

Abstract
Human telomeres can form DNA G-quadruplex (G4), an attractive target for anticancer drugs. Human telomeric G4s bear inherent structure polymorphism, challenging for understanding specific recognition by ligands or proteins. Protoberberines are medicinal natural-products known to stabilize telomeric G4s and inhibit telomerase. Here we report epiberberine (EPI) specifically recognizes the hybrid-2 telomeric G4 predominant in physiologically relevant K+ solution and converts other telomeric G4 forms to hybrid-2, the first such example reported. Our NMR structure in K+ solution shows EPI binding induces extensive rearrangement of the previously disordered 5’-flanking and loop segments to form an unprecedented four-layer binding pocket specific to the hybrid-2 telomeric G4; EPI recruits the (1) adenine to form a “quasi-triad” intercalated between the@external tetrad and a T:T:A triad, capped by a T:T base pair. Our study provides structural basis for small-molecule drug design targeting the human telomeric G4.
Keywords: anticancer drug targets, G4-drug complexes, human telomeres, G-quadruplexes, NMR spectroscopy, berberine
Human telomeres at the chromosome ends consist of guanine(G)-rich tandem DNA repeats of d(TTAGGG)n 5–20 kb in length, terminating with a 3’ single-stranded overhang of 35–600 bases.[1] With associated proteins, human telomeres play critical roles in cancer, aging, and genetic stability.[2] Human telomeres can form G-quadruplexes (G4s),[3] non-canonical DNA secondary structures built upon Hoogsteen hydrogen-bonded (H-bonded) planar G-tetrads (Figure 1a) stabilized by monovalent cations such as K+ or Na+.[4] Telomerase is a reverse transcriptase activated in 80–85% of human cancers to maintain telomeres.[5] Small molecules that stabilize the telomeric G4 have been demonstrated to inhibit telomerase and disrupt telomere capping and maintenance, resulting in cancer cell apoptosis.[6] G4s formed in human telomeres are structurally polymorphic;[7] including two equilibrating hybrid-type structures[8] and a 2-tetrad structure[9] in K+ solution and a basket-type structure in Na+ solution[10] (see Figure S1 in the Supporting Information). In physiologically relevant K+ solution, the hybrid-2 G4 (Figure 1b) is the major form in the wild-type (wt) four repeats human telomeric DNA[8d,e,11] and thus represents a main molecular target for small molecule drugs. Protoberberines, best known by berberine, are a class of natural products with anticancer and other pharmacological activ ities[12] related to their nucleic acids interactions.[13] Berberine (Figure S2) and its derivatives have been shown to stabilize telomeric G4s and inhibit telomerase.[14] We recently found that EPI (Figure 1c) exhibits great fluorescence enhancement upon binding to human telomeric G4 in K+ solution, up to 45 times stronger than other G4s and double-stranded and single-stranded DNA.[15] Here, we report that EPI specifically recognizes the hybrid-2 telomeric G4 and we elucidate the molecular mechanism for this specific recognition by NMR structure determination of the 1:1 EPI-wtTel26 complex in K+ solution. Furthermore, EPI can convert other telomeric G4s, for example, hybrid-1 and basket-type, to the hybrid-2 structure and is the first such compound reported.
Figure 1.

a) A G-tetrad with observable H1-H1 (red) and H1-H8 (green) NOE connectivities. b) Schematic of the EPI-wtTel26 complex, with the wtTel26 sequence shown. c) EPI structure. d,e) 1H NMR imino regions of (d) EPI titration to wtTel26 with assignments of complex (asterisk) and free DNA (dot), and e) 1:1 EPI-complexes of 2-AP substituted wtTel26.
