ DC migration and Th2 differentiation. Spontaneous migration of CD301b+ DCs appears to be unaffected by CCR8 deficiency, raising the question of whether spontaneous migration is functionally different. Additionally, it remains to be determined whether CCR8-mediated migration is a global mechanism used to control the migration of Th2-skewing DCs from non-cutaneous tissues.
Instead of acting to directly promote chemotaxis, we found that CCL8 augments CCL21-induced migration by two potential mechanisms. First, dual exposure of CD301b+ DCs to CCL8 and CCL21 led to Ca2+ flux at chemokine concentrations that were individually incapable of inducing a Ca2+ flux, suggesting that CCR8 signaling may increase CD301b+ DC sensitivity to CCR7-ligand induced migration. Alternatively, we found that CCL8 directly induced Src-SHP2-mediated signaling in a CCR8 transfected cell line, and that the augmentation of CCL21-mediated CD301b+ DC chemotaxis by CCL8 required Src signaling. Prostaglandin E2-induced oligomerization of CCR7 has been shown to promote biased signaling through the Src-SHP2 pathway, which is required for optimal migration of human monocyte derived DCs in response to CCL21 signaling (Hauser et al., 2016). By promoting Src-SHP2 signaling in CCR8+ CD301b+ DCs, CCL8 may facilitate the ability of CCR7 ligands to induce the migration of CD301b+ DCs across the lymphatic endothelium and into the dLN after Th2-immunization.
Why is another signal necessary for CD301b+ DCs to cross the border of the LN sinuses into the LN parenchyma? One possibility is based on the observation that chemokine receptors internalize after signaling, thus muting their responsiveness to ligands after signaling. This internalization has been postulated to underlie the role of the chemokine scavenger receptor ACKR4 in permitting conventional DCs to re-express CCR7, allowing them to cross the SCS (Ulvmar et al., 2014). Due to their relatively low level of CCR7 expression, CD301b+ DCs may not be able to recover from such ligand mediated internalization and require an additional chemokine signal. Alternatively, induction of the Src-SHP2 signaling pathway may be a crucial step for traversing the lymphatic endothelium at IFRs. In the absence of CCR8, CCR7 oligomerization can induce this signaling pathway, but oligomerization may require higher levels of surface CCR7 than those expressed by CD301b+ DCs. Although the precise molecular mechanism remains to be uncovered, we have shown a specific and necessary role for CCR8 in promoting CCR7-mediated migration in subset-specific DC migration.
Beyond showing a role for CCL8-CCR8 in CD301b+ DC migration, our data also reveal a role for CD169+SIGN-R1+ LN macrophages in controlling innate and adaptive immune interactions. SCS macrophages have been shown to efficiently capture lymph borne virus particles and present them to B lymphocytes, thereby preventing lymphatic spread of virus and promoting antigen specific immunity (Junt et al., 2007). But the role of other LN macrophage populations during the primary immune response is largely unknown. Through their positive effects on CD301b+ DC migration, CD169+SIGN-R1+ macrophages may play an important role in promoting a Th2-biased response. However, the precise mechanism by which they respond to protease allergen exposure remains unclear. Papain freely flows to the dLN and so may be able to directly activate LN macrophages. Alternatively, papain immunization has been shown to promote epithelial production of reactive oxygen species, thymic stromal lymphopoietin, and IL-33, all of which have been shown to play roles in the induction of the allergic immune response (Halim et al., 2014; Tang et al., 2010). Whether one of these epithelial derived factors activates CD169+SIGN-R1+ macrophages remains to be elucidated.
We propose a stepwise model for the allergen-induced migration of CD301b+ DCs. In the first step, DCs use CCR7 mediated signals to migrate into the draining lymphatics and enter the SCS space. In the second step, CCR8+ CD301b+ DCs respond to CCL8 produced by CD169+SIGN-R1+ macrophages, which allows them to transmigrate across the SCS floor and into the LN parenchyma. Such a process was previously identified in a melanoma metastasis model in which tumor cells entered the draining lymphatics and were only able to cross the SCS of the dLN if they expressed CCR8 on their surface (Das et al., 2013). This mirrors the accumulation of CD301b+ DCs seen in the LN sinuses of Ccr8−/− mice. We propose that this stepwise migration process evolved to specifically promote CD301b+ DC migration and adaptive immune bias, but in melanoma metastasis this control point is hijacked by cancer cells. Thus, we believe that our model may provide insights beyond DC migration that may be generalizable to cell migration and cancer metastasis.
