

Abstract
In silico methods for detecting functionally relevant genetic variants are important for identifying genetic markers of human inherited disease. Much research has focused on protein-coding variants since coding-regions have well-defined physicochemical and functional properties. However, many bioinformatics tools are not applicable to variants outside coding-regions. Here, we increase the classification performance of our Regulatory Single Nucleotide Variant Predictor (RSVP) for variants that cause regulatory abnormalities from an AUC of 0.90 to 0.97 by incorporating genomic regions identified by the ENCODE project into RSVP. RSVP is comparable to a recently published tool, Genome-Wide Annotation of Variants (GWAVA); both RSVP and GWAVA perform better on regulatory variants than a traditional variant predictor, Combined Annotation Dependent Depletion (CADD). However, our method outperforms GWAVA on variants located at similar distances to the transcription start site as the positive set (AUC: 0.96) compared to GWAVA (AUC: 0.71). Much of this disparity is due to RSVP’s incorporation of features pertaining to the nearest gene (expression, GO terms, etc), which are not included in GWAVA. Our findings hold out the promise of a framework for the assessment of all functional regulatory variants, providing a means to predict which rare or de novo variants are of pathogenic significance.
Keywords: RSVP, ENCODE, regulatory variant, intergenic variant, variant prediction, variant predictor
Introduction
Although it is likely that most single nucleotide variants (SNVs) lack functional significance (Sachidanandam, Weissman et al. 2001), SNVs are the most common form of human genetic variation (Gibbs, Belmont et al. 2003) and many have been shown to be associated with, or even causative of, human disease (Buckland, Hoogendoorn et al. 2004; Pastinen and Hudson 2004; Prokunina and Alarcon-Riquelme 2004; Campino, Forton et al. 2008; Savinkova, Ponomarenko et al. 2009; Ward and Kellis 2012). A substantial body of research has been devoted to characterizing functional SNVs in human genes (Mottagui-Tabar, Faghihi et al. 2005; Buckland 2006; Pampin and Rodriguez-Rey 2007; Chorley, Wang et al. 2008). However, it is currently impractical to investigate each variant in vitro given the very large number that have been identified in the human genome. Thus, computational (in silico) approaches for the prediction of functional SNVs represent a promising alternative to laborious large-scale in vitro analyses (Mooney 2005; Peterson, Doughty et al. 2013).
Several studies have been conducted to predict functional SNVs within the human genome using information that could be biologically relevant to gene transcriptional regulation. A number of such studies, focusing exclusively on promoter regions, have considered only the disruption of transcription factor binding sites (TFBS) for prediction (Ponomarenko, Orlova et al. 2002; Andersen, Engstrom et al. 2008; Lapidot, Mizrahi-Man et al. 2008). However, these methods rely on the completeness of genome-wide TFBS annotation and blithely ignore coding variants which may also influence gene transcriptional regulation. Moreover, these methods lack information pertaining to the distance to the nearest transcription start site (TSS), the presence of a CpG island, and local sequence repetitivity, which were found by Montgomery et al. (Montgomery, Griffith et al. 2007) to be important features for the prediction of functional regulatory variants. In addition, the integration of information from the Encyclopedia of DNA Elements (ENCODE) has previously been reported to improve the prediction of functional regulatory elements (Torkamani and Schork 2008), a finding that was recently confirmed by the Genome-Wide Annotation of Variants (GWAVA) tool (Ritchie, Dunham et al. 2014). However, all the aforementioned tools utilize only SNV-based information about the variant itself and ignore important information about the gene being regulated (gene-based features) such as gene expression, gene function, interaction complexity of gene products, frequency of optimal codons (FOP), and effective number of codons (ENC). In our previous study, where we developed a classifier to distinguish functional regulatory variants from putatively neutral variants, (Zhao, Clark et al. 2011), we demonstrated that these gene-based features are very important. The GWAVA tool did not incorporate replication timing information from the ENCODE project, which we found to be a very informative feature (Zhao, Clark et al. 2011).
In our Regulatory Single nucleotide Variant Predictor (RSVP), we have employed a supervised machine learning method using a set of 1,999 known functional regulatory SNVs from the Human Gene Mutation Database (HGMD) (Stenson, Mort et al. 2014) together with a dataset of putatively neutral SNVs that commonly occur in human genomes. The integration of both SNV-based and gene-based features enabled comparable performance to GWAVA when predicting all types of variant, with both methods achieving an area under the receiver operating characteristic (ROC) curve (AUC) value of 0.97. However, RSVP exhibits improved performance when classifying using only negative variants in close proximity to the true regulatory variants (0.95 AUC as compared to GWAVA’s 0.71 AUC). Additionally, we demonstrate that RSVP’s performance improves when analyzing only disease-causing (DM) variants from HGMD that are supported by functional evidence from in vitro expression assays (achieving an AUC of 0.98).