We first examined the binding of EPI to the 26-mer wt human telomeric sequence wtTel26, in K+ solution using 1H NMR titration. Free wtTel26 forms predominantly the hybrid-2 G4 structure in K+ solution (Figure 1b)[8d] and shows twelve major guanine imino proton peaks at 10.5–12.0 ppm (Figure 1d), characteristic of G4s. At 1:1 ratio, a new set of twelve guanine imino protons was observed with good spectral quality, indicating the formation of a well-defined 1:1 EPI-wtTel26 complex (Figures 1d, S3). Notably, upon EPI addition, a new imino peak emerged at 12.6 ppm, in the characteristic region of Watson–Crick-type H-bonded thy-mine imino protons, suggesting the formation of a thymine-involved base pair corresponding to complex formation. We unambiguously assigned the twelve guanine imino protons (Figures 1d and S3), and imino protons of the four 5’ end thymines, that is, T1, T2, T13, and T14 (Figure S4), using site-specifically 15N-labeled DNA and 15N-edited experiments. The strong imino proton at ca. 12.6 ppm observed in the EPI complex was assigned to T13. Subsequently, complete proton assignment of the EPI-bound DNA was achieved (Figures 2 and S5, S6 and Tables S1 and S2). The same five syn guanines as in the free wtTel26 G4 (Figure S1)[8d] were observed in the complex (Figure 1b), as shown by strong H8-H1’ NOEs (Figures S5a and S7). T1, T13, and T19 also adopted syn configurations in the complex. Exchange NOE (Nuclear Overhauser Effect) peaks between the free and bound wtTel26 indicated a medium-to-slow exchange rate of EPI binding on the NMR time scale (Figure S6). Protons of the free (Figure S8) and bound EPI (Figure S5b) were assigned using 1H 1D NMR and 2D NOESY (Table S3). The proton chemical shift differences between free and bound forms for EPI and DNA are summarized in Figure S9. Similar CD profiles were observed for the bound and free wtTel26 DNA (Figure S10a), and EPI increased the melting temperature of wtTel26 by 5 and 108C at 1 and 2 equivalents, respectively (Figure S10b).
Figure 2.
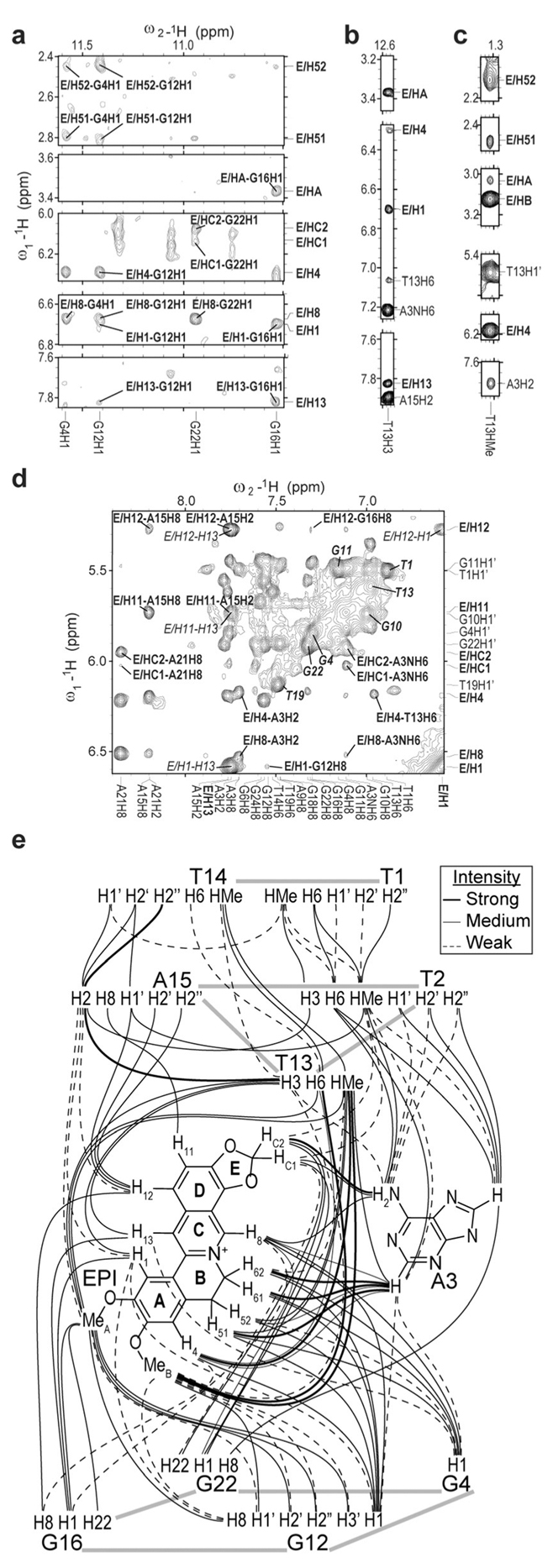
Select regions of the 2D-NOESY of 1:1 EPI-wtTel26 complex in pH 7, 100 mm K+. a–c) with intermolecular NOE assignments (a,b: 158C, c: 58C), d) the aromatic-H1’ region (58C) with intermolecular NOEs in bold, syn-DNA H8/H6-H1’ NOEs in bold-italics, intra-EPI NOEs in italics. e) Schematic summary of the observed intermolecular and inter-residue NOEs of the EPI binding pocket in the complex.