STAR METHODS
Experimental methods
Key Resources Table
| Reagent/Material | Source | Identifier |
|---|---|---|
| Antibodies: | ||
| Alexa Fluora 594 Streptavidin | Biolegend | Cat #: 405240 |
| Alexa Fluroa 647 Streptavidin | Biolegend | Cat #: 405237 |
| Anti-Mouse CD8α, Clone: 53-6.7 | Biolegend | Cat #: 100725 |
| Anti-Mouse CD11b, Clone: M1/70 | Biolegend | Cat #: 101251 |
| Anti-Mouse CD11c, Clone: HL3 | BD Biosciences | Cat #: 553801 |
| Anti-Mouse CD11c, Clone: N418 | Biolegend | Cat #: 117346 |
| Anti-Mouse CD45.1, Clone: A20 | Biolegend | Cat #: 110726 |
| Anti-Mouse CD45.2, Clone: 104 | Biolegend | Cat #: 109807 |
| Anti-Mouse CD169, Clone: 3D6.112 | Biolegend | Cat #: 142419 |
| Anti-Mouse CD197 (CCR7), Clone: 4B12 | Biolegend | Cat #: 120116 |
| Anti-Mouse CD273 (PDL2), Clone: TY25 | BD Biosciences | Cat #: 560086 |
| Anti-mouse CD301b (MGL2), Clone: URA-1 | Biolegend | Cat #: 146806 |
| Anti-Mouse F4/80, Clone: BM8 | Biolegend | Cat #: 123131 |
| Anti-Mouse Lyve-1, Clone: ALY7 | eBioscience | Cat #: 53-0443-80 |
| Anti-Mouse Podoplanin, Clone: 8.1.1 | Biolegend | Cat #: 127406 |
| Anti-rabbit IgG, HRP | Cell Signaling | Cat #: 7074S |
| Anti-SHP2 | Cell Signaling | Cat #: 3397T |
| Anti-phospho SHP2 (Tyr 542) | Cell Signaling | Cat #: 3751S |
| Anti-Siglec1, biotinylated | R&D Systems | Cat #: BAF5610 |
| Anti-SIGN-R1, biotinylated | R&D Systems | Cat #: BAF1836 |
| Anti-Src | Cell Signaling | Cat #: 2108S |
| Anti-phospho Src (Tyr 416) | Cell Signaling | Cat #: 2101S |
| Biotinylated Anti-mouse MCP-2/CCL8 Antibody | R&D Systems | Cat #: BAF790 |
| Human CCL1-AlexaFluor647 | Almac | Cat #: CAF-7 |
| Fixable Viability Dye eFluor 780 | Invitrogen | Cat #: 65-0865-14 |
| Mouse CCL1/I-309/TCA-3 Biotinylated Antibody | R&D Systems | Cat #: BAF845 |
| TruStain fcX anti-mouse CD16/32, Clone: 93 | Biolegend | Cat #: 101320 |
| Critical Commercial Assays: | ||
| Mouse IL-4 ELISA MAX Standard | Biolegend | Cat #: 431103 |
| Mouse IFN-g ELISA MAX Standard | Biolegend | Cat #: 430803 |
| Mouse IL-13 ELISA Ready-SET-Go! | eBioscience | Ref #: 88-7137-77 |
| RNeasy Plus Micro Kit (50) | Qiagen | Cat #: 74034 |
| RNeasy Plus Mini Kit (50) | Qiagen | Cat #: 74134 |
| Naïve CD4+ T Cell Isolation Kit, mouse | Miltenyi Biotec | Cat #: 130-104-453 |
| CD11c Microbeads UltraPure mouse | Miltenyi Biotec | Cat #: 130-108-338 |
| Experimental Models: Organisms and Strains: | ||
| Mouse: C57Bl/6 | Charles River Laboratory | Strain No.: 027 |
| Mouse: C57Bl/6 | The Jackson Laboratory | Strain No.: 000664 |
| Mouse: B6(FVB)-Mgl2tm1.1(HBEGF/EGFP)Aiwsk/J (CD301b-DTR) | The Jackson Laboratory | Strain No.: 023822 |
| Mouse: Ccr8−/− | Chensue et al., 2001 | N/A |
| Mouse: Ccl8−/−Ccl12−/− | Tsou et al., 2007 | N/A |
| Mouse: Kaede+ | Tomura et al., 2008 | N/A |
| Mouse: Ccr7−/− | The Jackson Laboratory | Strain No.: 006621 |
| Mouse: B6.Cg-Tg (TcraTcrb)425Cbn/J (OT2m) | The Jackson Laboratory | Strain No.: 004194 |
| Mouse: B6.SJL-Ptprc Pepc/BoyJ (CD45.1) | The Jackson Laboratory | Strain No.: 002014 |
| Mouse: REC8 | Cyagen Biosciences, this manuscript | N/A |
| Oligionucleotides | ||
| mCcl1 FWD (5’-GGCTGCCGTGTGGATACAG-3’) | This manuscript | N/A |
| mCcl1 REV (5’-AGGTGATTTTGAACCCACGTTT-3’) | This manuscript | N/A |
| mCcl2 FWD (5’-TGGCTCAGCCAGATGCAGT-3’) | This manuscript | N/A |
| mCcl2 REV (5’-TTGGGATCATCTTGCTGGTG-3’) | This manuscript | N/A |
| mCcl7 FWD (5’-TGGGAAGCTGTTATCTTCAAGACA’3’) | This manuscript | N/A |
| mCcl7 REV (5’-CTCGACCCACTTCTGATGGG-3’) | This manuscript | N/A |
| mCcl8 FWD (5’-GCGAGTGCTTCTTTGCCTG-3’) | This manuscript | N/A |
| mCcl8 REV (5’ TCTGGCCCAGTCAGCTTCTC-3’) | This manuscript | N/A |
| mCcl12 FWD (5’-GCTGGACCAGATGCGGTG-3’) | This manuscript | N/A |
| mCcl12 REV (5’-CCGGACGTGAATCTTCTGCT-3’) | This manuscript | N/A |
| REC8 genotyping primer P1 forward (5’-GAACACAGCAGCAGAAAGAACAC-3’) | This manuscript | N/A |
| REC8 genotyping primer P1 reverse (5’-CATGGCGTGTAATATGAGAGACGC-3’) | This manuscript | N/A |
| REC8 genotyping primer P2 forward (5’-GTAATAAATGGATGCCCTGCGTAAG-3’) | This manuscript | N/A |
| REC8 genotyping primer P2 reverse (5’-ACCTGTCAAGAATGGTGCCAG-3’) | This manuscript | N/A |
| REC8 genotyping primer P3 forward (5’-ATCAGTCAGGTACATAACCTCGAG-3’) | This manuscript | N/A |