In this analysis, we determined that genomic features discovered in the ENCODE project are powerful predictors of regulatory variants while emphasizing the importance of gene-based features and the variant’s distance to the transcription start site. Our findings hold out the promise of an in silico framework for the assessment of all functional regulatory variants and, more importantly, provide a means to predict which rare or de novo variants may be involved in genetic disease. To annotate new variants with the RSVP predictor, a script may be obtained from our FTP site (http://bioinf.umbc.edu/RSVP/ftp/) or precomputed scores for all variants in dbNSFP (Liu, Jian et al. 2011) can be obtained upon request.
Materials & Methods
Data Preparation
In order to evaluate features (attributes) with the potential to be useful in identifying sites that play a functional role in gene regulation, two datasets were collected. First, a positive set was obtained from the HGMD database (Stenson, Ball et al. 2012) on Jan. 30th, 2014 and this was filtered so as to include only “disease-causing mutations” (DM), “functional polymorphisms” (FP), and “disease-associated polymorphisms” with additional supporting functional evidence (DFP), resulting in a total of 1,999 regulatory variants. Second, variants were obtained from the dbSNP database and were filtered so as to include only validated variants with an allele frequency greater than or equal to 5%, yielding a set of 4,804,913 negative variants. To make the analysis computationally viable, down-sampling was employed on the dbSNP database with a random selection of 1% of these variants being used, yielding a set of 51,147 variants, which we shall henceforth refer to as the “full” negative set. To control for the different distribution of distances relative to the transcription start sites between the positive and negative datasets, we created two additional negative datasets to assess the possible impact of this feature. The “±1,000bp” and “±500bp” region sets comprise all variants from the dbSNP database set within a window around any positive variant of length 1,000 and 500 base-pairs (bp), respectively. This resulted in a total of 90,628 variants for the ±1,000bp region set and 18,872 for the ±500bp region set. To analyze the predictor’s performance on independent datasets, we obtained the full genome sequences of 454 individuals from the Wellderly Project (downloaded Dec. 26th, 2014), 2,513 individuals from the 1,000 Genomes Project (Abecasis, Auton et al. 2012) (v5a.20130502), somatic mutations from the tumors of 1,043 patients with Breast Invasive Carcinoma (BRCA) from the TCGA project (Collins and Barker 2007) (downloaded July 2nd, 2014), and a list of genes with a known relation to cancer from the Cancer Gene Census (CGC) (Futreal, Coin et al. 2004). In addition, an update to the HGMD regulatory variant dataset was obtained on May 20th, 2015 which was used as an unseen dataset for further validation of the method.
Features
Features used in this study were divided into two distinct sets: those directly annotated to the SNV under consideration (SNV-based) and those annotated to the gene within whose transcription regulatory region the SNV lies (gene-based). A summary of all features used in this analysis can be found in Supplementary Table S1. SNV-based features include SNV distance to the nearest transcription start site (TSS), flanking nucleotide GC-content, flanking nucleotide conservation, and SNV occurrence within known functional elements. The SNV-based known functional elements used were collected from several independent studies as well as the ENCODE project (Kellis, Wold et al. 2014). These binary SNV-based features describe the variant as being located within functional elements collected from independent studies for enhancers (Pennacchio, Loots et al. 2007), insulators (Bell, West et al. 2001), RNA polymerase II-enriched regions (Barski, Cuddapah et al. 2007), conserved noncoding sequences (Wang, Zhang et al. 2006), and nuclease hypersensitive sites (Crawford, Holt et al. 2006). In addition to the nuclease hypersensitive sites from the independent study, “DNase peaks” were obtained from the ENCODE project and were treated separately from the other nuclease hypersensitive sites. Also obtained from the ENCODE project were continuous-value features for histone peaks, FAIRE peaks, DNA methylation sites, and replication timing. Where a genomic region had not been annotated for replication timing, the average between the two nearest annotated regions was used. Two binary features from ENCODE were used to specify if the variant was located within a transcription factor binding site or a CpG island. Furthermore, two other binary features, one for exonic and one for intronic, were used to describe the location of the variant in the context of the gene. Finally, a database of sequences known to be exonic splicing enhancers (ESE) and exonic splicing silencers (ESS) were obtained from Sterne-Weiler et al. (Sterne-Weiler, Howard et al. 2011) and one feature for each was used, indicating a gain or loss of an ESE or ESS site.
Gene-based features for a given variant refer to features that are associated with the closest gene on the chromosome and all variants found near that gene would share this annotation. Gene-based features were further divided into two sets: those pertaining to the function of the associated gene (function-based) and those relating to the mRNA expression of the associated gene (expression-based). For function-based features, a set of prediction scores for GO biological process (1,788 features) and molecular function terms (344 features) was generated using the FANN-GO (functional annotator that uses multi-output artificial neural networks) predictor of protein GO term annotations (Clark and Radivojac 2011). The use of predicted GO terms instead of experimentally determined annotations allowed us to obtain values for all data points and a set of features that was less likely to be biased toward genes frequently studied by biomedical researchers (which could have resulted in an overestimation of performance accuracy). We also included interaction complexity (node degree in a protein–protein interaction network), which is derived from high-throughput experiments in this subset of functional features. Expression-based features were generated using microarray platforms GPL1074 and GPL96 (Su, Wiltshire et al. 2004). A set of 158 features was generated that represent the normalized expression levels of each gene across 79 tissues. Features pertaining to the mean, standard deviation, coefficient of variation, and maximum and minimum expression levels of each gene across tissues were also generated. Finally, two codon-usage features that were not classified as being either expression-based or function-based were generated using CodonW’s (Sharp and Li 1986) frequency of optimal codons (FOP) and effective number of codons (ENC).