Numerous intermolecular and inter-residue NOEs were observed (Figure 2, Tables S4–S6), clearly defining the EPI binding site. Using NOE-restrained molecular dynamics simulated annealing approaches,[8d,16] we determined the molecular structure of the 1:1 EPI-wtTel26 complex in K+ solution (PDB ID: 6CCW, Figures 3 and S11). A total of 631 NOE distance restraints were used, including 62 inter-EPIDNA restraints (Table S7).
Figure 3.

a) Cartoon representation of the 1:1 EPI-wtTel26 complex (PDB ID: 6CCW). b–d) Top views of the EPI:A3 quasi-triad plane (b), T2:T13:A15 triad plane (c), and T1:T14 pair (d). Potential H-bonds are shown in dashed lines.
The NMR structure in K+ solution showed that the EPI binding induced complete rearrangement of the previously disordered[8d] 5’-flanking segment T1-T2-A3 and the second lateral loop T13-T14-A15 so that a new extensive binding-pocket is formed in the hybrid-2 G4 (Figure 3). EPI recruits A3, the flanking (@1) adenine, to form a “quasi-triad plane” profoundly intercalated between the 5’-external G-tetrad and two additional layers of H-bonded capping structures, a T2:T13:A15 triad plane and a T1:T14 pair (Figure 3). In the EPI:A3 “quasi-triad” plane, EPI covers two bases, G12 and G16 of the 5’-external tetrad, while the recruited A3 covers the center of the G4-G22 edge (Figure 3b). The EPI:A3 pair is stabilized by a H-bond (Figure 3b), as shown by the observed A3/NH6 and strong NOEs between EPI protons and A3/H2 and A3/NH6 (Figure 2d, Table S4B). This explains why specific EPI binding requires the flanking (1) adenine in the human telomeric sequences.[15] Interestingly,@ the positively charged EPI/N7 (Figure 1c) is positioned over the central channel above the 5’ external tetrad (Figure 3b), likely interacting with the tetrad-guanine carbonyls analogous to a K+ ion. The EPI:A3 plane is capped by a T2:T13:A15 triad (Figure 3c), with EPI covered by T13/A15 and A3 covered by T2, as supported by numerous intermolecular and inter-residue NOEs (Figure 2, Tables S4,S5). The T13:A15 pair is stabilized by two H-bonds (Figure 3c), with strong NOE observed between T13/H3 and A15/H2 (Figure 2b). T13 is also H-bonded with T2; T2 is positioned more centrally (Figure 3c), with imino T2/H3 observed at 10.6 ppm (Figure S4) and showing NOEs with A15/H2 and T1/HMe (Figure S13). This stable H-bonded T2:T13:A15 triad explains the unusually strong T13/H3 peak (Figure 1d). The T2:T13:A15 triad is further capped by a H-bonded T1:T14 pair (Figure 3d), as demonstrated by inter-residue NOEs between T14, T1, and T2/T13/A15 (Figure 2e, Table S6), and the observed imino protons of T1 and T14 at ca. 10.5 ppm (Figure S4). Additionally, A21 of the strand-reversal loop covers the EPI E ring side (Figure 3a), as supported by EPIA21 NOEs (Table S5A). The same 3’ end triad T8:A9:T25 of the free wtTel26[8d] is observed in the complex.