| REC8 genotyping primer P3 reverse (5’-AGTCCATAACCTTCCTCCACG-3’) | This manuscript | N/A |
| REC8 genotyping primer P4 forward (5’-GATTGGTATCTGCTTCTGCACTTG=3’) | This manuscript | N/A |
| REC8 genotyping primer P4 reverse (5’-GTAGATGAACTCGCCGTCCTG-3’) | This manuscript | N/A |
| REC8 genotyping primer P5 forward (5’-CCTTCTCACCACAGCCACTTC-3’) | This manuscript | N/A |
| REC8 genotyping primer P5 reverse (5’-TGAGTTTGGACAAACCACAAC-3’) | This manuscript | N/A |
| REC8 genotyping primer P6 forward (5’-TGCTTCATGTGGTCGGGGTA-3’) | This manuscript | N/A |
| REC8 genotyping primer P6 reverse (5’-GCTTTCATTTCCCATTAGGCACA-3’) | This manuscript | N/A |
| Chemicals, Peptides, & Recombinant Proteins: | ||
| Macrophage depletion kit of Clodrosome and Encapsome | Encapsula Nano Sciences | Cat #: 8901 |
| Saponin from quillaja bark | Sigma-Aldrich | Cat #: S7900-25G |
| Recombinant Mouse CCL1/TCA-3 | R&D Systems | Cat #: 845-TC |
| Recombinant Mouse CCL21/6Ckine | R&D Systems | Cat #: 457-6C-025 |
| Recombinant Murine MCP-3 (CCL7) | PeproTech | Cat #: 250-08 |
| Recombinant Murine MCP-2 (CCL8) | PeproTech | Cat #: 250-14 |
| Recombinant Mouse CCL2/JE/MCP-1 | R&D Systems | Cat #: 479-JE-010 |
| Recombinant Mouse CCL11/Eotaxin | R&D Systems | Cat 3: 420-ME |
| TRIzol Reagent | Invitrogen | Ref #: 15596018 |
| Collagenase P | Sigma-Aldrich | Cat #: 11213857001 |
| Dispase II | Sigma-Aldrich | Cat #: D4693-1G |
| DNase I, grade II | Sigma-Aldrich | Cat #: 10104159001 |
| Liberase DH | Sigma-Aldrich | Cat #: 5401054001 |
| Streptavidin/Biotin Blocking Kit | Vector Lab | Cat #: SP-2002 |
| Papain | Calbiochem | Cat: 5125 |
| Ovalbumin | Sigma-Aldrich | Cat: A5503 |
| LPS | Invivogen | Cat: tlrl-eklps |
| Polyinosinic-polycytidylic acid | Invivogen | Cat:tlrl-picw |
| PBS | Corning | Ref #: 21-031-CV |
| PP2 | Sigma-Aldrich | Cat #: 529573-1MG |
| Indo-1, AM | Invitrogen | Cat #: I1223 |
| RBC Lysis buffer | Sigma-Aldrich | Cat #: R7757 |
| 0.5 M EDTA, pH 8.0 | Gibco | Cat #: 15575-038 |
| OCT Compound | Tissue-Tek | Cat #: 4583 |
| RPMI 1640, 1X | Corning | Ref #: 15-040-CV |
| Fetal Calf Serum (FCS) | Atlanta Biologicals | Cat #: S11550 |
| MultiScribe Reverse Transcriptase | Thermo Fisher Scientific | Ref #: 4311235 |
| GeneAMP dNTP mix with dTTP | Thermo Fisher Scientific | Ref #: N8080260 |
| MgCl2 Solution 25 mM | Thermo Fisher Scientific | Ref #: 4486224 |
| 10× PCR Buffer II | Thermo Fisher Scientific | Ref #: 4486220 |
| Oligio d(T)16 | Thermo Fisher Scientific | Ref #: 100023441 |
| Random Hexamers 50 μM | Thermo Fisher Scientific | Ref #: 100026484 |
| RNase Inhibitor | Thermo Fisher Scientific | Ref #: 100021540 |
| FastStart Essential DNA Green Master | Roche | Cat #: 25595200 |
| Software: | ||
| Prism v 7.0c | GraphPad | https://www.graphpad.com/demos |
| Zen Blue | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html |
| Imaris | Bitplane | http://www.bitplane.com/download |
| Softmax Pro | Molecular Devices | https://www.moleculardevices.com/systems/microplate-readers/softmax-pro-7-software |
| BD FACS Diva 8 | BD Biosciences | http://www.bdbiosciences.com/us/instruments/clinical/software/flow-cytometry-acquisition/bd-facsdiva-software/m/333333/overview |
| FlowJo (version 10) | Tree Star | https://www.flowjo.com/solutions/flowjo/downloads |
Contact for Reagent and Resource Sharing:
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Andrew Luster (aluster@mgh.harvard.edu).
Experimental Model and Subject Details:
Mice
Animals were bred and maintained under specific pathogen free (SPF) conditions at the Massachusetts General Hospital Center for Comparative Medicine. Ccr8−/− mice were bred under SPF plus Helicobacter and Pasteurella Pneumotropica free conditions and maintained in SPF conditions. All procedures were approved and carried out according to the standards and guidelines set forth by the Institutional Animal Care and Use Committee of Massachusetts General Hospital. All mouse strains were fully backcrossed (>9 generations) to C57Bl/6. Mice were used in experiments at 5-14 weeks of age and age matched littermates or purchased C57Bl/6 mice were used as wild type controls. Male mice were used for all DC migration assays unless otherwise noted. Male and female mice were used interchangeably in other assays, but sex was matched for all conditions within experiments.