Machine Learning & Cross Validation
An ensemble of decision trees was employed using the ‘treefit’ function in MATLAB with default parameters; 10-fold cross-validation was used to assess our prediction performance with respect to unseen variants not found in our training set. An ensemble of 1,000 trees was used, which was found to have better performance than ensembles of 100 or 500. A random selection of features was used for each tree with a minimum of 5 and a maximum of 1,000 features per tree, ensuring that the ensemble is not dominated by a few powerful features. For cross-validation, our dataset was divided into ten partitions each containing a random selection of 10% of the positive variants and 10% of the negative variants. For each of the ten folds, one partition was left out for testing and 9 partitions were used for training. During this process, each partition was used only once for testing. At each fold, a predictor was trained using 1,000 decision trees. For each tree, we balanced our training data by down-sampling our negative variants using random selection until our positive and negative variants were of equal size. The final prediction score was an average of all 1,000 scores output by the ensemble of decision trees; we calculated the area under the ROC (AUC) of all predicted scores using 1,000 evenly spaced thresholds between 0 and 1. Separately, to examine the effect of training on some of the same genes that are in the test set, we performed a cross-validation using “gene co-location”, meaning that each gene could be found in either only the test set or only the training set. In this analysis, the predictor will never train on any gene that is in the test set or vice versa. Classification performance was calculated using the Area Under the Receiver Operator Characteristic curve (AUC). Spanning the interval between 0 and 1 with 1 being the best possible predictor, the AUC measures the true positive rate (sensitivity) as a function of the false positive rate (1 – specificity).
Results
Cross-Validation Performance
When evaluating the ability of RSVP to discriminate between putatively neutral variants and variants that cause regulatory abnormalities, we achieved an AUC of 0.97. This indicates a marked improvement over our previous model, Zhao et al., which achieved an AUC of 0.90. To demonstrate that RSVP out-performs a traditional approach for regulatory variants , we compared it to Combined Annotation Dependent Depletion (CADD) (Kircher, Witten et al. 2014), a meta-predictor that also uses ENCODE features in addition to scores from PolyPhen, SIFT and several other predictors as features for a SVM. We scored the entire positive and negative dataset created in this study with the CADD webserver and normalized the PHRED scores between 0 and 1. This resulted in an AUC of 0.84 for the CADD predictor, indicating that RSVP will perform better with respect to identifying variants that play a role in gene regulation. Additionally, we compared RSVP’s results to DeepSEA (Zhou and Troyanskaya 2015), a deep learning-based tool for predicting chromatin effects of disease-associated HGMD variants. We found that DeepSEA achieves an AUC of 0.81 on our training data, suggesting a need for tools that are trained specifically on variants that cause regulatory abnormalities since such variants are expected to have different properties than disease-associated variants with no impact on gene regulation. Finally, we compared RSVP’s results to the GWAVA tool, which achieved a similar AUC (0.97) using the same HGMD regulatory variant dataset and a similar negative dataset. However, RSVP performs significantly better when compared to GWAVA’s AUC of 0.71 for their “region” negative dataset, which comprises all negative variants in the region of ±500 bases around the positive variants. When training and testing our model on our ±1,000bp and ±500bp region sets, our model achieved AUCs of 0.96 and 0.95 respectively (Figure 1). In addition to analyzing our performance on all variants from the HGMD regulatory variant dataset, we also performed our analysis on the DM, DFP and FP subsets of the positive variants (Figure 2). Of these subsets, training and testing using only the DM subset showed the best performance (AUC of 0.98), followed by DFP (AUC of 0.96), and finally FP (AUC of 0.96). This suggests that RSVP performs better on variants that are causatively involved with disease but the tool still performs well on known functional variants with no known disease association. Furthermore, since the performance of our tool will inevitably rely on the chosen threshold, we provide users with the F-measure and accuracy performance at different thresholds corresponding to estimated 1%, 5% and 10% false positive rates (Table 1).