To confirm the binding pocket formation, we performed 2-aminopurine (2-AP) mutational studies at each adenine of wtTel26. In the complex structure, A3 is H-bonded with EPI, while A15 H-bonded with T13 (Figure 3). 2-AP contains an amino group on C2 instead of C6, and can be used to determine the requirement of the H-bonded conformation involving adenine. Mutation of A3 and A15 completely disrupted the EPI-wtTel26 complex as shown by 1H NMR (Figure 1e), demonstrating the critical role of the H-bonded EPI:A3 pair and T2:T13:A15 triad in the specific EPI binding pocket. In contrast, the 2-AP mutations of A9 or A21 had little effect on the complex formation (Figure 1e).
Importantly, the EPI binding pocket can only form in the human telomeric hybrid-2 G4 (Figures 1b and S1). The unusually strong T13/H3 peak at 12.6 ppm results from the H-bonded conformation of T13 with both A15 and T2 in the T2:T13:A15 triad (Figure 3d), which is specific to the hybrid-2 topology. Thus the T13/H3 imino proton is a direct indicator of the EPI-hybrid-2 complex. To investigate the binding of EPI to other human telomeric sequences, we conducted 1H NMR titration experiments in K+ solution (Figures 4 and S14). wtTel25 forms predominantly the hybrid-2 structure (Figure S14a) [8e] and upon EPI titration showed very similar spectra to wtTel26, with slightly broader peaks (Figure S14c). In contrast, wtTel24 normally adopts a mixture of hybrid-1 and hybrid-2 topologies (Figure 4a).[8d] Upon EPI addition, one set of imino peaks resembling that of the wtTel26 complex appeared, indicating the formation of a single hybrid-2 structure (Figure 4b). Remarkably, titration of EPI to wtTel23 (Figure 4a), wtTel24b and Tel26 (Figure S14a), which predominantly form the hybrid-1 structure in the free state,[8a,b] all showed the imino peak at 12.6 ppm indicative of the EPI-hybrid-2 complex (Figures 4c and S14d,e), suggesting a similar EPI binding mode in all human telomeric sequences. In contrast, NMR titration of other known G4s with EPI showed less specific binding and no signature imino peak (Figure S15). Altogether, this indicates that EPI binding converts human telomeric hybrid-1 structure to hybrid-2 in order to form its specific binding pocket.
Figure 4.

a) Human telomeric sequences wtTel26, wtTel24, and wtTel23, and their folding topologies. b,c) 1H NMR titration of EPI in K+ with wtTel24, (b) and wtTel23, (c), with the signature T-imino peak of the EPI-hybrid-2 complex marked by asterisk. d) CD titration spectra of EPI with wtTel26 in Na+ solution. e) Taq DNA polymerase stop assay using a DNA template containing a wt human telomeric sequence (bottom) showing EPI can induce telomeric G4 formation. Left 4 lanes are sequencing reactions.
We then examined EPI binding of wtTel26 in Na+ solution. wtTel26 appeared to form a basket-type structure in 100 mm Na+, as indicated by characteristic maxima at 295 nm and 250 nm and a minimum at 270 nm in the CD spectrum[8a,c] (Figure 4d). Upon EPI addition, a new maximum at 260 nm and a new minimum at 240 nm emerged, reaching saturation at 6 equivalents EPI. The CD titration spectra exhibited a single isodichroic point, suggesting the conversion from one conformation to another. Notably, the saturated CD spectrum of the 6:1 EPI-wtTel26 complex in Na+ closely resembles that of the 1:1 EPI-wtTel26 (hybrid-2) complex in K+ solution (Figure S16), suggesting the formation of the hybrid-2 structure. Moreover, the 1H NMR titration of EPI to wtTel26 in Na+ showed the signature thymine imino proton peak indicative of the EPI-hybrid-2 binding pocket in 6:1 EPI complex (Figure S17). However, the NMR spectrum of the complex in Na+ was not as well-defined as that in K+ (Figure 1d), suggesting that Na+ cations are less favored for the hybrid-type G4 structure. Therefore, EPI appears to convert basket-type human telomeric G4 to hybrid-2 structure in Na+. Native EMSA gels confirmed that wtTel26 in the EPI complexes was monomeric in both K+ and Na+ solution (Figure S18).