Method Details
Generation of REC8 mice:
REC8 mice were generated on the C57Bl/6 background by Cyagen Biosciences using a PiggyBac on BAC technique by BAC modification in E. coli. RP23-409C7 was selected in pBACe3.6 because it contained both Ccl1 and Ccl8 target genes. First, a fragment containing 5’ and 3’ inverted terminal repeat sequences (ITRs) was inserted into pBACe3.6 to allow for eventual integration into TTAA short repeat element sites. eGFP-SV40 polyA was placed upstream of an FRT flanked kanamycin resistance gene and this construct was introduced immediately following the start codon of the Ccl8 gene via recombineering of the modified BAC clone RP23-409C7. After selection of positive colonies, the kanamycin resistance cassette was then removed via Flp-mediated recombination. mCherry-SV40 polyA was placed upstream of rox flanked ampicillin resistance gene and this construct was introduced immediately following the start codon of the Ccl1 gene via recombineering of the Ccl8-eGFP modified BAC clone. Ampicillin resistant clones were selected and then the ampicillin resistance cassette was deleted via Dre-mediated recombination. Recombinant clones were confirmed and monitored by PCR (primers P1-P6) and end-sequencing. The verified circular PiggyBac on BAC clone and helper plasmid was injected into fertilized C57Bl/6 eggs and implanted into surrogate mothers. Pups were genotyped by PCR (primers P1-P8) to identify transgene positive pups. Positive pups were screened negative for integration of the helper plasmid (Forward primer 5’-CTGGACGAGCAGAACGTGATCG-3’ and Reverse primer 5’-CGAAGAAGGCGTAGATCTCGTCCTC-3’). This yielded 11 founders. Of those, one did not produce offspring and two did not have transgene positive offspring. Of the 8 remaining founders, two had no detectable reporter expression, four had only detectable mCherry expression, and two had detectable expression of both reporters. One line had brighter reporter expression and so was used primarily for data generation, although experiments were confirmed with the alternative line.
Immunizations & Cell transfers:
Mice were immunized subcutaneously in the rear footpads and/or base-of-tail with 50 μg of both papain (Calbiochem) and ovalbumin or 50 μg ovalbumin in 20 μg Imject Alum (ThermoFisher) for Th2-skewing immunizations, 10 μg of both lipopolysaccharide (InvivoGen) and polyinosinic-polycytidylic acid (PolyI:C) (InvivoGen) and 50 μg of ovalbumin for Th1-skewing immunizations, and 50 μg of ovalbumin for Th0 immunizations. All immunizations except OVA/Alum were diluted in sterile PBS and subcutaneously injected at a final volume of 25 μl. N. brasiliensis infection was performed by injecting mice subcutaneously in the base-of-tail with 500 L3 stage larvae with 50 μg ovalbumin. Naïve CD4+ T cells were negatively selected for using Naïve CD4+ T cell isolation kit (Miltenyi) followed by magnetic cell separation (Miltenyi). Purified naïve CD4+ T cells were transferred (2.5-5.0 × 105 cells in 100 μl sterile PBS) into recipient mice via retro-orbital injection. SIGN-R1+ cells underwent magnetic cell separation using SIGN-R1-biotin and anti-biotin magnetic beads (Miltenyi).
Tissue Preparation:
For CD4+ T cell harvest for in vitro restimulation assays, whole LNs were placed in 70 μm sterile filters and mechanically disrupted. Otherwise, for standard LN digestion, whole LNs were harvested, minced, and placed into RPMI media containing Liberase DH (0.26 U/ml) and DNase I (0.1 mg/ml) and incubated at 37°C for 20 mins. Following incubation, LNs were mechanically disrupted by passing through a pipette tip before being incubated for another 20 mins at 37°C. For extended LN digest to ensure release of LN sinus stroma and entrapped cells, whole LNs were harvested into RPMI media and subjected to digestion in DNase I (100 μg/ml), Collagenase P (200 μg/ml), Dispase II (800 μg/ml), and 1% fetal calf serum (FCS) in RPMI. LNs were placed in the pre-warmed enzyme mixture and incubated at 37°C. At 8 min intervals, any supernatant was removed and added to stop buffer (RPMI/2mM EDTA/1% FCS), and the removed supernatant was replaced with fresh enzyme media. This was repeated at 8 min intervals until no tissue fragments remained. For both protocols, upon completion of digestion, cell suspensions were filtered with a 70 μm sterile filter.
Generation of mixed bone marrow chimeras:
To generate bone marrow chimeric mice, CD45.1 recipient mice were irradiated with 1000 rads prior to injection with isolated bone marrow. Bone marrow was harvested from 5-8 week old mice by mechanical disruption using a mortar and pestle followed by incubation with RBC lysis buffer. Lysis was stopped by adding excess PBS, centrifugation, and resuspension of pellet in sterile PBS. Within 6 hrs of irradiation, recipient mice received a total of 1 × 107 cells in a 1:1 mix of CD301b-DTR (CD45.2) and CD45.1 × CD45.2 cells, CD301b-DTR (CD45.2) and Kaede+Ccr8−/− cells or all Kaede+Ccr8−/− cells. Bone marrow cells were injected via retro-orbital injection in 100 μl. After eight- to ten-weeks, mice received purified CD4+ T cells (2.5 × 105) via retroorbital injection as well as 0.5 μg of Diphtheria toxin intraperitoneally. 16-24 hrs following T cell transfer, mice underwent footpad immunization and received a second dose of Diphtheria toxin at 0.5 μg. 48 hrs following immunizations, mice were given a final injection of Diphtheria toxin at 0.5 μg. 5 days post immunization, popliteal LNs were collected. Reconstitution and depletion of CD301b+ DCs were assessed in a subset of mice one day after immunization and in all other mice at time of final harvest.