Figure 1:
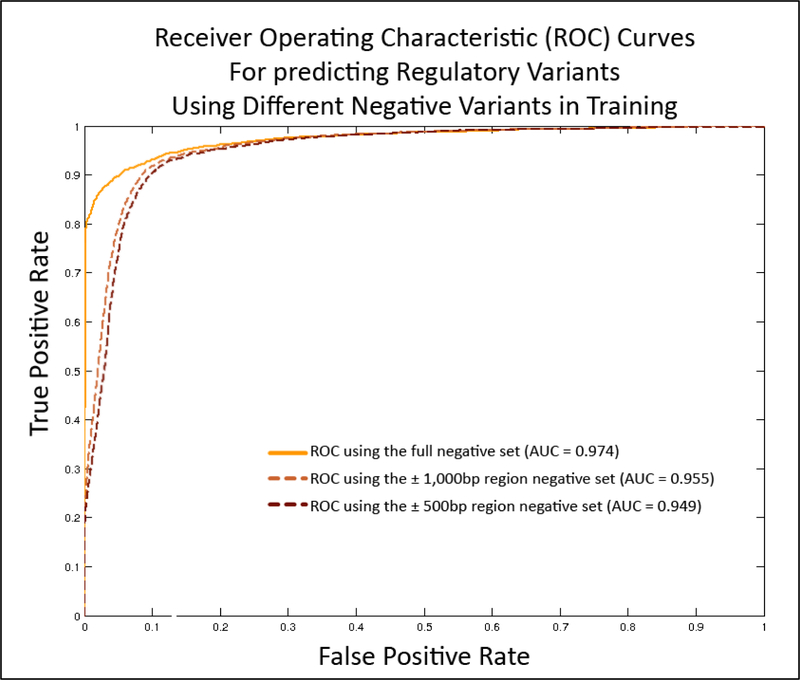
Comparison of cross validation classification performance for three different models each trained with a different negative set. The model trained using randomly sampled variants from dbSNP with a minor allele frequency of ≥ 5% (full negative set), had an Area Under the Receiver Operator Characteristic curve (AUC) of 0.974. Restricting the negative variants to those within ±1,000bp and ±500bp regions flanking any positive variant, resulted in models which achieved AUCs of 0.955 and 0.949 in cross validation respectively.
Figure 2:
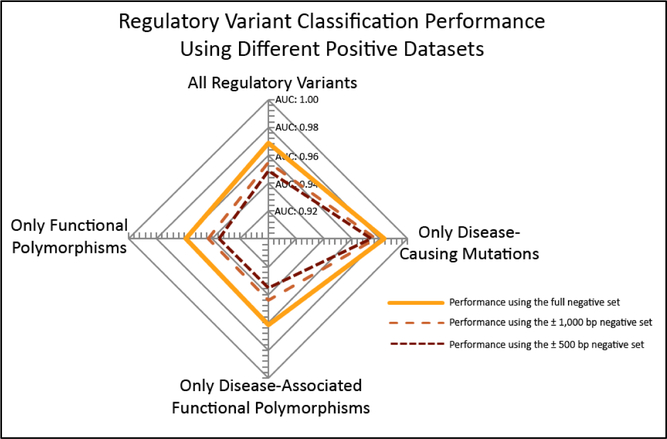
Regulatory variant classification performance using different mutation classes in the positive datasets during cross validation. The Area Under the Receiver Operator Characteristic curve (AUC) was calculated during cross validation for all regulatory variants (AUCs of 0.969, 0.955, and 0.949 for the full, ±1,000bp, and ±500bp negative sets) as well as subsets containing only disease-causing mutations (AUCs of 0.983, 0.977, and 0.974 for the full, ±1,000bp, and ±500bp negative sets), only disease-associated functional polymorphisms (AUCs of 0.962, 0.944, and 0.935 for the full, ±1,000bp, and ±500bp negative sets), and only functional polymorphisms with no disease-association (AUCs of 0.959, 0.943, and 0.935 for the full, ±1,000bp, and ±500bp negative sets).
Table 1:
RSVP performance at different false positive rates as measured by F-measure and accuracy.
| False Positive Rate | Threshold | F-Measure | Accuracy |
|---|---|---|---|
| 1 % | 0.90 | 0.69 | 0.98 |
| 5 % | 0.57 | 0.46 | 0.95 |
| 10 % | 0.40 | 0.32 | 0.90 |
In practice, our predictor will see many of the same genes during training and testing. However, since our predictor uses several thousand gene-based features which can be identical for many variants, it is important that our model performs well on genes that were not found in the training set. Additionally, since our model uses many gene-based features, it is also important that we do not overfit for genes that are already in the HGMD database. Thus, we performed a separate cross-validation using “gene co-location” where no genes appeared in both the training and testing sets in any fold. This analysis resulted in an AUC of 0.94 for all regulatory variants, 0.96 for the DM subset, 0.93 for the DFP subset, and 0.92 for the FP subset. This suggests that our model will still perform well on genes for which there is no known disease-association or functional evidence, and does not overfit in relation to genes already logged in the HGMD database.