To determine if EPI can induce G4 formation in the extended wild-type human telomeric DNA under physiologically relevant conditions, we performed a DNA polymerase stop assay using a template containing the human telomeric DNA with four TTAGGG repeats (Figure 4e). A G4 structure formed in the template strand can block the Taq DNA polymerase synthesis of the complementary strand.[17] At higher concentration of K+ (100 mm), a slight pausing (stop-site) of DNA polymerase was observed at the 3’-end of the telomeric sequence in the template strand, indicating the G4 formation. Remarkably, even in the absence of K+, EPI addition blocked DNA polymerase in a dose-dependent manner, as shown by the stop-sites observed at the same location (Figure 4e). This suggests EPI can induce G4 formation in the extended human telomeric DNA sequence in the absence of salt. Hence our results showed that EPI specifically binds and induces the physiologically relevant hybrid-2 human telomeric G4, and converts other telomeric G4 structures to the hybrid-2 G4 independent of cation type (Figure 5). The strong recognition of hybrid-2 G4 determines the EPI’s ability to convert other telomeric G4s, for example, hybrid-1 and basket, to the hybrid-2 structure in order to form its specific binding pocket (Figure 5 top left).
Figure 5.

EPI binding induces the formation of a four-layer binding pocket specific to hybrid-2 telomeric G4 (a) by rearranging the previously disordered 5’-flanking and loop segments of hyrid-2 in K+ (b), or by converting from the hybrid-1 in K+ (c), basket in Na+ (d), or from telomeric DNA in the absence of salt (e).
A striking aspect of the EPI binding is the extensiveness of the EPI-induced binding pocket, consisting of four layers, specific to the hybrid-2 telomeric G4. The deep intercalation of EPI in this multi-layer binding pocket explains the significant fluorescence enhancement of EPI induced by hybrid-2 telomeric G4.[15,18] Such an extensive 4-layered binding pocket is unprecedented in G4-ligand complexes. A solution structure in K+ was reported for the complex of hybrid-1 telomeric G4 and a telomestatin derivative, in which the macrocyclic L2H2–6M(2)OTD covered the external G-tetrad, but without formation of a distinctive binding pocket.[19] Very recently, a solution structure in K+ was reported for the complex of the hybrid-2 telomeric G4 and the binuclear gold(III)-metal complex Auoxo6, with Auoxo6 sandwiched between the 5’ G-tetrad and the flanking adenine.[20] In contrast, all of the intramolecular telomeric G4-ligand complexes reported in the crystalline state were parallel-stranded structures,[6d,21] which are different from the hybrid-type[8a,b,d,e] and 2-tetrad basket-type[9] structures observed in solution.
Solving the NMR structure of the 1:1 EPI-wtTel26 complex provides insights into the specific molecular recognition of the physiologically relevant hybrid-2 human telomeric G4 by EPI. EPI, a small molecule natural product from a family of medicinal alkaloids, binds the hybrid-2 G4 in a very specific manner, although it exhibits weak binding to dsDNA (Figure S19). Unlike symmetrical cyclic ligands typified by Telomestatin or the Au–metal complex, the asymmetric crescent-shaped EPI is more drug-like and provides an optimal stacking of two guanines, and by recruiting a base from the target DNA, forms a specifically oriented “quasi-triad” plane covering a G-tetrad. This is somewhat similar to the complex structure of quindoline with the parallel-stranded c-MYC promoter G4,[16] however, the binding pocket in the EPI-wtTel26 complex is far more extensive. EPI exhibits significantly greater binding to telomeric G4 than other structurally similar alkaloids including berberine (BER), palmatine (PAL), coptisine (COP), which only differ in the positions of the methylenedioxy group (Figure S2).[15] The NMR structure explains the superior binding of EPI, as the methylenedioxy ring E needed for the H-bonded EPI:A3 pair is missing in PAL and BER, whereas the methylenedioxy ring at the other end in BER and COP would cause steric clashes with the DNA backbone (Figure 3). This molecular level recognition information is important for rational design of improved analogs.