Flow cytometry:
Enzymatically digested cell suspensions were incubated for 15 min at 4°C in PBS with 0.5% FCS with the following antibodies: anti-CD16/32, anti-CD8α, anti-CD11b, anti-CD11c, anti-CD45.1, anti-CD45.2, anti-CD103, anti-CD169, anti-CD301b, anti-Thy1.1 (all from Biolegend), anti-PDL2 (BD Biosciences), or anti-SIGN-R1 (R&D Systems). Viability was determined using Fixable Viability Dye eFluor 780 (Invitrogen). Experiments examining CCR7 were done by first incubating cells with anti-CCR7 (BD Biosciences) for 30 min at 37°C prior to additional surface staining as above. Intracellular cytokine staining was done by first incubating cells in PMA/Ionomycin (Sigma) in the presence of GolgiPlug for 4 hours prior to surface staining followed by intracellular staining using BD Cytofix/Cytoperm kit (BD Biosciences). hCCL1-AF647 (Almac) binding was accomplished by first incubating 2×106 cells in 50 μl of 0.25 μg/ml hCCL1-AF647 in PBS/0.5% FCS/20mM HEPES for 1 hr at 37°C. Cells then underwent additional antibody staining as above. All samples were run on BD Fortessa X-20 or BD LSRII and analyzed using FACS Diva 8 (BD Biosciences) and FlowJo (Version 10) (TreeStar).
Enzyme-linked immunosorbent assay (ELISA):
Mouse IL-4 and IFN-γ were detected using a mouse IL-4 and IFN-γ ELISA MAX standard set (both from Biolegend). Mouse IL-13 was detected using a Mouse IL-13 ELISA Ready-SET-Go! kit (eBioscience). All procedures were carried out according to the manufacturer’s protocols. Briefly, samples were incubated overnight at 4°C on antibody-coated plates. Detection antibodies were allowed to incubate for 1 hr at room temperature, followed by addition of streptavidin-horseradish peroxidase for 30 mins. Finally, TMB substrate solution was added and the reaction was stopped with 2M sulfuric acid. Samples were assayed on Softmax Pro Software in triplicates and concentrations were determined from a standard curve.
In vitro restimulation assays:
To achieve restimulation of antigen-specific T cells, single cell suspensions of 5 × 105 isolated leukocytes of whole LNs were added to 96-well round bottom microwell plates with 100 μg/ml ovalbumin in 200 μl of media. Plates were incubated at 37°C in 5% CO2 between four and five days at which time cytokine production was analyzed.
In vitro chemotaxis assays:
Chemotaxis of sorted CD11c+CD11b+CD301b− (CD11c+) DCs and CD11c+CD11b+CD301b+ DCs was performed using 5-μm pore size Transwell membranes (Neuro Probe). Briefly, 29 μl of diluted chemokines in media, 0.5% (wt/vol) BSA in RPMI, or media alone were added to the lower chamber. To the upper chamber, 25,000 cells were added in triplicate in 50 μl of media with or without chemokine. Plates were incubated for 2 hrs at 37°C in 5% CO2 after which point the upper membrane was washed with PBS. A brief spin was performed to collect any migrated adherent cells, the upper chamber was removed, and migrated cells were counted. When applicable, cells were pretreated for 30 minutes with 10 uM of the Src inhibitor PP2 and chemotaxis was performed in the presence of this or DMSO as a control.
RNA purification & QPCR analysis:
Total RNA was collected from whole LN or skin using a Trizol (Invitrogen) reagent protocol in accordance with the manufacturer. Total RNA from sorted cells was collected using the Qiagen RNeasy Micro or Mini Kit, based on the manufacturer’s protocol. For both methods, RNA was diluted in RNase-free water and stored at - 80°C until prepared for cDNA using reverse transcription. 50 μl of cDNA per reaction was generated using random hexamers, Oligio(dT), magnesium chloride, dNTPs (10mM), Reverse Transcriptase, and RNase Inhibitor (all from Fisher). A Roche LightCycler 96 Real-Time PCR System was utilized to quantitate gene expression using SYBR Green Master Mix (Roche). The reaction cycles were carried out in the following manner: 95°C for 600 sec, then 95°C, 60°C, and 72°C for 10 sec each for 45 total cycles, followed by 95°C for 10 sec, 65°C for 60 sec, and finally 97°C for 1 sec. Fluorescence was measured during each amplification step and Roche LightCycler 96 Software 1.1 was utilized to determine quantification cycle (Cq) values for each sample. In Excel, copy values were determined based off of the Cq values which were then divided by values from the housekeeping genes β2-microglobulin or Gapdh to generate ratios.