Individual Feature Performance
For each feature (or group of features, as is the case with the FANN-GO and expression datasets), the classification performance was tested individually to assess predictive power. Here, we obtained AUCs for each feature or group of features by building the ensemble of decision trees using only that feature subset. The results for individual features are reported in Table 2 with similar results found when using both the disease-causing mutation positive subset (Supplementary Table S2) and when using the ±500bp region negative set (Supplementary Table S3). These results indicate that the distance to the transcription start site is important for identifying variants that play a functional role in gene regulation, a conclusion which supports the findings of previous research. Also highlighted is the importance of incorporating gene-based features (FANN-GO, expression data, FOP, ENC, etc.) in the analysis, a key finding of our previous study. Furthermore, incorporating newly discovered functional genomic sites from the ENCODE project improved the ability of our method to identify variants that play a functional role in gene regulation. Thus, replication timing, histone peaks, transcription factor binding sites, DNase sensitivity peaks and FAIRE peaks were found to be ‘power features’ for identifying functional regulatory variants.
Table 2:
Performance of individual features ranked by the Area Under the Receiver Operator Characteristic curve (AUC) achieved by using each feature or set of features (in the case of FANN-GO and expression level features) individually.
| Feature Name | Type | AUC | Feature Name (cont.) | Type | AUC |
|---|---|---|---|---|---|
| Distance to the nearest transcription start site |
SNV-based | 0.945 | PPINT complexity | Gene-based | 0.715 |
| FANN-GO Category | Gene-based | 0.881 | Flanking nucleotide GC content |
SNV-based | 0.710 |
| Gene expression across 79 tissues | Gene-based | 0.856 | FAIRE peak | SNV-based | 0.703 |
| Replication timing | SNV-based | 0.820 | Exonic Splicing Enhancers | SNV-based | 0.681 |
| Frequency of optimal codons (FOP) |
Gene-based | 0.805 | Exonic Splicing Silencers | SNV-based | 0.681 |
| Mean of gene expression sets | Gene-based | 0.800 | CpG island | SNV-based | 0.623 |
| Maximum of expression sets | Gene-based | 0.800 | Conserved noncoding region |
SNV-based | 0.580 |
| Covariance of expression sets | Gene-based | 0.798 | Insulator | SNV-based | 0.563 |
| StDev of expression sets | Gene-based | 0.794 | DNAse/nuclease region | SNV-based | 0.533 |
| Histone peak | SNV-based | 0.786 | Methylation peak | SNV-based | 0.518 |
| Minimum of expression sets | Gene-based | 0.771 | Enhancer | SNV-based | 0.501 |
| Effective number of codons (ENC) |
Gene-based | 0.757 | phastcons conserved region | SNV-based | 0.500 |
| TFBS | SNV-based | 0.752 | RNA Pol II Enriched site | SNV-based | 0.500 |
| DNase peak | SNV-based | 0.746 |
Independent Dataset Validation
To assess the model’s performance on data not used in training, the genomes of healthy individuals from the 1,000 Genomes Project and the Wellderly project were scored with the RSVP predictor in order to provide a comparison with an independent set of variants that cause regulatory abnormalities (Figure 3). Labeled “HGMD Update” in Figure 3A are scores for 199 new variants in 169 genes that were recently added to the HGMD regulatory variant subset of the database, and hence were not originally used to train the classifier. The second dataset, termed “Wellderly Project Germline Variants” (Figure 3B), were 18,157 variants from the same 169 genes as the HGMD update set found in the genomes of individuals from the Wellderly Project, which are assumed to be depleted in deleterious variants compared to other populations. Finally, the third dataset, labeled “1,000 Genomes Germline Variants” (Figure 3C), comprised 21,203 variants that are present in the 1,000 Genomes Project for these 169 genes. We found that variants from healthy individuals in the Wellderly Project and the 1,000 Genomes Project exhibit a significant difference in RSVP scores as compared to variants in the independent HGMD dataset (t-test p-value: <1×10−15 for both comparisons).
Figure 3:

Distribution of RSVP prediction scores for regulatory variants from three unseen and independent variant datasets. (A) 199 variants in 169 genes from the HGMD, which were added to HGMD after Jan. 30th, 2014 (HGMD Update) (B) 18,157 variants from patients sequenced in the Wellderly Project from the same 169 genes as the HGMD update. (C) 21,203 variants from patients sequenced in the 1,000 Genomes Project, also from the same 169 genes as the HGMD Update.
Somatic Variants Associated with Regulatory Abnormalities in Breast Invasive Carcinoma Patients
In Figure 4, RSVP scores are compared between somatic variants found in tumor samples from TCGA breast invasive carcinoma patients, somatic variants in regulatory/intergenic regions near genes with known cancer relevance from the Cancer Gene Census (CGC) database (Figure 4A), somatic variants in regulatory/intergenic regions near genes that were not in the CGC (Figure 4B), and germline variants in regulatory/intergenic regions from the 1,000 Genomes Project (Figure 4C). Overall, in comparison to germline variants located in regulatory/intergenic regions from the 1,000 Genomes Project, somatic variants in regulatory/intergenic regions from TCGA breast invasive carcinoma patients tend to have higher RSVP scores (t-test p value: 1.9×10−12) and similar results were found using variants from the Wellderly Project (t-test p-value: 1.3 ×10−11). Moreover, we find that somatic variants near genes with known cancer relevance from the CGC tend to have higher RSVP scores (t-test p-value: 2.3×10−2) than genes with no known cancer relevance.