In conclusion, our NMR solution structure elucidates the molecular basis for specific recognition of the hybrid-2 human telomeric G4 predominant in physiologically relevant K+ solution by EPI, a small molecule from a medicinal natural product family. An unprecedented extensive binding pocket consisting of four layers, specific to the hybrid-2 G4, is induced by EPI binding. Our study thus provides structural basis for rational design of small-molecule drugs targeting the human telomeric G4. Furthermore, the human telomeric G4 is structurally polymorphic;[7] its dynamic nature presents a challenge for understanding the specific recognition by ligands or proteins.[22] The selective binding of EPI converts other telomeric G4 forms to the hybrid-2 G4, the first such example reported.
Supplementary Material
Acknowledgements
This research was supported by the U.S. National Institutes of Health (R01CA122952 (D.Y.), R01CA177585 (D.Y.), and P30CA023168 (Purdue Center for Cancer Research)) and the China National Natural Science Foundation(21675142 (Y.S.)). We thank Drs. Jonathan Dickerhoff and Yawen Bai for insightful discussion and proofreading the manuscript.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Supporting information and the ORCID identification number(s) for the author(s) of this article can be found under: https://doi.org/10.1002/anie.201804667.
References
- [1].Blackburn EH, Annu. Rev. Biochem 1984, 53, 163–194. [DOI] [PubMed] [Google Scholar]
- [2].a) Hackett JA, Feldser DM, Greider CW, Cell 2001, 106, 275–286; [DOI] [PubMed] [Google Scholar]; b) Blackburn EH, Nature 2000, 408, 53–56. [DOI] [PubMed] [Google Scholar]
- [3].a) Sundquist WI, Klug A, Nature 1989, 342, 825–829; [DOI] [PubMed] [Google Scholar]; b) Williamson JR, Raghuraman MK, Cech TR, Cell 1989, 59, 871–880; [DOI] [PubMed] [Google Scholar]; c) Biffi G, Tannahill D, McCafferty J, Balasubramanian S, Nat. Chem 2013, 5, 182–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Sen D, Gilbert W, Nature 1988, 334, 364–366; [DOI] [PubMed] [Google Scholar]; b) Yang D, Okamoto K, Future Med. Chem 2010, 2, 619–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Greider CW, Blackburn EH, Cell 1985, 43, 405–413; [DOI] [PubMed] [Google Scholar]; b) Kim NW, et al. , Science 1994, 266, 2011–2015. [DOI] [PubMed] [Google Scholar]
- [6].a) Zahler AM, Williamson JR, Cech TR, Prescott DM, Nature 1991, 350, 718–720; [DOI] [PubMed] [Google Scholar]; b) Rezler EM, Bearss DJ, Hurley LH, Curr. Opin. Pharmacol 2002, 2, 415–423; [DOI] [PubMed] [Google Scholar]; c) Onel B, Lin C, Yang DZ, Sci. China Chem 2014, 57, 1605–1614; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Neidle S, Nat. Rev. Chem 2017, 1, 0041. [DOI] [PubMed] [Google Scholar]