Confocal immunofluorescence:
Lymph nodes were fixed at room temperature for 2-3 hrs in periodate-lysine-paraformaldehyde fixative. Organs then were placed sequentially in 10%, 20% and 30% sucrose (wt/vol) in PBS for cryoprotection, then embedded in optimum cutting temperature (OCT) compound (Tissue-Tek) and frozen in 2-methybutane over liquid nitrogen. A Cryostat (Microm HM 505 E, GMI) was used to generate 6-9 μm sections, which were kept at −20°C. On the day of staining, sections were thawed, rehydrated in PBS for 20 mins at room temperature, blocked in PBS containing 0.1% (vol/vol) Tween-20 and 5% (vol/vol) Normal Goat Serum (Jackson Immunoresearch), and were stained with biotinylated anti-Siglec1 or anti-SIGN-R1 (both from R&D Systems) or fluorochrome-conjugated anti-mouse CD11c (BD Biosciences), anti-mouse LYVE-1 (eBioscience), anti-mouse CD301b, anti-mouse podoplanin, anti-mouse CD169, or anti-mouse F4/80 (all from Biolegend). Streptavidin-Alexa Fluor 647 or Alexa Fluor 594 (both from Biolegend) were used as secondary antibodies. Secondary antibodies were allowed to incubate for 1 h at room temperature. Sections were also stained with biotinylated anti-mouse MCP-2/CCL8 or CCL1 (both from R&D Systems) which were blocked using a streptavidin/biotin blocking kit (Vector Laboratories) to block endogenous biotin. All dilutions were made in PBS containing 0.1% (wt/vol) Saponin and 5% rabbit serum (vol/vol), with the exception of primary antibody which was diluted in PBS containing 0.1% (w/v) Saponin only. Primary antibody was allowed to incubate overnight at 4°C. A biotinylated anti-goat IgG secondary antibody (Vector Laboratories) incubated at room temperature for 30 mins followed by addition of fluorescein streptavidin (1:200) for 15 mins at 4°C. All slides were mounted in Prolong Gold to prevent fading of staining. A Zeiss LSM confocal microscope was used to acquire images that were generated by collecting consecutive scans with 1 laser line active per scan to prevent excitation crosstalk.
Kaede photoconversion:
All Kaede transgenic mice were anesthetized to shave a ~2 cm x 2 cm patch of skin above the base-of-tail. A chemical depilatory agent (Veet) was applied to the shaved patch for 1 min, followed by multiple washings with PBS. To perform photoconversion, the exposed skin was subjected to violet light (420 nm) using a Bluewave LED visible light curing unit (Dymax) that expressed a 420 nm bandpass filter (Andover Corp). Skin was exposed to this wavelength with the light source at a maximum power, approximately 7.5 cm away from the skin for 5 mins. Following photoconversion, mice were immunized subcutaneously in the shaved region as described above. 24 hrs following immunization, dLNs were isolated and analyzed for skin DC migration using flow cytometry.
Clodronate-mediated cell depletion:
Mice were injected subcutaneously in the rear footpads with 50 μl of either Clodrosome Liposomal Clodronate or Encapsome Control Liposome (Encapsula Nano Sciences). Footpad immunizations were performed four days following liposome administration, as described above, and draining lymph nodes were isolated 24 hrs following immunization.
Immunoblotting:
4DE4 cells stably transfected with human CCR8 as previously described were serum starved for 4 hours and then stimulated with CCL8 (Islam et al., 2011). Cells were lysed and lysates were immunoblotted for presence of phosphorylated Src (Tyr416) and SHP2 (Tyr542) (Cell Signaling).
Calcium flux assay:
To access intracellular calcium flux in primary dendritic cells, MACS isolated CD11c positive cells from the dLN of Th2-immunized mice were washed with PBS (with Ca/Mg) supplemented with 1% FCS. Indo-1, AM (Invitrogen) was prepared immediately before use by dissolving each vial according to the manufacturer’s recommendations. 5 μ;l of freshly diluted Indo-1 was added to 500 μ;l of PBS 1% FCS (with Ca/Mg) per every 2 million cells. Cells were allowed to incubate for 45 mins at 37ºC in the dark and then washed with cold PBS (with Ca/Mg) and re-suspended in 500 μ;l cold PBS (with Ca/Mg). Calcium flux was then assessed by flow cytometry measuring Indo-1 ratio over a time course of 90 seconds following chemokine stimulation.
Quantitation and Statistical Analysis
Statistical analyses, as specified in figure legends, were performed using Prism 7 (GraphPad software). Data were analyzed with the non-paired Student’s t test. A p value of <0.05 was considered significant (*p<0.05, **p<0.01, ***p<0.001).
Supplementary Material
ACKNOWLEDGEMENTS
Work was supported by the NIH grants R37AI040618 (to A.D.L) and K08AI121421 (to C.L.S.). C.L.S. was supported by the NIH (T32HL116275) and the Asthma and Allergy Foundation of America. Flow cytometry was performed in the MGH Department of Pathology Flow and Image Cytometry Research Core, with support from the NIH Shared Instrumentation program (1S10OD012027-01A1, 1S10OD016372-01, 1S10RR020936-01, and 1S10RR023440-01A1). We thank Drs. Sergio Lira, Israel Charo, and Osami Kanagawa for providing breeding pairs of Ccr8−/−, Ccl8−/−/Ccl12−/−, and Kaede transgenic mice, respectively. We thank Drs. Robert Anthony and Joseph F. Urban for providing Nippostrongylus brasiliensis.
Footnotes
DECLARATION OF INTEREST
The authors declare no competing interests.