Figure 4:
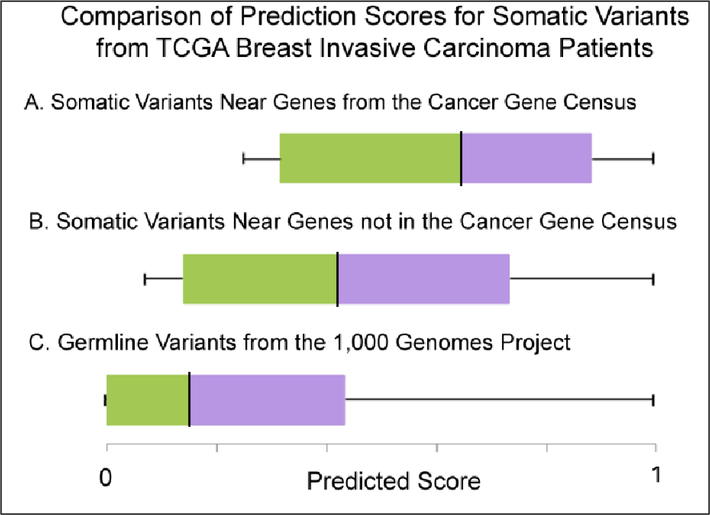
Comparison of RSVP prediction score distributions for somatic variants from TCGA breast invasive carcinoma patients. (A) RSVP scores for regulatory/intergenic somatic variants near genes with known cancer relevance from the Cancer Gene Census. (B) RSVP scores for regulatory/intergenic somatic variants near genes that were not in the Cancer Gene Census. (C) RSVP scores for regulatory/intergenic germline variants from the 1,000 genomes project.
Discussion
In this paper, we have evaluated the performance achieved by our Regulatory Single nucleotide Variant Predictor (RSVP) by incorporating the wealth of information generated in recent years by the ENCODE project. In comparison to our previous study which did not include features from the ENCODE project, Zhao et al., RSVP’s performance (measured in AUC) improved from 0.90 to 0.97 during cross-validation when trained using the full HGMD regulatory variant dataset. Additionally, we demonstrate that our tool also performs well on all types of functional regulatory variant recorded in the HGMD database, with “disease-causing mutations” (DMs) achieving an AUC of 0.98, “disease-associated polymorphisms” with additional supporting functional evidence (DFPs) achieving an AUC of 0.96, and “functional polymorphisms” (FPs) achieving an AUC of 0.96.
To benchmark our tool, we compared RSVP’s results to a recently published tool, Genome Wide Annotation of Variants (GWAVA), which also employs a machine learning approach that incorporates data from the ENCODE project as features to predict regulatory variants, and to a traditional variant predictor used to classify coding variants, the Combined Annotation Dependent Depletion (CADD) tool. We found that our overall performance is comparable to GWAVA, which also achieved an AUC of 0.97, and that both tools perform better on regulatory variants than a traditional tool, CADD, which only achieved an AUC of 0.84. RSVP and GWAVA also outperform a predictor trained with similar ENCODE-based features but which utilized the entire HGMD database, DeepSEA (AUC of 0.81), thereby underscoring the utility of a predictor trained specifically for variants that cause regulatory abnormalities. However, as highlighted in the publication describing the GWAVA tool, it is important to evaluate the predictor’s performance on variants displaying similar distances to the transcription start site as those in the positive set, since this could be considered to be a ‘power feature’ as a single-feature classifier, achieving an AUC of 0.95. Thus, to determine how the GWAVA tool performs on these variants, we performed a separate analysis using a subset of the negative variants during cross-validation that comprised all negative variants within a 1,000bp window around any HGMD variant. Here, the GWAVA tool achieved an AUC of only 0.71 whereas RSVP achieved an AUC of 0.96. In addition, our method performs well when only considering those negative variants within a 500bp window around any HGMD variant, achieving an AUC of 0.95. This disparity likely stems from the choice of features used in RSVP since the GWAVA tool does not include any gene-based features, which were determined to be highly predictive features in our previous study, Zhao et al., as well as our current study. Intuitively, gene-based features such as predicted GO terms and expression level in various tissues should be key to predicting the effect of variants on the regulation of genes in relation to disease. Indeed, these were found to be highly predictive features with individually assessed AUCs of 0.88 and 0.86 respectively. Not surprisingly, as seen in Supplementary Table S4, many of the most predictive FANN-GO features are involved with regulatory activity (e.g., response to stimulus, phosphorylation, transcription factor activity, etc.). Likewise, all other gene-based features also displayed good discriminatory power with AUCs of 0.81 for Frequency of Optimal Codons, 0.76 for Effective Number of Codons, and 0.72 for PPINT complexity).