- [7].Dai J, Carver M, Yang D, Biochimie 2008, 90, 1172–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Ambrus A, Chen D, Dai J, Bialis T, Jones RA, Yang D, Nucleic Acids Res 2006, 34, 2723–2735; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Luu KN, Phan AT, Kuryavyi V, Lacroix L, Patel DJ, J. Am. Chem. Soc 2006, 128, 9963–9970; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Xu Y, Noguchi Y, Sugiyama H, Bioorg. Med. Chem 2006, 14, 5584–5591; [DOI] [PubMed] [Google Scholar]; d) Dai JX, Carver M, Punchihewa C, Jones RA, Yang DZ, Nucleic Acids Res 2007, 35, 4927–4940; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Phan AT, Kuryavyi V, Luu KN, Patel DJ, Nucleic Acids Res. 2007, 35, 6517–6525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Lim KW, Amrane S, Bouaziz S, Xu W, Mu Y, Patel DJ, Luu KN, Phan AT, J. Am. Chem. Soc 2009, 131, 4301–4309; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang Z, Dai J, Veliath E, Jones RA, Yang D, Nucleic Acids Res. 2010, 38, 1009–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wang Y, Patel DJ, J. Mol. Biol 1993, 234, 1171–1183. [DOI] [PubMed] [Google Scholar]
- [11].Hänsel R, Lçhr F, Trantirek LS, Dçtsch V, J. Am. Chem. Soc 2013, 135, 2816–2824. [DOI] [PubMed] [Google Scholar]
- [12].a) Tang J, Feng Y, Tsao S, Wang N, Curtain R, Wang Y, J. Ethnopharmacol 2009, 126, 5–17; [DOI] [PubMed] [Google Scholar]; b) Ortiz L. M. Guamán, Lombardi P, Tillhon M, Scovassi AI, Molecules 2014, 19, 12349–12367; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lee C-H, Chen J-C, Hsiang C-Y, Wu S-L, Wu H-C, Ho T-Y, Pharmacol. Res 2007, 56, 193–201; [DOI] [PubMed] [Google Scholar]; d) Hayashi K, Minoda K, Nagaoka Y, Hayashi T, Uesato S, Bioorg. Med. Chem. Lett 2007, 17, 1562–1564. [DOI] [PubMed] [Google Scholar]
- [13].Bhadra K, Kumar GS, Med. Res. Rev 2011, 31, 821–862. [DOI] [PubMed] [Google Scholar]
- [14].a) Franceschin M, Rossetti L, D’Ambrosio A, Schirripa S, Bianco A, Ortaggi G, Savino M, Schultes C, Neidle S, Bioorg. Med. Chem. Lett 2006, 16, 1707–1711; [DOI] [PubMed] [Google Scholar]; b) Noureini SK, et al. , Biochim. Biophys. Acta Gen. Subj 2017, 1861, 2020–2030. [DOI] [PubMed] [Google Scholar]
- [15].Zhang L, Liu H, Shao Y, Lin C, Jia H, Chen G, Yang D, Wang Y, Anal. Chem 2015, 87, 730–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Dai J, Carver M, Hurley LH, Yang D, J. Am. Chem. Soc 2011, 133, 17673–17680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Han H, Hurley LH, Salazar M, Nucleic Acids Res. 1999, 27, 537–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Olmsted J III, Kearns DR, Biochemistry 1977, 16, 3647–3654. [DOI] [PubMed] [Google Scholar]
- [19].Chung WJ, Heddi B, Tera M, Iida K, Nagasawa K, Phan AT, J. Am. Chem. Soc 2013, 135, 13495–13501. [DOI] [PubMed] [Google Scholar]
- [20].Wirmer-Bartoschek J, Bendel LE, Jonker HR, Grün JT, Papi F, Bazzicalupi C, Messori L, Gratteri P, Schwalbe H, Angew. Chem. Int. Ed 2017, 56, 7102–7106; Angew. Chem. 2017, 129, 7208 – 7212. [DOI] [PubMed] [Google Scholar]
- [21].a) Parkinson GN, Cuenca F, Neidle S, J. Mol. Biol 2008, 381, 1145–1156; [DOI] [PubMed] [Google Scholar]; b) Bazzicalupi C, Ferraroni M, Bilia AR, Scheggi F, Gratteri P, Nucleic Acids Res. 2013, 41, 632–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].a) Mullins MR, Rajavel M, Hernandez-Sanchez W, De La Fuente M, Biendarra SM, Harris ME, Taylor DJ, J. Mol. Biol 2016, 428, 2695–2708; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hänsel-Hertsch R, Antonio M. Di, Balasubramanian S, Nat. Rev. Mol. Cell Biol 2017, 18, 279–284. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.