REFERENCES
- Asano K, Takahashi N, Ushiki M, Monya M, Aihara F, Kuboki E, Moriyama S, Iida M, Kitamura H, Qiu CH, et al. (2015). Intestinal CD169(+) macrophages initiate mucosal inflammation by secreting CCL8 that recruits inflammatory monocytes. Nat Commun 6, 7802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun A, Worbs T, Moschovakis GL, Halle S, Hoffmann K, Bolter J, Munk A, and Forster R (2011). Afferent lymph-derived T cells and DCs use different chemokine receptor CCR7-dependent routes for entry into the lymph node and intranodal migration. Nat Immunol 12, 879–887. [DOI] [PubMed] [Google Scholar]
- Bromley SK, Yan S, Tomura M, Kanagawa O, and Luster AD (2013). Recirculating memory T cells are a unique subset of CD4+ T cells with a distinct phenotype and migratory pattern. J Immunol 190, 970–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chensue SW, Lukacs NW, Yang TY, Shang X, Frait KA, Kunkel SL, Kung T, Wiekowski MT, Hedrick JA, Cook DN, et al. (2001). Aberrant in vivo T helper type 2 cell response and impaired eosinophil recruitment in CC chemokine receptor 8 knockout mice. J Exp Med 193, 573–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CD, Kuo F, Kumer J, Motani AS, Lawrence CE, Henderson WR Jr., and Venkataraman C (2003). CCR8 is not essential for the development of inflammation in a mouse model of allergic airway disease. J Immunol 170, 581–587. [DOI] [PubMed] [Google Scholar]
- Das S, Sarrou E, Podgrabinska S, Cassella M, Mungamuri SK, Feirt N, Gordon R, Nagi CS, Wang Y, Entenberg D, et al. (2013). Tumor cell entry into the lymph node is controlled by CCL1 chemokine expressed by lymph node lymphatic sinuses. J Exp Med 210, 1509–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieu MC, Vanbervliet B, Vicari A, Bridon JM, Oldham E, Ait-Yahia S, Briere F, Zlotnik A, Lebecque S, and Caux C (1998). Selective recruitment of immature and mature dendritic cells by distinct chemokines expressed in different anatomic sites. J Exp Med 188, 373–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher AL, Malhotra D, Acton SE, Lukacs-Kornek V, Bellemare-Pelletier A, Curry M, Armant M, and Turley SJ (2011). Reproducible isolation of lymph node stromal cells reveals site-dependent differences in fibroblastic reticular cells. Front Immunol 2, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Nish SA, Jiang R, Hou L, Licona-Limon P, Weinstein JS, Zhao H, and Medzhitov R (2013). Control of T helper 2 responses by transcription factor IRF4-dependent dendritic cells. Immunity 39, 722–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard JP, Moussion C, and Forster R (2012). HEVs, lymphatics and homeostatic immune cell trafficking in lymph nodes. Nat Rev Immunol 12, 762–773. [DOI] [PubMed] [Google Scholar]
- Goya I, Villares R, Zaballos A, Gutierrez J, Kremer L, Gonzalo JA, Varona R, Carramolino L, Serrano A, Pallares P, et al. (2003). Absence of CCR8 does not impair the response to ovalbumin-induced allergic airway disease. J Immunol 170, 2138–2146. [DOI] [PubMed] [Google Scholar]
- Gray EE, and Cyster JG (2012). Lymph node macrophages. J Innate Immun 4, 424–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray EE, Friend S, Suzuki K, Phan TG, and Cyster JG (2012). Subcapsular sinus macrophage fragmentation and CD169+ bleb acquisition by closely associated IL-17-committed innate-like lymphocytes. PLoS One 7, e38258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith JW, Sokol CL, and Luster AD (2014). Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol 32, 659–702. [DOI] [PubMed] [Google Scholar]
- Halim TY, Steer CA, Matha L, Gold MJ, Martinez-Gonzalez I, McNagny KM, McKenzie AN, and Takei F (2014). Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity 40, 425–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser MA, Schaeuble K, Kindinger I, Impellizzieri D, Krueger WA, Hauck CR, Boyman O, and Legler DF (2016). Inflammation-Induced CCR7 Oligomers Form Scaffolds to Integrate Distinct Signaling Pathways for Efficient Cell Migration. Immunity 44, 59–72. [DOI] [PubMed] [Google Scholar]
- Islam SA, Chang DS, Colvin RA, Byrne MH, McCully ML, Moser B, Lira SA, Charo IF, and Luster AD (2011). Mouse CCL8, a CCR8 agonist, promotes atopic dermatitis by recruiting IL-5+ T(H)2 cells. Nat Immunol 12, 167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam SA, Ling MF, Leung J, Shreffler WG, and Luster AD (2013). Identification of human CCR8 as a CCL18 receptor. J Exp Med 210, 1889–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki A, and Medzhitov R (2015). Control of adaptive immunity by the innate immune system. Nat Immunol 16, 343–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakubzick C, Tacke F, Llodra J, van Rooijen N, and Randolph GJ (2006). Modulation of dendritic cell trafficking to and from the airways. J Immunol 176, 3578–3584. [DOI] [PubMed] [Google Scholar]
- Junt T, Moseman EA, Iannacone M, Massberg S, Lang PA, Boes M, Fink K, Henrickson SE, Shayakhmetov DM, Di Paolo NC, et al. (2007). Subcapsular sinus macrophages in lymph nodes clear lymph-borne viruses and present them to antiviral B cells. Nature 450, 110–114. [DOI] [PubMed] [Google Scholar]