Since our method employed multiple gene-based features, one possible source of bias could be introduced if our predictor overfits for genes that are already in the HGMD database. To assess this potential bias, we performed a separate cross-validation using “gene co-location” where no genes appeared in both the training and testing sets in any fold. Resulting in an AUC of 0.94 for all regulatory variants, 0.96 for the DM subset, 0.93 for the DFP subset, and 0.92 for the FP subset (Figure 2), we determined that our model is unaffected by this bias and that it will perform well for new genes for which there are no known disease-associations or functional evidence. Overfitting can also become a concern when using large feature vectors like the FANN-GO feature set. Thus, in addition to using 10-fold cross-validation, we also address this problem in our random forest algorithm, where we randomly select between 5 and 1,000 features for each tree. This ensures that each tree will be trained using different features and that no tree will have more features than variants in the positive dataset. This also ensures that power features such as distance to the transcription start site do not dominate the entire ensemble of trees.
The selection of a negative training dataset is crucial for assessing performance and comparing models. Thus, in this study, we employed several negative datasets that were similar to the ones used in our previous study, Zhao et. al., and to the negative datasets used in the GWAVA tool. Like GWAVA, our “unmatched” dataset was randomly sampled from variants that are not known to cause regulatory abnormalities. Although our performance was unchanged from an AUC of 0.97 for three random samples of variants from dbSNP (data available upon request), we did notice a marked change when analyzing only negative variants within a 1,000 or 500 bp window around any positive variant. Indeed, we find that the most predictive feature, distance to the nearest transcription start site, achieves an AUC of 0.95 in cross-validation alone for the unmatched dataset although the AUC decreases to 0.91 and 0.86 for the 1,000 and 500 bp negative datasets respectively. Thus, it is vital for any comparison between methods to consider the unequal distribution of distances to the transcription start site in the negative datasets chosen.
To assess RSVP’s performance on an independent dataset, we scored and compared variants from a recent update to the HGMD regulatory variant database that were not used to train the model to and germline variants from the Wellderly and 1,000 Genomes Projects. As expected, variants from the independent HGMD update dataset that were not used to train the model were scored significantly higher than variants from the same genes found in the Wellderly and 1,000 Genomes Projects, indicating that RSVP has the power to detect novel variants that will cause regulatory abnormalities. Additionally, to assess the potential application of the RSVP tool to identifying somatic variants with the potential to drive tumor progression, we performed an analysis of RSVP scores for a dataset of somatic variants from the tumors of patients with breast invasive carcinoma from The Cancer Genome Atlas project. We found that, overall, somatic variants in regulatory/intergenic regions are predicted by RSVP to be more likely to be associated with regulatory abnormalities than similar variants from the Wellderly and 1,000 Genomes Projects. Moreover, we find that somatic variants located near genes known to play a role in cancer from the Cancer Gene Census tend to have higher RSVP scores than somatic variants near genes with no known cancer relevance. This indicates that the RSVP tool could also be used to identify variants that likely play a role in the regulation of pathways involved in tumor progression.
Supplementary Material
Acknowledgments
Funding
The National Institutes of Health (NIH) R01LM009722 to SDM (PI), PR (co-investigator) and MGK (co-investigator), NIH R01MH105524 to PR (PI) and SM (co-PI), and 1K22CA143148 to MGK (PI) supported this work. MM and DNC acknowledge financial support from Qiagen Inc through a License Agreement with Cardiff University.
References
- Abecasis GR, Auton A, et al. (2012). “An integrated map of genetic variation from 1,092 human genomes.” Nature 491(7422): 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen MC, Engstrom PG, et al. (2008). “In silico detection of sequence variations modifying transcriptional regulation.” PLoS Comput Biol 4(1): e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, et al. (2007). “High-resolution profiling of histone methylations in the human genome.” Cell 129(4): 823–837. [DOI] [PubMed] [Google Scholar]
- Bell AC, West AG, et al. (2001). “Insulators and boundaries: versatile regulatory elements in the eukaryotic genome.” Science 291(5503): 447–450. [DOI] [PubMed] [Google Scholar]
- Buckland PR (2006). “The importance and identification of regulatory polymorphisms and their mechanisms of action.” Biochim Biophys Acta 1762(1): 17–28. [DOI] [PubMed] [Google Scholar]
- Buckland PR, Hoogendoorn B, et al. (2004). “A high proportion of polymorphisms in the promoters of brain expressed genes influences transcriptional activity.” Biochim Biophys Acta 1690(3): 238–249. [DOI] [PubMed] [Google Scholar]
- Campino S, Forton J, et al. (2008). “Validating discovered Cis-acting regulatory genetic variants: application of an allele specific expression approach to HapMap populations.” PLoS One 3(12): e4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chorley BN, Wang X, et al. (2008). “Discovery and verification of functional single nucleotide polymorphisms in regulatory genomic regions: current and developing technologies.” Mutat Res 659(1–2): 147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark WT and Radivojac P (2011). “Analysis of protein function and its prediction from amino acid sequence.” Proteins 79(7): 2086–2096. [DOI] [PubMed] [Google Scholar]