- King IL, Kroenke MA, and Segal BM (2010). GM-CSF-dependent, CD103+ dermal dendritic cells play a critical role in Th effector cell differentiation after subcutaneous immunization. J Exp Med 207, 953–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumamoto Y, Hirai T, Wong PW, Kaplan DH, and Iwasaki A (2016). CD301b(+) dendritic cells suppress T follicular helper cells and antibody responses to protein antigens. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumamoto Y, Linehan M, Weinstein JS, Laidlaw BJ, Craft JE, and Iwasaki A (2013). CD301b(+) dermal dendritic cells drive T helper 2 cell-mediated immunity. Immunity 39, 733–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leon B, Ballesteros-Tato A, Browning JL, Dunn R, Randall TD, and Lund FE (2012). Regulation of T(H)2 development by CXCR5+ dendritic cells and lymphotoxin-expressing B cells. Nat Immunol 13, 681–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis KL, Caton ML, Bogunovic M, Greter M, Grajkowska LT, Ng D, Klinakis A, Charo IF, Jung S, Gommerman JL, et al. (2011). Notch2 receptor signaling controls functional differentiation of dendritic cells in the spleen and intestine. Immunity 35, 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian J, and Luster AD (2015). Chemokine-guided cell positioning in the lymph node orchestrates the generation of adaptive immune responses. Curr Opin Cell Biol 36, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MD, and Krangel MS (1992). The human cytokine I-309 is a monocyte chemoattractant. Proc Natl Acad Sci U S A 89, 2950–2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami R, Denda-Nagai K, Hashimoto S, Nagai S, Hattori M, and Irimura T (2013). A unique dermal dendritic cell subset that skews the immune response toward Th2. PLoS One 8, e73270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti S, Petkovic V, Sebastiani S, Danelon MG, Uguccioni M, and Gerber BO (2005). A rich chemokine environment strongly enhances leukocyte migration and activities. Blood 105, 3405–3412. [DOI] [PubMed] [Google Scholar]
- Qi H, Kastenmuller W, and Germain RN (2014). Spatiotemporal basis of innate and adaptive immunity in secondary lymphoid tissue. Annu Rev Cell Dev Biol 30, 141–167. [DOI] [PubMed] [Google Scholar]
- Qu C, Edwards EW, Tacke F, Angeli V, Llodra J, Sanchez-Schmitz G, Garin A, Haque NS, Peters W, van Rooijen N, et al. (2004). Role of CCR8 and other chemokine pathways in the migration of monocyte-derived dendritic cells to lymph nodes. J Exp Med 200, 1231–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallusto F, Schaerli P, Loetscher P, Schaniel C, Lenig D, Mackay CR, Qin S, and Lanzavecchia A (1998). Rapid and coordinated switch in chemokine receptor expression during dendritic cell maturation. Eur J Immunol 28, 2760–2769. [DOI] [PubMed] [Google Scholar]
- Schlitzer A, McGovern N, Teo P, Zelante T, Atarashi K, Low D, Ho AW, See P, Shin A, Wasan PS, et al. (2013). IRF4 transcription factor-dependent CD11b+ dendritic cells in human and mouse control mucosal IL-17 cytokine responses. Immunity 38, 970–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokol CL, Barton GM, Farr AG, and Medzhitov R (2008). A mechanism for the initiation of allergen-induced T helper type 2 responses. Nat Immunol 9, 310–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tal O, Lim HY, Gurevich I, Milo I, Shipony Z, Ng LG, Angeli V, and Shakhar G (2011). DC mobilization from the skin requires docking to immobilized CCL21 on lymphatic endothelium and intralymphatic crawling. J Exp Med 208, 2141–2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Cao W, Kasturi SP, Ravindran R, Nakaya HI, Kundu K, Murthy N, Kepler TB, Malissen B, and Pulendran B (2010). The T helper type 2 response to cysteine proteases requires dendritic cell-basophil cooperation via ROS-mediated signaling. Nat Immunol 11, 608–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomura M, Yoshida N, Tanaka J, Karasawa S, Miwa Y, Miyawaki A, and Kanagawa O (2008). Monitoring cellular movement in vivo with photoconvertible fluorescence protein “Kaede” transgenic mice. Proc Natl Acad Sci U S A 105, 10871–10876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou CL, Peters W, Si Y, Slaymaker S, Aslanian AM, Weisberg SP, Mack M, and Charo IF (2007). Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest 117, 902–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tussiwand R, Everts B, Grajales-Reyes GE, Kretzer NM, Iwata A, Bagaitkar J, Wu X, Wong R, Anderson DA, Murphy TL, et al. (2015). Klf4 expression in conventional dendritic cells is required for T helper 2 cell responses. Immunity 42, 916–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulvmar MH, Werth K, Braun A, Kelay P, Hub E, Eller K, Chan L, Lucas B, Novitzky-Basso I, Nakamura K, et al. (2014). The atypical chemokine receptor CCRL1 shapes functional CCL21 gradients in lymph nodes. Nat Immunol 15, 623–630. [DOI] [PubMed] [Google Scholar]
- Weber M, Hauschild R, Schwarz J, Moussion C, de Vries I, Legler DF, Luther SA, Bollenbach T, and Sixt M (2013). Interstitial dendritic cell guidance by haptotactic chemokine gradients. Science 339, 328–332. [DOI] [PubMed] [Google Scholar]
- Williams JW, Tjota MY, Clay BS, Vander Lugt B, Bandukwala HS, Hrusch CL, Decker DC, Blaine KM, Fixsen BR, Singh H, et al. (2013). Transcription factor IRF4 drives dendritic cells to promote Th2 differentiation. Nature communications 4, 2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff MC, Heesters BA, Herndon CN, Groom JR, Thomas PG, Luster AD, Turley SJ, and Carroll MC (2014). Trans-nodal migration of resident dendritic cells into medullary interfollicular regions initiates immunity to influenza vaccine. J Exp Med 211, 1611–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worbs T, Hammerschmidt SI, and Forster R (2017). Dendritic cell migration in health and disease. Nat Rev Immunol 17, 30–48. [DOI] [PubMed] [Google Scholar]
- Yabe R, Shimizu K, Shimizu S, Azechi S, Choi BI, Sudo K, Kubo S, Nakae S, Ishigame H, Kakuta S, and Iwakura Y (2015). CCR8 regulates contact hypersensitivity by restricting cutaneous dendritic cell migration to the draining lymph nodes. Int Immunol 27, 169–181. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.