- Collins FS and Barker AD (2007). “Mapping the cancer genome. Pinpointing the genes involved in cancer will help chart a new course across the complex landscape of human malignancies.” Sci Am 296(3): 50–57. [PubMed] [Google Scholar]
- Crawford GE, Holt IE, et al. (2006). “Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS).” Genome Res 16(1): 123–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futreal PA, Coin L, et al. (2004). “A census of human cancer genes.” Nat Rev Cancer 4(3): 177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs RA, Belmont JW, et al. (2003). “The international HapMap project.” Nature 426(6968): 789–796. [DOI] [PubMed] [Google Scholar]
- Kellis M, Wold B, et al. (2014). “Defining functional DNA elements in the human genome.” Proc Natl Acad Sci U S A 111(17): 6131–6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Witten DM, et al. (2014). “A general framework for estimating the relative pathogenicity of human genetic variants.” Nat Genet 46(3): 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapidot M, Mizrahi-Man O, et al. (2008). “Functional characterization of variations on regulatory motifs.” PLoS Genet 4(3): e1000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Jian X, et al. (2011). “dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions.” Hum Mutat 32(8): 894–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery SB, Griffith OL, et al. (2007). “A survey of genomic properties for the detection of regulatory polymorphisms.” PLoS Comput Biol 3(6): e106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooney S (2005). “Bioinformatics approaches and resources for single nucleotide polymorphism functional analysis.” Brief Bioinform 6(1): 44–56. [DOI] [PubMed] [Google Scholar]
- Mottagui-Tabar S, Faghihi MA, et al. (2005). “Identification of functional SNPs in the 5-prime flanking sequences of human genes.” BMC Genomics 6: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pampin S and Rodriguez-Rey JC (2007). “Functional analysis of regulatory single-nucleotide polymorphisms.” Curr Opin Lipidol 18(2): 194–198. [DOI] [PubMed] [Google Scholar]
- Pastinen T and Hudson TJ (2004). “Cis-acting regulatory variation in the human genome.” Science 306(5696): 647–650. [DOI] [PubMed] [Google Scholar]
- Pennacchio LA, Loots GG, et al. (2007). “Predicting tissue-specific enhancers in the human genome.” Genome Res 17(2): 201–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson TA, Doughty E, et al. (2013). “Towards precision medicine: advances in computational approaches for the analysis of human variants.” J Mol Biol 425(21): 4047–4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponomarenko JV, Orlova GV, et al. (2002). “rSNP_Guide: an integrated database-tools system for studying SNPs and site-directed mutations in transcription factor binding sites.” Hum Mutat 20(4): 239–248. [DOI] [PubMed] [Google Scholar]
- Prokunina L and Alarcon-Riquelme ME (2004). “Regulatory SNPs in complex diseases: their identification and functional validation.” Expert Rev Mol Med 6(10): 1–15. [DOI] [PubMed] [Google Scholar]
- Ritchie GR, Dunham I, et al. (2014). “Functional annotation of noncoding sequence variants.” Nat Methods 11(3): 294–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachidanandam R, Weissman D, et al. (2001). “A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms.” Nature 409(6822): 928–933. [DOI] [PubMed] [Google Scholar]
- Savinkova LK, Ponomarenko MP, et al. (2009). “TATA box polymorphisms in human gene promoters and associated hereditary pathologies.” Biochemistry (Mosc) 74(2): 117–129. [DOI] [PubMed] [Google Scholar]
- Sharp PM and Li WH (1986). “Codon usage in regulatory genes in Escherichia coli does not reflect selection for ‘rare’ codons.” Nucleic Acids Res 14(19): 7737–7749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson PD, Ball EV, et al. (2012). “The Human Gene Mutation Database (HGMD) and its exploitation in the fields of personalized genomics and molecular evolution.” Curr Protoc Bioinformatics Chapter 1: Unit1 13. [DOI] [PubMed]
- Stenson PD, Mort M, et al. (2014). “The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine.” Hum Genet 133(1): 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterne-Weiler T, Howard J, et al. (2011). “Loss of exon identity is a common mechanism of human inherited disease.” Genome Res 21(10): 1563–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su AI, Wiltshire T, et al. (2004). “A gene atlas of the mouse and human protein-encoding transcriptomes.” Proc Natl Acad Sci U S A 101(16): 6062–6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torkamani A and Schork NJ (2008). “Predicting functional regulatory polymorphisms.” Bioinformatics 24(16): 1787–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Zhang Y, et al. (2006). “Experimental validation of predicted mammalian erythroid cis-regulatory modules.” Genome Res 16(12): 1480–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward LD and Kellis M (2012). “Interpreting noncoding genetic variation in complex traits and human disease.” Nat Biotech 30(11): 1095–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Clark WT, et al. (2011). “Prediction of functional regulatory SNPs in monogenic and complex disease.” Hum Mutat 32(10): 1183–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J and Troyanskaya OG (2015). “Predicting effects of noncoding variants with deep learning-based sequence model.” Nat Methods 12(10): 931–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.